

Abstract
Rosiglitazone and pioglitazone are thiazolidinedione (TZD) compounds that have been used clinically as insulin-sensitizing drugs and are generally believed to mediate their effects via activation of the peroxisome proliferator-activated receptor γ (PPARγ). Recent work has shown that it is possible to synthesize TZD compounds with potent insulin-sensitizing effects and markedly-diminished affinity for PPARγ. Both clinically-used TZDs and investigational PPARγ-sparing TZDs, such as MSDC-0602, interact with the mitochondrial pyruvate carrier (MPC) and inhibit its activity. Two proteins, MPC1 and MPC2, comprise the MPC complex. Herein, we used mice expressing a hypomorphic MPC2 protein missing 16 amino acids in the N terminus (Mpc2Δ16 mice) to determine the effects of these residues in mediating the insulin-sensitizing effects of TZDs in diet-induced obese mice. We found that both pioglitazone and MSDC-0602 elicited their beneficial metabolic effects, including improving glucose tolerance, attenuating hepatic steatosis, reducing adipose tissue inflammation, and stimulating adipocyte browning, in both WT and Mpc2Δ16 mice after high fat diet feeding. In addition, truncation of MPC2 failed to attenuate the interaction between TZDs and the MPC in a BRET-based assay or to affect the suppression of pyruvate-stimulated respiration in cells. Collectively, these data suggest that the interaction between TZDs and MPC2 is not affected by loss of the N-terminal 16 amino acids nor are these residues required for the insulin-sensitizing effects of these compounds.
Keywords: mitochondria, pyruvate, diabetes, thiazolidinediones
INTRODUCTION
Thiazolidinediones (TZDs) have been in clinical use as a treatment for diabetes since the 1990s. TZDs are effective insulin-sensitizing and anti-inflammatory agents, but their use has been curtailed by several associated side effects (Colca & Kletzien, 2006). The identified and canonical mechanism of action for TZDs is via their effects as ligands for a nuclear receptor, the peroxisome proliferator-activated receptor γ (PPARγ) (Lehmann et al., 1995; Soccio et al., 2014). Activation of PPARγ drives fat deposition in adipocytes, is anti-inflammatory, and stimulates production of adipokines, including adiponectin, with beneficial metabolic effects (Halberg et al., 2008; Riera-Guardia & Rothenbacher, 2008). In addition, through use of mice with tissue-specific knockout of PPARγ, it has been demonstrated that several of the known side effects of TZDs are also mediated through this nuclear receptor (Duan et al., 2005; Guan et al., 2005; Zhang et al., 2005). However, there is strong evidence that TZDs also have PPARγ-independent actions that might have beneficial effects on metabolism (Adams et al., 1998; Raman et al., 1998; Raman & Judd, 2000; Chen et al., 2012). We and others have postulated that if the insulin-sensitizing effects of TZDs could be separated from the pure agonism of PPARγ, the resulting compounds might be effective anti-diabetic agents with a better safety profile. To this end, several TZDs with very low affinity for PPARγ have been developed and used in Phase 2 clinical trials as insulin sensitizers (Colca et al., 2013b).
One alternative mode of action for TZDs is through binding to the mitochondrial pyruvate carrier (MPC) in the inner mitochondrial membrane (Colca et al., 2013a). The MPC is made up of two proteins, MPC1 and MPC2, which form a heteroligomeric complex (Bricker et al., 2012; Herzig et al., 2012). This complex mediates the transport of pyruvate into the mitochondrial matrix, which is required for its metabolism. Several studies now suggest that TZDs bind to the MPC directly and inhibit pyruvate transport to elicit effects on mitochondrial oxidative metabolism (Colca et al., 2013a; Divakaruni et al., 2013; McCommis et al., 2015). Binding and acute inhibitory effects on downstream pyruvate metabolism have been demonstrated for both current clinically-used TZDs (rosiglitazone and pioglitazone), as well as next generation PPARγ-sparing TZDs like MSDC-0602 (Colca et al., 2013a; Divakaruni et al., 2013; McCommis et al., 2015). However, it is not yet clear whether binding to the MPC and the resultant effects on mitochondrial metabolism are involved in the PPARγ-independent effects of TZDs on insulin sensitivity and systemic metabolism.
In order to test whether the interaction with the MPC is required for the insulin-sensitizing effects of TZDs, we recently generated a constitutive knockout for MPC2 (Vigueira et al., 2014). Since the stability of the MPC complex requires both MPC1 and MPC2, this essentially results in a double or complete MPC knockout. Global and constitutive MPC deficiency is lethal in mice at early embryonic stages (Vigueira et al., 2014; Bowman et al., 2016; Li et al., 2016; Vanderperre et al., 2016), which is incompatible with studying the relationship between the MPC and TZDs in vivo. Fortuitously, due to an alternative translational start site in MPC2, we also unexpectedly generated mice expressing a protein lacking the first 16 amino acids (Mpc2Δ16 mice) (Vigueira et al., 2014). Mpc2Δ16 mice have a reduced abundance of both MPC1 and MPC2 protein compared to WT mice, but the remaining truncated MPC2 protein does properly localize to the inner mitochondrial membrane (Vigueira et al., 2014). These mice are viable, outwardly normal, and exhibit moderate (~30%) reductions in mitochondrial pyruvate metabolism (Vigueira et al., 2014). Besides increased plasma lactate concentrations, the primary phenotype of the Mpc2Δ16 mice is glucose intolerance that is due to defective insulin secretion by pancreatic β cells (Vigueira et al., 2014).
Given the viability of this model with partial MPC deficiency, we sought to evaluate the requirement of these 16 amino acids of MPC2 and the effects of MPC2 hypomorphism on TZD responses. In vivo studies conducted with WT and Mpc2Δ16 mice showed that the ability of pioglitazone (PIO) and MSDC-0602 to confer insulin sensitization in high fat diet-fed mice was not affected by MPC2 hypomorphism. Moreover, studies conducted in vitro show that N-terminal 16-amino acid-truncation of MPC2 fails to abolish the interaction between TZDs and the MPC or affect the ability of these drugs to modulate mitochondrial pyruvate metabolism. This suggests that loss of the N-terminal 16 amino acids of MPC2 does not affect the interaction with or insulin-sensitizing actions of TZDs.
MATERIALS AND METHODS
Ethical Approval
All vertebrate animal experiments were approved by the Animal Studies Committee of the Washington University School of Medicine and carried out according to the approved protocol (#20110101) and the principles and regulations described by Grundy (Grundy, 2015).
