

Abstract
Purpose
A hallmark of neoplasia is increased ribosome biogenesis, and targeting this process with RNA polymerase I (Pol I) inhibitors has shown some efficacy. We examined the contribution and potential targeting of ribosomal machinery in chemotherapy resistant and sensitive models of ovarian cancer.
Experimental Design
Pol I machinery expression was examined, and subsequently targeted with the Pol I inhibitor CX-5461, in ovarian cancer cell lines, an immortalized surface epithelial line, and patient derived xenograft (PDX) models with and without chemotherapy. Effects on viability, Pol I occupancy of rDNA, ribosomal content, and chemosensitivity were examined.
Results
In PDX models, ribosomal machinery components were increased in chemotherapy-treated tumors compared to controls. 13 cell lines were sensitive to CX-5461, with IC50s 25nM – 2μM. Interestingly two chemoresistant lines were 10.5- and 5.5-fold more sensitive than parental lines. CX-5461 induced DNA damage checkpoint activation and G2/M arrest with increased γH2AX staining. Chemoresistant cells had 2-4-fold increased rDNA Pol I occupancy and increased rRNA synthesis, despite having slower proliferation rates, while ribosome abundance and translational efficiency were not impaired. In five PDX models treated with CX-5461, one showed a complete response, one a 55% reduction in tumor volume, and one maintained stable disease for 45 days.
Conclusions
Pol I inhibition with CX-5461 shows high activity in ovarian cancer cell lines and PDX models, with an enhanced effect on chemoresistant cells. Effects occur independent of proliferation rates or dormancy. This represents a novel therapeutic approach that may have preferential activity in chemoresistant populations.
Keywords: ovarian cancer, ribosomal synthesis, RNA Polymerase I, CX-5461, chemoresistance, patient-derived xenograft
INTRODUCTION
Although ovarian cancer patients usually present with advanced disease, most will have a positive response to initial therapy consisting of surgical debulking and combined platinum/taxane chemotherapy 1. Nonetheless, as many as 80% of patients will ultimately develop recurrence with chemoresistant disease, suggesting that a small chemoresistant population must be identified and targeted in order to achieve durable cures 2. Although in recent years evaluation of multiple chemotherapeutic and targeted agents, alone or in combination, has yielded a modest improvement in survival, drug resistance remains the major cause of death of ovarian cancer patients 3–5.
An increase in size and number of nucleoli was one of the earliest hallmarks of cancer identified and is still a useful prognostic indicator today 6. The nucleolus is the primary site of ribosomal biogenesis and enlarged nucleoli correlate with accelerated ribosomal RNA (rRNA) synthesis by RNA polymerases 6,7. Synthesis of ribosomes is one of the most complex and energy demanding processes in cells. RNA polymerase I (Pol I) transcribes the ribosomal DNA (rDNA) to produce the pre-rRNA precursor that is processed into 18S, 5.8S and 28S rRNAs through a number of nucleolytic steps 8. RNA polymerase III (Pol III) synthesizes the 5S rRNA, which is also a structural component of the ribosome. Together Pol I and Pol III account for ~80 % of total nuclear transcription 8. An increase in ribosomal RNA synthesis in the nucleolus by Pol I has been correlated with an adverse prognosis in cancer 9.
A number of clinically approved chemotherapeutic drugs act, at least in part, through inhibition of ribosomal RNA synthesis 10. However, none of these drugs directly target Pol I 10–12. Recently, small molecule inhibitors have been developed, such as CX-5461 and CX-3543, which have apparent specificity for RNA Pol I 13–16. CX-5461 does not inhibit mRNA synthesis by RNA polymerase II, does not inhibit DNA replication or protein synthesis, and has preferential activity for cancer cells over non-transformed cells 13. Bywater et al. showed that CX-5461 exhibits inhibition of rRNA transcription dependent on TP53 mutational status and induces TP53-dependent apoptotic cell death of hematologic malignancies, while maintaining normal cells 17.
Our interest in CX-5461 came from the discovery that in heterogeneous PDX models of ovarian cancer treated with chemotherapy, high-throughput analysis suggested that ribosomal machinery was upregulated in the surviving population. Other investigators have suggested that ribosomal biogenesis is increased in gynecologic cancers and 18 might be a key component in the efficacy of some targeted therapies, such as RUNX factors 19 and c-MYC’s role in both cancer and global gene expression. However, an association of this process with chemoresistance and a mechanism to directly target ribosomal biogenesis in ovarian cancer has not been examined. In this study, we further investigate the activity of RNA Pol-I in treated tumors and the effects and mechanisms of CX-5461 on the viability of both chemosensitive and chemoresistant ovarian cancer, in cell lines and PDX models. We show that CX-5461 has activity alone on ovarian cancer cells and demonstrates increased efficacy in taxane-resistant ovarian cancer cells, independent of TP53 mutational status. Chemo-responsive ovarian PDX tumors show an increase in Pol I activity after chemotherapy, and have a variable response to Pol I inhibition, achieving a complete response in one model. These findings suggest that targeting Pol I is an exciting opportunity as a therapeutic strategy in treating ovarian cancer, particularly in targeting the chemoresistant population.
MATERIALS AND METHODS
Reagents and Cell Culture
CX-5461 was purchased from ChemScene and dissolved in 50 mmol/L NaH2PO4 (pH 4.5) to make 10mmol/L stock solution. The chemosensitive and chemoresistant ovarian cancer cell line matched pairs A2780ip2, A2780cp20, HeyA8, HeyA8MDR, SKOV3ip1, and SKOV3TRip2 as well as PEO1, PEO4, ES2, OVCAR3, CaOV3, OV90, OAW42, OVSAHO and HIO-180 20–25 were maintained in RPMI-1640 medium supplemented with 10% FBS (Hyclone). A2780cp20 (platinum resistant), HeyA8MDR (paclitaxel resistant), and SKOV3TRip2 (paclitaxel resistant, a kind gift of Dr. Michael Seiden) 26 were previously generated by sequential exposure to increasing concentrations of chemotherapy. All ovarian cancer cell lines were routinely screened for Mycoplasma species (GenProbe Detection Kit; Fisher) with experiments carried out at 70% to 80% confluent cultures. Purity of stock cell lines was confirmed with short tandem repeat genomic analysis, and all cell lines were used within 20 passages from stocks.
Ovarian Cancer Patient Derived Xenograft Model
Multiple ovarian cancer PDX models were developed and previously characterized by our group 27. Briefly, freshly-collected untreated patient tumors collected at the time of primary debulking were implanted subcutaneously into SCID mice and allowed to propagate. For 6 models, mice with established PDX tumors (approximately 1cm in greatest diameter) were treated with weekly doses of carboplatin (90 mg/kg) and paclitaxel (20 mg/kg), intraperitoneally, for 4 weeks. All tumors had greater than 50% reduction in size, and were collected in multiple formats. RNA-Seq was performed on treated and untreated tumors, and expression profiles in matched pairs were compared using IPA software.
To evaluate changes in nucleoli after chemotherapy staff pathologists took four random images of cancer cell rich areas of H&E stained PDX tissue samples before and after combined carboplatin/paclitaxel therapy. Cells were counted both manually and automatically with CellProfiler, quantitating nucleolar area changes and determining statistical significance using GraphPad Prism (La Jolla, CA).
For examination of the efficacy of CX-5461 in PDX models, previously-collected PDX tumors either 2 or 3 generations from original collection, from 5 separate patients, were reimplanted into 5 mice per model, with 2 tumors per mouse. After tumors reached 1cm in at least 1 dimension, treatment with single-agent CX-5461 (50 mg/kg q3d) was initiated and response was followed using caliper measurements. Tumor volume was calculated using the formula (Length × Width2)/2. Tumors response was averaged among all tumors and presented as change in volume compared to volume at initiation of treatment.
Assessment of cell viability and cell cycle analysis
2,000 cells/well were plated on 96-well plates and treated the next day with increasing concentrations of CX-5461, alone or in combination with carboplatin or paclitaxel, in triplicate. Viability was assessed by 2-hour incubation with 0.15% MTT (Sigma) and spectrophotometric analysis at OD570 (optical density at 570 nm). IC50 calculation was performed in GraphPad Prism® (version 6.0, La Jolla, CA) using variable slope, four-parameter logistic curve where the model is: Y=Bottom + (Top-Bottom)/(1+10ˆ((LogIC50-X)*HillSlope)). Drug concentration data (log transformed) and the percent inhibition data compared to vehicle control were fitted to the four-parameter model.
For cell-cycle analysis, cells were treated with vehicle alone or CX-5461 at the IC50 and IC90 dose for 48 hours, trypsinized, and fixed in 100% ethanol overnight. Cells were then centrifuged, washed in PBS, and suspended in PBS containing 0.1% Triton X-100 (v/v) 200 μg/ml DNase-free RNase A, and 20 μg/mL Propidium iodide (PI). PI fluorescence was assessed by flow cytometry and the percentage of cells in sub-G0, G0-G1, S-, and G2-M phases was calculated by the cell-cycle analysis module for Flow Cytometry Analysis Software (FlowJo v7.6.1, Ashland, OR).
Immunoblotting and Immunofluorescence
For immunoblotting: 15 μg of cell lysate was run on 10% Bis-Tris NuPage gels (ThermoFisher), transferred to 0.45 μM PVDF using a semi-dry Midi transfer system (ThermoFisher), blocked in 5% (w/v) non-fat dry milk and incubated overnight with primary antibodies at 4 degrees C. After detection with the appropriate HRP-conjugated secondary antibody, blots were developed using SuperSignal™ West Pico Chemiluminescent Substrate (ThermoFisher) and imaged using X-ray film. Antibodies used were incubated at manufacturer’s recommended dilutions: pCHK1(ser317)(1:1000), pCHK2(T68)(1:1000), pCDC2(Tyr15)(1:1000), γH2AX(ser139)(1:2000), Cyclin-B(1:500), α-tubulin(1:5000), and p21(1:1000) from Cell Signaling (Danvers, MA).
For immunofluorescence, cells were plated in 8-well chamber slides (Nunc Lab-Tek II CC2, Thermo Scientific) at densities of 10,000 cells per well. Cells were fixed with 4% paraformaldehyde and blocked with 2% BSA for one hour. After primary antibody incubation, wells were detected with anti-rabbit/mouse Alexa-Flour 568 (Cell Signaling, Danvers, MA) at 1:500 dilution for one hour at room temperature and counterstained/mounted with DAPI in Prolong Gold Antifade (Invitrogen). A random selection of multiple images was captured using a Photometrix CoolSnapHQ CCD camera with a 20x objective and analyzed both manually and with the image analysis software CellProfiler (Broad Institute, Cambridge, MA).
RNA Extraction and Reverse Transcription
Total RNA was extracted from the ovarian cancer cell lines and ovarian cancer PDX tumors using the RNeasy Mini kit per the manufacturer’s instructions (Qiagen, Frederick, MD). The concentrations of all RNA samples were quantified using spectrophotometric absorbance at 260/280 nm. cDNA was prepared using the High Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA).
Quantitative PCR
Primer and probe sets for UBTF (Hs01115792_g1), 18S (Hs99999901_s1), RNA28S (Hs03654441_s1), β-Actin (Hs01060665_g1, Housekeeping Gene), POLR1B (Hs00219263_m1), and RRN3 (Hs01592557_m1) were obtained from Applied Biosystems and used according to manufacturer’s instructions. ITS1, pre-rRNA, was custom ordered from Applied Biosystems (Forward Primer: CCGCGCTCTACCTTACCTACCT, 3′-Primer: GCATGGCTTAATCTTTGAGACAAG, Probe: TTGATCCTGCCAGTAGC). PCR amplification was performed on an ABI Prism 7900HT and gene expression was calculated using the comparative CT method as previously described 28.
Polysome fraction assay
For an assessment of ribosomal subunit populations and efficiency of translation, sucrose gradient fractionation was performed. Cells were grown to ~70% confluence in RPMI (10% FBS), treated with cycloheximide (100 μg/ml), washed in PBS, and cytoplasmic extracts were layered onto 10% to 50% linear sucrose gradients and centrifuged at 30,000 rpm in a Beckman SW41 ultracentrifuge rotor for 5 hours. To visualize ribosome populations, 60% sucrose was pumped into the bottom of each column and absorbance at 254 nm was monitored during elution from the top. Three different biological replicates were performed for each cell line, and representative traces are shown.
Chromatin immunoprecipitation
SKOV3ip1 and SKOV3TRip2 cells were grown to ~80% confluence and treated with formaldehyde (1% final concentration) for 10 minutes and then incubated in 0.125M glycine for an additional 5 minutes. Cells were washed in cold 1x PBS, and then processed for ChIP as described previously 29. Immunoprecipitation was performed with an anti-RPA194 antibody (Santa Cruz Biotechnology; SC-48385).
Isotopic labeling of cellular RNA
Cells were grown to ~80% confluence as described above in six well dishes. At time zero, 32P-orthophosphate was added to each well (20 μCi/ml) and incubated for the indicated times. Medium was removed and Triazole was added directly to the cells for harvest. RNA was purified and run on a 1% formaldehyde:agarose denaturing gel. RNA was transferred from the gel onto Zeta-Probe blotting membrane (BioRad, Hercules, CA), dried and analyzed by phosphoimaging.
RESULTS
Increase in expression of ribosomal machinery by chemotherapy
As previously reported 27, six PDX models were established immediately after resection from advanced high-grade serous ovarian cancer patients, with 10 mice per model. When tumors were 0.75cm in at least one dimension, mice were treated with either vehicle or combined carboplatin/paclitaxel, weekly for 4 weeks. 7 days after the final dose (to allow acute effects of chemotherapy to dissipate), tumors were collected and preserved in multiple formats. RNA was extracted and subjected to RNA-Seq analysis. IPA pathway analysis comparing matched treated and untreated PDX, described more thoroughly in our previous report, found that ribosomal synthesis machinery was significantly different in all pairs, and was the most commonly upregulated pathway after treatment in 4 of the 6 pairs. Our first priority after this preliminary global analysis was to confirm whether findings related to increases in ribosomal machinery with treatment could be verified. To confirm these high-throughput data, qPCR was conducted on the matched treated-untreated ovarian cancer PDX for UBTF, POLR1B, and RRN3. These genes encode transcription factors for rRNA synthesis (UBTF and RRN3) as well as the second largest subunit of Pol I (POLR1B), and can influence the rate of rRNA synthesis when differentially expressed 15,30. In all 6 pairs, either UBTF, POLR1B, or both were significantly upregulated in the post-therapy samples compared to their untreated tumor, with RRN3 upregulated in two (Figure 1A, B, C). The degree of increase was, however, highly variable in the 6 models. Additionally, the amount of 18S rRNA and 28S rRNA was determined, as a measure of overall ribosomal content. There was a surprising increase in the relative expression of ribosomes after chemotherapy treatment. 18S levels increased 6.59-fold (p=0.010) and 28S levels increased 5.53-fold (p=0.019) (Figure 1D). This was especially surprising, given our previous finding that immunohistochemical staining of these same samples for Ki-67 showed that proliferation was significantly reduced, on average from 67% in untreated and 12% in treated samples. Dogma would suggest that ribosomal production should correlate with proliferation rates, but clearly does not in the setting of chemotherapy. The significant upregulation of ribosomal activity and total ribosomes in surviving cells led us to hypothesize that inhibiting ribosomal synthesis may be a method for targeting ovarian cancer, and specifically the cell populations surviving primary chemotherapy. We previously presented a principle component analysis of 6 pairs of untreated/treated PDX tumors, showing that although tumors had a different baseline expression profile, as would be expected from 6 different patients, the changes when comparing matched treated and untreated tumors were similar. This relationship suggests that although tumors are different initially, globally they are having very similar responses to chemotherapy.
Figure 1. Expression of RNA Polymerase I initiation factors in ovarian cancer PDX models. Comparison of 6 pairs of untreated/treated (Carbo/taxol) PDX tumors showed similar changes in expression profiles. One common pathway was ribosomal synthesis.
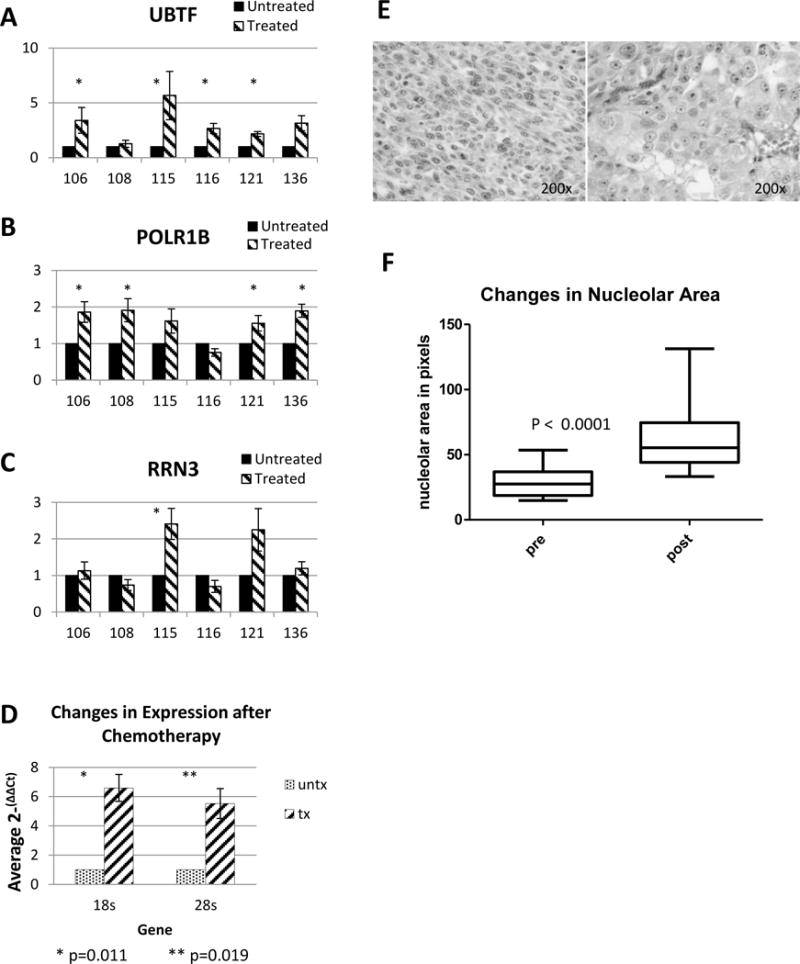
(A,B,C) qPCR was conducted on 6 pairs of ovarian cancer PDX treated with carboplatin and paclitaxel or control for UBTF, POLR1B, and RRN3 and gene expression was compared to the untreated matched PDX. The tumor cell population surviving initial chemotherapy generally had a greater expression of UBTF, POLR1B, and RRN3. (* = p < 0.05) (D) Total level of ribosomal subunits 18S and 28S was determined. Treated ovarian cancer PDXs had a 37.9-fold increase of 18S (p=0.010) and a 39.0-fold increase of 28S (p=0.019). (E) Comparison of H&E sections with and without treatment, demonstrating marked macronucleoli by (F) Quantification of macronucleoli before and after chemotherapy treatment in PDX models.
The increase in ribosomal synthesis activity led us to pathologically examine nucleoli of untreated and treated tumors. We found that nucleoli were much more prominent in PDX tumors after treatment (Figure 1F). The examining pathologist noted that this was a common finding seen in patient samples collected after neoadjuvant chemotherapy, and has been previously published in breast and ovarian cancer 31. In a blinded manner, the pathologist scored the percentage of malignant cells that would be considered to have prominent “macronucleoli”. In 3 of the 4 paired sections in which enough tumor was available to examine, there were significantly more macronucleoli after chemotherapy treatment (Figure 1G).
RNA Pol-I inhibition diminishes cell viability in ovarian cancer cell lines
CX-5461 has recently been reported as a selective inhibitor of Pol 1 with the ability to inhibit solid tumor growth 15, although in some settings it has been reported to be more effective in cells with wild-type TP53. In high-grade serous ovarian cancer, TP53 mutations have been identified in 96% of tumors 32. Nonetheless, based on the RNA-seq and follow up qPCR data, we examined the response of multiple ovarian cancer cell lines, and in 3 instances had matched chemoresistant lines: A2780ip/A2780cp20, SKOV3ip1/SKOV3TR, and HeyA8/HeyA8MDR. Viability as measured by the MTT assay demonstrated an IC50 range of 32 nM-5.5 μM (Table 1). Interestingly, in the matched chemosensitive/chemoresistant pairs, the two taxane-resistant lines SKOV3TRip2 and HeyA8MDR were more sensitive to CX-5461 than their matched chemosensitive SKOV3ip1 and HeyA8 lines, by 5.5 and 10.5-fold, respectively (Figure 2A, 2B). This difference was independent of TP53 status, as both SKOV3 lines have a missense mutation with no detectable expression at the protein level; and both HeyA8 lines are TP53 wildtype 28 and 33. These genotypes were confirmed in our cell lines (data not shown). Interestingly, A2780ip2 was more sensitive to CX-5461 than the platinum-resistant A2780cp20, with an IC50 of 32 nM compared to 146 nM (Figure 2C). TP53 status was examined in these lines, and it was discovered that A2780cp20 at some point in its evolution acquired a TP53 mutation (missense previously published 34 and confirmed in our cells). This mutation in TP53 likely explains the difference in CX-5461 sensitivity. Together, these differential effects demonstrate that although TP53 mutations can affect CX-5461 sensitivity, TP53-mutated cells in many cases have excellent sensitivity. These data also suggest that the mechanism of action of CX-5461 goes beyond that mediated by TP53. Additionally, the increased sensitivity in taxane resistant cells suggests that this might be an outstanding therapy in the setting of resistant disease, consistent with the in vivo observation that treated tumors had increased ribosomal synthesis.
Table 1.
IC50’s of CX-5461 in ovarian cell lines
| Translational Relevance | Translational Relevance | Translational Relevance |
|---|---|---|
| A2780ip2 | WT | 0.032 |
| A2780cp20 | MT | 0.146 |
| SKOV3ip1 | MT | 0.595 |
| SKOV3TRip2 | MT | 0.109 |
| HeyA8 | WT | 2.05 |
| HeyA8MDR | WT | 0.191 |
| Other Cancer Cell lines | ||
| PEO1 | MT | 0.525 |
| PEO4 | MT | 1.20 |
| OV90 | MT | 1.06 |
| OVCAR3 | MT | 2.00 |
| OAW42 | WT | 0.300 |
| OVSAHO | MT | 0.636 |
| CAOV3 | MT | 0.955 |
| COV362 | MT | 0.645 |
| ES2 | MT | 0.294 |
| Normal immortalized cells | ||
| HIO-180 | WT | 5.49 |
Figure 2. Response of ovarian cancer cell lines to CX-5461.
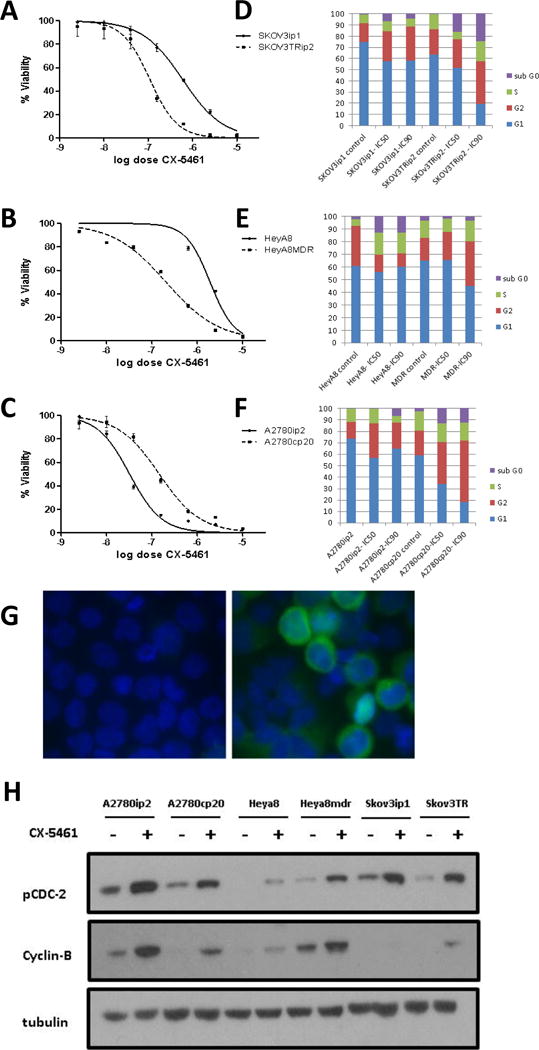
Cell viability and cell cycle analysis was conducted on 3 pairs of chemosensitive and chemoresistant ovarian cancer cell lines. (A–C) Dose response curves from MTT assay of each pair. Results show both SKOV3TRip2 and HeyA8MDR resistant lines being more sensitive to CX-5461 treatment than their chemosensitive parental line. (D-F) Cell cycle analysis of SKOV3ip1, SKOV3TRip2, HeyA8, HeyA8MDR, A2780ip2, and A2780cp20 using Propidium Iodide after 48 hr treatment with the control, IC50, or IC90 dose of CX-5461. In general, treatment with CX-5461 resulted in an increase in sub-G0 fraction and a portion of cells in the S/G2 phase. (G) Immunofluorescence assay example verifying G2/M phase cell cycle arrest in A2780cp20 treated cells with 1μM CX-5461 versus vehicle control. Results show an increase in Cyclin-B perinuclear staining in arrested cells compared to untreated control. (H) Western blot analysis verifying G2/M phase arrest in 6 cell lines. Treatment with IC50 dosages of CX-5461 showed an increase in expression of pCDC2(tyr15) and Cyclin-B versus untreated controls at 72 hours.
CX-5461 causes cell cycle arrest in G2/M
In order to examine the biologic effects of CX-5461, we first examined cell cycle composition with treatment, employing PI incorporation and flow cytometric analysis. The six chemosensitive/chemoresistant paired cell lines were treated with either vehicle control, the IC50 of CX-5461, or the IC90 of CX-5461 for 48 hours. Interestingly, there was pronounced G2/M arrest in both SKOV3 cell lines with the IC90 dose, from 24.48% to 37.89% in SKOV3ip1, and 36.45% to 56.16% in SKOV3TRip2 (Figure 2E). This was also observed with the IC90 dose in A2780cp20 (38.86% to 69.61%), HeyA8MDR (31.75% to 51.49%) and the IC50 dose in A2780ip2 (26.31% to 42.71%) (Figure 2D,F). An arrest in G2/M is consistent with prior observations that Pol I activity is the highest in the G2/M phase 35.
To confirm arrest at G2/M, cyclin B was measured by immunofluorescence. The G2/M transition is tightly regulated by the interplay of cyclin B and CDC2, with the dephosphorylation of CDC2 being required for movement into mitosis by the phosphatase CDC25. G2-arrested cells should show a buildup of cyclin B and pCDC2 (tyr15) in arrested cells. Immunofluorescence after treatment with CX-5461 showed intense staining of cyclin B appearing perinuclear after treatment (Figure 2G). Furthermore, immunoblotting indicated an increase in pCDC2 (tyr15) and cyclin B after treatment in all cell lines (Figure 2H), verifying significant arrest in late G2.
Transcription of rDNA is increased in chemoresistant cell lines despite lower overall growth rate
Since protein synthesis rate is directly proportional to cell growth rate, ribosomal production is traditionally thought to mirror proliferation rates. However this paradigm is not consistent with our in vivo observation that treated tumors had upregulation of Pol I machinery despite low proliferation rates, and the SKOV3TRip2 and HeyA8MDR cell lines were more sensitive to CX-5461 despite their lower doubling times. To identify the potential mechanism by which chemoresistant tumor cells are more sensitive to inhibition of rRNA synthesis, we characterized rRNA synthesis in SKOV3ip1 and SKOV3TRip2 cells. Based on isotopic labeling (Figure 3A) we observed a dramatic increase in rRNA synthesis in the chemoresistant line compared to the control cells. To confirm our isotopic labelling results, we measured pre-rRNA abundance using RT-qPCR, probing for the 5′-external transcribed sequence (5′-ETS; Figure 3B). Since this RNA species is short-lived, its abundance is used as a measure of rRNA synthesis rate. We observed a significant increase in HeyA8MDR compared to parental HeyA8; in SKOV3TRip2 compared to SKOV3ip1; but a nonsignificant decrease in A2780cp20 compared to A2780ip2. Therefore relative sensitivity to CX-5461 does follow with rRNA synthesis rate, at least when comparing the pairs. It does not follow with sensitivity when comparing different cell lines, and therefore cannot be used as an absolute marker of response alone. However, it does confirm that rRNA production does not follow precisely with proliferation rate, and is increased in the taxane-resistant lines when compared to parental controls. Increased total rRNA can be achieved by increasing the rate of transcription by Pol I, by inhibition of rRNA decay, or by a combination of these mechanisms. To test whether transcription by Pol I was induced, we measured polymerase occupancy on the rDNA repeat using chromatin immunoprecipitation (ChIP). We observed a two- to three-fold increase in polymerase occupancy of the gene in the SKOV3TRip2 cells compared to the parental line (Figure 3C). Together, these data suggest that synthesis of rRNA by Pol I is increased in the taxane-resistant line, despite its slower growth rate.
Figure 3. rRNA synthesis and polymerase occupancy is increased in SKOV3TRip2 cells compared to SKOV3ip1 cells.
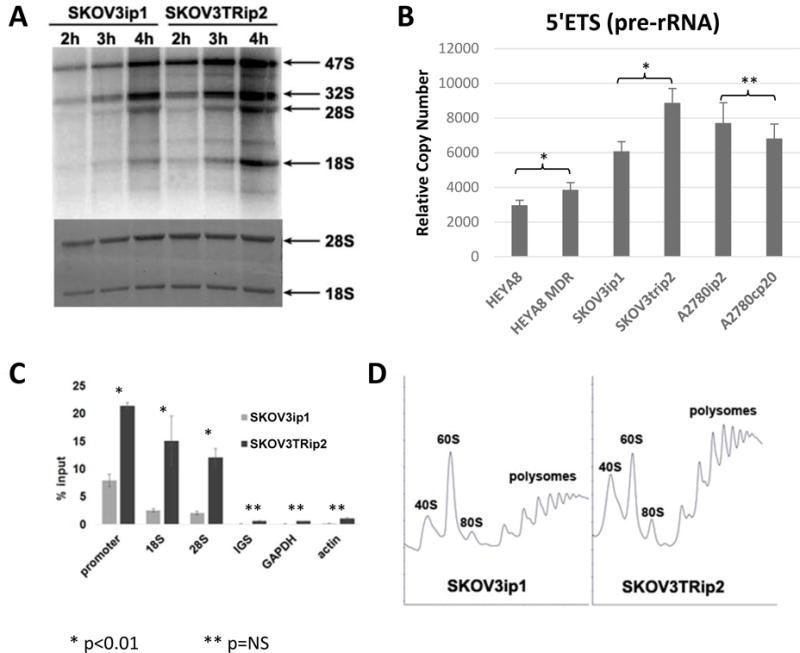
(A) Cells were pulse labeled with 32P-orthophosphate as a function of time. Total RNA was purified by Trizol, run on a formaldehyde denaturing agarose gel, dried, and visualized by autoradiography. The rate of 32P incorporation into rRNA in SKOV3TRip2 cells was 2.1 fold (+/- 0.3) higher than in SKOV3ip1 cells. (B) rDNA transcription was analyzed by RT-qPCR by targeting short-lived 5′ETS, and the rRNA signal was normalized to the control GAPDH mRNA. (C) CHIP analysis of RPA194 association with rDNA. Pol I molecules were immunoprecipitated by anti-A194 monoclonal antibody. The mean and SD are shown. (D) Polysome profiles were performed by sucrose gradient centrifugation.
Enhanced production of rRNA could suggest that more ribosomes are present and functional in the taxane-resistant cells, or may represent an overproduction in response to faulty ribosomal functionality. In order to semi-quantitatively examine engagement of ribosomes onto mRNA, we performed sucrose gradient fractionation of cytoplasmic fractions from cycloheximide-treated cells. As shown in figure 3D, ribosomes profiles from equal cell numbers reveal competent ability of ribosomes to bind to mRNA in both cell lines. Each peak following the 40S, 60S, and 80S peaks represents an mRNA strand with at least 2 ribosomes attached. The first peak is composed of an mRNA with 2 ribosomes attached, the next with 3 ribosomes, and so on. There is a suggestion that there are actually more actively-translating ribosomes/mRNA units, though this method is semi-quantitative and that conclusion is not definitive. However the prominent polysome peaks do confirm that there is not a decrease in the number of translating ribosome/mRNA units in the SKOV3TRip2 line, and therefore increased production of ribosomes is not due to defective ribosomal functionality. It does suggest that the increased ribosome production leads to increased translation. Therefore chemoresistant cells appear to be using increasing translation as a mechanism of chemoresistance.
Together, all of these data suggest that rRNA synthesis by Pol I is dramatically increased in the taxane resistant cells without a complementary increase in ribosome concentration, or a deficiency in ribosomal functionality, despite its slower growth rate. This is contrary to the dogma that proliferation and ribosomal production/activity are universally directly correlated.
Treatment with CX-5461 increases DNA damage markers in ovarian cancer cell lines
G2/M checkpoint arrest is a frequent consequence of genotoxic insult and is used to prevent entry into mitosis after DNA damage has occurred by a variety of mechanisms 36. Early sensing of DNA damage is done by the kinases ATM and ATR and further activation of the checkpoint is mediated through a variety of signals, both TP53 dependent and independent 37–39. An indicator of multiple forms of DNA damage is marked via phosphorylation of the histone γH2AX at Ser139 by the kinases ATM and ATR 40. After treatment with CX-5461, ovarian cancer cell lines showed a significant increase in γH2AX(Ser139) foci in the nucleus (Figure 4A). After DNA damage is detected, the ATM/ATR kinases also activate their downstream effectors CHK1 and CHK2, beginning a broad range of signaling, from inactivation of CDC25 leading to G2 arrest to TP53-mediated apoptosis. Immunoblotting after CX-5461 treatment showed an increase in the phosphorylated forms of CHK1(Ser317) and CHK2(Tyr68) as well as a dramatic increase in the TP53-mediated tumor suppressor p21 in the TP53 wild type cell line A2780ip2 (Figure 4B).
Figure 4. CX-5461 induces DNA damage response via ATM/ATR in ovarian cancer cell lines.
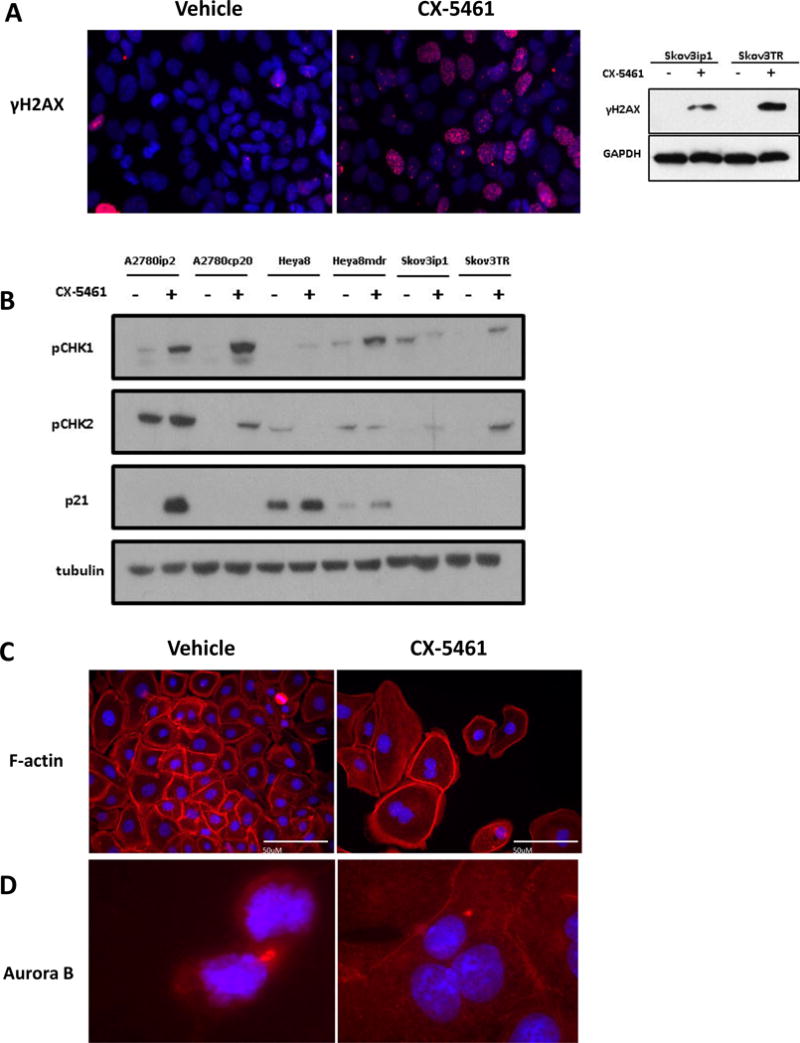
(A) Immunofluorescent detection of the pH2AX in control SKOV3ip1 cells (red) and cells exposed to 500nM of CX-5461. The punctate nuclear staining illustrated an accumulation of DNA damage foci. Pairs of chemosensitive/chemoresistant ovarian cancer cell lines were treated with their respective IC50 dosages of CX-5461. (B) Western blot analysis shows activation of the pCHK1(ser317) and pCHK2(T68), downstream substrates of ATM and ATR kinases. TP53 wild type cell lines showed a subsequent activation of p21 but drug response was independent of TP53 status. (C) DAPI (blue) labeling showed failed cytokinesis and an accumulation of multinucleated cells after CX-5461 treatment, with F-actin (red) indicating shared cytoplasm. (D) Aurora-B kinase (red) detection showing normal localization to the cleavage furrow during cytokinesis in control SKOV3ip1 cells. After treatment with CX-5461 Aurora-B mislocalization was apparent in an accumulation of multinucleated cells.
CX-5461 induces mitotic catastrophe
In performing immunofluorescent stains, it was noted that there was a significant increase in the size and presence of multinucleated cells with CX-5461 treatment. During the final stages of functional mitosis, a cleavage furrow is formed and a contractile ring pinches off the nascent daughter cells. Thus cells were stained with F-actin to identify cell membranes, and multinucleated cells were quantified (Figure 4C). Surprisingly, these images are collected at equal power, which can be appreciated if one focuses on the size of the nuclei, which are not significantly different, whereas the total body size is increased dramatically. Characterization and quantitation of binuclei, using both manual and image analysis based methods (Supplemental Figure 1), showed a dramatic increase in cell volume after treatment with CX-5461, as well as an increase in the number of multinucleated cells. We also performed immunofluorescent staining for Aurora-B kinase, as correct localization of this protein is required for cytokinesis. Normally Aurora-B is localized at the condensing contractile ring in between the two daughter cells. In CX-5461-treated cells, however, Aurora-B kinase showed mislocalization during cytokinesis, lateralized relative to the nucleus, suggesting mitotic catastrophe (Figure 4D).
Ovarian Cancer PDXs have variable response to Pol I inhibition
Based on the activity of CX-5461 in vitro even in the presence of TP53 mutations, five different ovarian cancer PDX models (from 5 advanced-stage papillary serous ovarian cancer patients) were treated with single-agent CX-5461 at 50mg/kg every three days for 45 days. Treatment was initiated when tumors were approximately 1.0 cm in at least one dimension, and responses are reported as maximal change after initiation of treatment. Response was varied among the 5 models, with PDX 208 and 182 growing on treatment, PDX 127 having stable disease, and PDX 153 and 225 showing response, with PDX 225 having a complete regression of tumor (Figure 5A). As a measure of health of the mice during treatment, weight was collected with each treatment, and behaviors carefully monitored. There were no behavioral changes, or functional abnormalities such as diarrhea. There was an appropriate weight gain throughout the treatment course (Figure 5B), as has been demonstrated at this dose level in prior studies 17. Tissue sections of liver, kidney, heart, and lung were examined pathologically and there was no gross toxicity to these major organs.
Figure 5. Response of ribosomal translation factors after CX-5461 treatment.
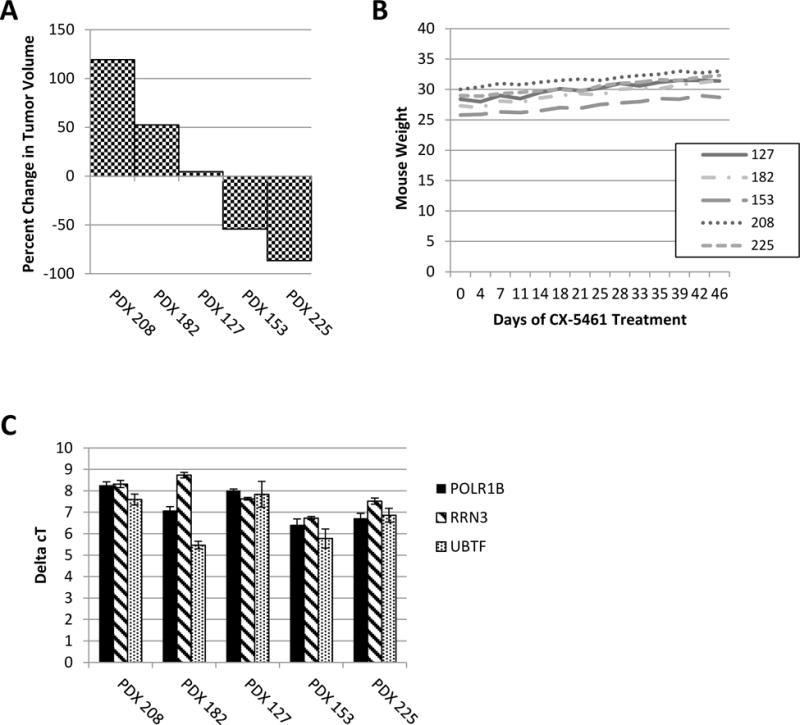
(A) Waterfall plot displaying percent change of tumor volume in PDXs 208, 182, 127, 153, and 225 after 45 days of treatment with 50mg/kg CX-5461 q3D. (B) Weight of mice through duration of treatment with CX-5461. (C) qPCR of Pol I initiation factors RRN3, POLR1B, and UBTF. ΔCT was calculated. There was not an association between baseline expression of these genes and response to CX-5461.
In order to determine if baseline expression of Pol I-associated genes correlated with the response to treatment, in an effort to identify a potential biomarker for response, previously-collected untreated tumors from each PDX model were analyzed using qPCR for POLR1B, RRN3, and UBTF (Figure 5C). There was no significant association between expression of these factors and response to treatment.
Next, to determine the effects of CX-5461 on expression of these genes, qPCR was performed on the treated PDXs 208, 182, 127, and 153 for POLR1B, RRN3, and UBTF; additionally, ITS1 was examined as a quantification of pre-rRNA. Since PDX 225 had a complete response to CX-5461, there was no tumor to analyze and compare to the untreated sample. Of these genes, the only correlative marker for model 153, which had a positive response, was a decrease in POLR1B, which was not noted in the other 3 models (data not shown). Interestingly, ITS1 decreased with treatment of the 208 and 182 models. These data suggests that while CX-5461 may be targeting and inhibiting ribosome synthesis, the lack of effect in some models may be due to other unknown mechanisms. Additional models and more thorough interrogation of the full expression profiles of these tumors will be required to fully understand the variability in response and potentially identify biomarkers predictive of response.
DISCUSSION
Ovarian cancer unfortunately has limited chemotherapeutic options after patients develop resistance to the standard regiment of platinum- and taxane-based therapy. Therefore, it is imperative to identify novel targets that could be used in conjunction with standard therapy to hopefully improve patient outcomes. Previously, our group has developed and characterized an ovarian cancer PDX model that recapitulates the complexity of a patient’s tumor in terms of heterogeneity and biological activity 27. When we conducted RNA-seq analysis comparing matched chemotherapy treated and untreated PDXs, we found that genes related to ribosomal synthesis were significantly up-regulated in the tumor cells surviving a platinum and taxane based therapy. In this study, we confirm that ribosomal machinery, particularly basal ribosomal RNA transcription components, are increased in surviving tumors, and that targeting Pol I is effective in cell lines and PDX models in vivo. We show that efficacy is not entirely dependent on TP53 status or proliferation rates, and is more effective in taxane-resistant cell lines. The mechanism of its toxicity is in part through induction of arrest in G2/M of the cell cycle with frequent mitotic catastrophe, and significant DNA damage.
Transcription of rRNA genes by Pol I and subsequent processing of the rRNA are fundamental control steps in the synthesis of functional ribosomes 6,7,11. Chemotherapeutic agents have been known for years to cause an increase in nucleolar size and reorganization of nucleolar morphology 41. Although it is generally thought that increases in nucleolar size are associated with increased ribosomal biogenesis, it is a matter of debate as to what the increase means after chemotherapy. He et al. have presented evidence suggesting a ribosomal autophagy (ribophagy) pathway can be activated in cancer cells after chemotherapy as part of the survival process 42. This pathway is distinct from the general autophagy pathway, and it is thought that this can be both a mechanism the cell can use to lower energy demands in times of nutrient deprivation, and also a way to globally shut down translation rapidly by consuming mature 60s and 40s ribosomal subunits. If the latter is active then the generation of nascent ribosomes to replace the degraded mature subunits could account for the overall increase in nucleolar size 35. This could account for the overall increased sensitivity of the chemoresistant population to CX-5461 treatment. If rRNA transcription in the nucleolus is inhibited, cells undergo cell cycle arrest associated with apoptosis, senescence or autophagy 43. Aberrant regulation of rRNA synthesis by Pol I and ribosome biogenesis (the complex and highly coordinated cellular process leading to the production of ribosomes) is associated with the etiology of a broad range of human diseases and is especially pervasive in cancer 9,44,45.
Admittedly our discovered association between ribosomal biogenesis and survival after chemotherapy treatment does not delineate whether we are seeing changes that are induced by exposure to chemotherapy, or if treatment with chemotherapy selects cells with inherently different ribosomal biogenesis processes ongoing that allow for their survival, and thus are revealed by our selection method. Furthermore our analysis does not distinguish between different types of ribosomes destined to mediate transcription of particular genes. It has been shown that there are specialized subpopulations of ribosomes 46. Further studies are needed to distinguish between these possibilities, and discover how cells respond to chemotherapy that may allow survival.
There have been numerous chemotherapeutic drugs developed that have an impact on ribosomal biogenesis, even if the effects are nonspecific, including cisplatin, actinomycin D, camptothecin (irinotecan/topotecan), mitomycin C, 5-fluorouracil, and doxorubicin 10–12. However, none of these chemotherapeutic drugs selectively targets Pol I to allow definitive conclusions on what degree their therapeutic effect is mediated through ribosome biogenesis 45. CX-5461 was recently developed by Cylene Pharmaceuticals and is an orally available small molecule that targets transcription by Pol I selectively 13,15. CX-5461 is thought to impair initiation of Pol I transcription by disrupting the binding of the Pol I transcription initiation factor SL-1 to the ribosomal DNA promoter 13,15. The marking of double strand breaks by γH2AX is a well characterized phenomenon 36–38,47,48. Double strand breaks do induce γH2AX but this phosphorylation is not limited only to DNA strand breakage 49. The consistent, late (48-72hr) marking of γH2AX after CX-5461 treatment in ovarian cancer cells could indicate a senescent state after an overwhelming amount of nucleolar stress. Hannan et al. have found a similar non-canonical ATM/ATR response phenotype, and our results agree with theirs 50. Panov et al. put forth evidence that the stabilization of g-quadruplex formation may play a role in CX-5461’s mechanism of action at the rDNA promoter, and our results show DNA damage response machinery as the most probable route of efficacy, with TP53 activation being a later event that is dependent on DNA damage response machinery as opposed to nucleolar stress 51.
Bywater et al. demonstrated that human hematologic cancer cells (leukemia and lymphoma) with TP53 wild type are more sensitive to CX-5461 than TP53 mutant cells in vitro and in vivo. As a result, they suggested that CX-5461 has therapeutic effect for hematologic malignancies by TP53 mutational status-dependent apoptosis 17. But, in solid tumor cell lines, CX-5461 induces both senescence and autophagy by a TP53-independent process 15,52. These differential responses between the nucleolar stress and cell death according to different types of cancer may mean that hematological malignancies have unique nucleolar biology susceptible to activation of TP53-dependent apoptosis following acute perturbations of ribosome biogenesis. We have seen that CX-5461 is more potent against A2780ip2 cells with TP53 wild type than against A2780cp20 with a TP53 mutation. However, we found that the taxane-resistant lines SKOV3TRip2 and HeyA8MDR were more sensitive to Pol I inhibition than their parental lines, even though SKOV3 has a mutation in TP53 in both lines, while HeyA8 is wild-type for TP53 in both lines. These data demonstrate that there are also important TP53-independent mechanisms induced by CX-5461 which still need to be elucidated. Interestingly, another inhibitor of RNA Pol I has been developed, BMH-21. This molecule binds preferentially to rRNA-encoding DNA, and reportedly behaves in a TP53-independent manner16. Further studies are required to determine if this molecule also has preferential activity against chemoresistant populations.
Based on our RNAseq data and the in vitro cell line data, we moved forward with treating 5 PDX lines with CX-5461 in a single agent setting. If up-regulation of ribosomal synthesis is a hallmark of aggressive cancer and is required for cellular proliferation, inhibition may lead to a novel treatment approach. In a single agent setting, we found a 60% clinically significant benefit rate (complete or partial response, or stable disease). An appropriate concern is whether inhibition of such an important process will be tolerable in patients. We and others have not seen gross organ damage at the pathologic level, and no weight loss in treatment mice. It may be that, similar to cytotoxic agents, cancer cells have higher susceptibility with inhibition of ribosomal biogenesis, and a therapeutic window can be found which is safe in patients. Many more extensive studies are required in this arena, and phase I trials with this medication are underway in Canada (https://clinicaltrials.gov/ct2/show/NCT02719977). Regarding a predictive biomarker, in our investigation, the only unifying feature is that higher basal levels of Pol I initiation factors RRN3 and POLR1B correlated with a greater response to therapy. More data will be required in a larger cohort to confirm where these are effective biomarkers for response. However, overall these data demonstrate the potential of CX-5461, and potentially other inhibitors of ribosomal synthesis, as a new approach to ovarian cancer, both in chemosensitive and chemoresistant tumors.
Supplementary Material
Translational Relevance.
Ovarian cancer remains a devastating diagnosis, with serous epithelial ovarian cancer (EOC) patients succumbing to recurrent chemoresistant disease in 80% of cases. There is a great need for novel therapeutics that specifically target the chemoresistant populations. Our results indicate that in patient-derived xenograft model’s cancer cell populations surviving chemotherapy, the basal ribosomal machinery is upregulated. This evidence led us to ask whether attacking ribosomal biogenesis could be a viable therapeutic target in EOC. We show that the Pol I inhibitor CX-5461 displays efficacy in EOC both in vitro and in vivo, induces significant G2 arrest through mitotic catastrophe, and may have preferential activity in the chemoresistant population. Cancer cells are especially sensitive to CX-5461, which was well-tolerated in mice. Inhibiting ribosomal synthesis with Pol I inhibitors may be a promising strategy in targeting the deadly chemoresistant population in ovarian cancer.
Acknowledgments
Funding support provided in part by UAB Medical Scientist Training Program (NIGMS T32GM008361) to ZCD; GM084946 to DAS; and by the Norma Livingston Ovarian Cancer Foundation, the Research Scientist Development Program (K12 HD00849), the Department of Defense Ovarian Cancer Research Academy (OC093443), and UVA Cancer Center Support Grant (P30 CA44579) to CNL.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Berek JS, Crum C, Friedlander M. Cancer of the ovary, fallopian tube, and peritoneum. Int J Gynaecol Obstet. 2012;119(Suppl):S118–29. doi: 10.1016/S0020-7292(12)60025-3. [DOI] [PubMed] [Google Scholar]
- 2.Bowtell DD, Böhm S, Ahmed AA, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668–679. doi: 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong DK. Relapsed ovarian cancer: challenges and management strategies for a chronic disease. Oncologist. 2002;7(Suppl 5):20–8. doi: 10.1634/theoncologist.7-suppl_5-20. [DOI] [PubMed] [Google Scholar]
- 4.Borst P. Cancer drug pan-resistance: pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol. 2012;2:120066. doi: 10.1098/rsob.120066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubin SC, Randall TC, Armstrong KA, et al. Ten-year follow-up of ovarian cancer patients after second-look laparotomy with negative findings. Obstet Gynecol. 1999;93:21–4. doi: 10.1016/s0029-7844(98)00334-2. [DOI] [PubMed] [Google Scholar]
- 6.Shiue C-N, Arabi A, Wright APH. Nucleolar organization, growth control and cancer. Epigenetics. 2010;5:200–5. doi: 10.4161/epi.5.3.11376. [DOI] [PubMed] [Google Scholar]
- 7.Thomson E, Ferreira-Cerca S, Hurt E. Eukaryotic ribosome biogenesis at a glance. J Cell Sci. 2013;126:4815–21. doi: 10.1242/jcs.111948. [DOI] [PubMed] [Google Scholar]
- 8.White RJ. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008;24:622–9. doi: 10.1016/j.tig.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Williamson D, Lu Y-J, Fang C, et al. Nascent pre-rRNA overexpression correlates with an adverse prognosis in alveolar rhabdomyosarcoma. Genes Chromosomes Cancer. 2006;45:839–45. doi: 10.1002/gcc.20347. [DOI] [PubMed] [Google Scholar]
- 10.Burger K, Mühl B, Harasim T, et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 2010;285:12416–12425. doi: 10.1074/jbc.M109.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannan KM, Sanij E, Rothblum LI, et al. Dysregulation of RNA polymerase I transcription during disease. Biochim Biophys Acta – Gene Regul Mech. 2013;1829:342–360. doi: 10.1016/j.bbagrm.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drygin D, Rice WG, Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu Rev Pharmacol Toxicol. 2010;50:131–156. doi: 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- 13.Haddach M, Schwaebe MK, Michaux J, et al. Discovery of CX-5461, the first direct and selective inhibitor of RNA polymerase I, for cancer therapeutics. ACS Med Chem Lett. 2012;3:602–606. doi: 10.1021/ml300110s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drygin D, Siddiqui-Jain A, O’Brien S, et al. Anticancer Activity of CX-3543: A Direct Inhibitor of rRNA Biogenesis. Cancer Res. 2009;69:7653–7661. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- 15.Drygin D, Lin A, Bliesath J, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- 16.Peltonen K, Colis L, Liu H, et al. Small Molecule BMH-Compounds That Inhibit RNA Polymerase I and Cause Nucleolar Stress. Mol Cancer Ther. 2014;13:2537–2546. doi: 10.1158/1535-7163.MCT-14-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bywater MJ, Poortinga G, Sanij E, et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Artero-Castro A, Castellvi J, García A, et al. Expression of the ribosomal proteins Rplp0, Rplp1, and Rplp2 in gynecologic tumors. Hum Pathol. 2011;42:194–203. doi: 10.1016/j.humpath.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 19.Deltcheva E, Nimmo R. RUNX transcription factors at the interface of stem cells and cancer. Biochem J. 474 doi: 10.1042/BCJ20160632. [DOI] [PubMed] [Google Scholar]
- 20.Louie KG, Behrens BC, Kinsella TJ, et al. Radiation survival parameters of antineoplastic drug-sensitive and -resistant human ovarian cancer cell lines and their modification by buthionine sulfoximine. Cancer Res. 1985;45:2110–5. [PubMed] [Google Scholar]
- 21.Landen CN, Kim T-J, Lin YG, et al. Tumor-selective response to antibody-mediated targeting of alphavbeta3 integrin in ovarian cancer. Neoplasia. 2008;10:1259–67. doi: 10.1593/neo.08740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halder J, Kamat AA, Landen CN, et al. Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin Cancer Res. 2006;12:4916–24. doi: 10.1158/1078-0432.CCR-06-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buick RN, Pullano R, Trent JM. Comparative properties of five human ovarian adenocarcinoma cell lines. Cancer Res. 1985;45:3668–76. [PubMed] [Google Scholar]
- 24.Moore DH, Allison B, Look KY, et al. Collagenase expression in ovarian cancer cell lines. Gynecol Oncol. 1997;65:78–82. doi: 10.1006/gyno.1997.4628. [DOI] [PubMed] [Google Scholar]
- 25.Yu D, Wolf JK, Scanlon M, et al. Enhanced c-erbB-2/neu expression in human ovarian cancer cells correlates with more severe malignancy that can be suppressed by E1A. Cancer Res. 1993;53:891–8. [PubMed] [Google Scholar]
- 26.Duan Z, Feller AJ, Toh HC, et al. TRAG-3, a novel gene, isolated from a taxol-resistant ovarian carcinoma cell line. Gene. 1999;229:75–81. doi: 10.1016/s0378-1119(99)00042-6. [DOI] [PubMed] [Google Scholar]
- 27.Dobbin ZC, Katre AA, Steg AD, et al. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget. 2014;5:8750–64. doi: 10.18632/oncotarget.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steg A, Wang W, Blanquicett C, et al. Multiple gene expression analyses in paraffin-embedded tissues by TaqMan low-density array: Application to hedgehog and Wnt pathway analysis in ovarian endometrioid adenocarcinoma. J Mol Diagn. 2006;8:76–83. doi: 10.2353/jmoldx.2006.040402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peltonen K, Colis L, Liu H, et al. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell. 2014;25:77–90. doi: 10.1016/j.ccr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider DA. RNA polymerase I activity is regulated at multiple steps in the transcription cycle: Recent insights into factors that influence transcription elongation. Gene. 2012;493:176–184. doi: 10.1016/j.gene.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Wang Y, Zheng W. Cytologic changes of ovarian epithelial cancer induced by neoadjuvant chemotherapy. Int J Clin Exp Pathol. 2013;6:2121–8. [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research Network. Bell D, Berchuck A, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crane EK, Kwan S-Y, Izaguirre DI, et al. Nutlin-3a: A Potential Therapeutic Opportunity for TP53 Wild-Type Ovarian Carcinomas. PLoS One. 2015;10:e0135101. doi: 10.1371/journal.pone.0135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skilling JS, Squatrito RC, Connor JP, et al. p53 Gene Mutation Analysis and Antisense-Mediated Growth Inhibition of Human Ovarian Carcinoma Cell Lines. Gynecol Oncol. 1996;60:72–80. doi: 10.1006/gyno.1996.0014. [DOI] [PubMed] [Google Scholar]
- 35.Klein J, Grummt I. Cell cycle-dependent regulation of RNA polymerase I transcription: the nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc Natl Acad Sci U S A. 1999;96:6096–101. doi: 10.1073/pnas.96.11.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang Y, Lin S-Y, Brunicardi FC, et al. DNA damage response pathways in tumor suppression and cancer treatment. World J Surg. 2009;33:661–6. doi: 10.1007/s00268-008-9840-1. [DOI] [PubMed] [Google Scholar]
- 37.Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 38.Yuan J, Adamski R, Chen J. Focus on histone variant H2AX: to be or not to be. FEBS Lett. 2010;584:3717–24. doi: 10.1016/j.febslet.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burma S, Chen BP, Murphy M, et al. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 40.Huen MSY, Chen J. The DNA damage response pathways: at the crossroad of protein modifications. Cell Res. 2008;18:8–16. doi: 10.1038/cr.2007.109. [DOI] [PubMed] [Google Scholar]
- 41.McCluggage WG, Lyness RW, Atkinson RJ, et al. Morphological effects of chemotherapy on ovarian carcinoma. J Clin Pathol. 2002;55:27–31. doi: 10.1136/jcp.55.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He J, Yu JJ, Xu Q, et al. Downregulation of ATG14 by EGR1-MIR152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophagy. Autophagy. 2015;11:373–384. doi: 10.1080/15548627.2015.1009781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hannan RD, Drygin D, Pearson RB. Targeting RNA polymerase I transcription and the nucleolus for cancer therapy. Expert Opin Ther Targets. 2013;17:873–878. doi: 10.1517/14728222.2013.818658. [DOI] [PubMed] [Google Scholar]
- 44.Narla A, Ebert BL. Translational medicine: ribosomopathies. Blood. 2011;118:4300–1. doi: 10.1182/blood-2011-08-372250. [DOI] [PubMed] [Google Scholar]
- 45.Bywater MJ, Pearson RB, McArthur GA, et al. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat Rev Cancer. 2013;13:299–314. doi: 10.1038/nrc3496. [DOI] [PubMed] [Google Scholar]
- 46.Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol. 2012;13:355–369. doi: 10.1038/nrm3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bewersdorf J, Bennett BT, Knight KL. H2AX chromatin structures and their response to DNA damage revealed by 4Pi microscopy. Proc Natl Acad Sci U S A. 2006;103:18137–42. doi: 10.1073/pnas.0608709103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinner A, Wu W, Staudt C, et al. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turinetto V, Giachino C. Multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 2015;43:2489–98. doi: 10.1093/nar/gkv061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quin J, Chan KT, Devlin JR, et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget. doi: 10.18632/oncotarget.10452. Epub ahead of print 6 July 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andrews WJ, Panova T, Normand C, et al. Old drug, new target: Ellipticines selectively inhibit RNA polymerase I transcription. J Biol Chem. 2013;288:4567–4582. doi: 10.1074/jbc.M112.411611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li L, Li Y, Zhao J, et al. CX-5461 induces autophagy and inhibits tumor growth via mammalian target of rapamycin-related signaling pathways in osteosarcoma. Onco Targets Ther. 2016;9:5985–5997. doi: 10.2147/OTT.S104513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.