

Abstract
Tumor-stromal communications impact tumorigenesis in ways that are incompletely understood. Here we show that Glioma Associated-human Mesenchymal Stem Cells (GA-hMSCs), a newly identified stromal component of glioblastoma, release exosomes that increase the proliferation and clonogenicity of tumor-initiating Glioma Stem-like Cells (GSCs). This event leads to a significantly greater tumor burden and decreased host survival compared to untreated GSCs in orthotopic xenografts. Analysis of the exosomal content identified miR-1587 as a mediator of the exosomal effects on GSCs, in part via down-regulation of the tumor suppressive nuclear receptor co-repressor NCOR1. Our results illuminate the tumor-supporting role for GA-hMSCs by identifying GA-hMSC-derived exosomes in the intercellular transfer of specific miRNA that enhance the aggressiveness of glioblastoma.
Keywords: exosomes, microRNA, glioma stem cells, mesenchymal stem cells, glioblastoma
Introduction
Glioblastoma (GBM) is the most common and aggressive primary malignant adult brain tumor, and despite surgery, radiation, and chemotherapy the median survival of patients with GBM is just over one year [1]. This poor outcome is due in part to the cell-autonomous functions of therapeutically resistant tumor initiating cells (TICs), also called glioma stem or stem-like cells (GSCs). The surrounding stroma maintains a GSC-supportive niche that likely enhances GSC aggressiveness [2]. Although classically the stroma of GBMs was thought to be composed of reactive astrocytes, endothelial cells, and immune cells, each of which has been implicated in creating pro-glioma conditions [3–5], we and others have recently shown that GBMs harbor stromal cells resembling Mesenchymal Stem Cells (MSCs), which we called Glioma associated human MSCs (GA-hMSCs) [6, 7]. GA-hMSCs are not merely bystanders in the tumor niche, but increase proliferation and self-renewal of GSCs in vitro, and enhance GSC tumorigenicity and mesenchymal features in in vivo intracranial models, confirming that GA-hMSCs are functionally important cells within the GSC niche [6]. Secretion of Interleukin-6 (IL-6) by GA-hMSCs is one major intercellular communication mechanism that mediates the proliferation and stemness of GSCs [6]. However, the mechanisms underlying the communication between the stroma and tumor cells are complex, and additional factors are likely involved.
Recently, evidence is building for a form of communication between neighboring cells that relies on nanosized (50–110 nm diameter), lipid bilayer vesicles, called exosomes, that are secreted by many cell types and taken up by neighboring or distant cells [8]. This type of communication may be especially exploited in pathological conditions such as cancer, where TICs and surrounding stromal cells may develop mutually supportive positive feedback loops of cellular communication [9, 10]. Studies show that exosome-mediated transfer of oncogenic miRNA from cancer cells can alter the biology of non-cancer cells, while the transfer of tumor-suppressor miRNA can inhibit tumor growth [11–13].
Exosomal communication has not been extensively evaluated in glioma. Most recent reports focus on the impact on tumor cell-derived exosomes on stromal components. Glioma cells secrete exosomes that contain mRNA and miRNA and promote blood vessel formation [14]. Extracellular vesicles from gliomas were shown to deliver miR-1 to recipient cells to modify glioma cell invasion and proliferation as well as impact stromal cells to promote endothelial tube formation [15]. Another report demonstrated that GBM-derived exosomes could transfer miRNA to microglia [16].
Studies in other cancer types have provided some clues to the role of stromal-derived exosomes. Exosomes released from cancer-associated fibroblasts (CAFs) in breast cancer increased the invasion and motility of breast cancer cells [17]. More recently, exosomes from gastric carcinoma-mesenchymal stem cells were shown to increase the growth and migration of human gastric carcinoma cells [18]. Although no mechanism by which the exosomal contents mediate these tumor-enhancing effects was established, together these two studies demonstrate the tumor-promoting role of stroma-derived exosomes.
The role of tumor stroma-derived exosomes in the development and evolution of gliomas and the mechanisms by which exosomal communication impacts tumor cells are poorly studied, highlighting a major gap in knowledge. Thus, we sought to investigate exosomal communication within the glioma microenvironment, utilizing GA-hMSCs and GSCs isolated from patient tumors. We hypothesized that the transfer of specific cell regulatory elements via GA-hMSC-derived exosomes could alter the biology of recipient GSCs, resulting in increased proliferation and clonogenicity.
Materials and Methods
GSC and GA-hMSC isolation and characterization
GA-hMSCs were isolated from surgical specimens as previously described [6, 19]. GA-hMSCs lines met the criteria for MSCs as outlined by the International Society for Cellular Therapy (ISCT) [19]. GA-hMSCs have spindle shape morphology, are adherent in culture, and express the mesenchymal surface markers CD73, CD90 and CD105. All GA-hMSCs possessed the ability to tri-differentiate into adipocytes, chondrocytes and osteocytes. GA-hMSCs were cultured in MSC medium: Eagle’s Minimum Essential Medium Alpha (Sigma-Aldrich), 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% glutamine. GSCs were isolated from human glioma surgical specimens as previously described and cultured in serum-free NSC medium: Dulbecco’s Modification of Eagle’s Medium/Ham’s F-12 50/50 mix with L-Glutamine (Corning CellGro), 2% B-27 supplement (Gibco), 20ng/mL EGF, 20ng/mL bFGF, and 1% penicillin-streptomycin [6, 20]. GSCs and GA-hMSCs were validated with short-tandem repeat (STR) fingerprinting in 2012. U251 cells were reauthenticated with STR fingerprinting in 2015. All cells tested Mycoplasma-free prior to freezing and cells were thawed and used for experiments within 5 passages.
Exosome Isolation
GA-hMSCs or GSCs were washed and incubated for 48hrs in serum-free and supplement-free NSC medium. GSC or GA-hMSC-derived exosomes were isolated by differential ultracentrifugation [21]. Briefly, GA-hMSC-derived conditioned medium was filtered and centrifuged at 10,000xg to remove large microvesicles (non-exosomal). The supernatant was ultra-centrifuged at 100,000xg. The raw exosome pellet was resuspended in PBS and ultra-centrifuged at 100,000xg, with or without a 30% sucrose cushion depending on the experimental usage. Exosome pellets were resuspended in PBS, NSC medium or lysis buffer where indicated.
Western Blot Analysis of GA-hMSC-Derived Exosomes
Protein from GA-hMSC-derived exosomes was extracted using membrane lysis buffer. Western blot for the exosomal surface markers CD63 and GAPDH on MSC-derived exosomes, and the corresponding parental cell, were performed using antibodies against CD63 (Santa Cruz Biotechnology, Inc.), GAPDH (Abcam #ab37168), CD16 (Abcam #ab94773) and CD32 (Abcam #ab45143).
Electron Microscopic Analysis of Exosomes
Bone marrow- human MSC (BM-hMSC)-derived and GA-hMSC-derived exosomes were prepared for electron microscopy (EM) according to Théry, et al. [21]. Exosomes were examined by electron microscopy on a JEM 1010 transmission electron microscope (JEOL) at an accelerating voltage of 80 Kv. Digital images were obtained using the AMT Imaging System (Advanced Microscopy Techniques). Exosome grids were subjected to immuno-gold staining of the exosomal surface marker CD63 and the non-exosomal markers CD16 and CD32, utilizing gold-anti-rabbit (Sigma Aldrich #G7277), anti-CD63 (Santa Cruz Biotechnology, Inc. #15363), anti-CD16 (Abcam #ab94773), and anti-CD32 (Abcam #ab45143).
GSC Internalization of GA-hMSC-Derived Exosomes
GA-hMSCs were transduced with a GFP-CD63 lentiviral construct (System Biosciences) and incubated for 48hrs in serum-free NSC medium. GFP-labeled GA-hMSC-derived exosomes were isolated and incubated with GSCs for 4 hours. Excess exosomes were removed by PBS wash, and internalization of GA-hMSC-derived exosomes by GSCs was analyzed by fluorescent confocal microscopy.
Proliferation Assays
GSCs were dissociated and placed in a 96 well plate at 2.5×103 cells/well. GA-hMSC-derived and GSC-derived exosomes quantified by CD63 ELISA were added to GSC cultures at doses of 5×104 and 10×104 exosomes/µL at time zero and at 48hrs, and incubated for a total of 96hrs. GSCs were also treated with NSC medium and GA-hMSC-derived ED-CM. For miRNA experiments, GSCs were treated with either NSC medium, GA-hMSC-derived exosomes (1×105 exosomes/µL), anti-miRNA (Qiagen), or GA-hMSC-derived exosomes (10×104 exosomes/µL) plus anti-miRNA. 1nM anti-miRNA was delivered to GSCs by lipofection, and Lipofectamine was added equally across all treatment groups to control for lipofection effects. After 96hrs, GSCs were assessed for viability using a colorimetric assay (water-soluble tetrazolium [WST-1], Roche). All experiments were performed in quadruplicate.
Clonogenic Assays
GA-hMSC-derived and GSC-derived exosomes were added to single-cell suspensions of GSCs (1 GSC/well verified prior to treatment) in 96-well plates at a dose of 10×104 exosomes/µL at time zero, and at 1 and 2 weeks. GSCs were also treated with NSC medium and GA-hMSC-derived ED-CM. For miRNA experiments, GSCs were treated with NSC medium, GA-hMSC-derived exosomes, anti-miRNA (Qiagen), or GA-hMSC-derived exosomes plus anti-miRNA. Anti-miRNA was delivered to GSCs by lipofection at 1nM. Neurosphere formation was assessed after 3 weeks. All experiments were performed in quadruplicate.
Effects of GA-hMSC-Derived Exosomes on GSC Tumorigenicity
Athymic nude mice (nu/nu) were purchased from the Department of Experimental Radiation Oncology, University of Texas MD Anderson Cancer Center (Houston, TX). All animal studies were performed under an IACUC approved protocol in accordance with federal, state and institutional regulations.
GSCs (5×106 cells) were pre-treated with exosomes (1×109 exosomes/ml), NSC medium, or GA-hMSC-derived ED-CM for 96hrs. Excess exosomes were eliminated by PBS wash, and GSC neurospheres were dissociated. Mice were anesthetized with ketamine (100 mg/kg)/xylazine (10 mg/kg). GSCs were injected into the right frontal lobe (n=12/group, 5.0 × 105 cells/mouse as previously described [22, 23]. Mice (n=9) were followed until moribund and then sacrificed. Another cohort (n=3) were sacrificed 40 days post implantation. Brains were removed, embedded in paraffin, and sectioned. Tumor volumes were calculated by adding multiple cross-sectional areas through the tumors after H&E staining.
Exosome Growth Factor and Cytokine Profiling
GA-hMSCs were expanded in MSC medium, washed, and cultured in NSC medium for 48hrs. GA-hMSC-derived conditioned medium (CM) was collected, ultracentrifuged, the exosome-depleted conditioned medium (ED-CM) supernatant removed and saved, and the MSC-derived exosome pellets resuspended and lysed. Immuno-blot was performed using human growth factor and cytokine antibody arrays (Bio Ray).
MicroRNA Characterization
Isolated GA-hMSC-derived exosomes were exposed to RNase (1nM) to eliminate free-floating extra-vesicular RNA elements. Total RNA was extracted using the mirVana RNA isolation kit (Ambion). Total RNA was extracted from the parental cell line, and the miRNA profile for each sample was obtained using µParaflo® microfluidic biochip technology, through LC Sciences (Houston, TX).
In order to identify miRNA in the enriched subpopulation that were also the most highly expressed in GA-hMSC-derived exosomes, an expression-to-enrichment ratio (E:R ratio) was calculated, by multiplying the change in expression between the exosome and the parental cell, by the ratio of expression levels between the exosome and the parental cell.
Gene Expression Profiling of GSCs Treated with GA-hMSC-Derived Exosomes
GSCs were treated with GA-hMSC-derived exosomes (1×109 exosomes/ml) or NSC medium for 48 hours. Excess exosomes were removed by PBS wash, and total RNA was isolated. Gene expression profiling was performed using Illumina next-generation sequencing technology, through LC Sciences.
Over-Expression of MicroRNAs in GSCs
miRNA were over-expressed in GSCs by lentiviral (LV) transduction. Each LV-GFP-miRNA viral vector was transduced in GSCs (1×106 cells) at a multiplicity of infection (MOI) of 3, and cultured under puromycin selection for 72hrs. Integration of the GFP-miRNA construct was assessed by fluorescent light microscopy.