Animal Studies
Mice homozygous for the Mpc2Δ16 allele have been previously described (Vigueira et al., 2014). Deletion of the original Mpc2 translational start codon led to enforcement of an alternative start codon that encoded an MPC2 protein lacking the N-terminal 16 amino acids (Vigueira et al., 2014). Mice were generated and studied in a pure C57BL/6 background and only male mice were studied in the experiments described. Six to 8 week-old mice were placed on purified diet providing 10% (Research Diets Inc., D12450B) or 60% (Research Diets Inc., D12492) of its calories as fat and remained on these diets for 10 weeks before initiation of TZD therapy. Subsets of mice were then randomized to remain untreated or to receive TZDs that were incorporated into the 60% fat chow (pioglitazone – 300 ppm and MSDC-0602 – 331 ppm) for an additional 4 weeks (Figure 1A). Previous work has shown that this concentration of MSDC-0602 in diet resulted in a blood concentration of 2–5 μM of drug in obese mice (Chen et al., 2012). Ad libitum access to food was provided for the duration of the experiment and body weights were monitored weekly.
Figure 1. Diet-induced obesity and glucose intolerance develops normally in MPC2 hypomorphic mice.

[A] A schematic depicting the experimental timeline is shown. [B] Body weight gain by WT and Mpc2Δ16 mice on diets that were low fat (LF) or high fat (HF). WT LF n=16; Mpc2Δ16 LF n=9; WT HF n=48; Mpc2Δ16 HF n=35 [C] Body fat percentage determined by DEXA analysis for mice fed LF or HF diet. [D] Blood glucose during glucose tolerance testing for WT and Mpc2Δ16 mice on diets that were low fat (LF) or high fat (HF). Area under the curve (AUC) values were calculated using the trapezoidal rule. Values are presented as the mean ± SD. WT LF n=15; Mpc2Δ16 LF n=9; WT HF n=40; Mpc2Δ16 HF n=25; * indicates p<0.05 vs LF-fed group. † indicates p<0.05 vs WT.
After 8 weeks of a 10% or 60% fat diet challenge and again after 3 weeks of treatment with TZDs, mice were briefly sedated by intraperitoneal injection of Ketamine (87 mg/kg) and Xylazine (13 mg/kg) cocktail and subjected to DEXA analysis to determine body fat percentage as previously described (Finck et al., 2005).
Glucose tolerance tests (GTT) were conducted in drug-naïve mice after 9 weeks of a 10% or 60% fat diet challenge and again following 2 weeks of treatment with TZDs (Figure 1A). Mice were fasted overnight for 16 h on aspen chip bedding and GTTs were performed as previously reported (Vigueira et al., 2014; McCommis et al., 2016a). Glucose levels were monitored using blood acquired from a single tail snip and gentle milking. Blood glucose area under the curve (AUC) was calculated using the trapezoidal rule.
Following a 3-day diet treatment and acclimation period, the activity of 18–19 week-old, male, wild-type C57BL6/J, diet-induced obese mice was measured for 5 days while fed control high fat diet or high fat diet containing MSDC-0602 or pioglitazone. Activity levels were determined using an InfraMot activity system (TSE systems). Light Period: 6am-6pm (0600-1800). Dark Period 6pm–6am (1800-0600).
Following four weeks of TZD treatment, mice were sacrificed by CO2 sedation and asphyxiation and blood was collected from the inferior vena cava into EDTA coated syringes. Plasma was collected after blood centrifugation. Tissue was collected and snap-frozen for later analysis.
Quantitative RT-PCR
Total RNA was isolated from epididymal white adipose tissue using the RNAzol method (Tel-Test), and cDNA was synthesized with Vilo reverse transcription kit (Invitrogen). Real-time PCR was performed using the ABI PRISM 7500 sequence detection system and the SYBR green kit (Applied Biosystems, Foster City, CA). Arbitrary units of target mRNA were corrected by measuring the levels of 36B4 RNA. Sequence of the oligonucleotides used in qRT-PCR analyses are available on request.
Plasma Measurements
Insulin content was analyzed by Singulex assay by the Washington University Immunoassay Core of the Diabetes Research Center. Plasma triglyceride and cholesterol concentrations were measured by Infinity colorimetric assay kits (Thermo Fisher Scientific). Plasma non-esterified fatty acids concentrations were measured using enzymatic assay (Wako Diagnostics). Plasma alanine transaminase (ALT) activity was measured by kinetic absorbance assays (Teco Diagnostics).
Liver Triglyceride Quantification
Liver triglyceride was measured by homogenizing 100 mg liver tissue in saline and solubilizing lipid with 0.5% sodium deoxycholate. Liver triglyceride was then determined by colorimetric assay (Thermo Fisher Scientific) as performed previously (Vigueira et al., 2014; McCommis et al., 2015).
REporter Sensitive to PYRuvate (RESPYR) Assay
pWPT lentiviral vectors expressing cDNA encoding human MPC2 fused to RLuc8 (BRET donor) and human MPC1 fused to Venus (BRET acceptor) have been described (Compan et al., 2015; McCommis et al., 2016a). hMPC2Δ16-L17M constructs were generated using a QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). The nucleotides encoding the first 16 residues of the MPC2 region of the protein were deleted first and then a second round of mutagenesis was performed to insert a new start codon (L17M) to mirror the MPC2Δ16 mouse protein using the manufacturer-recommended protocol. Site-directed mutagenesis primers used to delete the first 16 residues were 5′-CTCCACTTTATCGAGGGTGGCCTCGAGGC-3′ and 5′-GCCTCGAGGCCACCCTCGATAAAGTGGAG-3′. Site-directed mutagenesis primers used for the L17M mutation were 5′-CAGCTCCACTTTATCCATGGTGGCCTCGAGGCT-3′ and 5′-AGCCTCGAGGCCACCATGGATAAAGTGGAGCTG-3′. Successful mutagenesis was verified by DNA sequencing by the Protein and Nucleic Acid Chemistry Laboratory at Washington University in St. Louis. Viruses containing the mutant plasmids were generated and MPC2-KO HEK293T cells were transformed using the same methods as with the wild type plasmids. Briefly, MPC2-KO HEK293T cells (described below) were transiently transfected with WT and hMPC2Δ16-L17M RESPYR constructs by using Lipofectamine according to the instructions of the manufacturer. Twenty-four h post-transfection, HEK293T cells were seeded in white 96-well plates 48 h before recording using phenol red-free DMEM media supplemented with 10% FBS and 1x MEM non-essential amino acids. Assays were performed 48 h post-transfection and analyzed as described (Compan et al., 2015; McCommis et al., 2016a). Luminescence was measured using a plate reader (Biotek Synergy 2) at 460 and 528 nm at 37°C.
Oxygraph respiration
Mitochondrial respiration of HEK293T cells was conducted using an Oxygraph O2k (Oroboros Instruments). 2 × 106 cells were suspended in 2 mL Mir05 respiration buffer and permeabilized with 20 μg digitonin. Respiratory substrates were 5 mM pyruvate/2 mM malate in the presence or absence of 1 mM ADP. After establishment of maximal pyruvate/malate respiration with ADP, 20 μM MSDC-0602 was injected into the respiratory chamber to test acute effects on pyruvate-mediated respiration.
Cell Culture and Generation of MPC2 knockout and hypomorphic MPC2 cell lines using CRISPR/Cas9 systems
HEK293T cells were tested using the PlasmoTest™ - Mycoplasma Detection Kit according to the manufacturer’s instructions (Invivogen) and found to be mycoplasma-free. Lines were maintained in Dulbecco’s modified Eagle’s medium containing 4.5 g/L glucose supplemented with 10% FBS (Atlanta Biologicals), 1 mM pyruvate, 2 mM glutamine, 2 mM Glutamax, 1 mM Citrate, 50 μg/mL Uridine and 1X Penicillin/Streptomycin (Invitrogen) at 37°C in 5% CO2. CRISPR/Cas9 targeting constructs were generated by sub-cloning oligonucleotides (listed below) into the BbsI sites of pSpCas9(BB)-2A-GFP (PX458) (Addgene# 48138). HEK293T cells were obtained from American Type Culture Collection (ATCC). Two control non-targeted HEK293T cell lines were generated by co-transfection with pSpCas9(BB)-2A-GFP backbone lacking sgRNAs and PQCXIP backbone at 9:1 DNA ratio. MPC2KO-1, MPC2KO-2, and MPC2∆22 were generated by co-transfection with PX458 harboring either sgRNA pair #1 or pair #2, and PQCXIP at 4.5:4.5:1 ratio. MPC2KO-1 sgRNA1: 5′-CACCGGCTGCCAACGATCCCTCGG-3′ and 3′-CCGACGGTTGCTAGGGAGCCCAAA-5′ sgRNA2: 5′-CACCGGAGCTGACTTGAGATACGT-3′ and 3′-CCTCGACTGAACTCTATGCACAAA-5′. MPC2KO-2 sgRNA1: 5′-CACCGCTTTTCGGTTCACAGCGGGC-3′ and 3′-CGAAAAGCCAAGTGTCGCCCGCAAA-5′ sgRNA2: 5′-CACCGACTTGAGATACGTTGGCTTAGG-3′ and 3′-CTGAACTCTATGCAACCGAATCCCAAA-5′. MPC2∆22 sgRNA1: 5′-CACCGCGGACGCGAGTCGTCGTGGC-3′ and 3′-CGCCTGCGCTCAGCAGCACCGCAAA-5′ sgRNA2: 5′-CACCGCCACCGGCTCCTCGATAAAG-3′ and 3′-CGGTGGCCGAGGAGCTATTTCCAAA-5′.
At 72 h post-transfection, cells were selected using 2 μg/ml puromycin for 72hr. After 48 h recovery in growth media, cells were monoclonally diluted into 96-well plates for growth of monoclonal lines. Upon reaching 80% confluency, monoclonal lines were divided into two portions to be used for genotyping and maintenance. Genomic DNA was extracted using QuickExtract™ DNA Extraction Solution (Epicentre), according to the manufacture’s protocol. Cell lines were genotyped by PCR on extracted genomic DNA utilizing the following primers: 5′-CAGCGCAGACTTGGTGAGG-3′ and 3′-GTGCTGAGGTTGGGTTTGGA-5′.
Western Blots
CRISPR/Cas9 edited cells were lysed in a Laemmli’s Buffer containing 62.5mM Tris pH 6.8, 2% SDS (m/v), 10% glycerol (v/v) and DTT (0.1μM) and quantified using the Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific). Equal amount of protein lysates were prepared in a Laemmli’s buffer supplemented with bromophenol blue and boiled at 95°C for 10 minutes. Lysates were resolved on 10% Tricine-SDS-PAGE gels using a Mini-PROTEAN Tetra Cell running system (Bio-Rad). The Trans-Blot transfer cell system (Bio-Rad) was used to transfer samples to nitrocellulose membranes (GE Healthcare, 10600001). Membranes were blocked with 5% (m/v) nonfat dry milk in 50 mM Tris and 150 mM NaCl (TBS) for 1 h, incubated with primary antibodies against MPC2 (1:1,000; Cell Signaling Technology #46141), MPC1 (1:1,000; Cell Signaling Technology #14462), UCP1 (1:1000; Abcam #10983), αTubulin (1;1000; Sigma Aldrich #T6074), and VDAC (1:1,000; Cell Signaling Technology #4661) overnight at 4°C, and incubated with fluorescent goat anti-Rabbit DyLight 800 secondary antibody (1:10,000; Thermo Fisher 35571) for 1 h. Membranes were imaged using the Li-Cor Odyssey CLx system.
Statistical Analyses
P values for pairwise comparisons were calculated using a Student’s t-test. P values for RESPYR curves were calculated using repeated measures, 2-way ANOVA coupled to Tukey’s multiple comparison tests. In all experiments, P ≤ 0.05 was used to determine significant difference. All quantitative data is represented as mean ± SD.
RESULTS
WT and Mpc2Δ16 mice exhibit a similar response to high fat diet feeding
We evaluated the insulin-sensitizing effects of pioglitazone and MSDC-0602 in vivo in WT and Mpc2Δ16 mice to determine if the first 16 amino acids of MPC2 were required for drug effects. WT and Mpc2Δ16 mice were placed on a high fat (HF; 60% of calories from fat) or low fat control (LF; 10% of calories from fat) diet. At the start of the diet treatments Mpc2Δ16 mice weighed slightly less than WT controls, 21.3 ± 2.2 g versus 22.8 ± 1.9 g (p<0.001). No differences were observed for any parameter examined between genotypes in the mice fed LF diet. On high fat diet (HF), Mpc2Δ16 mice tended to gain less weight beginning at week 7 of the trial (Figure 1B). However, the body fat percentage, as assessed by DEXA, was not significantly different between the two genotypes (Figure 1C). In addition, HF feeding caused glucose intolerance in mice of both genotypes (Figure 1D). No effect of genotype on glucose tolerance was detected in mice on either dietary regimen.
Mpc2Δ16 mice are not refractory to the insulin-sensitizing effects of TZDs
We randomly assigned HF-fed WT and Mpc2Δ16 mice to either remain on the regular HF or be given ad libitum access to HF diet containing pioglitazone (+PIO) or MSDC-0602 (+0602) for an additional 4 weeks. Interestingly, while mice fed regular HF diet or HF diet containing PIO continued to gain weight or remained static, all mice fed MSDC-0602 lost a notable amount of weight (Figure 2A, B). This loss of body weight occurred within the first week of MSDC-0602 feeding and is similar to another recent study we conducted with MSDC-0602 (McCommis et al., 2016b). Weight loss was not associated with increased locomotor activity in WT mice (Figure 2C) or an overt reduction in food intake in mice of either genotype (Figure 2D) in MSDC-0602-treated mice compared to HF-fed controls. However, there was a non-significant tendency towards decreased activity counts in mice given PIO, compared to untreated controls. MSDC-0602 administration caused a reduction in body fat percentage in both WT and Mpc2Δ16 mice after 3 weeks of TZD treatment (Figure 2E). While PIO had no effect on adiposity in WT mice, Mpc2Δ16 mice in the pioglitazone treatment group exhibited decreased weight gain and adiposity compared to HF diet alone (Figure 2B, E). This reduction in adiposity could be due to reduced consumption of the PIO-containing diet compared to HF diet by the Mpc2Δ16 mice for reasons that are not clear. Although unlikely, this could be a gene-drug interaction leading to a conditioned taste aversion, but is more likely a type 1 statistical error.
Figure 2. MSDC-0602 causes weight loss and in WT and MPC2 hypomorphic mice.

[A,B] Body weight of WT and Mpc2Δ16 mice on diets that were low fat (LF), high fat (HF), HF containing pioglitazone (HF + PIO), or HF containing MSDC-0602 (HF + 0602). [C] Average activity (Area Under the Curve) of WT mice in each diet group: high fat diet (HF), HF diet supplemented with MSDC-602 (0602), HF diet supplemented with pioglitazone (PIO) determined using an infrared-sensing device (Inframot) during the dark phase of the diurnal cycle (1800-0600), light phase (0600-1800), and the sum of both. Values are presented as the mean ± SD. HF n=11; 0602 n=12; PIO n=12; * indicates p<0.05 vs LF-fed group. † indicates p<0.05 vs HF-fed group. [D] Consumption of food by WT and Mpc2Δ16 mice on diets that were low fat (LF), high fat (HF), HF containing pioglitazone (HF + PIO), or HF containing MSDC-0602 (HF + 0602) for the first 2 weeks of TZD administration. [E] Body fat percentage determined by DEXA analysis after 3 weeks of TZD treatment. WT LF n=15; Mpc2Δ16 LF n=9; WT HF n=14; Mpc2Δ16 HF n=9; WT HF + PIO n=13; Mpc2Δ16 HF + PIO n=8; WT HF + 0602 n=13; Mpc2Δ16 HF + 0602 n=9;
Two weeks of either PIO or MSDC-0602 feeding markedly improved the glucose tolerance of HF-fed mice and in fact, returned the blood glucose area under the curve values to levels similar to lean controls (Figure 3A, B). The effects of PIO and MSDC-0602 on glucose tolerance were again not affected by loss of the first 16 amino acids of MPC2 in Mpc2Δ16 mice (Figure 3A, B). These data suggest that the N-terminus of MPC2 does not mediate the effects of PIO or MSDC-0602 on glucose tolerance.
Figure 3. MSDC-0602 improves glucose tolerance and hepatic steatosis.
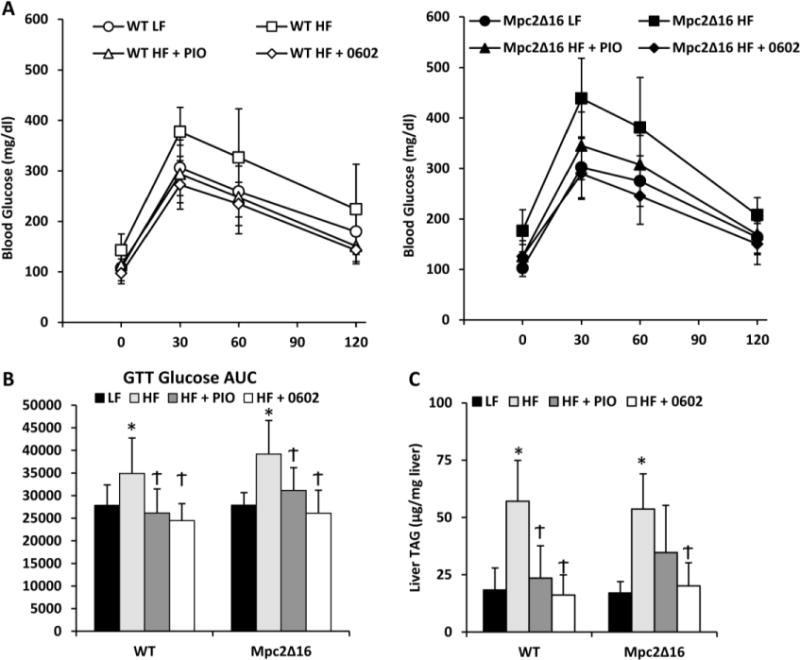
[A] Results of glucose tolerance testing in WT and Mpc2Δ16 mice on LF, HF, HF containing PIO, or HF containing MSDC-0602 diets. [B] Blood glucose area under the curve (AUC) values during glucose tolerance testing for WT and Mpc2Δ16 mice on diets that were low fat (LF) or high fat (HF) after treatment with PIO, 0602, or no drug calculated using the trapezoidal rule. WT LF n=15; Mpc2Δ16 LF n=9; WT HF n=14; Mpc2Δ16 HF n=9; WT HF + PIO n=13; Mpc2Δ16 HF + PIO n=8; WT HF + 0602 n=13; Mpc2Δ16 HF + 0602 n=9; [C] Liver triacylglycerol (TAG) content in WT and Mpc2Δ16 mice on LF, HF, HF containing pioglitazone (PIO), or HF containing MSDC-0602 diets. Values are presented as the mean ± SD. WT LF n=8; Mpc2Δ16 LF n=6; WT HF n=6; Mpc2Δ16 HF n=6; WT HF + PIO n=5; Mpc2Δ16 HF + PIO n=5; WT HF + 0602 n=7; Mpc2Δ16 HF + 0602 n=5; * indicates p<0.05 vs LF-fed group. † indicates p<0.05 vs HF-fed group.
Two weeks after the GTT, mice were sacrificed for tissue and plasma parameters. Both PIO and MSDC-0602 treatment led to improvements in a number of plasma parameters that were deranged in the HF-fed mice. Specifically, treatment with either TZD lowered plasma triglyceride, cholesterol, and free fatty acid concentrations compared to HF-fed comparators (Table 1). TZD treatment reduced plasma insulin concentration and increased adiponectin concentration (Table 1). These responses were all also observed in Mpc2Δ16 mice, again suggesting that the N-terminus of MPC2 does not mediate these beneficial metabolic effects of TZDs. Administration of either PIO or MSDC-0602 markedly reduced the hepatic steatosis observed in HF-fed mice and this was observed in mice of either genotype (Figure 3C). TZD administration also significantly lowered circulating transaminase (ALT) in mice of both genotypes (Table 1).
Table 1.
