

Abstract
Human melanocytic nevi driven by BRAFV600E are in a growth-arrested state referred as oncogene-induced senescence. FBXW7 tumor suppressor is mutated in melanomas, but not in benign precursor melanocytic nevi. In order to characterize whether inactivation of FBXW7 in cooperation with BRAFV600E (BrafV637E in the mouse) is sufficient to bypass from the growth-arrested state, we generated a murine model by conditionally silencing Fbxw7 in an established system, Tyr::CreER; BrafCA (or Tyr::CreER; BrafV600E). We show that loss of Fbxw7 in the presence of BrafV600E mutation is consequential and sufficient to drive tumorigenesis. This model provides further evidence that Fbxw7 is a tumor suppressor in the melanocytic lineage, and can serve as a tool for future pre-clinical studies.
Keywords: FBXW7, BRAF, nevus, melanoma, transformation, mouse model
Dear Editor
Oncogene-induced senescence (OIS) is a tumor suppressive mechanism that has been described in several cell types, including the melanocytic lineage (Michaloglou et al., 2005). In this phenomenon, cells initially undergo a proliferative phase due to an oncogenic insult followed by activation of a cell cycle arrest program (Peeper, 2011). Bypass from the senescent-like state can occur upon loss of key tumor suppressors such as CDKN2A, PTEN, TP53, or NF1 leading to malignant transformation (Vredeveld et al., 2012, Dankort et al., 2009, Viros et al., 2014, Maertens et al., 2013). It is critical to discern the combination of oncogene activation and tumor suppressor loss that is required for tumor initiation.
The best in vivo example of OIS or OIS-like state in humans is the melanocytic nevus (mole on the skin) (Michaloglou et al., 2005). Melanoma can arise from a pre-existing nevus, thus representing bypass from this condition (Shain et al., 2015). Approximately 85% of benign or precursor lesions (common acquired nevi) harbor the oncogenic mutation BRAFV600E, whereas 60% of melanomas harbor the same mutation, as well as other genetic and epigenetic events necessary for malignant transformation (Shain et al., 2015, Cancer Genome Atlas, 2015). BRAFV600E-induced senescence has been recapitulated in the murine system by the conditional expression of BrafV600E (BrafV637E in the mouse) in the melanocytic lineage, resulting in melanocytic hyperplasia resembling nevus formation (Dankort et al., 2009). By employing this model, inactivation of Pten, Nf1, and Tp53 has been demonstrated to bypass from an OIS-like state and result in tumor initiation (Dankort et al., 2009, Maertens et al., 2013, Viros et al., 2014).
FBXW7 is a tumor suppressor frequently altered in cancer (Aydin et al., 2014). The gene is deregulated by point mutations, mono- or bi-allelic deletion, promoter hypermethylation, microRNA-modulation, and transcriptional inhibition (Kourtis et al., 2015). FBXW7 encodes an E3 ubiquitin ligase responsible for regulating the stability of substrates containing a CPD domain (CDC4 phosphodegron motif). Several oncoproteins are substrates of FBXW7 including mTOR, CYCLIN E, NOTCH1, MYC, JUN, and PCG1α (Kourtis et al., 2015, Davis et al., 2014). We have previously reported FBXW7 mutations in melanoma identified through exome sequencing, its inactivation in over 40% of melanomas, and FBXW7-NOTCH1-mediated tumor promotion (Aydin et al., 2014). Here, in this study, we aimed to determine if conditional inactivation of Fbxw7 in cooperation with BrafV600E in melanocytes is sufficient to bypass from an OIS-like condition in an established murine system, Tyr::CreER; BrafCA (or Tyr::CreER; BrafV600E) (Dankort et al., 2009) and drive tumorigenesis.
Mice carrying BrafCA alleles were crossed with Fbxw7 floxed mice, followed by crossing to Tyr::CreERT2 to generate mice with genotypes Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox (Braf/Fbxw7) and Tyr::CreERT2; BrafCA/CA (Braf). Activation of CreER with topical 4-hydroxytamoxifen treatment at birth resulted in melanocytic-specific conversion of BrafCA to BrafV600E and the conversion of Fbxw7flox alleles to null alleles (Figure S1A). Approximately 3–4 weeks post-treatment, Tyr::CreER; BrafCA/CA mice showed hyperpigmentation of the skin and melanocytic hyperplasia within the dermis as determined by histology, resembling blue nevus-like clones as previously described (Figure S1B, C). Similarly, we observed hyperpigmentation of the skin in the Tyr::CreER; BrafCA/CA; Fbxw7flox/flox mice, but with significantly pronounced pigmentation and enhanced thickening of the skin as compared to Tyr::CreER; BrafCA/CA mice (Figure S1B). The thickening of the skin was mostly due to spindle cell tumor component beneath the epidermis and the nevus based on histologic examination (Figure S1C, upper panels).
