

Abstract
T-cell infiltration into tumors represents a critical bottleneck for immune-mediated control of cancer. We previously showed that this bottleneck can be overcome by depleting immunosuppressive Foxp3+ regulatory T cells (Tregs), a process which can increase frequencies of tumor-infiltrating lymphocytes (TILs) through promoting development of specialized portals for lymphocyte entry, namely high endothelial venules (HEVs). In this paper, we used a carcinogen-induced tumor model, that allows for co-evolution of the tumor microenvironment and the immune response, to demonstrate that Treg depletion not only results in widespread disruption to HEV networks in lymph nodes (LNs) but activates CD8+ T cells, which then drive intratumoral HEV development. Formation of these vessels contrasts with ontogenic HEV development in LNs in that the process is dependent on TNF receptor and independent of lymphotoxin β receptor-mediated signaling. These intratumoral HEVs do not express the chemokine CCL21, revealing a previously undescribed intratumoral blood vessel phenotype. We propose a model where Treg depletion enables a self-amplifying loop of T-cell activation, which promotes HEV development, T-cell infiltration, and ultimately, tumor destruction. The findings point to a need to test for HEV development as part of ongoing clinical studies in patients with cancer.
Keywords: Treg, HEV, T-cell, vasculature, TNF
Introduction
Evidence indicates that the extent of T-cell infiltration into tumors is a key parameter influencing cancer immunity (1–5). Our previous studies revealed an influence of Foxp3+ regulatory T cells (Treg) on the numbers of infiltrating T cells (6,7). Specifically, we found that Treg depletion promotes development of high endothelial venules (HEVs), whose presence is associated with significantly increased numbers of T cells and better control of tumor growth (6). HEVs develop during ontogenic secondary lymphoid organ development, which is initiated by interactions between lymphotoxin (LT) β receptor (LTβR)-expressing lymphoid tissue organizer (LTo) cells and LTα1β2-expressing lymphoid tissue inducer (LTi) cells (8). A role for canonical LTi cells in intratumoral HEV neogenesis following Treg depletion was excluded in our previous study, indicating that this process is governed by different cues than those dictating HEV development in lymph nodes (LNs) (6).
The current study set out to identify the mechanisms underpinning HEV development using a mouse model of carcinogen-induced fibrosarcoma that allows for evolving interactions between the immune system and cancer cells during the process of transformation. Such reciprocal interactions shape development of the tumor microenvironment, the nature of the immune response, and tumor immunogenicity (9). A key feature of this carcinogen-induced tumor model is that vessels expressing peripheral node addressin (PNAd; the hallmark of HEV), are not detected in tumors from Treg-replete mice, indicating that Treg depletion is a prerequisite to development of HEV (6). Therefore, the interactions of T and B lymphocytes and DCs, which become highly activated following Treg depletion (6,10), may be essential for coordinating HEV formation in the tumors described herein. This study reports the findings of experiments designed to test this hypothesis and their implications for cancer immunotherapy.
Materials and Methods
Mice
We are grateful to Professor Rudensky for supplying Foxp3DTR mice and to Professor Hammerling for supplying CD11c.DOG mice. Each have been described previously (10,11). Foxp3DTR mice express knocked-in human diphtheria toxin receptor (DTR) and enhanced green fluorescent protein (EGFP) genes under the control of the Foxp3 promoter, allowing specific elimination of Tregs in vivo. These mice were backcrossed with C57BL/6 mice for ≥5 generations. CD11c.DOG mice express the human DTR together with a fragment of the ovalbumin protein and EGFP on a bacterial artificial chromosome under the control of the CD11c promoter, allowing efficient elimination of CD11c-expressing cells in vivo. Male CD11c.DOG mice were bred with female Foxp3DTR mice. F2 and F3 progeny were genotyped by PCR for FoxP3DTR homozygosity (females) or hemizygosity (males) and the presence of the CD11cDTR gene. Those with both Foxp3DTR and CD11cDTR genes (CD11c.DOG-Foxp3DTR) were used in future experiments. Mice were housed in accordance with UK Home Office regulations, isolator-bred, and housed in filter-top cages for the duration of experiments.
Tumor induction and diphtheria toxin administration
Anesthetized mice (8–15 weeks old) were injected subcutaneously with 400 μg of 3-methylcholanthrene (MCA; Sigma-Aldrich) in 100 μl olive oil to induce tumors as previously described (12). Mice were monitored for tumor development weekly for up to 18 months. Diphtheria toxin (DT; Sigma-Aldrich) in 100 μl PBS was administered by intraperitoneal (i.p.) injection (5ng/g body weight to deplete Tregs; 8ng/g body weight to deplete Tregs and CD11c+ cells) every other day after palpable tumor development. Once tumors became palpable, they were measured using calipers every other day (tumor width, tumor height, tumor leg diameter and non-tumor leg diameter), and tumor growth rate (k, days−1) was calculated using the difference between tumor and nontumor leg diameters by the following equation: Y= Y0 × exp (k × X). Mice were sacrificed before tumors reached 1.5cm in diameter or if tumors caused apparent discomfort (irritation or decreased mobility).
In vivo treatments
All treatments were administered by i.p. injection. Depleting CD4– and CD8-specific antibodies were produced in-house (13). Briefly, hybridomas expressing depleting CD4– and CD8-specific antibodies were produced in vitro and antibodies were purified on protein-G affinity columns. 100 μg anti-CD4 (clones YTS-191 and YTA-3) and/or anti-CD8 (clones YTS-156 and YTS-169) mAbs were administered every other day beginning one day prior to DT. Mouse LTβR.Fc (10 mg/kg body weight; received from Dr. Grogan or Prof. Ware (14–16)) and Etanercept (5 mg/kg body weight; TNFRII.Ig; Enbrel®, Amgen/Wyeth) were administered every other day alongside DT. 2 mg anti-mouse TNF mAb (MP6-XT22; produced in-house as detailed above) was administered beginning one day before DT, after which 1 mg was given every other day. Anti-mouse LT-α mAb (clone S5H3), received from Dr Grogan (14), was administered (6 mg/kg body weight) every other day beginning one day prior to DT. Mice received 100 μg of agonistic anti-LTβR mAb (clone 4H8), received from Professor Ware (17,18) every 3–4 days.
Dissection of tissues
Spleen and inguinal LNs were removed, and tumors were resected avoiding muscle, other tissues, and the popliteal LN.
Flow cytometry
Spleens and LNs were mashed through a 70 μm cell strainer (BD Biosciences) using the back of a syringe plunger. Tumors were mechanically dissociated by dicing into small (~1–2mm) pieces using a scalpel and then mashed through a 70 μm cell strainer using the back of a syringe plunger. Cell suspensions were resuspended in complete RPMI (cRPMI; RPMI [Invitrogen] plus 2 mM L-glutamine, 1 mM sodium pyruvate, pen/strep [50 μg/ml], and 10% FCS) and passed through a 70 μm cell strainer. Cells were washed with PBS, and red blood cells in tumor and spleen pellets were lysed using RBC lysis buffer (Biolegend). Cells were washed with PBS, stained using LIVE/DEAD Aqua (Invitrogen), then washed and Fc receptors blocked with anti-CD16/32 (clone 93; eBioscience) before staining with surface antibodies (listed in Supplementary Table S1).
For intracellular TNF analysis, cells were stimulated in 24-well plates with 20 nM PMA (Sigma-Aldrich) and ionomycin (1 μg/ml; Sigma-Aldrich) at 37°C for 4 hours. After 1 hour, GolgiStop (1μl/ml; BD Biosciences) was added. Cells were stained for surface markers and then TNF following fixation/permeabilization following the manufacturer’s protocol (Foxp3-staining kit; eBiosciences). Data were acquired on a FACS Canto II (BD Biosciences) and analyzed using FlowJo (TreeStar, USA).
Immunohistochemistry
5 μm neutral buffered-formalin solution (NBFS) fixed, paraffin-embedded tumor sections were mounted, and then rehydrated in xylene, descending alcohol concentrations, and dH2O. Antigen retrieval was performed in Tris (10 mmol/L), EDTA (pH9, 1 mmol/L). Endogenous peroxidase activity was quenched using 1% H2O2/MeOH, and nonspecific binding was blocked with 2.5% normal horse serum (VectorLabs). Sections were incubated in rat anti-PNAd (clone MECA-79; Biolegend) overnight at 4°C, washed with PBS, and then incubated in anti-Rat ImmPRESS™ HRP Polymer Detection solution (VectorLabs). Slides were briefly incubated in Vector® chromagen DAB HRP substrate (VectorLabs), rinsed with dH2O, and counterstained in haematoxylin. Slides were then dehydrated via an ascending alcohol gradient and xylene and mounted in distyrene, plasticizer, xylene mountant (DPX; Sigma-Aldrich).
Paraffin-embedded tumors stained using anti-PNAd were scanned using a Zeiss Axio Scan.Z1 slide scanner. HEVs were indicated, including the vessel lumen, in Zen software to obtain vessel area calculated in μm2. Total HEV area was calculated as a proportion of the total tumor area.
Immunofluorescence
5 μm sections of frozen tissue embedded in OCT (RA Lamb) were fixed in ice-cold acetone or, in the case of CCL21 staining, in periodate-lysine-paraformaldehyde fixative (PLP; 0.075 M lysine, 0.37 M sodium phosphate pH 7.2, 1% formaldehyde, and 0.01 M NaIO4). Endogenous biotin was blocked with Avidin/Biotin blocking kit (VectorLabs), and nonspecific binding was blocked with 2.5% normal horse serum (VectorLabs). Sections were incubated in primary antibody overnight at 4°C, washed with PBS, incubated in secondary antibody, washed again, and then mounted in Vectashield mounting medium with DAPI (VectorLabs). Sections were imaged using a Zeiss LSM710 or LSM800 confocal microscope, and serial images were assembled in Adobe Photoshop. Antibodies are listed in Supplementary Table S1. T cells in stained, frozen tumors were counted per high power field of view, and an average of 10 fields of view was calculated per section.
Gene expression analysis
Data previously generated by MouseRef-8v2.0 whole genome expression bead chip (Illumina) profiling were reanalyzed for statistically significant differences between Treg+, Treg− HEVlo and Treg− HEVhi groups (6). Briefly, RNA was extracted from OCT embedded tissue using TRIzol reagent (Invitrogen). RNA integrity was assessed on the Agilent 2100 Bioanalyzer, and samples with an RNA integrity number of 9 or more were used for gene expression profiling using MouseRef-8v2.0 whole genome expression bead chip arrays and scanned on the iScan system (Illumina) as recommended by the manufacturer. Probe intensity values were corrected by background subtraction using Genome Studio software and subsequently log-2 and baseline (median) transformed using Genespring software (Agilent) before analysis of genes. Heat maps were generated using GENE-E software (https://software.broadinstitute.org/GENE-E/index.html).
Statistical analyses
The statistical relationships (correlations) between sets of measured variables were tested using the non-parametric Spearman’s correlation coefficient. The statistical difference between group(s) was determined by the non-parametric Mann Whitney t test or one-way ANOVA with Tukey’s post test to compare pairs of means. A P≤0.05 was considered significant. All statistical analyses were performed using GraphPad Prism software (GraphPad Prism Software, La Jolla, CA, USA).
Results
Treg depletion disrupts LN HEV networks and induces intratumoral HEVs
We examined the impact of Treg depletion on LN architecture, focusing on HEV networks. Significant disruption was observed following loss of Tregs (Fig. 1A and B). Demarcation between B-cell and T-cell zones was diminished, and T cells and HEVs could be found throughout the swollen LN (Fig. 1B). Despite preserved PNAd expression, HEV morphology was significantly altered with vessels displaying more open lumen relative to HEVs of Treg-replete mice (Fig. 1E and F). These data indicate large-scale disorganization and disruption to function of the HEV network. Expression of the lymphoid chemokine CCL21 was restricted to HEVs in Treg-replete LNs but upregulated in Treg-depleted LNs (Fig. 1I and J).
Figure 1. Treg depletion causes widespread disruption to LN HEV networks and induces intratumoral HEVs with a unique phenotype.
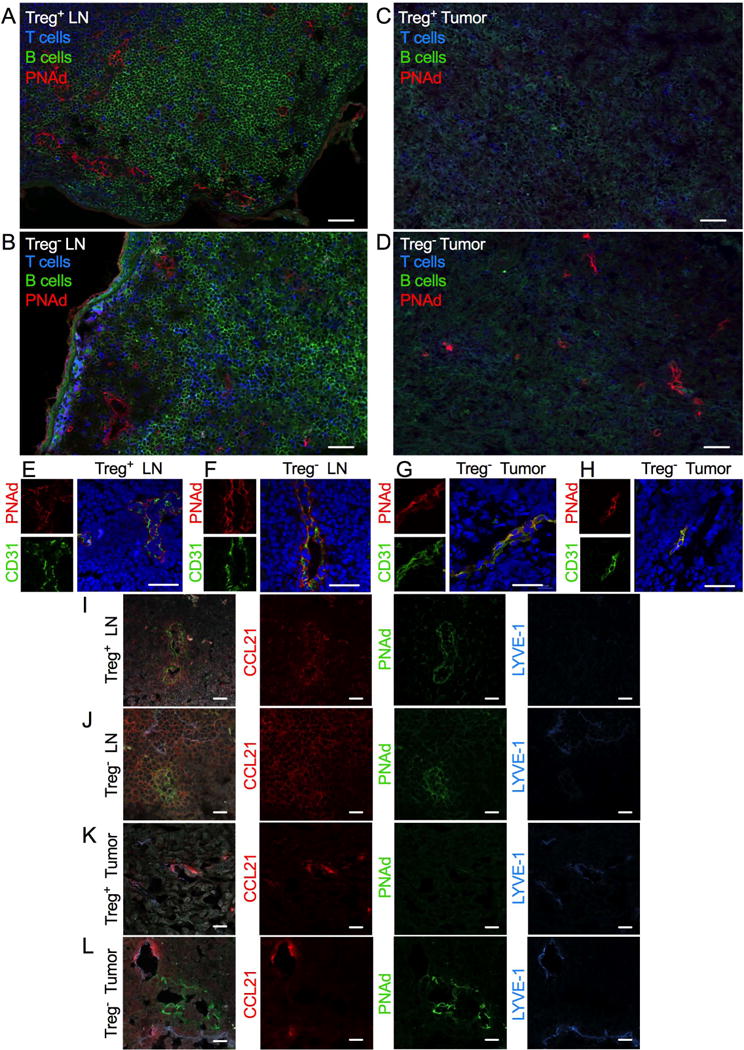
(A–D) Representative images of HEVs (PNAd+; red), T cells (CD3+; blue), and B cells (CD45R+; green) in LNs and tumors of Foxp3DTR animals. (A) Treg+ and (B) Treg− LNs; (C) Treg+ and (D) Treg− tumors. (E–H) High power representative images of HEVs (PNAd+; red) dual-stained for CD31 (green) in LNs and tumors of Foxp3DTR animals. (E) Treg+ and (F) Treg− LNs; (G and H) Treg− tumors. (I–L) High power representative images of LNs and tumors of Foxp3DTR animals stained for CCL21 (red), HEVs (PNAd+; green), and LYVE-1 (blue). (I) Treg+ and (J) Treg− LNs; (K) Treg+ and (L) Treg− tumors. Merged images include the nuclear stain DAPI (blue in E–H; grey in I–L). Scale bars represent 50 μm in A–D and 20 μm in E–L.
HEVs have been documented in multiple human malignancies, where their presence is detected by immunohistochemical staining for PNAd (19,20). It remains unknown whether tumor HEVs share other similarities with canonical LN HEVs. In LNs, HEVs are situated within T-cell zones and cortical ridges, which mark the regions between B-cell and T-cell zones (Fig. 1A) (21). In contrast, tumor HEVs, which only developed in the absence of Tregs, displayed no apparent pattern of distribution within the tumor mass (Fig. 1C and D). Although often observed in close proximity to lymphocytes, HEVs were not embedded within discrete lymphoid follicles (Fig. 1D). Endothelial cells lining LN HEVs co-expressed PNAd and CD31 and had a characteristic cuboidal morphology (Fig. 1E) (22,23). Expression of PNAd and CD31 did not entirely colocalize on LN HEVs, suggesting distinct patterns of expression at a cellular level. Although tumor HEVs also co-expressed PNAd and CD31, they lacked the distinctive plump morphology of LN HEV endothelial cells (Fig. 1G and H). In contrast to LN HEVs, CD31 and PNAd expression colocalized in closer proximity on tumor HEVs. However, the flat endothelium that lines tumor HEVs is likely to hide subtle differences in cellular expression. Tumor HEVs also co-expressed the adhesion molecule MAdCAM-1 (Supplementary Fig. S1). MAdCAM-1 is normally expressed by endothelial cells in all LN HEVs early in development, and then it is rapidly downregulated in peripheral LNs but not mucosal LNs where HEVs maintain MAdCAM-1 and PNAd expression (24,25). This observation indicates that tumor HEVs are newly formed vessels in the tumor microenvironment. In contrast to LN HEVs, tumor HEVs did not express CCL21. Instead, expression of this chemokine colocalized with lymphatic vessel endothelial hyaluronan receptor (LYVE)-1 expression in the tumor, suggesting restriction to lymphatic vasculature, which was the case for Treg-replete tumors (Fig. 1K and L). These findings indicate a phenotype which contrasts with previously described reports of intratumoral HEVs (26–28).
As described previously, HEV+ tumors from Treg-depleted mice contain significantly increased numbers of CD8+ T cells and display significantly reduced growth rates relative to HEV− tumors (6). As shown in Fig. 2, HEV area, calculated as a percentage of total tumor area following quantification of PNAd+ vessels (Fig. 2A), fitted a bifurcated pattern of distribution (Fig. 2B). Using a threshold defined by median HEV area, we delineated tumors into HEVhi and HEVlo categories (Fig. 2B). Because HEVs also co-expressed MAdCAM-1, HEVhi and HEVlo tumors also showed ‘high’ and ‘low’ MAdCAM-1 expression, respectively, for both quantity of MAdCAM-1+ vessels and intensity of MAdCAM-1 staining (Supplementary Fig. S1). However, PNAd positivity was used to quantify tumor HEVs because MAdCAM-1 can be expressed on other blood vessels and stromal cells (29). Although PNAd expression and not MAdCAM-1 defines the change in blood vessel phenotype and function after Treg depletion, MAdCAM-1, present on some blood vessels in Treg replete tumors, may be needed for predisposing vessels to upregulate PNAd upon depletion of Tregs. Following Treg depletion, PNAd+ HEV quantity significantly correlated with both the extent of T-cell infiltration and the degree of tumor control, which were themselves also linked (Fig. 2C, E, and F). Profiling data previously generated for Treg-replete tumors, Treg-depleted HEVhi tumors, and Treg-depleted HEVlo tumors (6) were reanalyzed and revealed that HEVhi and HEVlo tumors were distinguished by a genetic signature indicative of a Type 1 Helper T-cell/Cytotoxic T-cell (CTL) response (Fig. 2D). As a result of this refinement, tumors were categorized as HEVhi or HEVlo in all subsequent experiments.
Figure 2. HEV area correlates with increased T-cell infiltration and reduced growth rate in Treg-depleted tumors.
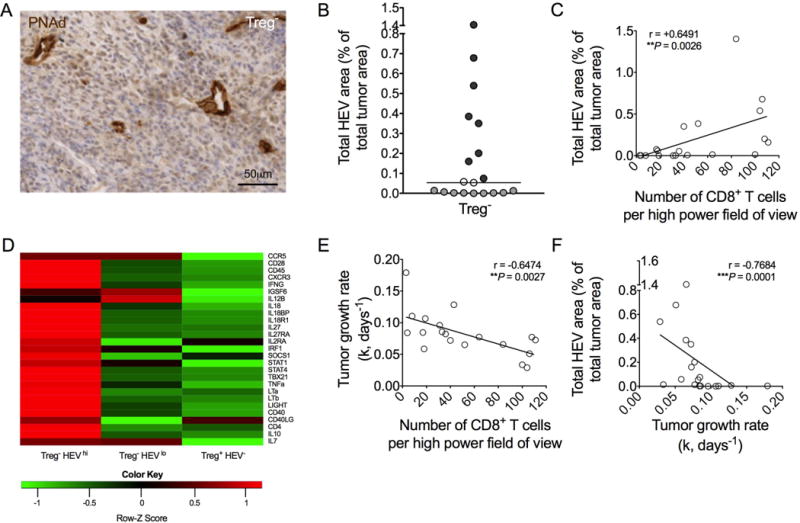
(A) Representative image of HEVs (PNAd+; brown) in paraffin embedded tumors. Counterstain: haematoxylin. (B) Total HEV area, as a percentage of total tumor area. Data are presented as individual data points (individual mice) plus median, which was used to define a cut-off. HEVlo (light grey); HEVhi (dark grey); borderline data points (open circles) were excluded thereafter. (C) Number of intratumoral CD8+ T cells plotted against total HEV area. (D) Transcriptional profiles revealed by microarray of Treg− HEVhi, Treg− HEVlo, and Treg+ HEV− tumors (n = 5 per group). Altered genes involved in Th1/CTL immune responses are displayed as a heat map of log2-fold change relative to the global median of genes. (E) Number of intratumoral CD8+ T cells plotted against tumor growth rates (k, days−1). (F) Total HEV area plotted against tumor growth rate (k, days−1). Statistical significance was determined by Spearman’s correlation coefficient test (r statistic and P values are shown). n = 19.
CD11c+ dendritic cells are not essential for HEV neogenesis in tumors
Evidence points to a role for CD11c+ DCs in the maintenance of LN HEVs (30), and DC abundance correlates positively with density of HEVs in human breast tumors (31,32). Considering that Treg depletion in Foxp3DTR mice increases the number and activation of DCs (10) and that DCs are observed in HEV-containing tumors (Fig. 3A), we tested whether DCs dictated HEV neogenesis in tumors by concurrently depleting CD11c+ DCs and Tregs in CD11c.DOG-Foxp3DTR mice. PNAd+ HEVs were detectable in tumors of DC-depleted mice (Fig. 3B), indicating that neogenesis of HEVs in tumors can proceed in the absence of DCs (Fig. 3C). However, we observed a trend towards fewer TILs and increased tumor growth rate relative to Treg− HEVhi DC-replete mice (Fig. 3D and E). Collectively, these data imply that although HEV development is not DC-dependent, the extent to which HEVs form, as well as impact on TIL frequencies and tumor growth, is compromised in the absence of DCs. It is possible that the effect of DCs on tumor growth is indirect through the driving of local activation and expansion of tumor-specific T cells.
Figure 3. CD11c+ dendritic cells are not essential for HEV neogenesis in tumors.
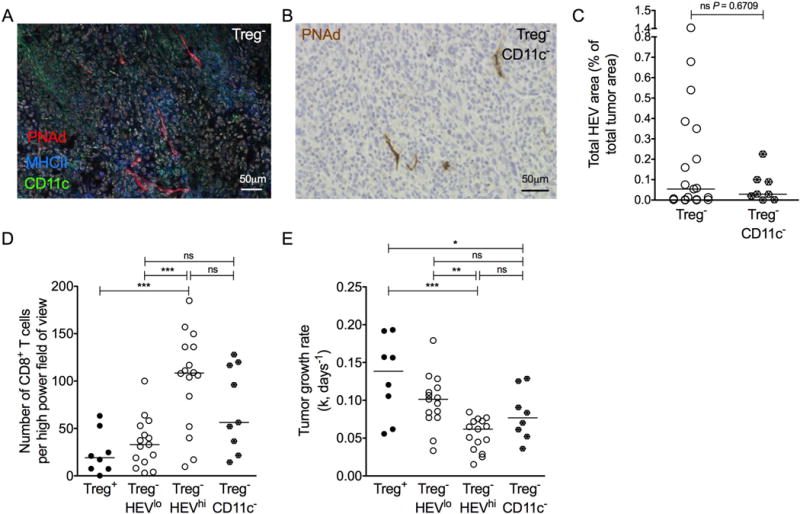
(A) Representative image of HEVs (PNAd+; red) alongside MHC Class II+ (blue) and CD11c+ (green) DCs in a tumor of a Treg− Foxp3DTR animal. (B) Representative image of HEVs (PNAd+; brown) in tumor of a Treg− CD11c− CD11c.DOG-Foxp3DTR animal. Counterstain: haematoxylin. (C) Total HEV area of Treg− CD11c− mice compared to Treg− controls. Data are presented as individual data points (individual mice) plus median. Statistical significance was determined by Mann Whitney t tests. n = 8 Treg− CD11c− animals; n = 19 Treg− animals. (D) Number of CD8+ T cells in tumors of Treg− HEVlo (n = 15) Foxp3DTR animals, Treg− HEVhi (n = 16) Foxp3DTR animals, and Treg− CD11c− CD11c.DOG-Foxp3DTR animals (n = 9). (E) Tumor growth rates (k, days−1) for Treg− HEVlo (n = 15) Foxp3DTR animals, Treg− HEVhi (n = 15) Foxp3DTR animals, and Treg− CD11c− CD11c.DOG-Foxp3DTR animals (n = 8). Statistical significance was determined by one-way ANOVA with Tukey’s test to compare pairs of means (* = P≤0.05, ** = P≤0.01, *** = P≤0.001).
T lymphocytes are essential for intratumoral neogenesis of HEVs
Given that Treg depletion results in a significant increase in number, proliferation, and activation of intratumoral T cells, particularly in HEV-containing tumors (6,10), we postulated that T cells influence intratumoral HEV neogenesis. To examine this, Treg-depleted tumor-bearing mice were injected with anti-CD4 and anti-CD8 (33–35). Treg-depleted mice treated simultaneously with anti-CD4 and anti-CD8 exhibited a significant reduction in tumor growth control (Fig. 4A). Almost all tumors recovered from these mice contained no HEVs. Remaining vessels had weaker PNAd+ staining (Fig. 4B and C), and a significant decrease in HEV area (Fig. 4F) relative to Treg-depleted tumors was seen. The most profound effect was observed upon CD8+ T cell–depletion (Fig. 4B–F). Considering T cell–depletion was suboptimal in Treg-depleted mice (Supplementary Fig. S2), these data show that a partial reduction in T-cell number, particularly CD8+ T cells, severely abrogates HEV neogenesis.
Figure 4. Depletion of T cells, in particular CD8+ T cells, significantly abrogates HEV neogenesis in tumors.
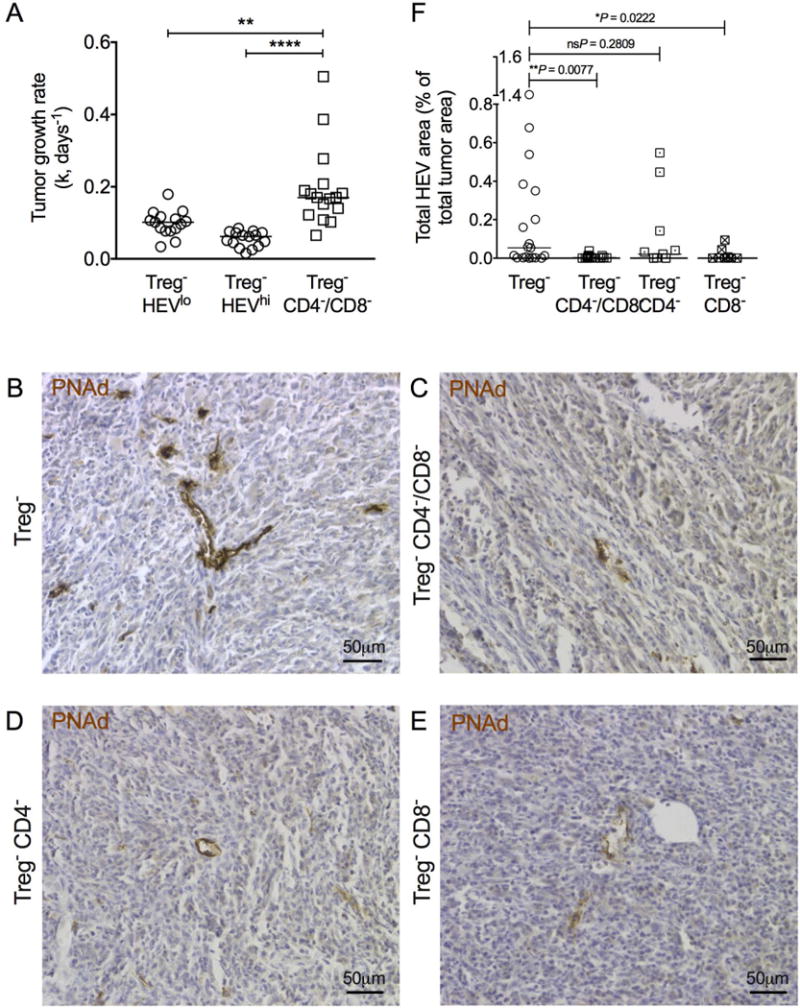
(A) Tumor growth rates (k, days−1) for Treg− HEVlo (n = 15), Treg− HEVhi (n = 15), and Treg− CD4−/CD8− (n = 16) Foxp3DTR animals. Statistical significance was determined by one-way ANOVA with Tukey’s test to compare pairs of means (** = P≤0.01, **** = P≤0.0001). (B–E) Representative images of HEVs (PNAd+; brown) in tumors of (B) Treg−, (C) Treg− CD4−/CD8−, (D) Treg− CD4−, and (E) Treg− CD8− Foxp3DTR mice. Counterstain: haematoxylin. (F) Total HEV area of Treg− CD4−/CD8− (n = 16), Treg− CD4− (n = 11), and Treg− CD8− (n = 8) animals compared to Treg− controls (n = 19). Data are presented as individual data points (individual mice) plus median. Statistical significance was determined by Mann Whitney t tests; P values are shown for each group compared to Treg− controls.
HEV neogenesis in tumors relies on TNFR but not LTβR signaling
To identify the molecular mechanisms underpinning HEV neogenesis in Treg-depleted tumors, we focused our attention on LTβR and TNFR signaling pathways based on their involvement in LN development (8,30,36–38). LTβR.Fc (binds mouse LTαβ and LIGHT) and TNFRII.Ig (Etanercept; binds mouse TNF and LTα) were used to block the LTβR and TNFR signaling pathways, respectively (39). In-line with reports of wildtype animals, LTβR.Fc treatment of Treg-depleted mice resulted in loss of marginal zone (MZ) B cells, follicular dendritic cells (FDC), and MAdCAM-1 expression on marginal sinus–lining stromal cells, confirming blockade of the LTβR signaling pathway (Supplementary Fig. S3 and S4) (14,40–43). Similarly, treatment of Treg-depleted mice with TNFRII.Ig decreased splenic FDCs and MAdCAM-1 expression, in-line with reports of wildtype animals (Supplementary Fig. S4) (44,45).
Disruption of LN HEV morphology was observed in Treg-depleted mice receiving either LTβR.Fc or TNFRII.Ig, with vessels appearing larger, often with more open lumen (Supplementary Fig. S5). PNAd+ HEV were readily detected in tumors of LTβR.Fc-treated mice, and quantification revealed a significant increase in HEV area relative to Treg-depleted controls (Fig. 5A, B, and F). In contrast, HEVs in tumors of mice receiving TNFRII.Ig were fewer in number (Fig. 5C), and quantification revealed a profound reduction in total HEV area compared to controls (Fig. 5F). Administration of monoclonal antibodies specific for either of the two ligands for TNFRII, TNF and LTα, to tumor-bearing mice concurrently with Treg depletion also resulted in a decrease in HEV area relative to Treg-depleted controls (Fig. 5D–F). In-line with published literature, LTβR.Fc treatment did not influence MAdCAM-1 expression, and both PNAd and MAdCAM-1 were detected on tumor HEVs following LTβR.Fc treatment. Blockade of TNFR signaling with TNFRII.Ig, anti-TNF, or anti-LTα treatment reduced both PNAd and MAdCAM-1 expression in tumors, with any remaining vessels maintaining expression of both markers (Supplementary Fig. S6). Although the LTα-specific antibody blocks soluble LTα3 and depletes cells expressing membrane bound LT (14), the effect of diminishing HEVs was not simply due to global T cell–depletion in this system. Overall, these data indicate that TNFR signaling, and not signaling via LTβR, drives intratumoral HEV neogenesis after Treg depletion.
Figure 5. Blockade of TNFR signaling, but not LymphotoxinβR signaling, severely abrogates HEV neogenesis in tumors.
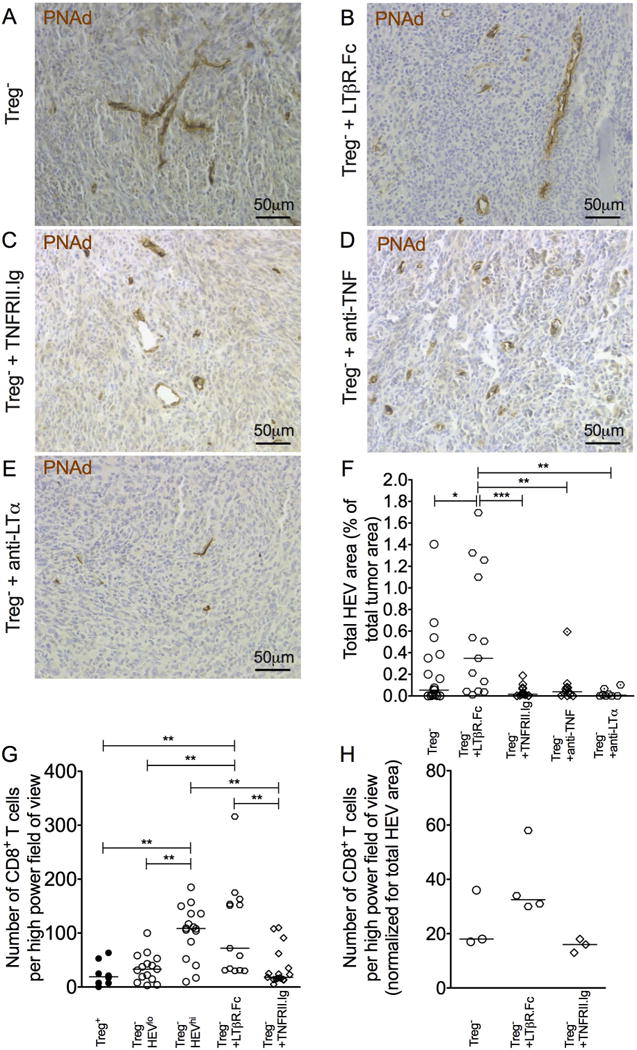
(A–D) Representative images of HEVs (PNAd+; brown) in tumors from Foxp3DTR animals. (A) Treg−; (B) Treg− plus LTβR.Fc; (C) Treg− plus TNFRII.Ig; (D) Treg− plus anti-TNF; and (E) Treg− plus anti-LTα. Counterstain: haematoxylin. (F) Total HEV area of tumors from mice treated with LTβR.Fc, TNRFII.Ig, anti-TNF, or anti-LTα compared to Treg− controls. Data are presented as individual data points (individual mice) plus median. n = 19 Treg− animals; n = 13 Treg− plus LTβR.Fc animals; n = 17 Treg− plus TNFRII.Ig animals; n = 11 Treg− plus anti-TNF; n = 8 Treg− plus anti-LTα. (G) Number of CD8+ T cells in tumors of Treg− HEVlo (n = 15), Treg− HEVhi (n = 16), Treg− plus LTβR.Fc (n = 13), and Treg− plus TNFRII.Ig (n = 17) Foxp3DTR animals. Statistical significance was determined by one-way ANOVA with Tukey’s test to compare pairs of means (* = P≤0.05, ** = P≤0.01, *** = P≤0.001). (H) Data as for (G) but normalized for total HEV area.
The increase in HEV area following LTβR.Fc treatment led to a significant increase in the number of TILs, whereas the decrease in HEV area observed after blockade of TNFR signaling resulted in no change in TIL frequency relative to Treg− HEVlo controls (Fig. 5G). When normalized for HEV area, these changes were more pronounced, suggesting a difference in HEV function following these treatments (Fig. 5H). Although administration of an agonist LTβR antibody to Treg-replete Foxp3DTR mice induced PNAd+ vessel formation in tumors, their presence was not accompanied by increased T-cell infiltration or reduced tumor growth relative to Treg+ controls (Supplementary Fig. S7). These data demonstrate that TNFR signaling is critical for development of functional HEV in tumors and indicate that blocking HEV development reduces the number of TILs and ability to control tumor growth.
HEV area and tumor growth correlate with TNF+ tumor-infiltrating T cells
Given our data, it seems likely that cytokines produced by T cells drive HEV development via TNFR signaling. To address this, proportions of CD8+ T cells in tumors and lymphoid organs expressing intracellular TNF were correlated with HEV area in tumors. A significant positive correlation was found between the proportion of intratumoral (but not LN-derived or splenic) CD8+ T cells producing TNF and intratumoral HEV area (Fig. 6A). These data directly index intratumoral HEV to TNF production by CD8+ TILs and support the premise that TNF-producing T cells are key drivers of HEV neogenesis in tumors. Additionally, proportion of TNF-producing CD8+ T cells also correlated with tumor growth rates (Fig. 6B). Although we could not distinguish between LTα3 and surface-bound LTαβ using anti-LTα, we found that T cells isolated from Treg-depleted tumors express LTα in addition to TNF at the mRNA level (Supplementary Fig. S8), consistent with the possibility that these TNF-producing T cells also produce LTα. Overall, the data are indicative of a pathway where Treg depletion leads to activation of T cells, which then produce TNF/LTα3. This, in turn, promotes blood vessel differentiation into HEVs, resulting in superior T-cell infiltration and tumor control (Fig. 6C).
Figure 6. HEV area and tumor growth rates correlate with the proportions of TNF+ tumor-infiltrating T cells.
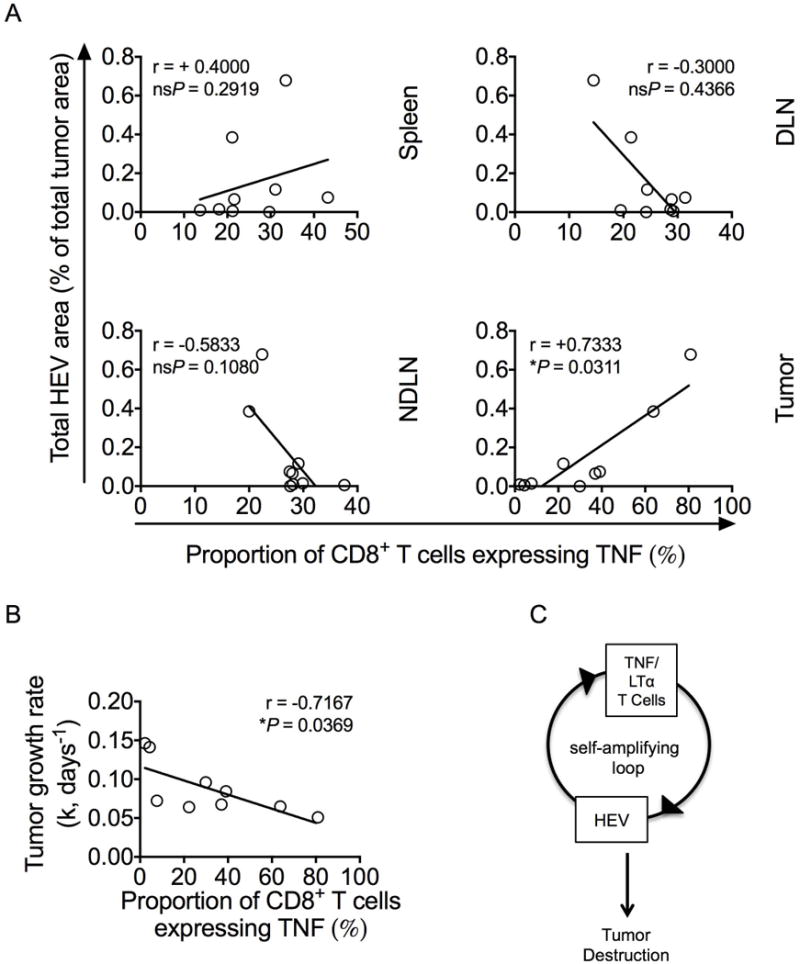
(A) Correlations between total HEV area within the tumor (%, y axis) and the proportion of CD8+ T cells expressing intracellular TNF in spleen, non-tumor draining lymph nodes (NDLNs), tumor draining lymph nodes (DLNs), or tumor as indicated (x axis). Statistical significance was determined by Spearman’s correlation coefficient test (r statistic and P values are shown). n = 9. (B) Correlation between tumor growth rate (k, days−1) and the proportion of CD8+ T cells expressing intracellular TNF in tumors. Statistical significance was determined by Spearman’s correlation coefficient test (r statistic and P values are shown). n = 9. (C) Schematic summary of the mechanism presented herein.
Discussion
In the presence of Tregs, T cells entering carcinogen-induced tumors express multiple and overlapping inflammatory chemokine receptor pairs to guide their migration to tumors (46). T cells also enter via aberrantly formed blood and lymphatic vessels (47). It is thought that Tregs become enriched within the TIL pool through selective proliferation and retention within the tumor microenvironment (48). In the absence of Tregs, a new route of entry presents itself in the form of intratumoral HEVs, which are significantly associated with TIL frequency and control of tumor growth (6). These vessels are morphologically distinct from those found in LNs and those reported previously in Treg-replete tumors arising either in the presence of strong antigenic stimulation (26) or as a result of administration of agonist LTβR antibodies (this study). Thus, in the case of the fibrosarcomas described herein, Treg depletion is the defining event in development of intratumoral HEVs and the consequent favorable antitumor immune response, as PNAd+ vessels are not observed in Treg-replete tumors.
The mechanisms identified herein as controlling intratumoral HEV neogenesis following Treg depletion are distinct from those occurring during ontogenic LN HEV development. Initial formation of HEV structure and abluminal PNAd expression on high endothelial cells in LNs proceeds in the absence of T or B cells (49), although the process of intratumoral HEV development is clearly T cell–dependent. A second departure from ontogenic HEV development is the reliance on TNFR signaling and dispensability of signaling via LTβR, strictly required for HEV formation and maintenance in LNs (8,36,37).
We also observed fundamental differences in our findings compared to those reported in a study of tumors induced by OVA-expressing cancer cell lines (26). First, although HEVs were readily observed in Treg-depleted carcinogen-induced tumors with clear implications for control of tumor growth, tyramide amplification was required to observe PNAd expression in cell line models, and its contribution to control of tumor growth was unclear. Second, in contrast to our findings, TNF played no role in HEV development in cell line–induced tumors, but a requirement for IFNγ-induced expression of CCL21 on high endothelial cells was reported (26). In the Treg-depleted tumors described herein, CCL21 expression is restricted to lymphatic vessels, indicating that it is not required for transmigration via HEVs. It has been suggested that CCL21+ lymphatic vessels may contribute to establishment of a memory response by enabling CCR7+ T cells to leave via the tumor lymphatics for re-entry into draining LNs (50), and our previous observation that CCL21 is significantly upregulated in whole HEV-containing Treg-depleted tumors (6) supports this.
Finally, we saw no evidence of organized lymphoid structures in HEV-containing Treg-depleted carcinogen-induced tumors, whereas organized aggregates comprising of B cells and gp38 fibroblasts were observed in cell line–induced tumors (26). The presence of such tertiary lymphoid structures (TLS) containing HEVs in murine tumors has been associated with immunosuppression rather than immune activation (28,51) unless Tregs are depleted (27). Therefore, expression of a strong neoantigen, such as OVA, resulting in activation of endogenous T cells or adoptively transferred transgenic T cells may allow immunosuppressive effects of TLS to be overcome. Such considerations might explain why evidence for a beneficial role of TLS/HEVs in human tumors is mixed, with some studies showing an association with a good prognosis whereas an increasing number of studies suggest a link to disease progression (19,20,52).
Models where tumors develop in vivo better represent the structural complexity of solid tumors with respect to the composition of the stroma, the myriad of cell-to-cell interactions occurring within, and tumor immunogenicity. Tumor cell lines already possess the mutational and epigenetic changes required to rapidly become palpable tumors, but injection of carcinogens can allow for evolving interactions between the immune system and cancer cells during the process of transformation. Sustained interactions between the immune system and tumor cells during this early phase leads to a gradual reduction in tumor immunogenicity (9). Effector lymphocyte–driven development of LN-like vasculature in tumors, which then promotes antitumor immunity, may only occur when antigen stimulation is sufficient to overcome local immunosuppression. Depleting Tregs can significantly reverse this immunosuppression, lowering the threshold for immune activation and enabling HEV development to occur in response to immune-edited tumors.
We observed that around 50% of tumors developed HEVs after Treg depletion. Depleting Tregs induced robust T-cell activation in all animals, indicating that although activation of these immune cell subsets is essential for HEV development, it is not sufficient to ensure that this occurs in all tumors. All tumors from Treg-replete mice contained some T cells. The range was small, and a similar proportion of these T cells produced TNF, indicating that the extent to which TNF-producing T cells infiltrate the tumor pre-DT treatment is not a determining factor in whether the tumors go on to develop HEVs post-treatment. Significant differences in T-cell numbers and in the proportion of T cells producing TNF were only observed after treatment, correlating with HEVs. Our studies have demonstrated the inability of activated T cells, purified from Treg-depleted tumor-bearing animals, to induce HEV development in tumors of Treg-replete mice upon adoptive transfer. These data also point to a yet unknown factor that renders tumors permissive to HEV neogenesis after Treg depletion. This may be a nonimmune component of the tumor microenvironment such as the endothelium, the extracellular matrix, or stromal components. Thus, future work should focus on defining key features that render some tumors refractive to HEV development after Treg depletion. Defining bottlenecks to HEV development in tumors may improve the design of new therapeutic approaches, combining immunotherapies targeting Tregs with regimens designed to alter tumor vasculature.
Supplementary Material
Acknowledgments
We are grateful to Professor Rudensky (Memorial-Sloan-Kettering Cancer Center) for the Foxp3DTR mice, Professor Hammerling (German Cancer Research Center (DKFZ)) for the CD11c.DOG mice, and Dr Grogan (Genentech) for the LTβR.Fc and anti-mouse LTα reagents. We thank Claudia Consoli (Central Biotechnology Services, Cardiff University) for her help with the qRT-PCR experiments, Dr Steinne (Sanford-Burnham-Prebys Medical Discovery Institute) for critical reading of this manuscript, and Cardiff University JBIOS staff for their continued support. We also wish to acknowledge the support of the Wales Cancer Research Centre.
Financial support: Emily J Colbeck is supported by a PhD studentship funded by Cancer Research UK. This work was supported by a University Award (Awen Gallimore) from the Wellcome Trust (086983/Z/08/Z) and a programme grant from Cancer Research UK (C16731/A21200). Production and use of the LTβR.Fc reagent was supported by an NIH grant (R01 CA164679; Carl F. Ware).
References
- 1.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Science. Vol. 313. American Association for the Advancement of Science; 2006. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome; pp. 1960–4. [DOI] [PubMed] [Google Scholar]
- 2.Madore J, Vilain RE, Menzies AM, Kakavand H, Wilmott JS, Hyman J, et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: implications for anti-PD-1/PD-L1 clinical trials. Pigment Cell Melanoma Res. 2015;28:245–53. doi: 10.1111/pcmr.12340. [DOI] [PubMed] [Google Scholar]
- 3.Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, et al. Nat Med. Vol. 20. Nature Publishing Group; 2014. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors; pp. 607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–4. [PubMed] [Google Scholar]
- 5.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Proc Natl Acad Sci USA. Vol. 102. National Acad Sciences; 2005. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer; pp. 18538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hindley JP, Jones E, Smart K, Bridgeman H, Lauder SN, Ondondo B, et al. Cancer Res. Vol. 72. American Association for Cancer Research; 2012. T-cell trafficking facilitated by high endothelial venules is required for tumor control after regulatory T-cell depletion; pp. 5473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallimore A, Godkin A. Immunology. Vol. 123. Blackwell Publishing Ltd; 2008. Regulatory T cells and tumour immunity - observations in mice and men; pp. 157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3:292–303. doi: 10.1038/nri1054. [DOI] [PubMed] [Google Scholar]
- 9.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. Nature. Vol. 410. Nature Publishing Group; 2001. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity; pp. 1107–11. [DOI] [PubMed] [Google Scholar]
- 10.Kim JM, Rasmussen JP, Rudensky AY. Nat Immunol. Vol. 8. Nature Publishing Group; 2007. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice; pp. 191–7. [DOI] [PubMed] [Google Scholar]
- 11.Hochweller K, Striegler J, Hämmerling GJ, Garbi N. Eur J Immunol. Vol. 38. WILEY‐VCH Verlag; 2008. A novel CD11c. DTR transgenic mouse for depletion of dendritic cells reveals their requirement for homeostatic proliferation of natural killer cells; pp. 2776–83. [DOI] [PubMed] [Google Scholar]
- 12.Hindley JP, Ferreira C, Jones E, Lauder SN, Ladell K, Wynn KK, et al. Cancer Res. Vol. 71. American Association for Cancer Research; 2011. Analysis of the T-cell receptor repertoires of tumor-infiltrating conventional and regulatory T cells reveals no evidence for conversion in carcinogen-induced tumors; pp. 736–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones E. Cancer Immunity. 2002;2:1. ARTICLE. 2002;: 1–12. [PubMed] [Google Scholar]
- 14.Chiang EY, Kolumam GA, Yu X, Francesco M, Ivelja S, Peng I, et al. Targeted depletion of lymphotoxin-alpha-expressing TH1 and TH17 cells inhibits autoimmune disease. Nat Med. 2009;15:766–73. doi: 10.1038/nm.1984. [DOI] [PubMed] [Google Scholar]
- 15.Benedict CA, Banks TA, Senderowicz L, Ko M, Britt WJ, Angulo A, et al. Lymphotoxins and cytomegalovirus cooperatively induce interferon-beta, establishing host-virus détente. Immunity. 2001;15:617–26. doi: 10.1016/s1074-7613(01)00222-9. [DOI] [PubMed] [Google Scholar]
- 16.Rooney I, Butrovich K, Ware CF. Expression of lymphotoxins and their receptor-Fc fusion proteins by baculovirus. Meth Enzymol. 2000;322:345–63. doi: 10.1016/s0076-6879(00)22032-6. [DOI] [PubMed] [Google Scholar]
- 17.Banks TA, Rickert S, Benedict CA, Ma L, Ko M, Meier J, et al. A lymphotoxin-IFN-beta axis essential for lymphocyte survival revealed during cytomegalovirus infection. J Immunol. 2005;174:7217–25. doi: 10.4049/jimmunol.174.11.7217. [DOI] [PubMed] [Google Scholar]
- 18.De Trez C, Schneider K, Potter K, Droin N, Fulton J, Norris PS, et al. J Immunol. Vol. 180. NIH Public Access; 2008. The inhibitory HVEM-BTLA pathway counter regulates lymphotoxin receptor signaling to achieve homeostasis of dendritic cells; pp. 238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dieu-Nosjean M-C, Giraldo NA, Kaplon H, Germain C, Fridman W-H, Sautès-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev. 2016;271:260–75. doi: 10.1111/imr.12405. [DOI] [PubMed] [Google Scholar]
- 20.Sautès-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman W-H, et al. Tertiary Lymphoid Structures in Cancers: Prognostic Value, Regulation, and Manipulation for Therapeutic Intervention. Front Immunol Frontiers. 2016;7:407. doi: 10.3389/fimmu.2016.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Vol. 9. Nature Publishing Group; 2009. pp. 618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gowans JL, Knight EJ. THE ROUTE OF RE-CIRCULATION OF LYMPHOCYTES IN THE RAT. Proc R Soc Lond, B, Biol Sci. 1964;159:257–82. doi: 10.1098/rspb.1964.0001. [DOI] [PubMed] [Google Scholar]
- 23.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol Annual Reviews. 2004;22:129–56. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 24.Mebius RE, Streeter PR, Michie S, Butcher EC, Weissman IL. A developmental switch in lymphocyte homing receptor and endothelial vascular addressin expression regulates lymphocyte homing and permits CD4+ CD3− cells to colonize lymph nodes. Proc Natl Acad Sci USA. 1996;93:11019–24. doi: 10.1073/pnas.93.20.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7:344–53. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 26.Peske JD, Thompson ED, Gemta L, Baylis RA, Fu Y-X, Engelhard VH. Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun. 2015;6:7114. doi: 10.1038/ncomms8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joshi NS, Akama-Garren EH, Lu Y, Lee D-Y, Chang GP, Li A, et al. Immunity. Vol. 43. Elsevier; 2015. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses; pp. 579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Science. Vol. 328. American Association for the Advancement of Science; 2010. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21; pp. 749–52. [DOI] [PubMed] [Google Scholar]
- 29.Ager A, May MJ. Oncoimmunology. Vol. 4. Taylor & Francis; 2015. Understanding high endothelial venules: Lessons for cancer immunology; p. e1008791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moussion C, Girard J-P. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature. 2011;479:542–6. doi: 10.1038/nature10540. [DOI] [PubMed] [Google Scholar]
- 31.Martinet L, Filleron T, Le Guellec S, Rochaix P, Garrido I, Girard J-P. J Immunol. Vol. 191. American Association of Immunologists; 2013. High endothelial venule blood vessels for tumor-infiltrating lymphocytes are associated with lymphotoxin β-producing dendritic cells in human breast cancer; pp. 2001–8. [DOI] [PubMed] [Google Scholar]
- 32.Martinet L, Girard J-P. Oncoimmunology. Vol. 2. Taylor & Francis; 2013. Regulation of tumor-associated high-endothelial venules by dendritic cells: A new opportunity to promote lymphocyte infiltration into breast cancer? p. e26470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin S, Cobbold S, Tighe H, Benjamin R, Waldmann H. Eur J Immunol. Vol. 17. WILEY‐VCH Verlag GmbH; 1987. CD4 monoclonal antibody pairs for immunosuppression and tolerance induction; pp. 1159–65. [DOI] [PubMed] [Google Scholar]
- 34.Qin SX, Cobbold S, Benjamin R, Waldmann H. J Exp Med. Vol. 169. The Rockefeller University Press; 1989. Induction of classical transplantation tolerance in the adult; pp. 779–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cobbold SP, Jayasuriya A, Nash A, Prospero TD, Waldmann H. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature. 1984;312:548–51. doi: 10.1038/312548a0. [DOI] [PubMed] [Google Scholar]
- 36.Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, et al. Immunity. Vol. 23. Elsevier; 2005. Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function; pp. 539–50. [DOI] [PubMed] [Google Scholar]
- 37.Onder L, Danuser R, Scandella E, Firner S, Chai Q, Hehlgans T, et al. J Exp Med. Vol. 210. Rockefeller Univ Press; 2013. Endothelial cell-specific lymphotoxin-β receptor signaling is critical for lymph node and high endothelial venule formation; pp. 465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity. 1998;9:71–9. doi: 10.1016/s1074-7613(00)80589-0. [DOI] [PubMed] [Google Scholar]
- 39.Bossen C, Ingold K, Tardivel A, Bodmer J-L, Gaide O, Hertig S, et al. J Biol Chem. Vol. 281. American Society for Biochemistry and Molecular Biology; 2006. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human; pp. 13964–71. [DOI] [PubMed] [Google Scholar]
- 40.Mackay F, Browning JL. Nature. Vol. 395. Nature Publishing Group; 1998. Turning off follicular dendritic cells; pp. 26–7. [DOI] [PubMed] [Google Scholar]
- 41.Mackay F, Majeau GR, Lawton P, Hochman PS, Browning JL. Eur J Immunol. Vol. 27. WILEY‐VCH Verlag GmbH; 1997. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice; pp. 2033–42. [DOI] [PubMed] [Google Scholar]
- 42.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. Annual Reviews 4139 El Camino Way, PO Box 10139, Palo Alto, CA 94303-0139, USA. [DOI] [PubMed] [Google Scholar]
- 43.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol Annual Reviews. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 44.Heesters BA, Myers RC, Carroll MC. Nature Publishing Group. Vol. 14. Nature Publishing Group; 2014. Follicular dendritic cells: dynamic antigen libraries; pp. 495–504. [DOI] [PubMed] [Google Scholar]
- 45.Allen CDC, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. 2008;20:14–25. doi: 10.1016/j.smim.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ondondo B, Colbeck E, Jones E, Smart K, Lauder SN, Hindley J, et al. A Distinct Chemokine Axis Does not Account for Enrichment of Foxp3(+) CD4(+) T cells in Carcinogen-Induced Fibrosarcomas. Immunology. 2014 doi: 10.1111/imm.12430. n/a–n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ondondo B, Jones E, Hindley J, Cutting S, Smart K, Bridgeman H, et al. Progression of carcinogen-induced fibrosarcomas is associated with the accumulation of naïve CD4+ T cells via blood vessels and lymphatics. Int J Cancer. 2014;134:2156–67. doi: 10.1002/ijc.28556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colbeck EJ, Hindley JP, Smart K, Jones E, Bloom A, Bridgeman H, et al. Eliminating roles for T-bet and IL-2 but revealing superior activation and proliferation as mechanisms underpinning dominance of regulatory T cells in tumors. Oncotarget. 2015;6:24649–59. doi: 10.18632/oncotarget.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao S, Ruddle NH. J Immunol. Vol. 177. American Association of Immunologists; 2006. Synchrony of high endothelial venules and lymphatic vessels revealed by immunization; pp. 3369–79. [DOI] [PubMed] [Google Scholar]
- 50.de Chaisemartin L, Goc J, Damotte D, Validire P, Magdeleinat P, Alifano M, et al. Cancer Res. Vol. 71. American Association for Cancer Research; 2011. Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer; pp. 6391–9. [DOI] [PubMed] [Google Scholar]
- 51.Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol. 2015;16:1235–44. doi: 10.1038/ni.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bento DC, Jones E, Junaid S, Tull J, Williams GT, Godkin A, et al. Oncoimmunology. Vol. 4. Taylor & Francis; 2015. High endothelial venules are rare in colorectal cancers but accumulate in extra-tumoral areas with disease progression; p. e974374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.