

SUMMARY
Classical lissencephaly is a genetic neurological disorder associated with mental retardation and intractable epilepsy, and Miller Dieker Syndrome (MDS) is the most severe form of the disease. In this study, to investigate effects of MDS on human progenitor subtypes that control neuronal output and influence brain topology, we analyzed cerebral organoids derived from control and MDS induced pluripotent stem cells (iPSCs) using timelapse imaging, immunostaining, and single cell RNA sequencing. We saw a cell migration defect that was rescued when we corrected the MDS causative chromosomal deletion, and severe apoptosis of the founder neuroepithelial stem cells accompanied by increased horizontal cell divisions. We also identified a mitotic defect in outer radial glia, a progenitor subtype that is largely absent from lissencephalic rodents but critical for human neocortical expansion. Our study therefore deepens understanding of MDS cellular pathogenesis and highlights the broad utility of cerebral organoids for modeling human neurodevelopmental disorders.
eTOC summary
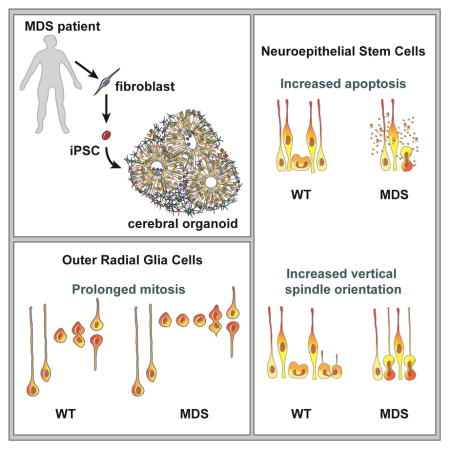
Bershteyn and colleagues show that cerebral organoid modeling of lissencephaly using iPSCs derived from Miller Dieker Syndrome patients can characterize cellular and neurodevelopmental disease phenotypes, and identify a mitotic defect in outer radial glia, a cell type that is particularly important for human cortical development.
INTRODUCTION
Human cerebral cortex develops from a pseudostratified layer of neuroepithelial stem cells (NESC) into a functionally complex six-layered structure with a folded (gyrencephalic) surface. The molecular underpinnings of brain size and topology are encoded by the genome and distinguish us from species with a small and smooth (lissencephalic) brain surface such as mice. Although brain folding in the human does not begin until the end of the second trimester (after gestation week 23 (GW23)) (Chi et al., 1977; Martin et al., 1988; Hansen et al., 1993; Armstrong et al., 1995), many of the key cellular events that influence this process, including expansion of the progenitor population and neuronal migration, occur starting around GW4 (Lui et al., 2011; Sidman, Rakic 1973; Stiles, Jernigan 2010). Genetic and infectious diseases that disrupt these processes underlie a number of cortical malformations and cause mental retardation, mortality, and morbidity (Guerrini, Dobyns 2014; Hu et al., 2014). Despite the prevalence and societal burden of cortical malformations, our understanding of how disease-linked mutations disrupt brain development is still limited.
Miller Dieker Syndrome (MDS) is a severe cortical malformation characterized by nearly absent cortical folding (lissencephaly) often associated with reduced brain size (microcephaly), craniofacial dysmorphisms, mental retardation, and intractable epilepsy (Dobyns et al., 1983; Dobyns et al., 1991; Nagamani et al., 2009). MDS is caused by large heterozygous deletions of human band 17p13.3, harboring dozens of genes, including PAFAH1B1 (LIS1 protein) and YWHAE (14-3-3ε protein) (Dobyns et al., 1983; Reiner et al., 1993; Hattori et al., 1994; Chong et al., 1997; Cardoso et al., 2003). Smaller deletions or mutations in PAFAH1B1 are the major cause of Isolated Lissencephaly Sequence (ILS), which exhibits less severe degrees of lissencephaly (Ledbetter et al., 1992; Lo Nigro et al., 1997; Pilz et al., 1998; Barkovich et al., 1991; Cardoso et al., 2003). Insight into lissencephaly pathogenesis is largely derived from mouse models and limited analyses of postmortem human brain. Reduction in LIS1 levels in Pafah1b1 mutant mice leads to defects in neuronal migration (Hirotsune et al., 1998; Smith et al., 2000), consistent with disrupted cortical layering and neuron dispersion seen in postmortem MDS brain (Sheen et al., 2006b; Saito et al., 2011). LIS1 is an atypical microtubule associated protein that regulates microtubule dynamics and nuclear-centrosomal coupling during neuronal migration (Borrell et al., 2000; Gambello et al., 2003; Shu et al., 2004; Tanaka et al., 2004; Youn et al., 2009). Collectively, these studies led to the prevailing model that lissencephaly is due to defective neuronal migration (Kato et al., 2003). However, the mouse brain is naturally lissencephalic, suggesting that certain aspects of cortical development may not be adequately assessed in mice.
Recent work has uncovered critical cellular and molecular differences between cortical development in humans and mice, further underscoring the need to develop human model systems. In the developing human cortex, the outer subventricular zone (OSVZ) is greatly expanded (Smart et al., 2002; Lukaszewicz et al., 2005). OSVZ progenitors, which include transit amplifying intermediate progenitor (IP) cells and outer or basal radial glia (oRG/bRG) (Hansen et al., 2010; Fietz et al., 2010; Betizeau et al., 2013), have been proposed to contribute to the majority of upper layer neurogenesis (Smart et al., 2002; Lukaszewicz et al., 2005). While IP cells are conserved between humans and mice, oRG cells are largely absent from the developing cortices of lissencephalic rodents (Shitamukai et al., 2011; Wang et al., 2011), which may explain why the phenotypes in Pafah1b1+/− mice are substantially milder than in human patients with heterozygous PAFAH1B1 mutations. Multiple lines of evidence suggest that the high abundance and proliferative capacity of oRG cells are critical for the vast developmental and evolutionary increase in cortical size (Stahl et al., 2013; Reillo et al., 2011).
To bridge the gap between mouse models and human disease, patient-derived induced pluripotent stem cells (iPSCs) (Takahashi et al., 2007; Yu et al., 2007; Park et al., 2008) represent a promising approach to study disease pathogenesis in a relevant genetic and cellular context. Human iPSCs provide a renewable source of neuronal progenitors and neurons, can be genetically and pharmacologically manipulated, and can recapitulate key cellular processes of in vivo brain development (Eiraku et al., 2008; Gaspard et al., 2008). Studies have shown that cortical-like oRG cells can be produced from human, but not mouse iPSCs (Shi et al., 2012; Kadoshima et al., 2013; Lancaster et al., 2013; Qian et al., 2016), highlighting the advantage of the species-specific cellular model systems. However, the extent to which in vitro-derived oRG cells recapitulate the functional properties and molecular identity of primary oRG cells has not been determined. Here, we implement a 3-D cerebral organoid culture method (Kadoshima et al., 2013) to analyze the impact of MDS lissencephaly mutations on distinct biological processes and cell types corresponding to the first trimester of cortical development. Using a multifaceted approach integrating histology, live cell imaging and single-cell transcriptomics, we uncover cell-type-specific defects in lissencephaly spanning from stages of neuroepithelial cell expansion, to neuronal migration, to the mitotic properties of oRG progenitors, providing deeper insight into human cortical development and lissencephaly.
RESULTS
Increased apoptosis of neuroepithelial stem cells (NESCs) in Miller-Dieker Syndrome (MDS) cortical organoids
To study MDS pathogenesis, we generated iPSCs from three MDS individuals (Figures 1A–C) using non-integrating episomal reprogramming vectors (Okita et al., 2011; Bershteyn et al., 2014). From each individual, we chose two independent iPSC clones with confirmed pluripotency, normal karyotype and stable maintenance of the 17p13.3 deletion for subsequent studies (Bershteyn et al., 2014 and data not shown). To assess the impact of MDS on NESCs, we implemented a published suspension culture method, whereby equal numbers of dissociated iPSCs are quickly re-aggregated in serum-free cortical differentiation medium (Kadoshima et al., 2013). Initially and on day 3, there were no significant differences in the size of WT and MDS aggregates (Figures S1A, S1C). However, by 2.5 weeks of suspension culture (day 18±1), the MDS organoids were significantly smaller (Figure 1D and Figures S1B–D). We observed that 25% of MDS organoids (N=3 individuals, 2 iPSC clones each) were very small (less than 0.5mm in circumference, Figure 1D) and invariably disintegrated before they could be analyzed. Of note, organoids derived from both of the MDS4 clones exhibited particularly poor growth in suspension and were therefore omitted from most of the subsequent assays.
Figure 1. Apoptosis of NESCs in Miller-Dieker Syndrome (MDS) organoids (see also Figures S1, S2 and Table S2).

(A) Schematic and coordinates of the deletions (red lines) on chromosome 17 in MDS cells used in this study. YWHAE and PAFAH1B1 genes delineate the minimal critical deletion that causes MDS. (B) Quantitative real-time PCR (qRT-PCR) analysis of PAFAH1B1 expression in the WT and MDS fibroblasts that were used to make iPSCs. Average values ± S.D. for each individual are shown. (C) Representative images of MDS iPSCs grown on Matrigel. Scale bar=200μm. (D) Analysis of organoid sizes at 2.5 weeks of differentiation representing an average ± S.E.M. from WT (n=4 independent iPSC clones) and MDS (n=6 iPSC clones) (also see table in Experimental Procedures). (E) Representative images of organoid sections after 5 weeks of differentiation. Scale bar=100μm. (F) Representative images of VZ-like regions in 5 week organoids immunostained with apoptotic marker cleaved-CASPASE-3 and proliferation marker Ki67. Scale bar=100μm. (G/H) Percent of SOX2+ cells in VZ-like regions expressing cleaved CASP3 (G) and Ki67 (H). Average values ± S.E.M. for WT (n=3) and MDS (n=3) are plotted (also see table in Experimental Procedures). Statistical analysis was done using t-test (p=0.04).
The larger organoids were fixed and sectioned after 5 weeks of differentiation for further characterization. At this time point, the organoids consisted of NESCs organized into multiple pseudo-stratified VZ-like progenitor regions expressing SOX2 (Figures 1E). Cortical patterning was verified by co-expression of the dorsal telencephalon progenitor marker PAX6 (Georgala et al., 2011) in the VZ-like regions, which were surrounded by DCX-positive neurons also expressing the deep layer subcortical projection neuron marker CTIP2 (Molyneaux et al., 2005; Leone et al., 2008) (Figure S2). Consistent with reduced organoid size, we found markedly increased apoptosis in the MDS cortical VZ-like regions, while marker of proliferation was not significantly altered (Figures 1F–H). Interestingly, while acute removal of Pafah1b1 in the mouse telencephalon at E8–8.5 leads to NESC apoptosis, genetic Pafah1b1 heterozygosity does not exhibit a strong phenotype in the mouse neuroepithelium (Hirotsune et al., 1998; Yingling et al., 2008). Our results suggest that the human genetic model may be more sensitive in recapitulating lissencephaly severity and highlight an early defect in the founder population of neuroepithelial stem cells independent of neurogenesis or neuronal migration.
Increased incidence of horizontal cleavage angles in MDS neuroepithelium at 5 weeks
NESCs undergo interkinetic nuclear migration (INM) coordinately with the cell cycle and expand through symmetrical divisions with vertically oriented cleavage planes (60–90 degrees relative to the ventricular surface) at the apical VZ surface (Gotz and Huttner, 2005). These cell intrinsic properties, including apical-basal polarity, dynamic behavior, and precise regulation of mitotic spindle orientation, are recapitulated in vitro in cortical organoids (Movie S1) (Kadoshima et al., 2013). We consistently found that both WT and MDS SOX1 and SOX2 NESC nuclei were distributed throughout the apical-basal VZ axis in a pseudo-stratified fashion, with mitotic cells expressing phospho-Vimentin and phospho-Histone H3 (Figures 2A, 2B), located at the apical surface. Other apical determinants, including PKC, N-Cadherin and Pericentrin, which marks the centrosomes, were generally localized to the apical surface in both genotypes (Figures 2A, B, C). Notably, we sometimes observed a less-organized cellular structure and a discontinuous VZ surface in MDS organoids (right panel in Figure 2B).
Figure 2. Increased incidence of horizontal cleavage angles in MDS VZ-like regions at 5 weeks (see also Movies S1, S2 and Table S2).

(A, B) Representative images of VZ-like regions in WT and MDS. Scale bar=100μm. (C) Frames from time-lapse imaging showing examples of vertical (top) and horizontal (bottom) divisions. Scale bar=20μm. White arrowheads mark the parent cell, red arrowheads mark the progeny. The apical surface and the axis of the cleavage plane are demarcated with dotted white lines. (D) Examples of vertical and horizontal divisions observed in VZ-like regions of fixed organoids. Anaphase mitoses were identified by chromosomal position and morphology, only anaphase cells were considered for analysis of the cleavage angle. (E) Schematic of how cleavage angles were measured from time-lapse and immunostaining data. (F–H) Quantification of cleavage angle data. Collectively, 70–92 dividing cells were analyzed from each of WT (n=2) and MDS (n=2) individuals. The red line in (G) represents the mean cleavage angle. (H) Average cleavage angles ± S.D for each individual are shown. Statistical analysis was done using t-test (p=0.0158).
LIS1 and ASPM (a microcephaly causative gene) were shown to regulate mitotic spindle orientation in the developing mouse forebrain, with loss of function leading to increased horizontal divisions associated with premature neurogenesis (Fish et al., 2006; Yingling et al., 2008; Pawlisz et al., 2008; Pramparo et al., 2010; Xie et al., 2013). To determine whether a similar defect is present in human lissencephaly, we analyzed mitotic cells in VZ-like regions of week five cortical organoids using time-lapse imaging (Figure 2C) and immunostaining (Figure 2D). In WT organoids, the majority of cleavage planes in the VZ were perpendicular to the apical surface, with an average angle of ~65° (Figures 2E–H). In contrast, dividing cells in the VZ regions of MDS organoids displayed more frequent horizontal cleavage planes, and a significantly reduced average cleavage angle of ~46° (Figures 2E–H, Movie S2). Collectively, our analyses suggest that expansion of the founder NESCs is greatly diminished in MDS due to increased apoptosis and decreased vertical divisions.
Defective neuronal migration in MDS organoids at 5 weeks
Defective radial migration of cortical neurons is a well-established developmental phenotype in lissencephaly (Hirotsune et al., 1998; Gambello et al., 2003; Youn et al., 2009; Toyo-oka et al., 2003; Sheen et al., 2006b; Saito et al., 2011). However, direct visualization and analysis of radial migration of human lissencephalic neurons have not been demonstrated. Thus, we sought to develop functional assays to analyze radial migration using the human organoid model system. Early born subcortical projection neurons expressing CTIP2 and DCX were present by five weeks of differentiation (Figures S2 and 3A). Within a day of attachment to a Matrigel-coated surface, numerous processes began extending from the organoids. These processes were marked by expression of neuronal proteins DCX and TUJ1, as well as radial glia proteins BLBP and NESTIN (Figure S3), suggesting that they belong to bundles of neurites and radial glia fibers. The abundance and lengths of processes were comparable between WT and MDS organoids (Figures 3C, Figures S3A and S4D). Over the next 48 hours, DCX-positive neurons were observed migrating out of the organoids along these processes (Figures 3B,C, Figures S3, S4D and Movie S3), akin to radial migration. We measured the displacement of neurons on fibers from the edge of the organoid three days after attachment to Matrigel. Migration of WT neurons was very robust, with more than 55% of migrating cells reaching 100–800μm from the edge of the organoids (Figures 3C–E and Figure S2A). In contrast, fewer MDS neurons migrated out of the organoids despite abundant processes, and the majority (65%) of migrating neurons were within 100μm from the edge (Figures 3C–E).
Figure 3. Defective neuronal migration in MDS is rescued by compensatory duplication of wild type chromosome 17 (see also Figures S3, S4 and Movies S3, S4 and Table S2).

(A) Representative images of WT and MDS cortical organoids after 5 weeks of differentiation. Scale bar=100μm. (B/C) Immunostaining of cells migrating on processes three days after intact organoids were attached to Matrigel. (B) Some of the processes seen in phase are not marked by DCX, suggesting that these may be radial glia fibers (see also Figure S3). (D) Quantification of cell displacement along the processes on day 3. On the y-axis is distance from the edge of the organoid (μm), on the x-axis is fraction of the total migrating cells at the corresponding distance bin. Migration assays were performed with WT (n=3), MDS (n=3) and MDS rescue (n=1) individuals. Data from the 3rd MDS individual is not represented in this quantification because there were very few (almost no) migrating cells (data not shown). (E) Summary of cell distribution across 200μm bins of displacement. (F) Representative tracks of speed over time for three different WT, MDS and MDS rescue cells. (G) Average migration speed. (H) Average track straightness. Statistical significance was determined by one way ANOVA followed by Tukey’s multiple comparison test. (I) Frames from time-lapse imaging of an MDS sample showing two cells migrating on processes. The top cell exhibits salutatory migration pattern of rounding up followed by leading process extension, while the bottom cell rounds up but fails to continue migration.
In addition to end point analyses, we tracked migrating neurons in real time using live imaging starting one day after organoid attachment for up to 48 hours. WT neurons migrated in a characteristic saltatory fashion (Figure 3F) with an average speed of 20μm/hr (Figure 3G, Movie S3), comparable to what has been recorded during radial neuronal migration in ferret cortical explants (Gertz, Kriegstein 2015). In addition, WT neurons nearly always migrated away from the organoid along the processes. As a result, the calculated total length of the migratory track divided by the net displacement, or track straightness, was close to 1 for WT neurons (Figure 3H). In contrast, many of the MDS neurons initiated but did not maintain saltatory migration on processes, spending much of the time tumbling in place or not moving (Figures 3F,G, Movie S4), which significantly reduced average migration speed (13μm/hr, p=0.0004) and track straightness (Figures 3G–I). In addition, migrating MDS neurons had less elongated nuclei than WT neurons (Movie S4), consistent with reduced tension during nucleokinesis (Tanaka et al., 2004).
LIS1 and 14-3-3ε proteins are part of a complex with cytoplasmic dynein that regulates neuronal migration (Toyo-oka et al., 2003). The corresponding genes, PAFAH1B1 and YWHAE, are several megabases apart on locus 17p13.3 (Figure 1A), and would be challenging to rescue in MDS. However, we previously reported a reprogramming-induced rescue of an MDS case, in which the 5.7Mbp deletion affecting locus 17p13.3 was in a ring chromosome (Bershteyn et al., 2014). Upon reprogramming fibroblasts from this individual (MDS1), the ring was lost and replaced by a second copy of the wild type chromosome 17 through compensatory uniparental isodisomy. As a consequence, the whole deletion was rescued in MDS1 iPSCs, and we showed that LIS1 and 14-3-3ε proteins were restored to WT levels (Bershteyn et al., 2014). Remarkably, when we performed endpoint and live imaging-based migration assays with organoids from these iPSCs, we observed that MDS1 neurons were indistinguishable from WT, completely rescuing neuronal displacement, migration speed, and track straightness (Figures 3C–H, Movie S4).
Finally, to ask whether migration defects in MDS neurons are cell autonomous, we developed a new assay using co-culture of organoids with human cortical tissue explants. Specifically, week 5 organoids were infected with non-replicating adeno-associated virus expressing td-Tomato fluorescent protein under the control of the CAG promoter, (AAV1-cag-Tomato), which preferentially labels neurons (Figure S4A–D). About 60% of cells labeled with this virus expressed the neuronal marker CTIP2 and about 25% of the labeled cells expressed the progenitor marker SOX1, in both WT and MDS organoids (Figures S4B, C). Separately, slices of human cortical tissue (GW18.5) were infected with a cmv-GFP adenovirus and then placed tightly against the organoids to allow attachment of the differentially labeled tissues. The human cortical tissue thus provided a physiological substrate for migration of iPSC-derived neurons (Figures S4E). After 4 days of co-culture, Tomato-expressing cells from the organoids were found throughout the cortical tissue explants, and their position relative to the ventricular and pial surfaces was quantified. Fifty percent of the WT iPSC-derived neurons that migrated had reached the pial surface after four days (Figure S4G). In contrast, only about 10% of the MDS iPSC-derived neurons that migrated had reached the pial surface on the cortical tissue explants (Figure S4G), suggesting that MDS migration defects are cell autonomous. Collectively, we established two novel in vitro migration assays that are sensitive to defects associated with human lissencephaly, and can be used in vitro to study multiple dynamic aspects of neuronal migration. Moreover, we demonstrated that this major developmental phenotype in MDS is functionally rescued through the duplication of the wild type homologue of chromosome 17, confirming that the migration defects were due to the heterozygous deletion.
Normal lamination with increased abundance of deep-layer neurons in MDS organoids at 10 weeks
While the long-term consequence of increased horizontal divisions is expected to be a reduction in total neuronal output through depletion of the progenitor pool (Pawlisz et al., 2008), the short-term consequence might be the opposite, i.e. overproduction of neurons. To examine this, we analyzed the distribution of progenitor cells and neurons in 10-week cortical organoids. At this time point, staining with the radial glia (RG) marker PAX6, the intermediate progenitor (IP) cell marker TBR2/EOMES (Hevner et al., 2006), and the deep-layer cortical neuron marker CTIP2, revealed three distinct regions resembling the developing VZ, SVZ, and cortical plate (CP). The boundaries between these regions could be consistently demarcated in WT and MDS organoids based on cell density, cell orientation, and expression of the three transcription factors. Thus, the VZ was defined based on dense vertical columns of PAX6-positive RG cells (Figure 4A). The SVZ was defined based on less-dense horizontally oriented cells, many of which expressed TBR2 (Figure 4A). Immediately superficial to the TBR2 boundary was the rudimentary CP, with strong expression of CTIP2 (Figure 4A). These well-organized structures were found close to the surface in every cortical organoid (typically 1–5 per organoid), with neurons always found on the outside, superficial to the progenitors.
Figure 4. Increased abundance of CTIP2-positive neurons in MDS organoids at 10 weeks (see also Table S2).

(A) Representative images of WT (1323), MDS2 and MDS3 cortical organoids. Scale bar=100μm. The inferred VZ-, SVZ- and CP-like regions are delineated based on the combination of DAPI, PAX6, TBR2 and CTIP2 staining patterns. White arrows in the MDS TBR2 and PAX6/TBR2 panels point to IP cells located in the apical portions of the VZ. (B–F) Analysis of marker distribution across the VZ/SVZ/CP (B–D) and cell composition (E,F) within the organized regions in WT (12 total organoids from 3 individuals, 4 iPSC clones) and MDS (13 organoids from 2 individuals, 3 iPSC clones) organoids. Average value ± S.E.M. is plotted after collapsing technical replicates from each clone (WT n=4, MDS n=3). Two way ANOVA followed by Bonferroni’s adjustment for multiple comparisons was performed to determined statistical significance. For cell distribution (B–C) the only significant source of variation was region. For cell composition, there was a significant difference between WT and MDS in the proportion of PAX6+TBR2− cells and CTIP2+ cells.
We analyzed the distribution of the PAX6, TBR2 and CTIP2-exressing cell populations in at least 12 organoids from WT (n=3) and MDS (n=2) individuals generated in multiple independent differentiations. The distribution of PAX6, TBR2 and CTIP2 was not significantly different between the two genotypes, with the majority of PAX6+ cells (~60%) found in the VZ, the majority of TBR2+ cells (~55%) in the SVZ, and the majority of CTIP2+ cells (~70%) in the CP, as expected (Figures 4B–D), indicating that the general laminar distribution of cell types is preserved. However, in MDS organoids, particularly in the more severe MDS3 case, there was a notable increase in the abundance of IP cells located in the apical portions of the VZ (arrows in Figure 4A). In addition, analysis of relative cell proportions in the organized cortical regions revealed a slight but significant decrease in PAX6+/TBR2− RG cells with a complementary increase in CTIP2+ neurons in MDS (Figures 4A, 4E, 4F). These findings are consistent with the increase in horizontal divisions that we observed at earlier time points in MDS organoids and indicate an overproduction of deep-layer neurons. Moreover these data indicate that migration defects are unlikely a secondary consequence of neuronal deficiency, as no such deficiency was observed up to this point.
Histological and molecular evidence of oRG-like cell production in WT and MDS organoids after 10 weeks
By 10 weeks, close to 30% of PAX6-positive cells within organized structures were located in the SVZ-like regions (Figures 4A, B) and expressed other canonical radial glia genes such as SOX2 and SOX1 (Figure 5A and data not shown). We sought to determine whether some of these cells might be oRGs. In contrast to the ventricular or apical radial glia (vRG/aRG), which extend bipolar processes and attach to the ventricular surface (Florio, Huttner 2014), oRG cells reside in the SVZ and are typically unipolar, extending a single basal process towards the pial surface (Hansen et al., 2010; Fietz et al., 2010). After 10–15 weeks of culture, the outer edge of the organoids contained many radially oriented cells with unipolar processes pointing away from the center of the organoids (white arrows in Figures 5B and Figure S5A). Many of these unipolar cells expressed the radial glia marker SOX2 (Figure 5C and Figure S5A), consistent with oRG identity. Within the same regions, IP-like cells with characteristic multipolar morphologies and expression of TBR2/EOMES could be clearly distinguished from oRG-like cells (blue arrow in Figure S5A).
Figure 5. Production of oRG cells in organoids after 10 weeks (see also Figures S5, S6 and Tables S1, S2 and dbGaP: phs000989.v3.p1).

(A) Representative images of WT and MDS progenitor zones in 10 week organoids. (B) GFP labeling with adenovirus reveals unipolar morphology and radial orientation of cells close to the edge of the organoids. Scale bar=100μm. (C) Immunostaining confirms radial glia fate of unipolar cells that express SOX2 and not the IP cell marker TBR2. (D) Heat map shows relative gene expression levels across 95 single radial glia cells captured from cerebral organoid samples (n=5 individuals from both WT and MDS). Genes represent canonical markers of radial glia (green bar) and forebrain identity (brown bar), as well as genes correlated with oRG identity (light and dark orange, the dark shade highlighting genes validated in vivo) (Pollen et al., 2015). The expression of oRG-correlated genes increases with age of the organoid (also see Supplementary Figure 5). (E) Violin plots representing distribution of the average expression of oRG marker genes across single cells captured from cerebral organoids at each stage of differentiation. (F) Validation of oRG markers’ (PTPRZ1 and TNC) co-expression with radial glia marker SOX2 in 10 week organoids. Note the similarity in the staining pattern between human OSVZ (GW17.3) and iPSC-derived organoids. (G) Network maps representing Pearson’s correlation between oRG marker genes across single cells in primary human brain tissue (Pollen et al., 2015) (left), and across cells captured from cerebral organoids (right). Correlations greater than 0.25 are highlighted with a pale green line, and correlations greater than 0.5 are highlighted with a dark green line. Genes that are most highly correlated across in vitro cells out of the oRG marker genes are highlighted in orange.
A recent study used transcriptome wide profiling of single cells to show that stem cell-derived cerebral organoids can recapitulate many of the molecular pathways that control normal human cortical neurogenesis (Camp et al., 2015). We reasoned that a subset of radial glia derived in vitro may adopt the molecular profile of oRG cells. Therefore, we analyzed single cell gene expression from WT and MDS organoids after 5, 10, and 15 weeks of differentiation. We identified a group of 95 cortical radial glia-like cells (Figure 5D) out of all the in vitro-derived cells (Figure S6A) (See Experimental Procedures for clustering methods). Among these radial glia-like cells, we found that the oRG gene expression signature (Pollen et al., 2015) emerged at week 10 and became stronger at week 15 (Figures 5E, Figures S6B, S6C), consistent with observations from histology. We confirmed the expression of a few oRG markers in a subset of organoid radial glia that showed consistent subcellular localization to human tissue (Figure 5F). In addition, the expression of many of the predicted and validated oRG-markers is well correlated across radial glia-like cells from organoids, indicating that these genes are also co-expressed in the in vitro system (Figure 5G). Together, these data establish the presence of oRG-like cells that share features of tissue-level distribution, morphology, and molecular identity with oRG cells found in the developing human cortex.
oRG cell-specific cytokinesis delay in human lissencephaly
The ability to recapitulate human oRG production and three-dimensional organization in vitro enabled us to analyze the impact of lissencephaly mutations on this class of progenitors. Towards that end we performed time-lapse imaging following infection of 10-week organoids with CMV-GFP adenovirus, which labels all the dividing progenitors including vRG, IP and oRG cells. The morphology, spatial organization and mitotic behavior of GFP-labeled progenitors were consistent with properties observed in vivo. Namely, IP-like cells had a larger cell body with multiple short processes (blue arrows in Figures 6A and S5A), and they divided in place without much movement (Figure S5B). vRG cells were typically bipolar with apical processes coalescing at a common VZ-like surface, and their nuclei underwent apically-oriented INM prior to division at the apical surface (arrowheads in Figures 6A1, A2 and D). In contrast, oRG cells had a single long basal process and a very short apical process, were located above the ventricular-like zone (star in Figure 6A1 and S5C), and underwent a rapid mitotic somal translocation (MST) in the direction of the basal process prior to division (Figure 6G; Movie S5).
Figure 6. Prolonged mitosis in MDS oRG but not vRG cells at 10 weeks (see also Figure S5, Movie S5 and Table S2).

(A) Frames from time-lapse imaging showing representative examples of distinct vRG and oRG cell morphology, orientation and dividing properties in the same imaging field. The oRG-like cell is marked with a white star. Note the long basal process, and its superficial location relative to the bipolar vRG-like cells (example: white arrowhead). The indicated bipolar vRG-like cell from panel A1 moves apically and divides in A2 with a cleavage angle parallel to its basal process. In contrast, the indicated oRG-like cell undergoes an MST in the basal direction (not shown) and divides in A3 with a cleavage angle perpendicular to its basal process. Scale bar=50μm. (B) Schematic of parameters used to analyze vRG mitotic behavior: time from cell rounding to the first appearance of two daughters, angle of cleavage relative to the cell body axis just prior to basal process retraction. (C) Schematic of parameters used to analyze oRG mitotic behavior: MST distance, time from cell rounding to the first appearance of two daughters, angle of cleavage relative to the basal process. (D,E) Frames from time-lapse imaging showing representative examples of WT (D) and MDS (E) vRG-like divisions. (F) divisions. (I,J) Quantification of oRG division time (p=0.0028). (K) Quantification of oRG MST distance (p=0.05). (L, M) Quantification of vRG (L) and oRG (M) cleavage angle relative to the basal process. Average values ± S.E.M. are plotted in F and J. Statistical analysis was done using t-test (WT n=3; MDS n=2). For all the phenotypes, 20–50 cells were analyzed from each individual.
To examine whether MDS mutations impact the behavior of vRG and oRG cells, we measured multiple dynamic properties. For both vRG and oRG cells, we measured the time from when the dividing cell was rounded up to when the two daughters first appeared and the angle of the cleavage plane relative to the basal fiber (Figures 6B, C). In addition, we measured MST distance for oRG cells (Figure 6C). We found that WT oRG and vRG cells divided within 50 minutes from the time that the cell rounded up (Figures 6D, F, G, J and Movie S5). Surprisingly, MDS oRG cells tended to translocate farther during MST (Figure 6K), but then remained in mitosis for prolonged periods (up to several hours) prior to cytokinesis (Figures 6H, J, Figure S5C and Movie S5). The delay in cell division appeared to be specific to MDS oRG cells, as a similar defect was not observed in vRG or IP cell divisions from the same imaging sessions (Figures 6E, F and Figure S5B). Of note, this behavior mimicked a phenotype previously observed in primary oRG cells treated with a microtubule depolymerizing agent, nocodazole (Ostrem et al., 2014). We also compared the division cleavage angle relative to the basal process in dividing vRG and oRG cells between WT and MDS groups using live imaging. This analysis confirmed in vivo observations that vRG divisions become less tightly controlled with developmental maturation and that the majority of oRG divisions occur perpendicular to the basal process (Figures 6L, M) (LaMonica et al., 2013). There were no significant differences between the two genotypes in terms of division cleavage angles (Figures 6K, L). Collectively, our findings implicate MDS-deleted genes in the regulation of oRG cell division and point to a possible involvement of oRG cells in the pathogenesis of human lissencephaly.
DISCUSSION
By implementing a patient iPSC-derived 3-D model of early corticogenesis, we were able to delineate cell type- and cell stage-specific defects in human lissencephaly caused by deletions of the distal tip of chromosome 17. Ultimately these defects need to be linked with the corresponding genetic regulators for a deeper understanding of lissencephaly pathogenesis and human cortical development. While Pafah1b1 (LIS1) and YWHAE (14-3-3ε) mutations have been modeled in the rodent (Hirotsune et al., 1998; Toyo-oka et al., 2003; Gambello et al., 2003; Youn et al., 2009), there has been a lack of laboratory models that recapitulate the complete genetic defects of MDS. Thus, the roles of most of the deleted genes in brain development or MDS pathogenesis have not been examined. Here we provide a platform and establish functional assays that will enable validation of additional gene candidates in distinct cell types and cellular processes and will lead to identification of novel regulators of cortical development and malformation.
Several of the conserved developmental phenotypes implicated in lissencephaly by mouse models, including dysregulation of the NESC mitotic spindle (Faulkner et al., 2000; Yingling et al., 2008; Pawlisz et al., 2008; Pramparo et al., 2010; Xie et al., 2013; Moon et al., 2014) and neuronal migration defects (Hirotsune et al., 1998; Gambello et al., 2003; Youn et al., 2009; Toyo-oka et al., 2003) are recapitulated here. Deficiencies in neural progenitor cell viability in human MDS were previously implicated by Sheen and colleagues based on the analysis of postmortem samples, but without distinguishing which progenitor subclasses were affected (Sheen et al., 2006a). Here, by following in vitro differentiation from patient-derived iPSCs, we were able to recapitulate developmental lineage progression from NESC, to vRG, to IP and oRG, with spatial and temporal resolution that could not have been achieved with previous methods. Our analyses suggest that the founder NESCs and oRG cells are particularly vulnerable in MDS, whereas vRG and IP cells seem to be less affected.
Outer RG progenitors are thought to support the developmental and evolutionary expansion of the human neocortex due to their tremendous proliferative potential (Lukaszewicz et al., 2005; Hansen et al., 2010; Fietz et al., 2010; Pollen et al., 2015). In addition, oRG cells have been proposed to promote cortical folding by producing large numbers of neurons and by providing divergent tracks for increased tangential dispersion of migrating neurons along their basal fibers (Lui et al., 2011; Fietz and Huttner, 2011; Hevner and Haydar, 2012; Florio and Huttner, 2014; Taverna et al., 2014; Borrell, Gotz, 2014; Nowakowski et al., 2016a). Our studies identified specific mitotic defects in oRG cells from a severe form of lissencephaly, providing another link between this cell type and human neocortical gyration.
Prolonged mitosis of mouse cortical radial glia in a Magoh+/− model of microcephaly was recently shown to induce preferential production of neurons rather than progenitors, as well as increased incidence of apoptotic progeny (Pilaz et al., 2016). Mitotic delay through pharmacological means also altered cell fate, producing more apoptotic progeny or leading to ectopic neuronal differentiation. Similarly to Pilaz et al. (2016), we observed examples of apoptotic oRG-like cells (data not shown), however, the technical challenges of prolonged time-lapse imaging of organoid slices precluded us from direct visualization of progeny fate. Interestingly, Magoh functions upstream of Pafah1b1, with loss of function leading to decreased LIS1 protein levels during neurogenesis (Silver et al., 2010). This connection suggests a possible mechanistic basis for the convergence of microcephaly and lissencephaly phenotypes in MDS.
The genetic factors that regulate oRG behavior have been difficult to study due to the scarcity of these cells in mice. Here, we find that cerebral organoids generate oRG-like cells with matching properties defined in vivo, including position, morphology, mitotic behavior, and molecular identity. In addition, we show that DNA sequence in the MDS deletion interval is necessary for normal MST behavior. MDS oRG cells exhibited increased MST distance, similar to the effect of nocodazole (Ostrem et al., 2014), which interferes with microtubule cytoskeleton polymerization. Among the deleted genes, PAFAH1B1 is known to control microtubule dynamics during mitosis (Yingling et al., 2009; Moon et al., 2014), pointing to a possible oRG-specific regulatory mechanism, which may be LIS1-dependent. However, LIS1 haploinsufficiency alone cannot account for the severity of MDS, as mutations or smaller deletions affecting only Pafah1b1 have less severe outcomes and are not associated with human microcephaly (Ledbetter et al., 1992; Lo Nigro et al., 1997; Pilz et al., 1998; Barkovich et al., 1991; Cardoso et al., 2003). Therefore, additional genes in the MDS-deleted locus are likely involved in the regulation of oRG cell division. Future studies can examine the role of specific candidate genes in this interval using the organoid model system, which recapitulates key features of oRG biology.
Finally, the involvement of oRG cells in the pathophysiology of MDS suggests a link between this radial glial subtype and the development of lissencephaly. In addition to genetic causes, cortical abnormalities, including lissencephaly and microcephaly may also result from infectious diseases including cytomegalovirus (CMV) and West Nile Virus (Lanari et al., 2012; Teissier et al., 2014; O’Leary et al., 2006;Gerardin et al., 2014). Recently, the Zika virus has been declared a public health emergency of international concern (Heymann et al., 2016) due to a strong correlation with increased incidence of microcephaly, which has been reported to involve lissencephaly (Oliveira Melo et al., 2016; Schuler-Faccini et al., 2016; Mlakar et al., 2016). These infectious diseases may preferentially affect the same cell types and developmental process that are vulnerable in genetic forms of microcephaly and lissencephaly. The Zika virus can infect human cortical neural progenitors, leading to cell death and cell cycle dysregulation (Tang et al., 2016; Qian et al., 2016) and has been shown to infect human skin cells through the phosphatidylserine receptor, AXL (Hamel et al., 2015). We have found that radial glia cells show very high expression of the candidate Zika virus entry factor AXL (Nowakowski et al., 2016b), and the current study highlights how stalled mitosis of oRG cells may relate to lissencephaly. Collectively, these observations support the hypothesis that oRG dysfunction may be a feature of cortical malformations associated with lissencephaly.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the lead contact, Dr. Arnold Kriegstein (KriegsteinA@stemcell.ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
hiPSC derivation and culture methods
All human iPSC studies were approved by the UCSF Committee on Human Research and the UCSF GESCR (Gamete, Embryo, and Stem Cell Research) Committee. All the iPSC lines were generated and characterized previously (Kreitzer et al., 2013; Bershteyn et al., 2014). Briefly, three micrograms of episomal expression plasmid mixture encoding OCT3/4, SOX2, KLF4, L-MYC, LIN28, and shRNA for TP53 (Y4 mixture, Okita et al., 2011) were electroporated into 3 × 105 fibroblasts with a Neon Electroporation Device (Invitrogen) with a 100-μl kit, according to the manufacturer’s instructions. Conditions for electroporations were 1,650V, 10ms, and three pulses. Cells were detached within 1 week after electroporation and seeded at 1–3× 105 cells per 10-cm dish onto irradiated or mitomycin-C treated SNL feeder cells. The culture medium was replaced the next day with primate ESC medium (Reprocell) containing βFGF. Colonies were counted 25 days after electroporation, and those colonies similar to hESCs were selected for further cultivation and evaluation. Established hiPSC lines were cultured in mTeSR1 medium (Stem Cell Technologies, 05850) supplemented with Penicillin/Streptomycin/Gentomycin on Matrigel-coated dishes. Accutase or ReleSR were used for cell passaging. For a complete list of iPSC lines and experiments, please refer to Table S2.
METHOD DETAILS
Cerebral organoid generation and culture
Cerebral organoids were generated according to previously published methods (Kadoshima et al., 2013). Briefly, hIPSCs were dissociated to single cells with Accutase and re-aggregated using cortical differentiation medium in lipidure-coated 96-well V-bottom plates at a density of 10,000 cells per aggregate, 100μl per well. The cortical differentiation medium (Glasgow-MEM, 20% KSR, 0.1mM NEAA, 1mM sodium pyruvate, 0.1mM β-ME, 100 U/mL penicillin/streptomycin) was supplemented with Rho Kinase Inhibitor (Y-27632, 20μM, days 0–3), WNT inhibitor (IWR1-ε, 3μM, days 0–18) and TGF-β inhibitor (SB431542, 5μM, days 0–18). Media changes were performed on days 3, 6 and then every 2–3 days until day 18. On day 18, the aggregates were transferred to ultra low adhesion 6-well plates in DMEM/F12 medium with Glutamax supplemented with N2, Lipid Concentrate, Fungizone (2.5μg/mL), and penicillin/streptomycin (100 U/mL) and grown under 40% O2 5% CO2 conditions. From day 35, FBS (10% v/v), Matrigel (1% v/v) and heparin (5μg/mL) were added to the medium.
Quantitative PCR (qPCR)
To isolate RNA, cells were lysed with TRIzol reagent. Following chloroform extraction, aqueous phase containing RNA was mixed with 70% ethanol, transferred onto mini spin column, isolated with RNeasy mini kit, and digested with DNase I. RNA quality and concentration were assessed using a Nanodrop. cDNA was prepared with iScript cDNA synthesis kit. qPCR was done with SsoFast EvaGreen Supermix using a Vii7 instrument (Applied Biosystems). Relative mRNA expression levels were determined by ΔΔCt method relative to B2M housekeeping control. qPCR data is reported as a mean ± standard deviation from 3 technical replicates for each of the WT and MDS lines. Primers sequences are available upon request.
Organoid slice culture, viral infection and time-lapse imaging
At 5 and 10 weeks of culture (days 35 and 70, ± 2 days, respectively), organoids were imbedded in blocks of 3.5% low melting point Agarose dissolved in DMEM. Subsequently, 200-μm vibratome sections were generated and incubated overnight at 37°C, 8% O2, 5% CO2 in organoid culture medium containing AV-CMV-GFP adenovirus (diluted 1:1000) for preferential labeling of dividing progenitors or AAV1-CAG-tdTomato adeno associated virus (diluted 1:100) for preferential labeling of migrating neurons. Following infection (1–2 days for AV, 2–3 days for AAV1), organoid slices were immobilized on glass-bottom dishes (MatTek) using concentrated Matrigel and then overlaid with culture medium. Slices were imaged for 2–5 days using 10× or 20× air objectives on an inverted Leica TCS SP5 microscope with an on-stage environment-controlled chamber, streaming 5% O2, 5% CO2, balanced N2. Imaging was done at 15–20 minute intervals for analyzing neuronal progenitor divisions or at 30min-1hr intervals for analyzing neuronal migration. Maximum intensity projections of the collected z-stacks were analyzed using Imaris software.
Neuronal migration assays
For the in vitro migration assay, intact organoids were resuspended in concentrated Matrigel using the wide orifice 200μl tips, and placed on a glass-bottom surface of 6- or 12-well culture plates (MatTek) (3–4 organoids per well in separate Matrigel drops). The Matrigel was allowed to solidify for 30min at 37°C, and culture medium was then carefully overlaid on top of Matrigel drops (day 0). Processes were observed coming out of the organoids 24 hours later (day 1). Time-lapse imaging was initiated on day 2, and post time-lapse immunostaining was performed on day 3. Cell tracking and end-point analyses were done using Imaris software.
For migration on cortical slice explants, intact organoids were incubated on day 0 with the AAV1-CAG-tdTomato virus at 37°C (1:100). Also on day 0, cortical tissue (GW18.5) was embedded in 3.5% low melting point Agarose in DMEM. Cortical vibratome sections (250-μm thick) were generated, cut into smaller pieces (of ventricular width similar to the diameter of spheres) and incubated at 37°C, 8% O2, 5% CO2 in slice culture medium (66% βME, 25% Hanks, 5% FBS, 1% N-2, Penicillin/Streptomycin, Glutamine (all from Invitrogen) and 0.66% D-(+)-glucose (Sigma) containing AV-CMV-GFP adenovirus (1:1000). On day 1, organoids and cortical slices were washed to remove residual viral particles and placed against each other on glass bottom dishes. Any residual liquid was removed to allow the pieces of tissues to attach to each other (about 3–5min) and subsequently overlaid with concentrated Matrigel. The Matrigel was allowed to solidify for 30min and 37°C, and organoid and slice culture media (50:50 mixture) was then carefully overlaid on top of Matrigel drops. The endpoint analysis was performed on day 4 using Imaris software.
Immunocytochemistry
Organoids were fixed with 4% PFA in PBS for 20min, washed with PBS, dehydrated with 30% sucrose in PBS overnight at 4°C, imbedded in blocks of 30% sucrose/OCT compound (50:50 mixture) and frozen at −80°C. 16–20μm cryosections were subjected to heat/citrate-based antigen retrieval for 20 min, permeabilized and blocked with 10% donkey serum in PBS, 0.1% Triton X-100, 0.2% gelatin. Primary antibody incubations were performed at 4°C overnight and secondary incubations were performed at room temperature for 1–3 hours. After primary and secondary antibody incubations, three 20-min washes in PBS were performed. Secondary antibodies were: AlexaFluor 488, 546, 594, or 647 -conjugated donkey anti-goat, -rabbit, -rat or -mouse IgG (Invitrogen, all used at 1:1000 dilution).
Single Cell RNA-Sequencing
For single cell analysis, organoids from five biological replicates: WT (2 individuals), MDS (3 individual) were dissociated to a single cell suspension for 30 to 60 minutes using the papain dissociation system (Worthington). Single cells were captured using small cell (5 – 10 μm) C1 Single-Cell Auto Prep Integrated Fluidic Circuit (IFC, Fluidigm). Reverse transcription and cDNA amplification were performed on the IFC using the SMARTer Ultra Low RNA Kit, and single cell libraries were prepared using the Nextera XT DNA Sample Preparation Kit. Sequencing was performed on a HiSeq 2500 Ultra-High-Throughput Sequencing System (Illumina). Reads were aligned using Tophat2, and the expression of RefSeq genes was quantified by the featureCounts routine.
Outlier removal and enrichment for cortical lineage cells
Chambers with multiple cells as visualized by phase and fluorescent microscopy were discarded from further analyses, leaving 1107 libraries. Gene expression values were normalized based on library size as counts per million reads (CPM). Libraries with fewer than 1,000 genes detected above 1 CPM were eliminated as outliers, leaving 643 libraries. These libraries were further filtered by performing the identifyOutliers routine with default setting from the Singular Analysis Toolset (Fluidigm) in R. Of these 469 single cell libraries passing bioinformatic quality control criteria, 358 libraries expressed the telencephalon marker FOXG1 above 1 CPM, and 274 also lacked inhibitory neuron lineage marker DLX6, suggesting cells belonging to the cortical lineage.
Identification of radial glia-like cells and analysis of heterogeneity
Principal component analysis (PCA) was performed using the prcomp function in R and centering the data to identify the major sources of variation across cells, and k-means clustering (optimal k = 6 was determined using ConsensusClusterPlus R package) of PC 1–5 sample scores was performed followed by multiscale bootstrapping using pvClust package (1000 bootstraps) to determine the approximately unbiased p value for every cluster. This analysis, combined with a supervised analysis of cell identity based on the expression of canonical marker genes revealed revealed 90 dorsal telencephalic radial glia-like in vitro cells. We previously identified genes enriched in all radial glia cells (pan-RG), and genes enriched in oRG and vRG cells (Pollen et al., 2015). We visualized the expression of these marker genes over time and across cells using GENE-E (Broad Institute) and R. To examine whether in vitro-derived radial glia-like cells shared patterns of heterogeneity with primary cells, we evaluated the extent to which gene co-expression relationship among radial glia subtype marker genes are preserved in vitro. We computed the mean pairwise correlation coefficient between oRG marker genes across primary and in vitro cells. We then examined the probability that this observed level of co-expression between oRG markers would appear by chance in a background set of oRG, vRG, and pan-RG markers by calculating the mean pairwise correlation of genes randomly sampled from this combined set of radial glia markers across 100,000 permutations. For in vitro cells, we narrowed the list of radial glia marker genes to genes expressed in at least 10% of cells above log2CPM of 1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Throughout the paper, independent iPSC clones are treated as biological replicates. For statistical analyses, a single average value is used for each clone after collapsing technical replicates arising from multiple observations in sister organoids and from repeat experiments. On information about which specific iPSC lines were used for each of the experiments, please refer to Table S2. All of the statistical details of experiments can be found in the figure legends.
DATA AND SOFTWARE AVAILABILITY
Data Resources
Normalized gene expression values from single cell RNA sequencing profiling are expressed as log2 counts per million (CPM) reads for all genes across cells analyzed with more than 1000 genes detected above 1 CPM. The accession number for the single cell sequencing data reported in this paper is dbGaP: phs000989.v3.p1.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PAX6 | Covance | PRB-278P |
| SOX2 | Santa Cruz | Sc-17320 |
| p-VIMENTIN | MBL (ser82) | D095-3 |
| p-VIMENTIN | Abcam (ser55) | Ab22651 |
| SOX1 | R&D Systems | AF3369 |
| N-CADHERIN | BD Bioscience | 610920 |
| Cleaved CASPASE 3 | Cell Signaling Technology | 9661S |
| Ki67 | BD Pharmingen | 550609 |
| PKCζ | Santa Cruz | Sc-216 |
| PERICENTRIN | Abcam | Ab4448 |
| p-HISTONE H3 | Abcam | Ab10543 |
| TBR2 | Millipore | AB15894 |
| CTIP2 | Abcam | Ab18465 |
| DCX | Millipore | AB2253 |
| β III TUBULIN | Covance | MMS-435P |
| PARD6b | Santa Cruz | Sc-166405 |
| GFP | Aves Labs | GFP-1020 |
| PTPRZ1 | Sigma | HPA015103 |
| TNC | Abcam | Ab108930 |
| SATB2 | Abcam | Ab51502 |
| BLBP | Millipore | ABN14 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FBS | HyClone | Cat# SH30071.03 |
| Accutase | Stem Cell Technologies | Cat# 07920 |
| ReleSR | Stem Cell Technologies | Cat# 05872 |
| Growth Factor-Reduced Matrigel | BD Biosciences | Cat# 354263 |
| Y-27632 | Tocris | Cat# 1254 |
| IWR1-ε | Cayman Chemical | Cat# 13659 |
| SB431542 | Tocris | Cat# 1614 |
| Glutamax | Lifetech | Cat# 10565-018 |
| N2 supplement | Lifetech | Cat# 17502-048 |
| Lipid Concentrate | Lifetech | Cat# 11905-031 |
| Heparin | Sigma | Cat# H3149 |
| Critical Commercial Assays | ||
| RNeasy mini kit | Qiagen | Cat# 74104 |
| RNAse-Free DNase Set | Qiagen | Cat# 79254 |
| iScript cDNA synthesis kit | Biorad | Cat# 1708891 |
| SsoFast EvaGreen Supermix | Biorad | Cat# 1725201 |
| SMARTer Ultra Low RNA Kit | Clontech | Cat# 634833 |
| Nextera XT DNA Sample Preparation Kit | Illumina | Cat# FC-131-1096 |
| Deposited Data | ||
| Single cell RNA sequencing data | This paper | dbGaP: phs000989.v3.p1 |
| Experimental Models: Cell Lines | ||
| Human WT iPSC line “BJ-4” derived from healthy newborn male fibroblasts | ATCC (fibroblasts); Conklin lab, UCSF (iPSC line) | BJ-4 |
| Human WT iPSC lines “1323-2” and “1323-4” derived from healthy 48-year-old Caucasian female fibroblasts | Cell Applications; 106-05a (fibroblasts); Conklin lab, UCSF (iPSC lines) | 1323-2/1323-4 |
| Human WT iPSC line “WTc” derived from healthy 30-year- old Japanese male fibroblasts | Conklin lab, UCSF (fibroblasts and iPSC line) | WTc-10 |
| Human MDS iPSC line “MDS Rescue” derived from 4-month old Caucasian male with a ring chromosome 17 | Coriell Institute for Medical Research (GM06047- fibroblasts); Bershteyn et al., 2014 (iPSC lines) | MDS1r(17)-1 |
| Human MDS iPSC lines “MDS2-9” and “MDS2-10” from 1 year old Caucasian female | Coriell Institute for Medical Research (GM06097-fibroblasts); Bershteyn et al., 2014 (iPSC lines) | MDS2-9/MDS2-10 |
| Human MDS iPSC lines “MDS3-3” and “MDS3-4” from 18 fetal weeks male | Coriell Institute for Medical Research (GM09208-fibroblasts); Bershteyn et al., 2014 (iPSC lines) | MDS3-3/MDS3-4 |
| Human MDS iPSC lines “MDS4-4” and “MDS4-15” from six month old Caucasian female | This paper | MDS4-4/MDS4-15 |
| Recombinant DNA | ||
| Ad-CMV-eGFP | Vector Biolabs | Cat# 1060 |
| AAV1-CAG-tdTomato | University of Pennsylvania Vectorcore | N/A |
| Software and Algorithms | ||
| Imaris | Bitplane | http://www.bitplane.com/Default.aspx?tabid=423#imaris |
| Singular Analysis Toolset | Fluidigm | https://www.fluidigm.com/software |
| GENE-E | Broad Institute | https://software.broadinstitute.org/GENE-E/download.html |
Supplementary Material
Highlights.
Cortical organoids from control and MDS patient-derived iPSCs model lissencephaly
Neural stem cells in MDS organoids show increased apoptosis and horizontal divisions
Deep-layer neurons are more abundant in MDS organoids than in controls
Outer radial glia-like cells forming in MDS organoids show a mitotic delay
Acknowledgments
The authors are grateful to Joseph Loturco, Catherine Priest, Haim Belinson, Carmen Sandoval Espinosa and members of the Kriegstein and Wynshaw-Boris labs for helpful feedback on the manuscript. We thank Melanie Bedolli, Lillian Adame and Yinging Wang for technical support. M.B. was supported by a postdoctoral fellowship from the California Institute for Regenerative Medicine, CIRM (Grant Number TG2-01153) and a K99 career development award from the National Institute for Neurological Disorders and Stroke, NINDS (Grant Number 5K99NS088572). This research was funded by NIH grants NS075998, MH105989 and CIRM award GCIR-06673 to A.R.K.
Footnotes
AUTHOR CONTRIBUTIONS
M.B. conceived the project with guidance from A.W.B. and A.R.K. All the experiments were designed and performed by M.B. with help from E.D.L. and A.N. Single cell capture was performed by M.B. and T.J.N. RNA sequencing data analysis was performed by T.J.N. and A.A.P. M.B. prepared the figures and wrote the manuscript with input from all the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Armstrong E, Schleicher A, Omran H, Curtis M, Zilles K. The ontogeny of human gyrification. Cereb Cortex. 1995;1:56–63. doi: 10.1093/cercor/5.1.56. [DOI] [PubMed] [Google Scholar]
- Bershteyn M, Hayashi Y, Desachy G, Hsiao EC, Sami S, Tsang KM, Weiss LA, Kriegstein AR, Yamanaka S, Wynshaw-Boris A. Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature. 2014;507:99–103. doi: 10.1038/nature12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkovich AJ, Koch TK, Carrol CL. The spectrum of lissencephaly: report of ten cases analyzed by magnetic resonance imaging. Ann Neurol. 1991;30:139–146. doi: 10.1002/ana.410300204. [DOI] [PubMed] [Google Scholar]
- Betizeau M, Cortay V, Patti D, Pfister S, Gautier E, Bellemin-Ménard A, Afanassieff M, Huissoud C, Douglas RJ, Kennedy H, Dehay C. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron. 2013;2:442–57. doi: 10.1016/j.neuron.2013.09.032. [DOI] [PubMed] [Google Scholar]
- Borrell V, Gotz M. Role of radial glial cells in cerebral cortex folding. Curr Opin Neurobiol. 2014;27:39–46. doi: 10.1016/j.conb.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Bräuninger M, Lewitus E, Sykes A, Hevers W, Lancaster M, Knoblich JA, Lachmann R, Pääbo S, Huttner WB, Treutlein B. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci U S A. 2015;51:15672–7. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, Martin CL, Allanson J, Pilz DT, Olney AH, Mutchinick OM, Hirotsune S, Wynshaw-Boris A, Dobyns WB, Ledbetter DH. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003;72:918–930. doi: 10.1086/374320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi JG, Dooling EC, Gilles FH. Gyral development of the human brain. Ann Neurol. 1977;1:86–93. doi: 10.1002/ana.410010109. [DOI] [PubMed] [Google Scholar]
- Chong SS, Pack SD, Roschke AV, Tanigami A, Carrozzo R, Smith ACM, Dobyns WB, Ledbetter DH. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet. 1997;6:147–155. doi: 10.1093/hmg/6.2.147. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Stratton RF, Parke JT, Greenberg F, Nussbaum RL, Ledbetter DH. Miller-Dieker syndrome: Lissencephaly and monosomy 17p. J Ped. 1983;102:552–558. doi: 10.1016/s0022-3476(83)80183-8. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Curry CJR, Hoyme HE, Turlington L, Ledbetter DH. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet. 1991;48:584–594. Reiner. [PMC free article] [PubMed] [Google Scholar]
- Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasai Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell. 2008;3:519–532. doi: 10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Faulkner NE, Dujardin DL, Tai CY, Vaughan KT, O’Connell CB, Wang Y, Vallee RB. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–91. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- Fietz SA, Kelava I, Vogt J, Wilsch-Bräuninger M, Stenzel D, Fish JL, Corbeil D, Riehn A, Distler W, Nitsch R, Huttner WB. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci. 2010;13:690–699. doi: 10.1038/nn.2553. [DOI] [PubMed] [Google Scholar]
- Fietz SA, Huttner WB. Cortical progenitor expansion, self-renewal and neurogenesis-a polarized perspective. Curr Opin Neurobiol. 2011;21:23–35. doi: 10.1016/j.conb.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Fish JL, Kosodo Y, Enard W, Pääbo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. PNAS. 2006;27:10438–10443. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Huttner WB. Neural progenitors, neurogenesis and the evolution of the neocortex. Development. 2014;141:2182–2194. doi: 10.1242/dev.090571. [DOI] [PubMed] [Google Scholar]
- Gambello MJ, Darling DL, Yingling J, Tanaka T, Gleeson JG, Wynshaw-Boris A. Multiple dose- dependent effects of Lis1 on cerebral cortical development. J Neurosci. 2003;23:1719–29. doi: 10.1523/JNEUROSCI.23-05-01719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspard N, Bouschet T, Hourez R, Dimidschstein I, Naeije G, Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN, Gaillard A, Vanderhaeghen P. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature. 2008;455:351–357. doi: 10.1038/nature07287. [DOI] [PubMed] [Google Scholar]
- Georgala PA, Carr CB, Price DJ. The role of Pax6 in forebrain development. Dev Neurobiol. 2011;8:690–709. doi: 10.1002/dneu.20895. [DOI] [PubMed] [Google Scholar]
- Gertz CC, Kriegstein AR. Neuronal Migration Dynamics in the Developing Ferret Cortex. J Neurosci. 2015;42:14307–15. doi: 10.1523/JNEUROSCI.2198-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;10:777–88. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Dobyns WB. Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 2014;7:710–26. doi: 10.1016/S1474-4422(14)70040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao-Lormeau VM, Choumet V, Briant L, Desprès P, Amara A, Yssel H, Missé D. Biology of Zika Virus Infection in Human Skin Cells. J Virol. 2015;17:8880–96. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;7288:554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- Hansen PE, Ballesteros MC, Soila K, Garcia L, Howard JM. MR imaging of the developing human brain, 1: prenatal development. Radiographics. 1993;13:21–36. doi: 10.1148/radiographics.13.1.8426929. [DOI] [PubMed] [Google Scholar]
- Hattori M, Adachi H, Tsujimoto M, Arai N, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase. Nature. 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- Heymann DL, Hodgson A, Sall AA, Freedman DO, Staples JE, Althabe F, Baruah K, Mahmud G, Kandun N, Vasconcelos PFC, Bino S, Menon KU. Zika virus and microcephaly: why is this situation a PHEIC? Lancet. 2016;387:719–21. doi: 10.1016/S0140-6736(16)00320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res. 2006;55:223–233. doi: 10.1016/j.neures.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Haydar TF. The (not necessarily) convoluted role of basal radial glia in cortical neurogenesis. Cereb Cortex. 2012;2:465–8. doi: 10.1093/cercor/bhr336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–9. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- Hu WF, Chahrour MH, Walsh CA. The diverse genetic landscape of neurodevelopmental disorders. Annu Rev Genomics Hum Genet. 2014;15:195–213. doi: 10.1146/annurev-genom-090413-025600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, Sasai Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci USA. 2013;50:20284–9. doi: 10.1073/pnas.1315710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Dobyns WB. Lissencephaly and the molecular basis of neuronal migration. Hum Mol Genet. 2003;12(1):R89–96. doi: 10.1093/hmg/ddg086. [DOI] [PubMed] [Google Scholar]
- LaMonica BE, Lui JH, Hansen DV, Kriegstein AR. Mitotic spindle orientation predicts outer radial glial cell generation in human neocortex. Nat Commun. 2013;4:1665. doi: 10.1038/ncomms2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanari M, Capretti MG, Lazzarotto T, Gabrielli L, Rizzollo S, Mostert M, Manzoni P. Neuroimaging in CMV congenital infected neonates: how and when. Early human development. 2012;88(Suppl 2):S3–5. doi: 10.1016/S0378-3782(12)70003-8. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–9. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledbetter SA, Kuwano A, Dobyns WB, Ledbetter DH. Micro-deletions of chromosome 17p13 as a cause of isolated lissencephaly. Am J Hum Genet. 1992;50:182–189. [PMC free article] [PubMed] [Google Scholar]
- Leone DP, Srinivasan K, Chen B, Alcamo E, McConnell SK. The determination of projection neuron identity in the developing cerebral cortex. Curr Opin Neurobiol. 2008;1:28–35. doi: 10.1016/j.conb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller- Dieker syndrome. Hum Mol Genet. 1997;6:157–64. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell. 2011;1:18–36. doi: 10.1016/j.cell.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewicz A, Savatier P, Cortay V, Giroud P, Huissoud C, Berland M, Kennedy H, Dehay C. G1 phase regulation, area-specific cell cycle control, and cytoarchitectonics in the primate cortex. Neuron. 2005;3:353–64. doi: 10.1016/j.neuron.2005.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Kikinis R, Zuerrer M, Boesch C, Briner J, Kewitz G, Kaelin P. Developmental stages of human brain: an MR study. J Comput Assist Tomogr. 1988;6:917–22. doi: 10.1097/00004728-198811000-00002. [DOI] [PubMed] [Google Scholar]
- Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodušek V, Vizjak A, Pižem J, Petrovec M, Avšič Županc T. Zika Virus Associated with Microcephaly. N Engl J Med. 2016;10:951–8. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;6:817–31. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Moon HM, Youn YH, Pemble H, Yingling J, Wittmann T, Wynshaw-Boris A. LIS1 controls mitosis and mitotic spindle organization via the LIS1-NDEL1-dynein complex. Hum Mol Genet. 2014;2:449–66. doi: 10.1093/hmg/ddt436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamani SC, Zhang F, Shchelochkov OA, Bi W, Ou Z, Scaglia F, Probst FJ, Shinawi M, Eng C, Hunter JV, Sparagana S, Lagoe E, Fong CT, Pearson M, Doco-Fenzy M, Landais E, Mozelle M, Chinault AC, Patel A, Bacino CA, Sahoo T, Kang SH, Cheung SW, Lupski JR, Stankiewicz P. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J Med Genet. 2009;46:825–833. doi: 10.1136/jmg.2009.067637. [DOI] [PubMed] [Google Scholar]
- Nowakowski TJ, Pollen AA, Sandoval-Espinosa C, Kriegstein AR. Transformation of the Radial Glia Scaffold Demarcates Two Stages of Human Cerebral Cortex Development. Neuron. 2016a;6:1219–27. doi: 10.1016/j.neuron.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Pollen AA, Di Lullo E, Sandoval-Espinosa C, Bershteyn M, Kriegstein AR. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell. 2016b;5:591–6. doi: 10.1016/j.stem.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary DR, Kuhn S, Kniss KL, Hinckley AF, Rasmussen SA, Pape WJ, Kightlinger LK, Beecham BD, Miller TK, Neitzel DF, et al. Birth outcomes following West Nile Virus infection of pregnant women in the United States: 2003–2004. Pediatrics. 2006;117:e537–545. doi: 10.1542/peds.2005-2024. [DOI] [PubMed] [Google Scholar]
- Oliveira Melo AS, Malinger G, Ximenes R, Szejnfeld PO, Alves Sampaio S, Bispo de Filippis AM. Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet Gynecol. 2016;1:6–7. doi: 10.1002/uog.15831. [DOI] [PubMed] [Google Scholar]
- Ostrem BE, Lui JH, Gertz CC, Kriegstein AR. Control of outer radial glial stem cell mitosis in the human brain. Cell Rep. 2014;3:656–64. doi: 10.1016/j.celrep.2014.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Pawlisz AS, Mutch C, Wynshaw-Boris A, Chenn A, Walsh CA, Feng Y. Lis1-Nde1-dependent neuronal fate control determines cerebral cortical size and lamination. Hum Mol Genet. 2008;17:2441–55. doi: 10.1093/hmg/ddn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilaz LJ, McMahon JJ, Miller EE, Lennox AL, Suzuki A, Salmon E, Silver DL. Prolonged Mitosis of Neural Progenitors Alters Cell Fate in the Developing Brain. Neuron. 2016;1:83–99. doi: 10.1016/j.neuron.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilz D, Macha M, Precht K, Smith A, Dobyns W, Ledbetter D. Fluorescence in situ hybridization analysis with LIS1 specific probes reveals a high deletion mutation rate in isolated lissencephaly sequence. Genet Med. 1998;1:29–33. doi: 10.1097/00125817-199811000-00007. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu SJ, Oldham MC, Diaz A, Lim DA, Leyrat AA, West JA, Kriegstein AR. Molecular identity of human outer radial glia during cortical development. Cell. 2015;1:55–67. doi: 10.1016/j.cell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramparo T, Youn YH, Yingling J, Hirotsune S, Wynshaw-Boris A. Novel embryonic neuronal migration and proliferation defects in Dcx mutant mice are exacerbated by Lis1 reduction. J Neurosci. 2010;8:3002–12. doi: 10.1523/JNEUROSCI.4851-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, Yoon KJ, Jeang W, Lin L, Li Y, Thakor J, Berg DA, Zhang C, Kang E, Chickering M, Nauen D, Ho CY, Wen Z, Christian KM, Shi PY, Maher BJ, Wu H, Jin P, Tang H, Song H, Ming GL. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;5:1238–54. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reillo I, de Juan Romero C, García-Cabezas MÁ, Borrell V. A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb Cortex. 2011;7:1674–94. doi: 10.1093/cercor/bhq238. [DOI] [PubMed] [Google Scholar]
- Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–21. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- Saito T, Hanai S, Takashima S, Nakagawa E, Okazaki S, Inoue T, Miyata R, Hoshino K, Akashi T, Sasaki M, Goto Y, Hayashi M, Itoh M. Neocortical layer formation of human developing brains and lissencephalies: consideration of layer-specific marker expression. Cereb Cortex. 2011;21:588–96. doi: 10.1093/cercor/bhq125. [DOI] [PubMed] [Google Scholar]
- Schuler-Faccini L, Ribeiro EM, Feitosa IM, Horovitz DD, Cavalcanti DP, Pessoa A, Doriqui MJ, Neri JI, Neto JM, Wanderley HY, Cernach M, El-Husny AS, Pone MV, Serao CL, Sanseverino MT. Possible Association Between Zika Virus Infection and Microcephaly - Brazil, 2015. MMWR Morbidity and mortality weekly report. 2016;65:59–62. doi: 10.15585/mmwr.mm6503e2. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Ferland RJ, Harney M, Hill RS, Neal J, Banham AH, Brown P, Chenn A, Corbo J, Hecht J, Folkerth R, Walsh CA. Impaired proliferation and migration in human Miller-Dieker neural precursors. Ann Neurol. 2006a;1:137–44. doi: 10.1002/ana.20843. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Ferland RJ, Neal J, Harney M, Hill RS, Banham A, Brown P, Chenn A, Corbo J, Hecht J, Folkerth R, Walsh CA. Neocortical neuronal arrangement in Miller Dieker syndrome. Acta Neuropathol. 2006b;111:489–96. doi: 10.1007/s00401-005-0010-3. [DOI] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, Robinson HPC, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nature Neurosci. 2012;15:477–486. doi: 10.1038/nn.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitamukai A, Konno D, Matsuzaki F. Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J Neurosci. 2011;10:3683–95. doi: 10.1523/JNEUROSCI.4773-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron. 2004;44:263–77. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62:1–35. doi: 10.1016/0006-8993(73)90617-3. [DOI] [PubMed] [Google Scholar]
- Smart IH, Dehay C, Giroud P, Berland M, Kennedy H. Unique morphological features of the proliferative zones and postmitotic compartments of the neural epithelium giving rise to striate and extrastriate cortex in the monkey. Cereb Cortex. 2002;12:37–53. doi: 10.1093/cercor/12.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–75. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- Stahl R, Walcher T, De Juan Romero C, Pilz GA, Cappello S, Irmler M, Sanz-Aquela JM, Beckers J, Blum R, Borrell V, Götz M. Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell. 2013;3:535–49. doi: 10.1016/j.cell.2013.03.027. [DOI] [PubMed] [Google Scholar]
- Stiles J, Jernigan TL. The basics of brain development. Neuropsychol Rev. 2010;4:327–348. doi: 10.1007/s11065-010-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol. 2004;165:709–21. doi: 10.1083/jcb.200309025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, Christian KM, Didier RA, Jin P, Song H, Ming GL. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell. 2016;16:106–5. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna E, Götz M, Huttner WB. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol. 2014;30:465–502. doi: 10.1146/annurev-cellbio-101011-155801. [DOI] [PubMed] [Google Scholar]
- Teissier N, Fallet-Bianco C, Delezoide AL, Laquerriere A, Marcorelles P, Khung-Savatovsky S, Nardelli J, Cipriani S, Csaba Z, Picone O, et al. Cytomegalovirus-induced brain malformations in fetuses. J Neuropath & Exp Neurol. 2014;73:143–158. doi: 10.1097/NEN.0000000000000038. [DOI] [PubMed] [Google Scholar]
- Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A. 14-3-3 epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274–85. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- Wang X, Tsai JW, LaMonica B, Kriegstein AR. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci. 2011;14:555–61. doi: 10.1038/nn.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Juschke C, Esk C, Hirotsune S, Knoblich JA. The phosphatase PP4c controls spindle orientation to maintain proliferative symmetric divisions in the developing neocortex. Neuron. 2013;79:254–65. doi: 10.1016/j.neuron.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling J, Youn YH, Darling D, Toyo-Oka K, Pramparo T, Hirotsune S, Wynshaw-Boris A. Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell. 2008;3:474–86. doi: 10.1016/j.cell.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn YH, Pramparo T, Hirotsune S, Wynshaw-Boris A. Distinct dose-dependent cortical neuronal migration and neurite extension defects in Lis1 and Ndel1 mutant mice. J Neurosci. 2009;29:15520–30. doi: 10.1523/JNEUROSCI.4630-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.