Tyr::CreER; BrafCA/CA; Fbxw7flox/flox mice developed tumors that were characterized as melanomas. In these mice, the tumor penetrance was 81% and the melanoma-free survival (latency) was 6.1 months (6.17 ± 2.25 months), whereas mice harboring the Tyr::CreER; BrafCA/CA genotype alone developed tumors with 22.7% penetrance and 7.1 month latency (7.1 ± 2.46 months) (Figure 1A, B). Our findings on the control group (Tyr::CreER; BrafCA/CA) were in line with previous studies that have described ~10% of mice harboring BrafV600E mutations developing tumors beyond melanocytic hyperplasia within the first 6 months of life, increasing up to 50% within the first year (Dankort et al., 2009, Maertens et al., 2013). Importantly, melanomas of Tyr::CreER; BrafCA/CA; Fbxw7flox/flox mice were unique and compellingly different than tumors observed in Tyr::CreER; BrafCA/CA mice. Histologically, they were characterized by increased cellularity, spindle cell morphology, fascicular growth pattern, and atypical cells extending deep into the subcutis (Figure 1C). While the tumor cells were non-pigmented (amelanotic), many tumors had clusters of coarse melanin granules in the upper and deeper parts of the dermis. Tumor cells were positive for S100, Ki67, and MITF (Figure 1C, D). Of note, tumor cells retained high levels of p16INKA expression (Figure S1C, lower panels).
Figure 1.
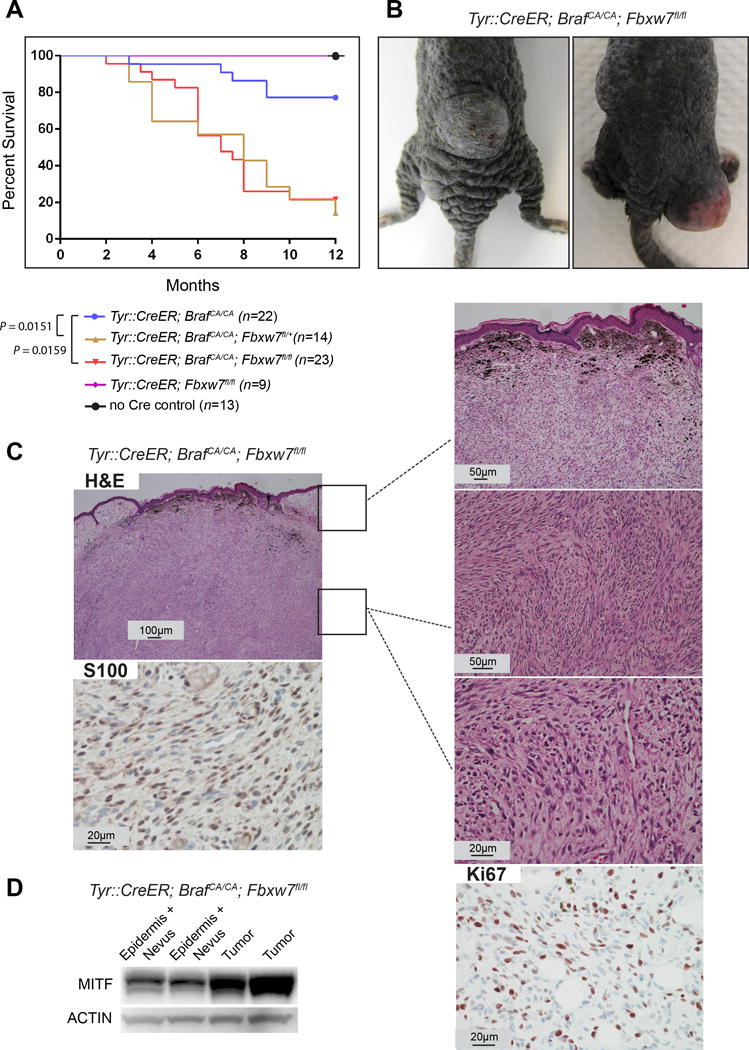
FBXW7 inactivation together with BrafV600E mutation cooperates in vivo to drive melanoma formation. (A) Survival curves of the mouse cohorts. Animals with tumor size greater than 1 cm3 or at 12 months after birth were euthanized. Log rank test was used for statistical analysis. (B) Representative examples of melanomas in the Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox mice are indicated. (C) Histology sections of the melanomas stained with hematoxylin and eosin (H&E) and immunohistochemistry for S100 and Ki67 are shown in the micrographs. (D) Tumors from Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox mice (without the overlying nevus cells and epidermis) were lysed and analyzed for MITF by western blotting. Actin was used as loading control.
To evaluate the signaling pathways involved in promoting tumorigenesis upon loss of FBXW7, we investigated the activity of mTOR, an established oncogene and target of FBXW7 (Kourtis et al., 2015). We examined phosphorylated ribosomal protein S6 (p-S6), a direct mTOR target (mTORC1 complex) and phosphorylated AKT (p-AKT) that is activated by the mTORC2 complex, in tumors of Tyr::CreER; BrafCA/CA; Fbxw7flox/flox mice. Growth-arrested nevus clones of the Tyr::CreER; BrafCA/CA mice were used as controls (Figure 2A). We noted significant up-regulation of p-S6 (S235/S236) as well as p-AKT (S473) in S100-expressing melanoma tumors throughout the dermis extending deep into the subcutis of the Tyr::CreER; BrafCA/CA; Fbxw7flox/flox mice (Figure 2B, C). These data suggest that both mTORC1 and mTORC2 signaling are activated in Fbxw7 inactivated and BrafV600E-mutant murine melanocytes. These results propose a potential therapeutic avenue for melanomas with FBXW7 inactivation.
Figure 2.
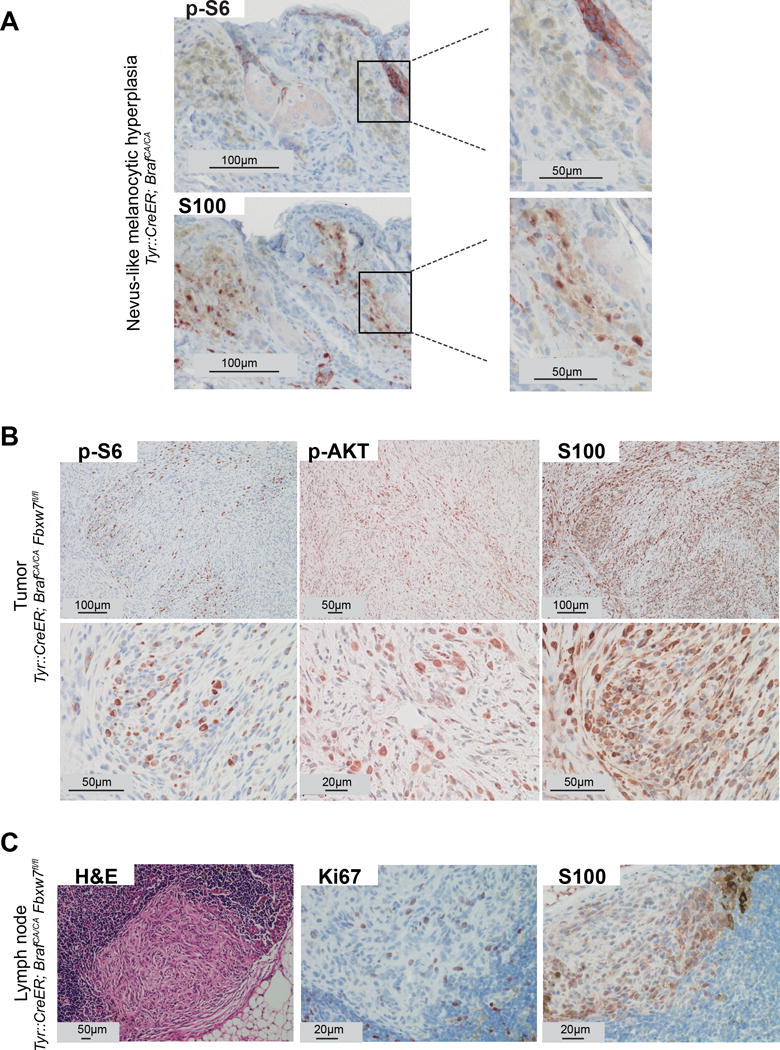
mTOR signaling is activated during loss of FBXW7-mediated tumorigenesis. (A) Melanocytic hyperplasia (blue nevus-like clones) from Tyr::CreERT2; BrafCA/CA mice stained with p-S6 (upper panel) and S100 (lower panel) using immunohistochemistry. Positive staining for p-S6 within the follicular epithelium is indicated as an internal control. (B) Melanomas (tumors within the deep portion of the dermis) from Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox mice stained with p-S6, p-AKT (S473), and S100. Low (upper panel) and high power (lower panel) magnifications are shown..(C) Histologic and immunohistochemistry analysis of lymph nodes from Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox mice stained with H&E, S100, and Ki67. Representative micrographs are indicated.
While we noted micro-metastases of melanoma to the lymph nodes in Tyr::CreER; BrafCA/CA; Fbxw7flox/flox mice, overt systemic metastasis could not be evaluated as the mice needed to be euthanized due to the large and fast growing primary tumors. The lymph node metastasis showed similar histologic features of the primary tumor and displayed spindle-shaped melanoma cells with coarse melanin granules growing in a fascicular pattern in the sub-capsular region invading into the lymph node parenchyma (Figure 2C). Tumor cells in the lymph nodes were positive for S100 and Ki67 (Figure 2C).
In summary, we report a mouse model that provides further evidence that Fbxw7 is a bona fide tumor suppressor in the melanocyte lineage. We show that loss of Fbxw7 in the presence of BrafV600E mutation (mitogen-activated protein kinase pathway activation) is consequential and sufficient to drive tumorigenesis, but does not lead to an aggressive phenotype with short latency period and overt systemic metastasis. As expected, the FBXW7 substrate, mTOR, and mTOR-mediated signaling were activated (mTORC1 and mTORC2). It remains to be established whether FBXW7-driven tumorigenesis is due to bypass from OIS. To this end, high levels of p16INK4A expression in these tumors suggest mechanisms other than those previously described where p16INK4A inactivation in the context of other mutations leads to bypass from OIS. It is possible that the non-aggressive phenotype observed may be due to retention of p16INK4A in this model. The model described here can serve as a tool for future pre-clinical studies.
Supplementary Material
Figure S1. Phenotypic characteristics. (A) PCR showing mouse genotypes before and after 4-hydroxytamoxifen treatment. These mice were induced at birth with 3 consecutive applications of 4-hydroxytamoxifen within the first three days of life. (B) Skin coat color, gross morphologic differences, and mouse size differences are depicted in the representative examples (Tyr::CreERT2; BrafCA/CA and Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox). (C) H&E sections of the epidermis and blue nevus-like melanocytic hyperplasia of the Tyr::CreERT2; BrafCA/CA mice, as well as epidermis and underlying blue nevus-like melanocytic hyperplasia and/or spindle cell melanoma cell proliferation of the Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox mice are shown (upper panels). Immunohistochemistry analysis of p16INK4A of the nevus and tumor sections is depicted (lower panels).
Acknowledgments
This work has been supported in part by the Icahn School of Medicine at Mount Sinai (J.T.C.), a pilot developmental research grant from the Tisch Cancer Institute (J.T.C.), a gift from the Dow Family Charitable Fund (J.T.C.), and research grants from the National Institutes of Health (CA158557 and CA177940 to J.T.C).
Footnotes
DR. JULIDE CELEBI (Orcid ID : 0000-0001-9926-3780)
References
- Aydin IT, Melamed RD, Adams SJ, Castillo-Martin M, Demir A, Bryk D, Brunner G, Cordon-Cardo C, Osman I, Rabadan R, Celebi JT. NOTCH1 Activation through FBXW7 Tumor Suppressor: A New Therapeutic Paradigm for Melanoma. J Natl Cancer Inst. 2014 doi: 10.1093/jnci/dju107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–96. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, Depinho RA, Mcmahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014;26:455–64. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N, Strikoudis A, Aifantis I. Emerging roles for the FBXW7 ubiquitin ligase in leukemia and beyond. Curr Opin Cell Biol. 2015;37:28–34. doi: 10.1016/j.ceb.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, Mcmahon M, Granter S, Flaherty K, Wargo JA, Marais R, Cichowski K. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3:338–49. doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, Van Der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Peeper DS. Oncogene-induced senescence and melanoma: where do we stand? Pigment Cell Melanoma Res. 2011;24:1107–11. doi: 10.1111/j.1755-148X.2011.00933.x. [DOI] [PubMed] [Google Scholar]
- Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, Rickaby W, D’arrigo C, Robson A, Bastian BC. The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med. 2015;373:1926–36. doi: 10.1056/NEJMoa1502583. [DOI] [PubMed] [Google Scholar]
- Viros A, Sanchez-Laorden B, Pedersen M, Furney SJ, Rae J, Hogan K, Ejiama S, Girotti MR, Cook M, Dhomen N, Marais R. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature. 2014;511:478–82. doi: 10.1038/nature13298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vredeveld LC, Possik PA, Smit MA, Meissl K, Michaloglou C, Horlings HM, Ajouaou A, Kortman PC, