

Summary
Fluid shear stress due to blood flow on the vascular endothelium regulates blood vessel development, remodeling, physiology and pathology [1, 2]. A complex consisting of PECAM-1, VE-cadherin and vascular endothelial growth factor receptors (VEGFRs) that resides at endothelial cell-cell junctions transduces signals important for flow-dependent vasodilation, blood vessel remodeling and atherosclerosis. PECAM-1 transduce forces to activate src family kinases (SFKs), which phosphorylate and transactivate VEGFRs [3-5]. By contrast, VE-cadherin functions as an adapter that interacts with VEGFRs through their respective cytoplasmic domains and promotes VEGFR activation in flow [6]. Indeed, shear stress triggers rapid increases in force across PECAM-1 but decreases the force across VE-cadherin, in close association with downstream signaling [5]. Interestingly, VE-cadherin cytoplasmic tyrosine Y658 can be phosphorylated by SFKs [7], which is maximally induced by low shear stress in vitro and in vivo [8]. These considerations prompted us to address the involvement of VE-cadherin cytoplasmic tyrosines in flow sensing. We found that phosphorylation of a small pool of VE-cadherin on Y658 is essential for flow sensing through the junctional complex. Y658 phosphorylation induces dissociation of p120ctn, which allows binding of the polarity protein LGN. LGN is then required for multiple flow responses in vitro and in vivo, including activation of inflammatory signaling at regions of disturbed flow, and flow-dependent vascular remodeling. Thus, endothelial flow mechanotransduction through the junctional complex is mediated by a specific pool of VE-cadherin that is phosphorylated on Y658 and bound to LGN.
Results and Discussion
Previous work from our lab used a VE-cadherin tension sensor (VEcadTS) in which the fluorescent tension sensor module [9] was inserted into the VEcad cytoplasmic domain just before the β-catenin binding site that anchors VEcad to the cytoskeleton [5] (Fig 1A). Tension across VEcad extends the elastic domain and reduces FRET efficiency in a predictable way, which allows calculation of the force across the molecule. We also made a control sensor in which the module was attached to the C-terminus. This construct localizes similarly but the module is always under zero tension, thus, has constitutively high FRET efficiency. Our previous work showed that under resting conditions, junctional VEcadTS had low FRET/high tension due to myosin-dependent contractility and that application of shear stress at 15 dynes/cm2, a level typical of human arteries, triggered a rapid increase in FRET indicating decreased tension [5].
Figure 1. VE-cadherin Tyr658 phosphorylation regulates EC shear responses.
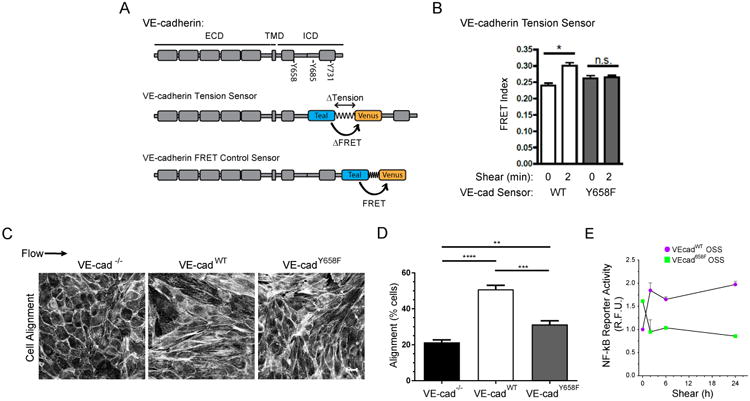
(A) VE-cadherin domains. VE-cadherin contains an extracellular domain (ECD), a transmembrane domain (TMD), and an intracellular domain (ICD) containing catenin binding sites and several tyrosines capable of phosphorylation. The FRET-based tension sensor module was inserted at the indicated sites in the active and control sensors. (B) BAECs expressing either VE-cadherin wild-type (WT) or Y658F tension sensors were exposed to 15dynes/cm2 shear stress for 2 min, at which time the change in FRET is maximal. Cells were fixed and junction FRET signals were measured as described in STAR Methods. Values are means ±SEM. n>20 junctions per condition. Similar results were obtained in an additional three experiments. * p ≤ 0.05. (C) VE-cadherin-/- cells stably expressing either WT or Y658F VE-cadherin were exposed to laminar shear stress at 15 dynes/cm2 for 24h. Cells were fixed, F-actin stained, and imaged. Scale bar = 20μm. (D) Quantification of (C). Percent of cells orientated within 23° relative to the axis of shear was determined. Values are means ± SD (n=3 experiments). ** p < 0.02, *** p < 0.001, **** p< 0.0001. (E) VE-cadherin-/- cells re-expressing WT or Y658F VE-cadherin plus a NF-κB response element-driven reporter were subjected to oscillatory shear stress (OSS). NF-κB activity was assayed by fluorimetry as described in STAR Methods. Three experiments gave similar results. See also Figure S1.
To address the possible role of SFK-dependent phosphorylation in shear stress mechanotransduction, we first carried out a preliminary experiment in which the ECs were pretreated with the SFK inhibitor PP2. This drug abolished the flow-induced change in FRET for VEcadTS (Fig S1), suggesting a requirement for tyrosine phosphorylation by an SFK. To test whether VE-cadherin was a direct target, we mutated the three principle candidate tyrosines within VEcadTS [7]. Measurement of FRET in the VEcadTS mutants at cell-cell junctions at its peak at 2 min showed that Y731F and Y685F mutations had no effect, whereas the Y658F mutation abolished the effect of flow on FRET efficiency (Figs 1B and S1B). When expressed in VE-cadherin null ECs, the Y658F mutant also impaired alignment in the direction of laminar flow (Fig 1C,D). As described below in more detail, this mutation also blocks flow activation of integrins and NF-κB signaling, which are downstream of the junctional complex (Figs 1E, 3B, C). The Y658 site is therefore critical for flow responses transduced through the junctional complex.
Fig. 3. VE-cadherin-p120ctn binding suppresses shear responses.
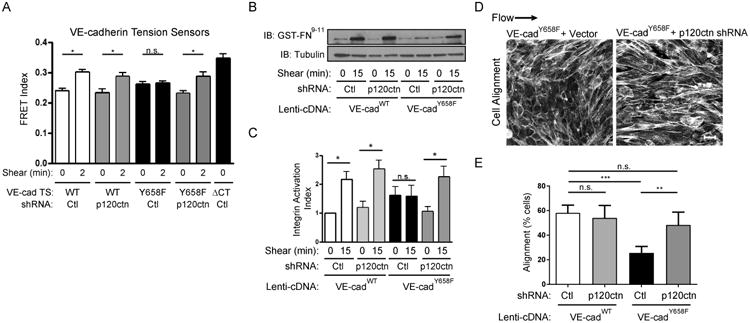
(A) BAECs expressing control or p120ctn shRNA and VE-cadherin tension sensors (WT, Y658F, or the ΔCT 0 tension/maximum FRET control) were subjected to 0 or 2min shear, and FRET measured. Values are means ±SEM. n>20 junctions per condition. Similar results were obtained in three independent experiments. * p ≤ 0.05. (B) VE-cadherin-/- cells expressing either WT or Y658F VE-cadherin plus either control or p120ctn shRNA were subjected to 0 or 15 min of shear; integrin activation was then measured by binding GST-FN9-11 and assaying by Western blotting, as described in STAR Methods. (C) Quantification of results in (B). Values are means ± SEM from 4 independent experiments. (D) VE-cadherin-/- cells expressing VE-cadherinY658F and control or p120ctn shRNA were exposed to 15dynes/cm2 of laminar shear stress for 24h, fixed and stained for F-actin. Scale bar = 20μm. (E) Images from (D) were quantified for cell alignment. Values are means ± SEM, n=4 For percent of cells orientated within 23° relative to the axis of shear. ** p ≤ 0.01, *** p ≤ 0.001. See also Figures S2.
Previous work showed that Y658 was maximally phosphorylated under fluid shear stress at ∼5 dynes/cm2, a level typical of veins, and was lower (though still detected) at the higher flow found in most regions of arteries; in agreement with this result, phosphorylation is higher in veins than in arteries in vivo [8]. However, atherosclerosis-susceptible regions of arteries are also under lower shear stress that is often oscillatory or multidirectional [1, 2]. In vitro application of flow to endothelial monolayers confirmed higher VEcad Y658 phosphorylation under oscillatory compared to high laminar flow (Fig 2A,B). Staining of aortic arches of WT C57Bl6 mice with a specific antibody to pY658 revealed higher phosphorylation in the athero-susceptible inner curvature where flow is low and oscillatory compared to the nearby outer curvature that is under high, laminar flow (Fig 2C,D).
Figure 2. VE-cadherin Y658 phosphorylation functions in vivo.
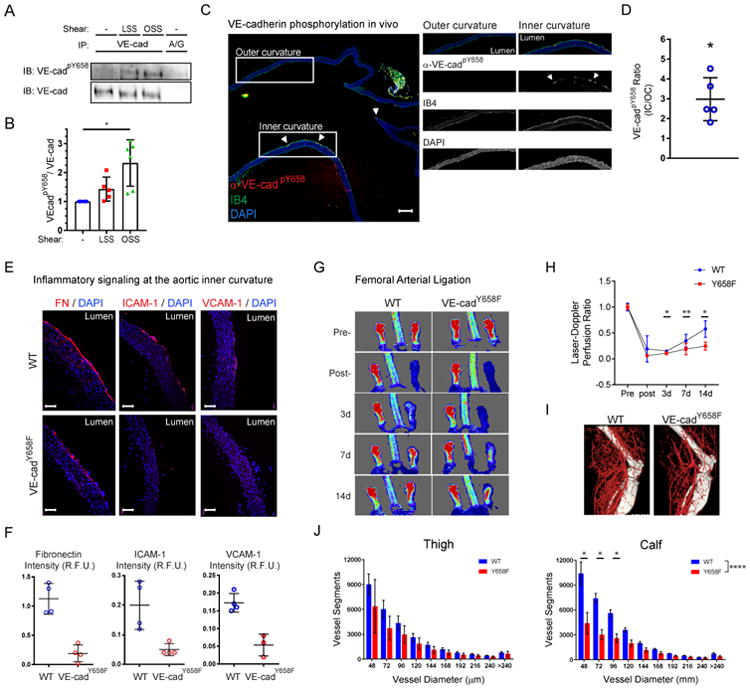
(A) Endothelial cells in vitro were exposed to no flow, laminar flow at 15 dynes/cm2 (LSS) or oscillatory flow at 1±3 dynes/cm2 (OSS) for 16h. VE-cadherin Y658 phosphorylation was then assayed by Western blotting as in STAR Methods. (B) quantification of (A). (C) Aortas from adult WT mice were sectioned longitudinally and stained with anti-VEcadpY658 and fluorescent Isolectin B4 (IB4) to label the endothelium. Scale bar = 200μm. (D) Quantification of (C). The ratio of endothelial inner curvature/outer curvature signal for each image was quantified (n=5 aortas). * p ≤ 0.05. (E) Aortas from 6-8wk WT or VE-cadherinY658F knock in mice were sectioned and immunostained as indicated. (F) Fluorescent signals from Inner curvature regions were quantified in 3-4 different aortas for each antibody. Scale bar = 100μm. * p ≤ 0.05. (G) WT or Y658F mice were subject to femoral artery ligation and blood flow in the hindlimb assayed by Doppler imaging on subsequent days. (H) Quantification of laser Doppler imaging results. * p ≤ 0.05, ** p ≤ 0.01. (I) MicroCT of vasculature in treated leg of WT and Y658F mice. (J) Quantification of vascular microCT from 4-5 mice. * p ≤ 0.001, **** p ≤ 0.0001 by ANOVA.
Low or oscillatory flow activates the inflammatory transcription factor NF-κB and induces expression of fibronectin and the leukocyte recruitment genes ICAM1 and VCAM1 [10, 11]. These events result in a “primed” state in which ECs are more sensitive to other inflammatory mediators such as oxidized LDL and cytokines that promote atherosclerosis [12]. The ability of Y658F VE-cadherin expressed in VE-cadherin-/- ECs to suppress oscillatory flow-induced NF-κB signaling in vitro (Fig 1E) prompted us to examine its effect in vivo in VE-cadherin Y658F knock in mice. The Y658F mutation essentially abolished expression of fibronectin, ICAM1 and VCAM1 in the inner curvature of the aortic arch mice (Fig 2E,F). To further examine the importance of Y658 phosphorylation in vivo, we examined physiological flow-dependent remodeling using a hindlimb ischemia model. In this system, ligation of the femoral artery triggers formation of collateral circulation in the thigh that is primarily driven by increased flow through vascular beds parallel to the blockage, restoring blood flow to the lower limb [13]. Whereas WT C57Bl6 mice showed substantial recovery of blood flow to the foot over the 2 week period of observation, VEcad Y658F mice responded poorly (Fig 2G,H). This effect correlated with decreased density of vessels by microCT (Fig 2I,J). Thus, VE-cadherin Y658 phosphorylation is required for flow signaling in vitro and in vivo.
VE-cadherin tyrosine 658 resides in the middle of the p120ctn binding region, where its phosphorylation inhibits p120ctn binding [14, 15]. We confirmed that a phosphomimetic mutation inhibits co-IP of VE-cadherin and p120ctn (Fig S2). We therefore considered whether phosphorylation of that site might affect flow signaling by inducing p120ctn dissociation. To test this idea, p120ctn was knocked down in cells expressing VEcadTS and its mutants. Remarkably, suppression of p120ctn restored the effect of flow on VE-cadherin tension (Fig 3A). Flow is known to trigger activation of integrins downstream of the junctional complex [4]; this pathway, assayed at its peak at around 15 min, was also inhibited in ECs that expressed Y658F VE-cadherin and rescued by p120ctn shRNA (Fig 3B,C). Lastly, p120 shRNA restored EC alignment in the direction of flow (Fig 3D,E). This was confirmed using CRISPR-Cas9 and a specific single guide RNA (sgRNA) against p120ctn (Fig S3A,B). Although p120ctn stabilizes cadherins on the plasma membrane by inhibiting endocytosis, the change in surface VE-cadherin was less that 2-fold and did not affect the function of WT VE-cadherin in our experiments. Together, these data raise the interesting possibility that Y658 phosphorylation is important in flow signaling because it inhibits p120ctn binding.
One possible explanation for these findings is that p120ctn inhibits the association of another protein that binds to the same region but is not inhibited by tyrosine phosphorylation. LGN has those characteristics [16]; we therefore examined LGN binding to VE-cadherin. LGN co-immunoprecipitated with VE-cadherin and the association was increased by p120ctn knockdown (Fig 4A). The VE-cadherin Y658F mutant bound LGN poorly but this effect was reversed by p120ctn depletion (Fig 4B). To test competition directly, over-expressed LGN in cell lysates was incubated with recombinant VE-cadherin cytoplasmic domain (Fig 4C) in the presence of increasing amounts of a second lysate containing over-expressed p120ctn. When binding to the immobilized VE-cadherin was assayed, p120ctn reduced LGN binding in a dose-dependent manner (Fig 4D). These results suggest that LGN binds preferentially to phosphorylated VE-cadherin. To test this idea, VE-cadherin was immunoprecipitated either directly or with antibodies to LGN or p120ctn. When these IPs were probed for pY658 VE-cadherin, this band was strongly enriched in the LGN immunoprecipitates and depleted in the p120 immunoprecipitates (Fig 4E). This result also indicates that only a small fraction of VE-cadherin is phosphorylated, despite its functional importance. Together, these results show that phosphorylation of Y658 leads to dissociation of p120ctn and binding of the polarity protein LGN to a small pool of VE-cadherin.
Figure 4. LGN competes with p120ctn for VE-cadherin binding and is important for shear mechanotransduction.
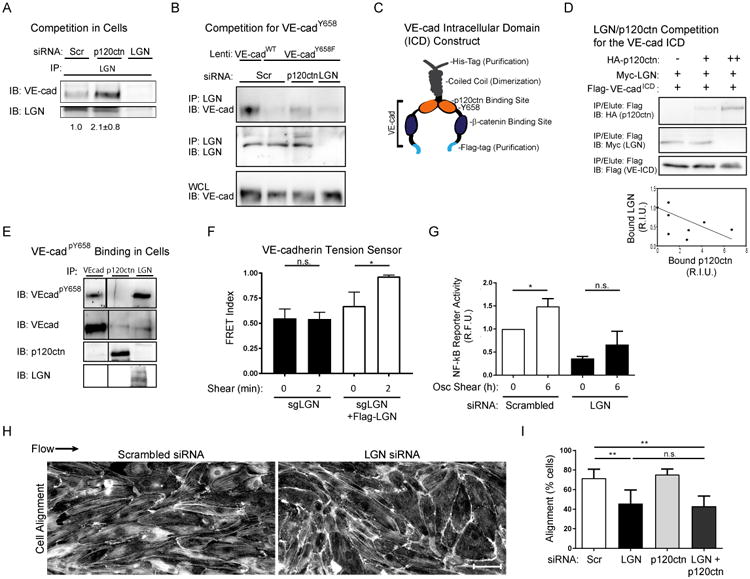
(A) HUVECs treated with either scrambled (scr), p120ctn, or LGN siRNAs were lysed and immunoprecipitated with either anti-LGN or control IgG. Immunoprecipitates were Western blotted for LGN and VE-cadherin. Three independent experiments gave similar results. (B) Cells were transfected with either scrambled control, anti-p120ctn, or anti-LGN siRNAs and additionally infected with either VE-cadherin WT or Y658F lentiviral constructs. LGN was immunoprecipitated and bound VE-cadherin was immunoblotted as in (A). Three independent experiments gave similar results. (C) Human VE-cadherin intracellular domain was fused to a C-terminal Flag, an N-terminal, coiled-coil dimerization domain and a His6 tag. (D) Recombinant VE-ICD-Flag protein on anti-Flag beads was incubated with lysates from cells over-expressing Myc-tagged LGN in the presence of variable amounts of cell lysate from control or HA-tagged p120ctn-expressing cells. Control lysate (-); LGN:p120ctn 1:5 (+); LGN:p120ctn 1:1 (++). Protein bound to VE-ICD-Flag resin was immunoblotted for HA, Myc, and Flag. The graph shows quantified results from 4 independent experiments. (E) HUVECs were lysed and immunoprecipitated for VE-cadherin, p120ctn, or LGN, then immunoblotted for VE-cadherin and anti-VE-cadpY658. Three independent experiments gave similar results. (F) HUVECs expressing the VE-cadWT tension sensor were treated with CRISPR cas9/LGN sgRNA to mutate LGN and then transduced with control virus or sgRNA-resistant Flag-tagged LGN. Cells were treated with or without shear stress for 2 min, and FRET measured. Values are means ± SEM, n>20 per condition. Similar results were obtained in two additional experiments. (G) HUVECs expressing the NF-κB reporter were transfected with scrambled control or anti-LGN siRNAs. Cells were treated with or without oscillatory shear stress for 6h, and lysates immunoblotted for the GFP reporter. Values are means ± SEM, n=3 independent experiments. (H) HUVECs treated with siRNA as in (G) were subjected to 12 dynes/cm2 laminar shear stress for 12-16h, then fixed and stained for F-actin. (I) Quantified cell alignment from 3 independent experiments. Values are means ± SEM, n=3 Scale bar = 20μm. * p ≤ 0.05. See also Figures S3.
To test directly its requirement in flow signaling, LGN was stably depleted from ECs using CRISPR-Cas9 and a specific single guide RNA (Fig S3C). VEcadTS was then expressed and FRET assayed. Deletion of LGN blocked the flow-induced change in tension on VE-cadherin, which was reversed by re-expression of LGN (Fig 4F). Depletion of LGN using siRNA also decreased the activation of NF-κB by flow (Fig 4G) and inhibited cell alignment in the direction of flow, which, unlike the VE-cadherin Y658F mutant, was not rescued by p120ctn siRNA (Fig 4H,I).
Taken together, these data demonstrate that phosphorylation of a small fraction of VE-cadherin on Y658 is required for flow signaling through the junctional complex. This effect is mediated by a mechanism involving dissociation of p120ctn and binding of LGN to the cadherin juxtamembrane region. Thus, a small pool of VE-cadherin bound to LGN mediates fluid shear stress sensing.
LGN is more widely recognized as a determinant of mitotic spindle orientation, especially during development [17, 18]. Its genetic deletion in mice leads to defects in brain development due to failure to orient the spindle during neurogenesis [19]. Its function in this pathway appears to be as an adapter protein in which the C-terminal GoLoCo motifs bind to Gαi in its free but GDP bound state, while the N-terminal TPR repeats bind to the microtubule binding protein NUMA and the Par3/Par6 partner inscuteable. The protein interactions that mediate its role in flow sensing are very likely distinct from those that participate in mitosis and remain to be elucidated. However, it is notable that the Par3/Par6 complex has been reported to associate with VE-cadherin and participate in flow signaling [20].
An unexpected implication of these results is that flow regulates VE-cadherin Y658 phosphorylation, which in turn regulates flow responses. These interactions create a feedback loop in which flow magnitude could then modulate endothelial cell responses to flow. This pathway might therefore play a role in tuning endothelial sensitivity to flow magnitude in different types of blood vessels that experience different flow magnitudes, providing a way to adjust the shear stress set point that governs vessel remodeling [21]. Further work will be required to determine the protein interactions and pathways that mediate LGN's role in flow signaling and vascular function in vivo.
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Martin A. Schwartz (Martin.Schwartz@yale.edu).
Experimental Model and Subject Details
The mice used in these studies were extensively backcrossed with the C57bl/6j background. Males between 6-14wk of age were used for experimentation. All experiments were approved by the Institutional Animal Care and Use Committee of Yale University.
BAEC cells (Coriell) were isolated from male adult bovine aortas. HUVEC cells from three donors were isolated at the Yale Vascular Biology and Therapeutics core and pooled. Mouse endothelial cells derived embryiod bodies were previously immortalized with polyoma middle T. All cells were maintained at 37°C and 5% CO2. Cells used in this study were not authenticated, but we observe that they show uniform expression of endothelial markers and appropriate responses to physiological shear stress. More precise culturing conditions are described in STAR Methods Details.
STAR Methods Details
Cell Culture and Reagents
VE-cadherin null embryoid body-derived endothelial cells (VEcad-/-) were maintained in DMEM, 20% FBS, 150mg/L ECGS, 100mg/L heparin, 1× Pen/Strep. HUVECs were obtained from the Yale tissue culture core, cultured in M199, 20% FBS, 150mg/L ECGS, 100mg/L heparin, 1× Pen/Strep, and used between passages 1-8. For FRET experiments, HUVEC were cultured and sheared in EGM media (Lonza). Figure 1B, 3A, Supp 1A and 1B, used bovine aortic endothelial cells (BAECs) obtained from the Coriell Institute and were maintained in DMEM, 20% FBS, 150mg/L ECGS, 100mg/L heparin, 1× Pen/Strep. Measurements were at 2 min because the change in FRET is maximal at that time [5].
RNAi and Crispr/Cas9
siRNA experiments were performed as described [6]. A psuper (Oligoengine) vector encoding shRNA against both mouse and bovine p120ctn (5′- GGAGCTATGAAGACATGAT-3′) was electroporated into mouse VEcad-/- cells or BAECs. For CRISPR/Cas9-mediated gene disruption experiments, sgRNAs against mouse p120ctn (sequence; 5′-CTTATGCTACCGCAATGACA-3′) or human LGN (sequence; 5′-GGTGGGAGAAGCAAGAGCACTTTACAATCT-3′) were cloned into LentiCrisprV2 (Addgene #52961) and produced as lentivirus in 293T cells co-transfected with psPAX2 and pVSVG. Three days after transduction in either mouse ECs (anti-mouse p120ctn) or HUVEC (anti-human LGN), infected cells were selected with 5 or 1μg/mL puromycin, respectively, for 3-5d. Experiments were performed within 1-4 passages.
Shear Stress
Shear stress experiments used a parallel plate flow chamber as described [6, 31]. Briefly, cells were seeded on slides coated with 10μg/mL bovine fibronectin. For alignment assays, cells were exposed to approximately 15dynes/cm2 laminar shear in complete media for 12h (HUVEC) or 24h (mouse ECs). Cells were fixed in 3.7% formaldehyde and stained with Alexa-phalloidin. Alignment within 45° of the axis of flow was quantified by fluorescence microscopy. Oscillatory shear stress was applied to cells at 1±3 dynes/cm2 at 1Hz frequency using syringe and peristaltic pumps for the indicated times. Cells were transduced with an NF-kB reporter as described [32] and GFP detected by Western blotting or fluorimetry (Synergy HT, BioTek), as indicated. Integrin activation was measured as described previously, using GST-FN9-11 which binds the high affinity conformation [6].
FRET Measurements
FRET were measured as described [5, 33]. Briefly, cells were fixed and imaged the same day using either a Zeiss 510 or 710 LSM confocal microscope with 458 and 514 nm excitation. Images were background subtracted in the donor, FRET and acceptor channels and saturated pixels were removed. Ratio images were normalized by dividing the FRET channel by the donor channel or acceptor channel.
Co-Immunoprecipitation
Co-immunoprecipitation assays were performed with HUVECs. Cells were transfected with siRNAs for 84-96h and extracted with 25mM Tris pH=7.4, 200mM NaCl, 1mM MgCl2, 1% TritonX-100, 0.01% SDS, 1.5× Complete EDTA-free Protease Inhibitor, and 1.5× PhosStop (Roche). Lysates were clarified at 20,000×g 4°C. 2μg of each antibody (rabbit anti-LGN from Sigma, goat anti-VE-cadherin C19 from Santa Cruz, and mouse anti-p120ctn from BD) pre-bound to Protein-A/G agarose (Santa Cruz), were added to the lysates, incubated at 4°C overnight, and beads washed 3 times with 4°C lysis buffer. For comparing VE-cadpY658 enrichment between LGN and p120ctn co-immunoprecipitates, the LGN IP required approximately 5× more lysate than the p120ctn IP. Bound protein was eluted with 2× protein sample buffer and subjected to SDS-PAGE. Western blots were probed with rabbit anti-LGN (Millipore), goat anti-VE-cadherin (Santa Cruz), mouse anti-p120ctn (BD), rabbit anti-phospho-VEcadY658 [16], and developed with Protein-A/G-HRP (Life Tech). For cells infected with VEcadWT or VEcadY658F constructs, lentiviruses encoding these cDNAs were transduced 18h before lysis and immunoprecipitated using anti-Flag beads (Sigma).
Pull-Down Assays
Recombinant proteins for pull-down experiments were produced by subcloning cDNAs encoding 162 amino acids of the complete human VEcadWT ICD, including an C-terminal Flag-tag, into pET15b, in frame with an N-terminal His6-tag and a coiled-coil domain which stably dimerizes [34]. Protein was induced in BL21 bacteria, purified with Ni++ NTA resin, and eluted with 200mM Imidizole in 25mM Tris pH=7.4, 200mM NaCl, 1mM MgCl2, 1% TritonX-100, and 1.5× protease inhibitors. In parallel, 293T cells transfected with pCDNA3.1 empty vector, HA-tagged p120ctn, or Myc-tagged LGN were lysed in the same buffer without the imidizole [35, 36]. Lysates were clarified at 20,000×g, 10min, at 4°C. The HA-p120ctn-transfected lysate was mixed with empty vector-transfected lysate at ratios of 1:1, 1:5, and 0:1. These was then mixed with an equal volume of Myc-LGN lysates plus 1μg of recombinant VEcadICD. Tubes were mixed for 2h at RT before addition of 5μL equilibrated Flag-bind beads for 30min. Beads were washed 3 times with ice cold lysis buffer and eluted with 2× protein sample buffer.
Animals and Immunostaining
For phospho-VEcadY658 staining, aortas from 6-8week C57bl/6 mice were perfusion fixed with 3.7% formaldehyde, cleared of periadventitial tissue, and further fixed overnight at 4°C. They were then embedded in paraffin and sectio ned longitudinally by the Yale Pathology Tissue Microarray facility. Sections were de-paraffinized and antigen retrieved with 0.5mM EDTA in a pressure cooker. Tissue sections were permeabilized with 0.2% TritonX-100 and blocked with Start-block (Thermo) before incubating with rabbit anti-phospho-VEcadY658 overnight at 4°C. Slides were labeled with DAPI, 488-conjugated isolectin B4, and Alexa647-conjugated donkey anti-rabbit. 10× images were collected on a Nikon 80i upright microscope. Diluted Alexa-647 was mounted on a glass slide and imaged to obtain a shade correction image. The mean fluorescence intensity of the inner curvature (IC) and outer curvature (OC) at a depth of about 15μm from the lumen, including the entire endothelial monolayer was measured by tracing in ImageJ. Background signal was measured within the lumen and subtracted. The ratio of IC/OC per mouse was calculated and graphed using PRSIM.
VE-cadherinY658F knock-in mice were created by the Vestweber lab using their recombination-mediated cassette exchange approach [37] and backcrossed 5 times into C57BL/6. For immune staining, aortas were dissected from 3-4 month old mice, embedded in OCT, and frozen-sectioned longitudinally (10-15μm). Staining was performed as described [38]. Briefly, cryosections were fixed in −20 °C acetone for 10 min. Sections were blocked in IHC-Tek antibody diluent for 1 h at room temperature, and were then incubated with primary antibodies at the indicated concentrations in IHC-Tek antibody diluent overnight at 4 °C. After washing 3 times in PBS, sections were inc ubated with Alexa Flour 598-conjugated Donkey anti rabbit/rat secondary antibody for 1 h at room temperature. After washing with PBS, sections were mounted in Vectashield with DAPI (Vector Laboratories) and images taken using a confocal microscope. Four male mice (3 month old) were used for each condition to monitor inflammatory markers on the inner curvature of the aortic arch.
Hindlimb Ischemia
For Hindlimb Ischemia, age matched (12- 14 weeks old mice) WT and VEcad Y658F knock-in mice were subjected to femoral artery ligation and analyzed by Laser Doppler imaging to measure blood flow in the hind limb and microCT to assess vessel remodeling as described [39]. Briefly, the femoral artery was ligated proximal of the femoral artery (underline of the inguinal ligament) and the superficial epigastric artery, and all branches between two sites. The arterial segment between the ligatures was then excised. Then for laser dopplar, the acquisition of flow images in the foot was performed using a Moor Infrared Laser Doppler Imager (LDI; Moor Instruments Ltd) at 37.4°C to 38.0°C under ketamine/xylazine (80/5 mg/kg) anesthe sia. The data were analyzed with Moor LDI image processing software V3.09 and reported as the ratio of flow in the right/left (R/L) hindlimb after background subtraction (values for dead mice).
Mice were then prepped for Micro-CT imaging. The vasculature was flushed with 0.9% normal saline containing heparin (1000 IU/L), papaverine (4 mg/L), and adenosine (1 g/L) for 3min before perfusion fixation for 5min with 2% PFA at 100mm Hg pressure. Contrast agent containing bismuth (0.5 ml) (Sigma-Aldrich Inc) in 5% gelatin was injected for 2min with a syringe pump. The mice were immersed in 2% PFA overnight at 4°C. The hindlimb vasculature was imaged with a high-resolution micro-CT imaging system (GE explore Locus SP), set to a 0.016-mm effective detector pixel size. Data were acquired in an axial mode, covering a volume of 2.0 cm in the z direction with a 1.04-cm field of view (covering a single hindlimb). Micro-CT was calibrated using standard wires of different sizes (10, 20, 30, 40, and 50 μm). During postprocessing of the image, a 40 000 gray-scale value was set as a threshold to eliminate bone with minimal sacrifice of vessel visualization. Microview software (GE Healthcare) was used to reconstruct three 2D maximum-intensity projection images (x, y, and z axes) from raw images. A volume of interest was reconstructed of the upper (500 pixels) and lower (250 pixels) hindlimb. Quantification was performed by use of a modified Image Pro-Plus 5.0 algorithm. The data are expressed as vascular segment number, representing total number of vessels, of specified diameter, counted in 500 z sections for thigh or 250 z sections for calf images.
Study design and data inclusion criteria
Each replicate shear stress experiment was performed with chambers assembled in series within a single station. FRET experiments were performed on cells selected for characteristic localization of VE-cadherin into adherens junctions. Shear-induced alignment experiments were considered successful when wild-type or reconstituted cells were 40-70% aligned. The shear-induced VEcadpY658 experiment described in Fig2a-b was tested for an outlier using PRISM (ROUT, Q=0.1%) and one laminar shear datapoint was excluded from Fig 2b. We pre-estimated a required sample size at about 3-6 experiments or 6-12 cells before performing experiments. We report summary results and variability from multiple independent experiments or, when necessary due to experimental variability, representative data from an individual experiment. Figure 4d displays normalized datapoints compiled from 4 experiments. We did not blind in vitro experiments.
Aortic VEcadpY658 experiments were performed using aortas that were previously shown to have characteristic distribution of the inflammatory markers VCAM-1 and Fibronectin within the inner curvature. Quantification of VEcadpY658 and inflammatory markers within the Y658F knockin mouse aortas was restricted to regions within the entire inner curvature and the outer curvature of the ascending aorta. Immunofluorescence experiments were not blinded. Hindlimb ischemia experiments (surgery, laser dopplar, and MicroCT) were performed blinded. All mice were male. No data were excluded from these experiments.
Quantification And Statistical Analysis
Statistics were analysed using Student's t-test or one-way ANOVA (multiple comparisons) as indicated in GraphPad Prism 6. Data are represented as means ± s.e.m. Statistical significance was taken as P < 0.05. All representative data were confirmed in three or more independent experiments.
Supplementary Material
Acknowledgments
This work was funded by the American Heart Association 16SDG27370007 (DEC) and 13POST16720007 (BGC); National Institutes of Health (NIH) GM119617 (DEC), RO1 HL75092 and PO1 HL107205 (MAS).
Footnotes
The authors declare no conflicts of interest.
Author Contributions: DEC, BGC, and MAS designed experiments and analyzed results. DEC, BGC, and PTA performed in vitro experimentation. BGC, MB, JZ and ZHZ performed animal experiments. FO, FW, ED, and DV prepared essential reagents. BGC and MAS prepared the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:2191–2198. doi: 10.1161/ATVBAHA.114.303422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conway DE, Schwartz MA. Flow-dependent cellular mechanotransduction in atherosclerosis. Journal of cell science. 2013;126:5101–5109. doi: 10.1242/jcs.138313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osawa M, Masuda M, Kusano K, Fujiwara K. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: is it a mechanoresponsive molecule? The Journal of cell biology. 2002;158:773–785. doi: 10.1083/jcb.200205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 5.Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Current biology : CB. 2013;23:1024–1030. doi: 10.1016/j.cub.2013.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coon BG, Baeyens N, Han J, Budatha M, Ross TD, Fang JS, Yun S, Thomas JL, Schwartz MA. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. The Journal of cell biology. 2015;208:975–986. doi: 10.1083/jcb.201408103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gavard J. Endothelial permeability and VE-cadherin: a wacky comradeship. Cell adhesion & migration. 2013;7:455–461. doi: 10.4161/cam.27330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nature communications. 2012;3:1208. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–264. doi: 10.1172/JCI10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10:53–62. doi: 10.1038/nrm2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaper W. Collateral circulation: past and present. Basic research in cardiology. 2009;104:5–21. doi: 10.1007/s00395-008-0760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatanaka K, Simons M, Murakami M. Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. American journal of physiology. Heart and circulatory physiology. 2011;300:H162–172. doi: 10.1152/ajpheart.00650.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 16.Gloerich M, Bianchini JM, Siemers KA, Cohen DJ, Nelson WJ. Cell division orientation is coupled to cell-cell adhesion by the E-cadherin/LGN complex. Nature communications. 2017;8:13996. doi: 10.1038/ncomms13996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lancaster MA, Knoblich JA. Spindle orientation in mammalian cerebral cortical development. Current opinion in neurobiology. 2012;22:737–746. doi: 10.1016/j.conb.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mapelli M, Gonzalez C. On the inscrutable role of Inscuteable: structural basis and functional implications for the competitive binding of NuMA and Inscuteable to LGN. Open biology. 2012;2:120102. doi: 10.1098/rsob.120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konno D, Shioi G, Shitamukai A, Mori A, Kiyonari H, Miyata T, Matsuzaki F. Neuroepithelial progenitors undergo LGN-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nature cell biology. 2008;10:93–101. doi: 10.1038/ncb1673. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Collins C, Kiosses WB, Murray AM, Joshi M, Shepherd TR, Fuentes EJ, Tzima E. A novel pathway spatiotemporally activates Rac1 and redox signaling in response to fluid shear stress. The Journal of cell biology. 2013;201:863–873. doi: 10.1083/jcb.201207115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baeyens N, Nicoli S, Coon BG, Ross TD, Van den Dries K, Han J, Lauridsen HM, Mejean CO, Eichmann A, Thomas JL, et al. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set point. eLife. 2015;4 doi: 10.7554/eLife.04645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frangos JA, Eskin SG, McIntire LV, Ives CL. Flow effects on prostacyclin production by cultured human endothelial cells. Science. 1985;227:1477–1479. doi: 10.1126/science.3883488. [DOI] [PubMed] [Google Scholar]
- 23.Wieder KJ, King KR, Thompson DM, Zia C, Yarmush ML, Jayaraman A. Optimization of reporter cells for expression profiling in a microfluidic device. Biomedical microdevices. 2005;7:213–222. doi: 10.1007/s10544-005-3028-3. [DOI] [PubMed] [Google Scholar]
- 24.Arsenovic PT, Ramachandran I, Bathula K, Zhu R, Narang JD, Noll NA, Lemmon CA, Gundersen GG, Conway DE. Nesprin-2G, a Component of the Nuclear LINC Complex, Is Subject to Myosin-Dependent Tension. Biophysical journal. 2016;110:34–43. doi: 10.1016/j.bpj.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfaff M, Liu S, Erle DJ, Ginsberg MH. Integrin beta cytoplasmic domains differentially bind to cytoskeletal proteins. The Journal of biological chemistry. 1998;273:6104–6109. doi: 10.1074/jbc.273.11.6104. [DOI] [PubMed] [Google Scholar]
- 26.Ouyang M, Lu S, Kim T, Chen CE, Seong J, Leckband DE, Wang F, Reynolds AB, Schwartz MA, Wang Y. N-cadherin regulates spatially polarized signals through distinct p120ctn and beta-catenin-dependent signalling pathways. Nature communications. 2013;4:1589. doi: 10.1038/ncomms2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du Q, Taylor L, Compton DA, Macara IG. LGN blocks the ability of NuMA to bind and stabilize microtubules. A mechanism for mitotic spindle assembly regulation. Current biology : CB. 2002;12:1928–1933. doi: 10.1016/s0960-9822(02)01298-8. [DOI] [PubMed] [Google Scholar]
- 28.Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF, et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. The Journal of experimental medicine. 2011;208:2393–2401. doi: 10.1084/jem.20110525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yun S, Budatha M, Dahlman JE, Coon BG, Cameron RT, Langer R, Anderson DG, Baillie G, Schwartz MA. Interaction between integrin alpha5 and PDE4D regulates endothelial inflammatory signalling. Nature cell biology. 2016;18:1043–1053. doi: 10.1038/ncb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tirziu D, Jaba IM, Yu P, Larrivee B, Coon BG, Cristofaro B, Zhuang ZW, Lanahan AA, Schwartz MA, Eichmann A, et al. Endothelial nuclear factor-kappaB-dependent regulation of arteriogenesis and branching. Circulation. 2012;126:2589–2600. doi: 10.1161/CIRCULATIONAHA.112.119321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.