Plasma hormones and metabolite concentrations.
| WT | Mpc2Δ16 | |||||||
|---|---|---|---|---|---|---|---|---|
| LF | HF | HF + Pio | HF + 0602 | LF | HF | HF + Pio | HF + 0602 | |
| TAG (mg/dL) |
81.7 (15.8) |
91.4 (18.3) |
54.3 (7.7)Ϯ |
63.6 (14.6)Ϯ |
78.2 (18.6) |
100.0 (24.9)* |
62.8 (13.3)Ϯ |
53.3 (10.3)Ϯ |
| Cholesterol (mg/dL) |
109.8 (34.7) |
187.3 (41.1)* |
91.1 (26.5)Ϯ |
77.2 (33.0)Ϯ |
102.9 (39.5) |
199.8 (42.4)* |
86.0 (23.4)Ϯ |
68.1 (23.2)Ϯ |
| Free Fatty Acids (mM) |
0.82 (0.19) |
1.08 (0.17)* |
0.68 (0.25)Ϯ |
0.53 (0.19)Ϯ |
0.82 (0.20) |
1.14 (0.33)* |
0.70 (0.32)Ϯ |
0.61 (0.29)Ϯ |
| Insulin (pg/mL) |
1082 (547) |
4024 (1297)* |
909 (227)Ϯ |
750 (479)Ϯ |
1192 (208) |
5045 (2419)* |
782 (314)Ϯ |
854 (520)Ϯ |
| Adiponectin (μg/ml) |
12.0 (1.9) |
12.2 (2.3) |
27.2 (7.8)Ϯ |
23.1 (6.7)Ϯ |
14.0 (6.0) |
12.5 (2.1) |
32.2 (7.3)Ϯ |
28.1 (9.0)Ϯ |
| ALT (U/L) |
15.0 (4.1) |
29.7 (14.8)* |
18.4 (8.7) |
12.0 (3.0) |
12.8 (3.3) |
28.8 (13.9)* |
12.7 (5.1) |
13.9 (2.4) |
p < 0.05 vs LF
p < 0.05 vs LF and HF
WT LF n=15; Mpc2Δ16 LF n=9; WT HF n=14; Mpc2Δ16 HF n=9; WT HF + PIO n=12; Mpc2Δ16 HF + PIO n=6; WT HF + 0602 n=13; Mpc2Δ16 HF + 0602 n=8; Data are presented as the mean (SD). Differences between WT and Mpc2Δ16 genotypes are not statistically significant.
Effects of TZDs on adipose tissue expression of inflammatory and mitochondrial biogenesis markers
TZDs are known to reduce macrophage infiltration and adipose tissue proinflammatory cytokine expression. The administration of HF diet robustly induced the expression of the macrophage markers Cd68 and F4/80 (Emr1), the chemotractant Mcp1, and the proinflammatory cytokine Tnfa in the epididymal white adipose tissue (Figure 4). Treatment with PIO or MSDC-0602 suppressed the elevated expression of most of these genes in mice of both genotypes. Consistent with the plasma data, the expression of adiponectin (Adipoq) was also rescued by treatment with either TZD and was not affected by genotype.
Figure 4. Diminished expression of markers of macrophage infiltration after treatment with PIO or MSDC-0602.
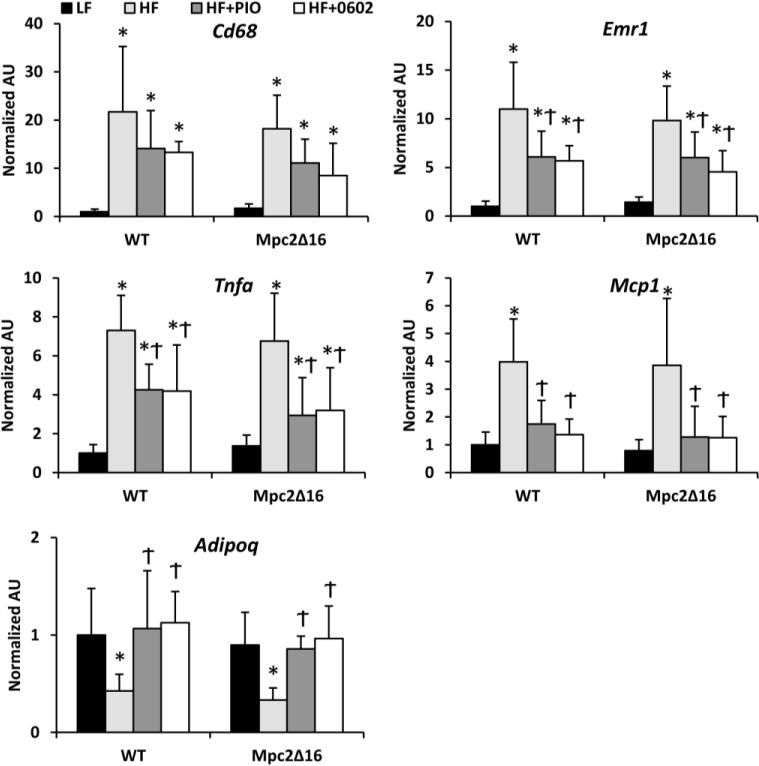
The expression of indicated genes encoding proinflammatory cytokines, macrophage markers, and adiponectin (Adipoq) in epididymal adipose tissue of mice fed LF, HF, or HF containing PIO or MSDC-0602 diets is shown. Values are presented as the mean ± SD. N=6 per group, * indicates p<0.05 vs LF-fed group. † indicates p<0.05 vs HF-fed group.
Among the genes whose expression was down regulated by obesity and stimulated by TZD treatment, were the genes encoding MPC1 and MPC2 (Figure 5A). Western blotting confirmed that these changes in gene expression resulted in corresponding changes in MPC1 and MPC2 protein abundance as well (Figure 5B). TZDs are also known to stimulate mitochondrial biogenesis and “browning” of white adipose tissue. In addition, TZDs stimulated a marked activation of Ucp1 expression in the epididymal fat pad (Figure 5C), which was also confirmed at the protein level (Figure 5B and 5C).
Figure 5. MPC expression is reduced in adipose tissue by HF diet and restored by TZD treatment.

[A] The expression of Mpc1 and Mpc2 in epididymal adipose tissue of mice fed LF, HF, or HF containing PIO or MSDC-0602 diets is shown. [B] Representative western blots for MPC1, MPC2, UCP1, and tubulin in epididymal adipose tissue of mice fed LF, HF, or HF containing PIO (HF+P) or MSDC-0602 (HF+M) diets is shown. Average band intensity is shown in graphs below. [C] The expression of Ucp1 mRNA (left) and UCP1 protein (right) is quantified in epididymal adipose tissue of mice treated as above. [For all panels] Values are presented as the mean ± SD. N=6 per group, * indicates p<0.05 vs LF-fed group. † indicates p<0.05 vs HF-fed group. 1 indicates p<0.05 vs comparable WT group.
Consistent with a “browning” of this depot, the expression of Prdm16 and Ppargc1a, which transcriptionally activate the browning program, were also induced (Figure 6). Lastly, expression of several other genes associated with a thermogenic transcriptional program was increased in epididymal fat by administration of pioglitazone or MSDC-0602 (Cidea, Cox2, and Acadm) (Figure 6). In all cases, the response to TZD administration was unaffected by mouse genotype.
Figure 6. WT and MPC2 hypomorphic mice exhibit transcriptional activation of browning markers in response to PIO or MSDC-0602.

The expression of indicated genes encoding mitochondrial markers or regulators of browning in epididymal adipose tissue of mice fed LF, HF, or HF containing PIO or MSDC-0602 diets is shown. Values are presented as the mean ± SD. N=6 per group, * indicates p<0.05 vs LF-fed group. † indicates p<0.05 vs HF-fed group. 1 indicates p<0.05 vs comparable WT group.
The N-terminus of MPC2 is dispensable for interaction with TZD and effects on pyruvate metabolism
The recently described, BRET-based, MPC activity system known as RESPYR (Compan et al., 2015) can also be used to detect interactions with small molecule competitive inhibitors of MPC activity (McCommis et al., 2016b). As we have previously described, addition of UK-5099, a potent and specific MPC inhibitor, or MSDC-0602 caused a dramatic and immediate increase in RESPYR activity in HEK293 cells (Figure 7A, left panel). To determine whether loss of the first 16 amino acids affected this interaction, an MPC2Δ16 mutant MPC2-Rluc8 fusion protein was generated. The mutant MPC2-Rluc8 fusion protein was expressed at levels similar to the WT fusion protein (Figure 7B). To eliminate the potential for contribution of endogenous MPC2 to the MPC complex, these studies were conducted in HEK293T cells that were completely MPC2 null (described in Figure 7C). Loss of the first 16 amino acids did not prevent activation by pyruvate, MSDC-0602, or UK5099 (Figure 7A).
Figure 7. TZDs stimulate the activity of a BRET-based RESPYR biosensor of the mitochondrial pyruvate carrier (MPC).

[A] BRET kinetics of MPC2-deficient HEK293T (MPC2KO) cells expressing RLuc8 fused to MPC2 and Venus yellow fluorescent protein fused to MPC1. Experiments conducted with human MPC2 fusion proteins that were either full length (wild type (WT)) or lacking the N-terminal 16 amino acids (MPC2Δ16-L17M). Cells were stimulated after 5 minutes with vehicle (DMSO), 5 mM pyruvate plus vehicle, 5 μM UK-5099, or 10 μM MSDC-0602. Values are presented as the mean ± SD. Three independent experiments were performed with 4 technical replicates per experiment. * indicates p<0.05 for pyruvate vs all other conditions. † indicates p<0.05 for UK-5099 versus all other conditions. ‡ indicates p<0.05 for MSDC-0602 versus all other conditions. [B] Western blot of HEK293T cell lysates depicting expression of MPC1 or MPC2 RESPYR fusion proteins and tubulin. [C] Western blot of HEK293T cell lysates depicting either complete loss of MPC2 (MPC2KO) or the “human MPC2Δ16 equivalent” MPC2Δ22 truncation induced by CRISPR. [D] Oxygen consumption rates (OCR) of permeabilized HEK293T cells expressing WT MPC2, MPC2Δ22, or MPC2KO induced by CRISPR. Respiration stimulated by 5 mM pyruvate and 2 mM malate (P/M), P/M with 1 mM ADP (P/M +ADP), and after addition of 20 μM MSDC-0602. * indicates p<0.05 vs WT cells. † indicates p<0.05 vs P/M +ADP respiration.
We also determined the effect of MPC2 truncation on the ability of MSDC-0602 to suppress pyruvate-mediated respiration by permeabilized cells. To this end, HEK293T cells that were wild-type, completely MPC2 null, or expressing the endogenous human equivalent (MPC2Δ22) of the mouse Mpc2Δ16 allele were generated by using CRISPR/Cas9 (Figure 7C). We found that complete loss of MPC2 markedly reduced maximal pyruvate-stimulated respiration and rendered cells insensitive to the inhibitory effects of MSDC-0602 on oxygen consumption (Figure 7D). In contrast, loss of the first 22 amino acids only moderately reduced maximal pyruvate respiration and cells expressing this allele fully responded to MSDC-0602 with reduced oxygen consumption (Figure 7D). Collectively, these data suggest that the interaction of TZDs to the MPC complex does not require the first 16 amino acids of MPC2, and that loss of the N-terminus of MPC2 does not abrogate TZD effects on pyruvate metabolism.
DISCUSSION
Recent work has suggested that TZDs may have insulin-sensitizing effects that are independent of their canonical roles as ligands for PPARγ. One potential alternative mechanism that has emerged recently is an interaction with the MPC complex and subsequent attenuation of mitochondrial pyruvate metabolism. However, the mechanistic details of this interaction remain unresolved. In this work, we used mice expressing an MPC2 protein that lacks the N-terminal 16 amino acids to evaluate the effects of this MPC2 truncation on the metabolic effects of PIO and the PPARγ-sparing TZD, MSDC-0602. We found that MPC2 hypomorphic mice were not refractory to the insulin-sensitizing effects of TZDs. Loss of the first 16 amino acids of MPC2 also did not affect the interaction of TZDs with the MPC complex nor did it affect the ability to modulate pyruvate-mediated respiration. On the other hand, complete deletion of MPC2 led to a loss of the inhibitory effect of MSDC-0602 on pyruvate-mediated respiration indicating that this effect on metabolism is MPC-dependent. Thus, the N-terminus of MPC2 is not required for the insulin-sensitizing effect of TZDs. However, it is still not clear whether the entire MPC complex is required for TZD-mediated insulin sensitization.
Because the inner mitochondrial matrix is impermeable to charged molecules like pyruvate, it has long been hypothesized that a carrier-based mechanism for pyruvate import into the mitochondrial matrix must exist (McCommis & Finck, 2015). However, the identity of this carrier was only recently discovered by two groups evaluating the functional roles of mitochondrial proteins that are highly conserved across deeply diverged lineages (Bricker et al., 2012; Herzig et al., 2012). The mitochondrial pyruvate carrier mediates an important step in intermediary metabolism. The majority of pyruvate generation occurs via glycolysis and by oxidation of lactate by lactate dehydrogenase in the cytosol of the cell. The enzymes that further metabolize pyruvate, pyruvate dehydrogenase and pyruvate carboxylase, are localized to the mitochondrial matrix. Thus, the MPC-mediated import of pyruvate into the mitochondria is required for high rates of metabolic flux through anabolic and catabolic pathways.
In some tissues, the mechanisms by which inhibition of mitochondrial pyruvate metabolism would lead to glucose lowering effects can be clearly identified. For example, MPC inhibition in hepatocytes leads to reduced flux through gluconeogenic pathways (Gray et al., 2015; McCommis et al., 2015), and we have shown that liver MPC2 deletion protects mice from the development of diabetes (McCommis et al., 2015). In other organs, it is not yet clear how inhibition of the MPC might lead to insulin-sensitizing effects. Potentially, modulating pyruvate flux leads to alterations in the cellular redox status and/or synthesis of NAD+ as a byproduct of lactate synthesis from pyruvate. It might also reduce mitochondrial reactive oxygen species production. On the other hand, interaction with the MPC and/or effects on pyruvate metabolism may not be involved in all of the mitochondrial and insulin-sensitizing effects of TZDs. Indeed, a number of potentially direct mitochondrial effects of these drugs have been identified and warrant additional exploration (Brunmair et al., 2004; Feinstein et al., 2005).
Data from this and previous studies suggest that it may be necessary to completely disrupt the MPC complex in order to determine the effect on insulin sensitization in vivo. Unfortunately, constitutive knockout of MPC2, which destabilizes the MPC complex and essentially leads to double deletion of MPC1 and MPC2 protein, leads to early embryonic lethality in mice (Vigueira et al., 2014). In addition, mice with whole-body, tamoxifen-inducible deletion of MPC2 (Lam et al., 2016) also succumb within a week of tamoxifen-induced MPC2 deletion (unpublished observation). Whereas mice with liver-specific (McCommis et al., 2015) or β-cell specific (McCommis et al., 2016a) MPC2 deletion are viable and relatively normal, their utility in determining the requirement for the MPC in the insulin-sensitizing effects of TZDs is limited due to effects in other tissues, including adipocytes. Thus, defining the role of MPC in TZD-mediated insulin sensitization is very challenging with existing molecular genetic tools. Identifying the residues required for TZD interaction with the MPC complex, as we attempted to do here, would be the first step towards making mutant alleles that might still be competent for pyruvate transport and support life, but would be refractory to TZD binding. These tools could allow for the testing of the requirement for the MPC in TZD effects, which might not be identical in all cell types.
We have previously shown that Mpc2Δ16 mice are glucose intolerant due to reduced insulin secretion (Vigueira et al., 2014), but that effect was not evident in the present study even in LF diet-fed mice. In that previous work, the absolute differences in blood glucose during GTT were significantly, but not dramatically, different with a high “n”. Our additional studies conducted with beta cell MPC2 KO mice (McCommis et al., 2016a) very much support the veracity of the glucose intolerance phenotype we observed in the Mpc2Δ16 mice and we believe that these published findings are accurate. However, an effect of the Mpc2Δ16 genotype on glucose tolerance was not observed in the LF group in this study. There are a number of potential explanations for this. The previous study used female mice and whereas the present studies used male mice and the mice used in this study were also somewhat older at the time of the GTT studies. Perhaps the most important difference was that the LF control diet used in the present study is a purified diet matched to the HF diet while mice in the previous study were fed standard mouse chow. The purified diet contains a good deal of simple sugar (sucrose) while the chow does not and this could affect the insulin sensitivity/glucose tolerance of the controls. We cannot pinpoint one of these differences as a definitive explanation, but these are all potentially factors.
In conclusion, the present study aimed to evaluate the effects of partial deletion of the MPC2 protein on the binding and insulin-sensitizing effects of TZD insulin sensitizers. The present results confirmed that a TZD with very low affinity for PPARγ still had potent insulin-sensitizing effects and led to browning of white adipose tissue in diet-induced obese mice. The data also indicate that pioglitazone and MSDC-0602 acutely suppress mitochondrial pyruvate metabolism and, using a BRET-based approach, that these compounds directly interact with the MPC complex. However, it was determined that deletion of the N terminus of MPC2 did not affect the response to TZDs nor did it disrupt the interaction between the compound and the MPC complex. Further study is required to fully elucidate whether/how the MPC complex is involved in the cellular-specific, non-genomic insulin sensitizing effects of TZDs.
NEW FINDINGS
What is the central question of this study?
The antidiabetic effects of thiazolidinedione (TZD) drugs may be mediated in part by a molecular interaction with the constituent proteins of the mitochondrial pyruvate carrier complex (MPC1 and MPC2). We examined the ability of a mutant mouse strain expressing an N-terminal truncation of MPC2 (Mpc2Δ16 mice) to respond to TZD treatment.
What is the main finding and its importance?
The response of Mpc2Δ16 mice to TZD treatment was not significantly different from wild-type C57BL6/J controls, suggesting that the 16 N-terminal amino acids of MPC2 are dispensable for the effects TZD treatment.
Acknowledgments
We thank J.C. Martinou for the gift of RESPYR plasmids.
GRANTS:
This research was supported by NIH grant R01 DK104735 (B.N.F.), R01 DK104998 (E.B.T), and R42 AA021228 (R.F.K.). The Core services of the Diabetes Research Center (P30 DK020579) and the Nutrition Obesity Research Center (P30 DK56341) at the Washington University School of Medicine also supported this work. P.A.V (T32 DK007296) and K.S.M. (DK007120) were Diabetes Research Postdoctoral Training Program fellows. G.G.S. was a Cardiovascular Research Training Program fellow (T32 HL007081).
LIST OF ABBREVIATIONS
- ALT
alanine aminotransferase
- AUC
area under the curve
- BRET
bioluminescence resonance energy transfer
- CRISPR
clustered regularly interspaced short palindromic repeats
- DEXA
dual-energy X-ray absorptiometry
- DMEM
Dulbecco’s modified eagle’s medium
- FBS
fetal bovine serum
- GTT
glucose tolerance test
- HEK293
human embryonic kidney 293 cells
- HF
high fat
- LF
low fat
- MPC
mitochondrial pyruvate carrier
- OCR
oxygen consumption rate
- PIO
pioglitazone
- PPARγ
peroxisome proliferator-activated receptor gamma
- RESPYR
reporter sensitive to pyruvate
- TZD
thiazolidinedione
- WT
wild type
Footnotes
Author contributions: PAV, KSM, WTH, GGS, SLC, and LO performed experiments, analyzed data, wrote, and edited the manuscript. EBT, WGM, RFK, JRC conceived experiments, and edited the manuscript. BNF conceived experiments, wrote, and edited manuscript.
DISCLOSURES:
Serena Cole, William McDonald, Rolf Kletzien, and Jerry Colca are employees and stockholders of Metabolic Solutions Development Company.
REFERENCES CITED
- Adams MD, Raman P, Judd RL. Comparative effects of englitazone and glyburide on gluconeogenesis and glycolysis in the isolated perfused rat liver. Biochem Pharmacol. 1998;55:1915–1920. doi: 10.1016/s0006-2952(98)00052-5. [DOI] [PubMed] [Google Scholar]
- Bowman CE, Zhao L, Hartung T, Wolfgang MJ. Requirement for the mitochondrial pyruvate carrier in mammalian development revealed by a hypomorphic allelic series. Mol Cell Biol. 2016;36:2089–2104. doi: 10.1128/MCB.00166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhausl W, Furnsinn C. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53:1052–1059. doi: 10.2337/diabetes.53.4.1052. [DOI] [PubMed] [Google Scholar]
- Chen Z, Vigueira PA, Chambers KT, Hall AM, Mitra MS, Qi N, McDonald WG, Colca JR, Kletzien RF, Finck BN. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor gamma-sparing thiazolidinedione. J Biol Chem. 2012;287:23537–23548. doi: 10.1074/jbc.M112.363960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colca JR, Kletzien RF. What has prevented the expansion of insulin sensitisers? Expert Opin Investig Drugs. 2006;15:205–210. doi: 10.1517/13543784.15.3.205. [DOI] [PubMed] [Google Scholar]
- Colca JR, McDonald WG, Cavey GS, Cole SL, Holewa DD, Brightwell-Conrad AS, Wolfe CL, Wheeler JS, Coulter KR, Kilkuskie PM, Gracheva E, Korshunova Y, Trusgnich M, Karr R, Wiley SE, Divakaruni AS, Murphy AN, Vigueira PA, Finck BN, Kletzien RF. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)–relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One. 2013a;8:e61551. doi: 10.1371/journal.pone.0061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colca JR, VanderLugt JT, Adams WJ, Shashlo A, McDonald WG, Liang J, Zhou R, Orloff DG. Clinical proof-of-concept study with MSDC-0160, a prototype mTOT-modulating insulin sensitizer. Clin Pharmacol Ther. 2013b;93:352–359. doi: 10.1038/clpt.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compan V, Pierredon S, Vanderperre B, Krznar P, Marchiq I, Zamboni N, Pouyssegur J, Martinou JC. Monitoring mitochondrial pyruvate carrier activity in real time using a BRET-based biosensor: Investigation of the Warburg effect. Mol Cell. 2015;59:491–501. doi: 10.1016/j.molcel.2015.06.035. [DOI] [PubMed] [Google Scholar]
- Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, Henry RR, McDonald WG, Colca JR, Simon MI, Ciaraldi TP, Murphy AN. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci U S A. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 2005;97:372–379. doi: 10.1161/01.RES.0000179226.34112.6d. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, Dello Russo C. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70:177–188. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Finck BN, Bernal-Mizrachi C, Han DH, Coleman T, Sambandam N, LaRiviere LL, Holloszy JO, Semenkovich CF, Kelly DP. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell Metab. 2005;1:133–144. doi: 10.1016/j.cmet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, Lane EE, Dohlman A, Zepeda-Orozco D, Xie J, Rutter J, Norris AW, Cox JE, Burgess SC, Potthoff MJ, Taylor EB. Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis. Cell Metab. 2015;22:669–681. doi: 10.1016/j.cmet.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D. Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol. 2015;593:2547–2549. doi: 10.1113/JP270818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat Med. 2005;11:861–866. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- Halberg N, Wernstedt-Asterholm I, Scherer PE. The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am. 2008;37:753–768. x–xi. doi: 10.1016/j.ecl.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, McCommis KS, Fahrmann J, Pizzato HA, Nunley RM, Lee J, Wolfgang MJ, Patti GJ, Finck BN, Pearce EL, Bhattacharya D. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity. 2016;45:60–73. doi: 10.1016/j.immuni.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Li X, Li Y, Han G, Li X, Ji Y, Fan Z, Zhong Y, Cao J, Zhao J, Mariusz G, Zhang M, Wen J, Nesland JM, Suo Z. Establishment of mitochondrial pyruvate carrier 1 (MPC1) gene knockout mice with preliminary gene function analyses. Oncotarget. 2016 doi: 10.18632/oncotarget.13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, Burgess SC, Finck BN. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 2015;22:682–694. doi: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCommis KS, Finck BN. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J. 2015;466:443–454. doi: 10.1042/BJ20141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCommis KS, Hodges WT, Bricker DK, Wisidagama DR, Compan V, Remedi MS, Thummel CS, Finck BN. An ancestral role for the mitochondrial pyruvate carrier in glucose-stimulated insulin secretion. Mol Metab. 2016a;5:602–614. doi: 10.1016/j.molmet.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCommis KS, Hodges WT, Brunt EM, Nalbantoglu I, McDonald WG, Holley C, Fujiwara H, Schaffer JE, Colca JR, Finck BN. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology. 2016b doi: 10.1002/hep.29025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman P, Foster SE, Stokes MC, Strenge JK, Judd RL. Effect of troglitazone (Rezulin) on fructose 2,6-bisphosphate concentration and glucose metabolism in isolated rat hepatocytes. Life Sci. 1998;62:PL89–94. doi: 10.1016/s0024-3205(97)01177-6. [DOI] [PubMed] [Google Scholar]
- Raman P, Judd RL. Role of glucose and insulin in thiazolidinedione-induced alterations in hepatic gluconeogenesis. Eur J Pharmacol. 2000;409:19–29. doi: 10.1016/s0014-2999(00)00806-2. [DOI] [PubMed] [Google Scholar]
- Riera-Guardia N, Rothenbacher D. The effect of thiazolidinediones on adiponectin serum level: a meta-analysis. Diabetes Obes Metab. 2008;10:367–375. doi: 10.1111/j.1463-1326.2007.00755.x. [DOI] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20:573–591. doi: 10.1016/j.cmet.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderperre B, Herzig S, Krznar P, Horl M, Ammar Z, Montessuit S, Pierredon S, Zamboni N, Martinou JC. Embryonic lethality of mitochondrial pyruvate carrier 1 deficient mouse can be rescued by a ketogenic diet. PLoS Genet. 2016;12:e1006056. doi: 10.1371/journal.pgen.1006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigueira PA, McCommis KS, Schweitzer GG, Remedi MS, Chambers KT, Fu X, McDonald WG, Cole SL, Colca JR, Kletzien RF, Burgess SC, Finck BN. Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep. 2014;7:2042–2053. doi: 10.1016/j.celrep.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang A, Kohan DE, Nelson RD, Gonzalez FJ, Yang T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proc Natl Acad Sci U S A. 2005;102:9406–9411. doi: 10.1073/pnas.0501744102. [DOI] [PMC free article] [PubMed] [Google Scholar]