Luciferase reporter assays
The pEZX-MT06 luciferase reporter plasmid containing the NCOR1 3’UTR was purchased (Genecopoeia). A mutant 3’UTR in which the 3 miR-1587 binding sites identified by TargetScan were mutated was generated by synthetic gene synthesis (ThermoFisher) and cloned into the same reporter plasmid. U251 glioma cells were transfected with the reporter plasmid and 10nM of either miR-1587 or control miRNA mimics (Dharmacon). Luciferase activity was measured 48hrs following transfection using the Dual-Glo Luciferase kit (Promega).
Immunohistochemistry
Following antigen retrieval with citrate buffer (pH6), immunostaining for NCOR1 was performed with the EnVision + System HRP (DAB) kit (Dako) according to kit instructions, with anti-NCOR1 (Abcam # ab58396). 4 fields from each tumor bearing mouse (GSC-262 alone, n=2; GSC-262 + GA-hMSC exosomes, n=3) were scored for positive NCOR1 cells and the average percent of NCOR1 positive cells per mouse reported.
NCOR1 knockdown and overexpression
GSC-20 and GSC-262 were infected with lentiviral shRNA against NCOR1 and cultured under puromycin selection for 72hrs. To generate GSCs overexpressing NCOR1, an expression vector containing the full length cDNA for NCOR1 was transfected into GSC-262 using Lipofectamine (ThermoFisher) followed by G418 selection. RNA was isolated from GSCs using the RNeasy kit (Qiagen), and NCOR1 expression was determined by quantitative RT-PCR (ABI, NCOR1 hs01094541_m1 and GAPDH hs02758991_g1). shRNA/ORF clones were purchased from GE Dharmacon through the MD Anderson shRNA and ORFeome Core and the clone numbers are: shRNA1, V3LHS_395515; shRNA2, V2LHS_91777; shRNA3, V2LHS_91779, shRNA4, V3LHS_395517; NCOR1 ORF, 100069131.
Statistical analysis
Survival analysis was performed using the log-rank (Mantel-Cox) test. All other comparisons were performed using t-tests and all statistical tests are noted in the figure legends. Statistical analysis was performed using GraphPad Prisim v6.
Results
Isolation and characterization of GA-hMSC-derived exosomes
We recently reported the isolation and characterization of GA-hMSCs from a large set of human glioma surgical specimens [6]. To determine the role of exosomes in the interaction of stromal GA-hMSCs with tumor cells, specifically patient-derived GSCs, we selected four GA-hMSC lines, all of which met the ISCT criteria for MSCs [19]. In addition, we chose four GSC lines isolated from surgical specimens. Two of these, GSC-262 and GSC-20, were isolated from the same tumor specimens as their respective GA-hMSCs (GA-hMSC-262 and GA-hMSC-20). The GA-hMSCs were not tumorigenic when implanted into the brains of athymic mice, consistent with their role as stromal cells. In contrast, all four GSC lines formed aggressive tumors when as few as 1000 cells were implanted into mouse brains, consistent with their role as tumor initiating cells. Three GSC lines and 3 GA-hMSC lines were previously characterized [6] and we characterized one additional line for both GSCs and GA-hMSCs (Table S1). Low passage whole genome sequencing confirmed that GA-hMSC-230 [6] and GA-hMSC-247 (Figure 1A) harbor few genetic alterations, similar to bone marrow-derived human MSCs (BM-hMSCs), and are likely normal stromal cells recruited into the tumor (type 1 GA-hMSCs). We previously reported that GA-hMSC-262 and GA-hMSC-20 do have some block copy number variations, but these differ from the alterations found in the paired GSCs, and may represent examples of recruited normal MSC-like cells that acquired unique genomic alterations (type 3 GA-hMSCs). The cell lines described here provide two matched and two un-matched GA-hMCS/GSC pairs and represent 2 subtypes of GA-hMSCs.
Figure 1. Characterization of GA-hMSCs and GA-hMSC-derived nano-vesicles.

(A) Circos plot of GA-hMSC-247 copy number variations relative to BM-hMSCs. The outside circle represents chromosomes and cytogenetic bands. The inner circle shows red DNA amplifications and green regions of genomic loss. (B) Western blot demonstrating the presence of exosomal markers CD63 and GAPDH, and the lack of non-exosomal markers CD32 and CD16, on nano-vesicles from GA-hMSCs. C-cell lysate, E-exosomes (C) Scanning electron microscopy demonstrates GA-hMSC-derived vesicles (top panels) have a classic cupped-shape morphology with a distinct lipid bilayer and are within the 40nm-100nm range. Immuno-gold transmission electron microscopy (bottom panels) identifying the CD63 exosomal marker but not non-exosomal markers CD32 and CD16, on the surface of nano-vesicles from GA-hMSCs. (D) NanoSight plot demonstrating the average diameter of GA-hMSC-derived nano-vesicles to be within the 40–100nm range of exosomes.
To determine whether GA-hMSCs secrete exosomes, BM-hMSCs and each of the GA-hMSCs were cultured in serum-free, exosome-free medium, and exosomes were isolated from the supernatant. Western blot of protein extracted from isolated vesicles from all four GA-hMSCs and from BM-hMSCs revealed the presence of the widely accepted exosome markers CD63 and GAPDH (Figure 1B) [24]. CD63 was enriched in the exosome fraction compared with the whole cell lysates of the parental cells. The non-exosomal markers CD16 and CD32 were absent from the vesicles isolates from all GA-hMSCs and BM-hMSCs, although they were present in the parental cells.
EM was used to further characterize the vesicles isolated from BM-hMSCs and GA-hMSCs. We identified abundant bilayer vesicles, 30–100nm in diameter, consistent with the known appearance of exosomes (Figure 1C). This size distribution was confirmed with NanoSight technology (Figure 1D). Immunostaining and EM revealed positive CD63 gold-labeling on the membrane of the vesicles from all GA-hMSCs and BM-hMSC (Figure 1C). In contrast, CD16 and CD32 gold-labeling was not observed, further supporting the classification of the isolated vesicles as exosomes.
GSCs internalize GA-hMSC-derived exosomes
To confirm the uptake of GA-hMSC-derived exosomes by GSCs, we labeled exosomes using a CD63-green fluorescent fusion protein (GFP). GSCs were co-cultured with CD63-GFP labeled GA-hMSC-derived exosomes, and fluorescent confocal microscopy revealed the presence of GFP-labeled GA-hMSC-derived exosomes exclusively in the cytoplasm of GSCs (Figure S1). These results indicate that GSCs can internalize GA-hMSC-derived exosomes.
GA-hMSC-derived exosomes enhance GSC proliferation and clonogenicity in vitro
To determine the growth effects of GA-hMSC-derived exosomes on GSCs, we assayed GSC proliferation and clonogenicity after exposure to GA-hMSC-derived exosomes. GSCs were treated with purified GA-hMSC-derived exosomes, GA-hMSC-derived exosome-depleted-conditioned media (ED-CM) as a positive control, or purified GSC-derived exosomes (“self exosomes”). The purified GA-hMSC-derived exosomes significantly increased the proliferation of GSCs in a dose-dependent manner (Figure 2A, B). Consistent with our previous observations [6], GA-hMSC-derived ED-CM also significantly increased GSC proliferation compared with untreated cells. At higher doses, the effect of purified exosomes significantly increased proliferation of all GSCs compared with ED-CM (Figure 2A,B). Similarly, GA-hMSC-derived exosomes significantly increased GSC clonogenicity (Figure 2C, D). Increased GSC proliferation and clonogenicity were observed upon treatment with exosomes derived from both matched and unmatched GA-hMSCs (Figure 2 and Figure S2A–H). In contrast, treatment of GSCs with GSC-derived self-exosomes did not have a significant effect on GSC proliferation or clonogenicity; confirming previous reports that exosomes do not exert any known autocrine effects [25].
Figure 2. GA-hMSC-derived exosomes enhance GSC proliferation and clonogenicity in vitro.

(A, B) Proliferation assay demonstrating a significant dose-dependent increase in GSC proliferation with the addition of matched (A) and un-matched (B) GA-hMSC-derived exosomes. * p<0.01, ** p<0.001, paired t-test (C, D) Clonogenicity assay demonstrating a significant (*p< 0.05) increase in GSC neurosphere formation with the addition of matched (C) and un-matched (D) GA-hMSC-derived exosomes. In un-matched experiments GSC-7-2 was treated with exosomes and ED-CM from GA-hMSC-247 and GSC-11 was treated with exosomes and ED-CM from GA-hMSC-230. ED-CM: exosome depleted-conditioned media. See also Figure S2.
GA-hMSC-derived exosomes enhance GSC tumorigenicity in vivo
To assess the effects of GA-hMSC-derived exosomes on GSCs in vivo, GSCs were pre-treated with GA-hMSC-derived exosomes, and subsequently implanted into the right frontal lobe of nude mice. Pre-treatment of GSC-262 with GA-hMSC-262-derived exosomes significantly decreased (p<0.05) median survival from 56 to 45 days (Figure 3A). Furthermore, pre-treatment of GSC-20 with GA-hMSC-20-derived exosomes significantly decreased (p<0.05) median survival from 49 to 37 days (Figure 3B). In agreement with in vitro results, pre-treatment of GSC xenografts with GA-hMSC-derived ED-CM also significantly decreased (p<0.05) median survival, while pre-treatment of GSCs with GSC-derived self-exosomes did not have a significant effect on median survival.
Figure 3. GA-hMSC-derived exosomes enhance GSC tumorigenicity in vivo.

(A,B) Survival curves showing a significant (p<0.05, log-rank test) decrease in median survival for GSC tumor bearing mice which were pre-treated with matched GA-hMSC-derived exosomes prior to implantation. Median survial was 45 days for GSC-262 (A) and 37 days for GSC-20 (B). (C,D) H&E staining of mice brain tumor sections demonstrating a significant (p<0.05, paired t-test) increase in tumor volume at 40 days. Arrows indicate tumor location. CM-conditioned media, ED-CM-exosome depleted-conditioned media.
Histologic analysis of brain specimens from mice implanted with GSCs pre-treated with GA-hMSC-derived exosomes demonstrated a significant increase in tumor volume at 40 days post-implantation (Figures 3C,D). As expected, pre-treatment of GSCs with GA-hMSC-derived ED-CM also significantly increased tumor volume. In contrast, and in agreement with in vitro results, treatment of GSCs with GSC-derived self-exosomes did not have a significant effect on tumor volume. These histologic results corroborate with survival data and findings from in vitro experiments. Together, these results indicate that GA-hMSC-derived exosomes significantly increase the tumorigenicity of GSCs.
GA-hMSC-derived exosomes do not contain major growth promoting proteins
We previously demonstrated that GA-hMSCs impact GSC proliferation via secretion of IL-6 [6]. To determine whether GA-hMSC-derived exosomes also contain growth promoting proteins or cytokines, we utilized protein array technology to examine 41 growth factors and receptors and 59 cytokines. Consistent with our previous study, GA-hMSC conditioned media and ED-CM contained several growth promoting factors (e.g. IL-6). However, cytokines and growth factors were below detection in the purified exosome fraction. (Figure S3A,B). These data indicate that, whereas GA-hMSC-derived CM and ED-CM contain growth factors secreted by the cell, these growth factors, receptors, and cytokines are not detected within purified GA-hMSC-derived exosomes.
Specific miRNA are enriched in GA-hMSC-derived exosomes
We next utilized miRNA micro-array technology to analyze 2,019 miRNAs (miRBase 19.0) for their level of expression in MSCs and MSC-derived exosomes. MicroRNA profiles for GA-hMSC-derived exosomes and BM-hMSC-derived exosomes contained numerous miRNAs, which were significantly different (p-values <0.001, paired t-test) from that of the parental cell line (Figure 4A). Cluster analysis of the top 20 differentially expressed miRNAs indicated that exosomal miRNA cluster separately from cellular miRNA (Figure 4B). GA-hMSC exosomes clustered together with the exception of GA-hMSC 247, which was more closely related to exosomes from BM-hMSCs. We identified 37 exosomal miRNAs with a ≥ 5000 hybridization intensity (top 0.2% most highly expressed miRNA), among exosomes derived from all 4 GA-hMSC, which we refer to as highly expressed miRNA (Table S2).
Figure 4. GA-hMSC-derived exosomes contain a unique miRNA profile.

(A, B) miRNA profiles for GA-hMSC and GA-hMSC-derived exosomes, demonstrating a significant (p<0.001) difference in miRNA content. (A) 320 miRNAs were selected by identifying the top 200 miRNAs for each pair of GA-hMSC and GA-hMSC-derived exosomes according to the absolute fold change and collapsing them into non-duplicated miRNAs. (B) The top 20 miRNAs and the 8 miRNA that were highly expressed and highly enriched in exosomes were selected as in A and subjected to cluster analysis. (C) Distribution of the average expression level for miRNA in exosomes four GA-HMSC lines, compared with the parental cell. Exosome enriched miRNA were both highly expressed (> 5000 hybridization intensity, and highly enriched (> 3.0 SD), compared with exosome depleted miRNA which were both highly expressed (> 5000 hybridization intensity) and highly enriched (< −1.0 SD) in parental cells. (D) Expression levels of predicted gene targets of the exosomal miRNA are significantly (p<0.01) decreased in GSCs after treatment with GA-hMSC-derived exosomes. Exo-Exosome.
We then identified a sub-population of miRNA that were highly enriched in MSC-derived exosomes when compared with the parental cell. Exosomal miRNA that had significantly different (p<0.05, paired t-test) average levels of expression between the GA-hMSC-derived exosomes and the parental GA-hMSCs are referred to as enriched miRNA. miRNA that had expression changes greater than 2 standard deviations from the mean (top 2.5%) were termed highly enriched miRNA (Table S2). We calculated the expression-to-enrichment ratio (E:R ratio) for each miRNA, and identified a group of 8 miRNAs that were the most highly expressed and highly enriched (> 3 SD from the mean) in MSC-derived exosomes when compared with the parental cells (Figure 4C, Table S2). This cutoff correlated with average miRNA expression levels > 5000 hybridization intensity. The expression of each of these 8 highly expressed and highly enriched miRNAs was significantly increased in GSCs after treatment with GA-hMSC-derived exosomes (Figure S4A). Conversely, we also identified a group of 8 miRNA that were the most highly expressed and highly enriched (> 1 SD from the mean) in the parental cells when compared with MSC-derived exosomes (Figure 4C, Table S2). This group of miRNA are thus relatively depleted in MSC-derived exosomes.
Using the miRTarget 2.0 database, we identified 251 unique predicted gene targets (> 90 target score) for 7 of the 8 highly expressed and highly enriched miRNA (no predicted gene targets for miR-4508) in MSC-derived exosomes (Table S3) [26]. Conversely, we also identified 245 unique predicted gene targets for all 8 of the miRNAs that are depleted from MSC-derived exosomes (Table S3). Expression levels for the predicted gene targets in the two groups were obtained from gene expression profiling performed on GSC-262 and GSC-20. The expression levels of the predicted gene targets in untreated GSCs were compared with that of GSCs treated with GA-hMSC-derived exosomes for 48 hours. The average fold change in expression of the 251 predicted gene targets for the 7 enriched miRNA, was significantly greater (p<0.01) than the average fold change in expression of the 245 predicted gene targets for the 8 depleted miRNA (Figure 4D). These results indicate that highly expressed and highly enriched miRNA in GA-hMSC-derived exosomes are capable of down-regulating their predicted gene targets in GSCs.
MicroRNA in GA-hMSC-derived exosomes increase GSC proliferation and clonogenicity
To determine the influence of the enriched miRNA in GA-hMSC-derived exosomes on GSC proliferation, we utilized lentiviral transduction to over-express each of the 7 enriched exosomal miRNAs in GSC-262 and GSC-20. Of the 7 miRNAs evaluated, only the over-expression of miR-1587 and miR-3620-5p produced significant increases in the proliferation of GSCs, which were similar to that of GSCs after treatment with GA-hMSC-derived exosomes (Figure 5A). MiR-1587 also promoted a significant increase in the clonogenicity of GSCs, similar to that of GSCs after treatment with GA-hMSC-derived exosomes (Figure 5B).
Figure 5. miRNA in GA-hMSC-derived exosomes promote GSC proliferation and clonogenicity.

(A, B) Proliferation (A) and clonogenic (B) assays demonstrating a significant increase in GSC proliferation after over-expression of miR-1587 and miR-3630 and an increase in GSC clonogenicity after over-expression of miR-1587, similar to the effects of GA-hMSC-derived exosomes. (C, D) Proliferation (C) and clonogenicity (D) assays showing a significant decrease in the proliferation of GSCs treated with matched GA-hMSC-derived exosomes after the addition of anti-miR-1587. (E) Western blot demonstrating an increase in NCOR1 expression in GSCs treated with matched GA-hMSC-dervied exosomes after the addition of anti-miR-1587. (F) Proliferation assay demonstrating a significant increase in GSC proliferation after knockdown of NCOR1 *p<0.01, t-test. LV-lentiviral, NSC-neural stem cell.
To evaluate the role of miR-1587 in exosome-mediated GSC proliferation and self-renewal, we treated GSCs with GA-hMSC-derived exosomes in the presence of anti-miR-1587. The effects of GA-hMSC-derived exosomes on GSC proliferation (Figure 5C) and self-renewal (Figure 5D) were significantly impaired with the addition of anti-miRNA-1587. This indicates that miR-1587 contained within GA-hMSC-derived exosomes has a causal role in increasing the proliferation and self-renewal of GSCs.
miR-1587 targets the tumor suppressor NCOR1 in GSCs
We assessed the functionality of miR-1587 by directly analyzing one of its predicted gene targets, the nuclear hormone receptor co-repressor-1 (NCOR1). Over-expression of miR-1587 decreased GSC expression of NCOR1 when compared with untreated GSC controls, similar to the effects of purified exosomes on GSCs (Figure 5E). Importantly, treating GSCs with anti-mir-1587 partially reversed the exosome-mediated downregulation of NCOR1. NCOR1 protein levels were also decreased in tumor sections from mice bearing GSC-262 tumors after treatment with GA-hMSC-262 exosomes (Figure S4B,C). To demonstrate a direct impact of NCOR1 levels on GSC proliferation, we used shRNA to knockdown NCOR1. NCOR1 inhibition was verified by quantitative RT-PCR (Figure S5A,B). Specific NCOR1 knockdown led to increased GSC proliferation (Figure 5F, S5C). In contrast, overexpression of NCOR1 in GSC-262 led to a decrease in proliferation (Figure S5C). A luciferase reporter assay demonstrates direct regulation of the NCOR1 3’UTR by miR-1587 (Figure S5D). These results indicate that regulation of NCOR1 in GSCs by miR-1587 in GA-hMSC-derived exosomes plays a role in promoting the proliferation and clonogenicity in GSCs.
Discussion
We took a unique approach to explore stroma-tumor communication by examining the interactions between matched and unmatched pairs of GA-hMSCs and GSCs. Here we show that GA-hMSCs release exosomes that are internalized by GSCs, resulting in a significant increase in GSC proliferation and clonogenicity in vitro, as well as a significant increase in GSC tumorigenicity in vivo. Moreover, we show that GA-hMSC-derived exosomes contain highly expressed and highly enriched miRNA, one of which, miR-1587, was able to significantly increase both GSC proliferation and clonogenicity in in vitro experiments. The delivery of miR-1587 by GA-hMSC-derived exosomes resulted in the down-regulation of the tumor-suppressor NCOR1 in the recipient GSCs (Figure 6).
Figure 6. Summary Schemata.
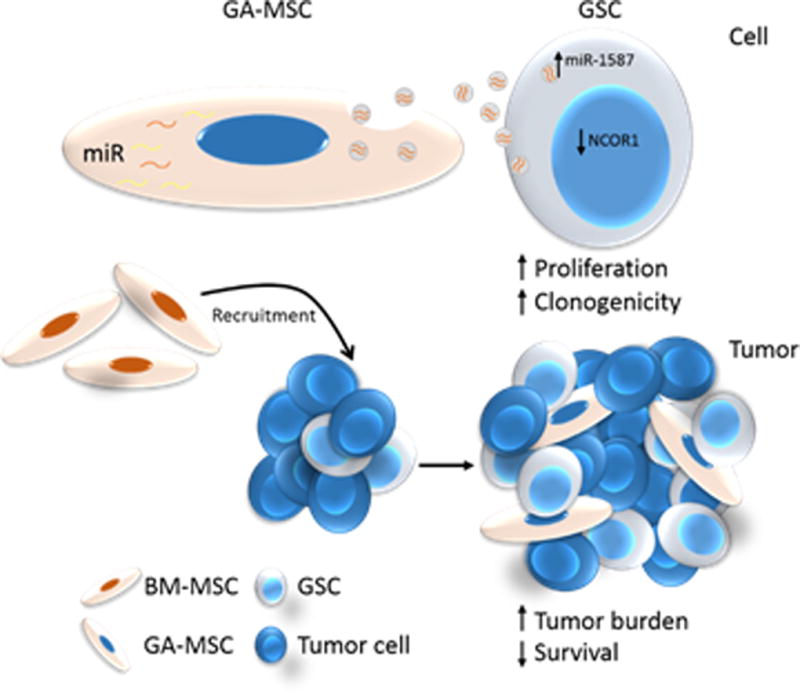
BM-hMSCs are attracted to gliomas and become GA-hMSCs within the tumor niche. These GA-hMSCs preferentially package miR-1587 into exosomes, which are released by the GA-hMSCs and taken up by neighboring GSCs. The increased level of miR-1587 down-regulates NCOR1 levels in GSCs. These effects ultimately result in increased GSC proliferation and clonogenicity, and subsequently increases tumor growth.
Although there are several studies exploring the intercellular communication between stromal cells and cancer stem cells (CSCs), few studies have investigated the role of stromal cell-derived exosomes in interactions with CSCs. Previous studies have indicated that stromal cell-derived exosomes can enhance the growth of CSCs in other cancer types, thereby supporting the results of our study [17, 18]. More recently, astrocyte-derived exosomes were shown to support brain metastasis via miRNA delivery [27]. We demonstrate that GA-hMSC-derived exosomes not only enhance the growth of GSCs, but also increase their clonogenicity, ultimately resulting in increased tumor burden and decreased survival in vivo. To our knowledge, this is the first study to examine the function of exosomes from tumor-derived MSCs on CSC growth and self-renewal in gliomas.
A recent report demonstrated that GSC-derived extracellular vesicles support intratumoral heterogeneity and that GSCs of the mesenchymal subtype had an increased proliferative effect on proneural subtype GSCs whereas treatment with self-exosomes had no effect [28]. Consistent with this report, we observed minimal effects on GSC growth when treated with self-derived exosomes. However, we demonstrate increased GSC proliferation and clonogenicity after exposure to exosomes derived from both matched and unmatched GA-hMSCs. Also in agreement with this report, the greatest increases in GSC proliferation were observed when GSCs were treated with exosomes from GA-hMSC-20, which was derived from a mesenchymal tumor. Notably, treatment with exosomes from GA-hMSC-20 (mesenchymal) produced the most significant increases in proliferation of GSC-262, a proneural GSC. We also observed that GSCs derived from classical GBM (GSC-7-2 and GSC-11) had a lower response to the GA-hMSC exosomes of all subtypes (Figure S2 C–D, G–H). Clustering analysis indicated that GA-hMSC-247 and the corresponding exosomes were more closely related to BM-hMSCs than the other GA-hMSCs in terms of miRNA profiles (Figure 4B). Further work with additional GSCs and GA-hMSCs from each GBM subtype is needed to confirm the impact of GBM subtype on the effects of GA-hMSC derived exosomes on tumor growth, but our current studies suggest that exosomal communication is impacted by the complex heterogeneity of GBM.
Our previous study demonstrated that GA-hMSCs support GSC growth through secretion of IL-6 [6]. This effect is verified in the present study by the effects of ED-CM on GSC proliferation and clonogenicity (Figure 2). GA-hMSC-derived exosomes lacked IL-6 and other cytokines and growth factors (Figure S3), prompting us to explore the miRNA content of exosomes. microRNA have the potential to produce longer lasting effects in recipient cells due their slow decay rate and their ability to regulate multiple genes. Consistent with other cell types, including BM-hMSCs [29–31], we showed that the GA-hMSC-derived exosomes contained unique miRNA profiles as compared with the parental cell, including a specific group of miRNA that were highly expressed.
Unlike the group of depleted miRNA from GA-hMSC-derived exosomes that are tumor-suppressive, the group of highly expressed and highly enriched miRNA in GA-hMSC-derived exosomes consists of newly identified miRNA whose targets and functions have not been clarified. The potential for these unstudied miRNA to be onco-miRNAs is suggested by our experimental results in which GA-hMSC-derived exosomes increased the tumorigenicity of GSCs. Given the wide range of genes that miRNAs target, and the wide range of miRNAs that can target the same gene, the effects of GA-hMSC-derived exosomal miRNA on GSCs are likely not attributable to a single miRNA. However, our results indicate that miR-1587 is at least one exosomal miRNA that appears to mediate the increase in proliferation and clonogenicity of GSCs. Although we focused our report on the miRNA that were both highly expressed and highly enriched in exosomes, it is possible that miRNA highly expressed in GA-hMSCs that were not overly enriched in exosomes may also be important for exosomal effects on tumor growth. Consistent with this concept, we observed high expression (without enrichment) of miR-21, a well-known oncomiR (Table S2). Our observations that the highly expressed/exosomal depleted miRNA contained several tumor suppressive miRNA and that the highly expressed/highly enriched miRNA have growth promoting roles suggests that there may be a specificity to exosomal miRNA packaging as has been reported for other cell types [32].
The capability of exosomal miRNA to regulate gene expression in recipient cells has been established in various tissues, and in the brain, this process has been described in both physiologic and pathologic conditions. The delivery of miR-124a via neuron-derived exosomes increased the expression of glutamate transporter-1 in recipient astrocytes. Furthermore, the disruption of this communication pathway is linked to key changes that occur during the progression of amyotrophic lateral sclerosis [33]. miRNA in exosomes derived from the U251 glioblastoma cell line were able to alter the transcriptome of recipient human brain microvascular endothelial cells [34]. The communication of exosomal miRNA significantly down-regulated the expression of 19 genes involved in the maintenance of the blood-brain barrier [34]. Our study also demonstrated that exosomal miRNA cargo can result in downregulation of target genes in GSCs (Figures 4, 5), further confirming that cells can utilize exosome-mediated regulation of gene expression to modify their surrounding microenvironment. Our in vitro studies indicate that the GA-hMSC exosomes effects on GSC proliferation and clonogenicity were at least in part via exosomal miRNA targeting gene expression in GSCs. Furthermore, levels of the miR-1587 target NCOR1 were decreased in the tumors of mice when GSCs were pretreated with exosomes from GA-hMSCs, suggesting that similar exosome-mediated gene regulation mechanisms occur in vivo.
This study is also the first to describe the targeting and down-regulation of NCOR1 in gliomas by miR-1587. Since the identification of NCOR1 by Horlein and colleagues in 1995, several other NCORs have been identified, and have been shown to function in both normal physiologic and pathologic conditions, including cancer [35–39]. NCOR was initially established as an oncogene [40]. However, this study evaluated the NCOR family and did not distinguish between effects caused by NCOR1 and NCOR2. In tumor cells from patients with astrocytic gliomas, low expression of NCOR1 in both cytoplasmic and nuclear fractions, and high nuclear expression of NCOR2, were observed. This study suggested that NCOR2 may have an oncogenic function, while NCOR1 has a tumor-suppressor function [41]. Consistent with this concept, knockdown of NCOR1 resulted in glioma growth and invasiveness both in vitro and in vivo, demonstrating a tumor-suppressive function [42]. Furthermore, decreased expression of NCOR1 correlated with epithelial-to-mesenchymal transition in glioblastoma [42]. Together, these studies indicate that down-regulation of the tumor-suppressor NCOR1 in gliomas promotes tumor growth.
This study describes exosome-mediated miRNA delivery as an additional stroma-tumor communication mechanism in glioma (Figure 6). GA-hMSCs therefore provide a tumor supportive environment both by the secretion of soluble, growth-promoting factors, such as IL-6, and via exosomal delivery of specific oncogenic miRNAs. We identified one specific miRNA and characterized one potential target, however further study will be required to clarify the specificity of miRNA loading into exosomes and to fully characterize these novel miRNAs and their targets. As both MSCs and exosomes are currently being explored as delivery modes for therapeutics, this and future work will be important considerations in the design and implementation of such delivery methods.
Supplementary Material
Acknowledgments
We thank David M. Wildrick, Ph.D., for editing the manuscript.
Grant Support
We acknowledge the following for support of this study: The National Institutes of Health [5R01 CA115729-05]; National Cancer Institute, SPORE in Brain Cancer (Project 1, Core B, and Core C) [1P50 CA127001-06]; The Broach Foundation for Brain Cancer Research; The Elias Family Fund; The Gene Pennebaker Brain Cancer Fund; The Ben and Catherine Ivy Foundation; The Anthony Bullock III Foundation; The Brian McCulloch Fund; The Uncle Kory Foundation; The Jason & Priscilla Hiley Fund to F.L. Lang. The NIH/NCI grants [1UH2TR00943-01] and [1 R01 CA182905-01] and the UT MD Anderson Cancer Center Brain SPORE [2P50CA127001] to G.A. Calin.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60(3):502–14. doi: 10.1002/glia.21264. [DOI] [PubMed] [Google Scholar]
- 3.Kim SJ, Kim JS, Park ES, Lee JS, Lin Q, Langley RR, et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia. 2011;13(3):286–98. doi: 10.1593/neo.11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12(11):1113–25. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71(18):6061–72. doi: 10.1158/0008-5472.CAN-10-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hossain A, Gumin J, Gao F, Figueroa J, Shinojima N, Takezaki T, et al. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells. 2015;33(8):2400–15. doi: 10.1002/stem.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behnan J, Isakson P, Joel M, Cilio C, Langmoen IA, Vik-Mo EO, et al. Recruited brain tumor-derived mesenchymal stem cells contribute to brain tumor progression. Stem Cells. 2014;32(5):1110–23. doi: 10.1002/stem.1614. [DOI] [PubMed] [Google Scholar]
- 8.Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J. Proteomics. 2010;73(10):1907–20. doi: 10.1016/j.jprot.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013;32(3–4):623–42. doi: 10.1007/s10555-013-9441-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fruhbeis C, Frohlich D, Kuo WP, Kramer-Albers EM. Extracellular vesicles as mediators of neuron-glia communication. Front. Cell. Neurosci. 2013;7:182. doi: 10.3389/fncel.2013.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng DQ, Huang B, Li J, Liu J, Chen XM, Xu YM, et al. Selective miRNA expression profile in chronic myeloid leukemia K562 cell-derived exosomes. Asian Pac. J. Cancer Prev. 2013;14(12):7501–8. doi: 10.7314/apjcp.2013.14.12.7501. [DOI] [PubMed] [Google Scholar]
- 12.Kruger S, Abd Elmageed ZY, Hawke DH, Worner PM, Jansen DA, Abdel-Mageed AB, et al. Molecular characterization of exosome-like vesicles from breast cancer cells. BMC Cancer. 2014;14:44. doi: 10.1186/1471-2407-14-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013;21(1):185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008;10(12):1470–6. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bronisz A, Wang Y, Nowicki MO, Peruzzi P, Ansari KI, Ogawa D, et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014;74(3):738–50. doi: 10.1158/0008-5472.CAN-13-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Vos KE, Abels ER, Zhang X, Lai C, Carrizosa E, Oakley D, et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 2016;18(1):58–69. doi: 10.1093/neuonc/nov244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012;151(7):1542–56. doi: 10.1016/j.cell.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 18.Wang M, Zhao C, Shi H, Zhang B, Zhang L, Zhang X, et al. Deregulated microRNAs in gastric cancer tissue-derived mesenchymal stem cells: novel biomarkers and a mechanism for gastric cancer. Br. J. Cancer. 2014;110(5):1199–210. doi: 10.1038/bjc.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 20.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–8. [PubMed] [Google Scholar]
- 21.Théry C, Amigorena S, Raposo G, Clayton A. Curr. Protoc. Cell Biol. John Wiley & Sons, Inc; 2001. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. [DOI] [PubMed] [Google Scholar]
- 22.Lal S, Lacroix M, Tofilon P, Fuller GN, Sawaya R, Lang FF. An implantable guide-screw system for brain tumor studies in small animals. J. Neurosurg. 2000;92(2):326–33. doi: 10.3171/jns.2000.92.2.0326. [DOI] [PubMed] [Google Scholar]
- 23.Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 24.Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 1998;273(32):20121–7. doi: 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- 25.Liang X, Ding Y, Zhang Y, Tse HF, Lian Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: current status and perspectives. Cell Transplant. 2014;23(9):1045–59. doi: 10.3727/096368913X667709. [DOI] [PubMed] [Google Scholar]
- 26.Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14(6):1012–7. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527(7576):100–4. doi: 10.1038/nature15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricklefs F, Mineo M, Rooj AK, Nakano I, Charest A, Weissleder R, et al. Extracellular Vesicles from High-Grade Glioma Exchange Diverse Pro-oncogenic Signals That Maintain Intratumoral Heterogeneity. Cancer Res. 2016;76(10):2876–81. doi: 10.1158/0008-5472.CAN-15-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen TS, Lim SK. Measurement of Precursor miRNA in Exosomes from Human ESC-Derived Mesenchymal Stem Cells. In: Kosaka N, editor. Circulating MicroRNAs: Methods and Protocols. Humana Press; Totowa, NJ: 2013. pp. 69–86. [DOI] [PubMed] [Google Scholar]
- 30.Koh W, Sheng CT, Tan B, Lee QY, Kuznetsov V, Kiang LS, et al. Analysis of deep sequencing microRNA expression profile from human embryonic stem cells derived mesenchymal stem cells reveals possible role of let-7 microRNA family in downstream targeting of hepatic nuclear factor 4 alpha. BMC Genomics. 2010;11(Suppl 1):S6. doi: 10.1186/1471-2164-11-S1-S6. Suppl 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xin H, Li Y, Buller B, Katakowski M, Zhang Y, Wang X, et al. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells. 2012;30(7):1556–64. doi: 10.1002/stem.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morel L, Regan M, Higashimori H, Ng SK, Esau C, Vidensky S, et al. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013;288(10):7105–16. doi: 10.1074/jbc.M112.410944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li CC, Eaton SA, Young PE, Lee M, Shuttleworth R, Humphreys DT, et al. Glioma microvesicles carry selectively packaged coding and non-coding RNAs which alter gene expression in recipient cells. RNA Biol. 2013;10(8):1333–44. doi: 10.4161/rna.25281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377(6548):397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 36.Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, et al. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. U. S. A. 1999;96(17):9873–8. doi: 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Battaglia S, Maguire O, Campbell MJ. Transcription factor co-repressors in cancer biology: roles and targeting. Int. J. Cancer. 2010;126(11):2511–9. doi: 10.1002/ijc.25181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen JD, Evans RM. A Transcriptional Co-Repressor That Interacts with Nuclear Hormone Receptors. Nature. 1995;377(6548):454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 39.Watson PJ, Fairall L, Schwabe JW. Nuclear hormone receptor co-repressors: structure and function. Mol. Cell. Endocrinol. 2012;348(2):440–9. doi: 10.1016/j.mce.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park DM, Li J, Okamoto H, Akeju O, Kim SH, Lubensky I, et al. N-CoR pathway targeting induces glioblastoma derived cancer stem cell differentiation. Cell cycle. 2007;6(4):467–70. doi: 10.4161/cc.6.4.3856. [DOI] [PubMed] [Google Scholar]
- 41.Campos B, Bermejo JL, Han L, Felsberg J, Ahmadi R, Grabe N, et al. Expression of nuclear receptor corepressors and class I histone deacetylases in astrocytic gliomas. Cancer Sci. 2011;102(2):387–92. doi: 10.1111/j.1349-7006.2010.01792.x. [DOI] [PubMed] [Google Scholar]
- 42.Heldring N, Nyman U, Lonnerberg P, Onnestam S, Herland A, Holmberg J, et al. NCoR controls glioblastoma tumor cell characteristics. Neuro Oncol. 2014;16(2):241–9. doi: 10.1093/neuonc/not214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.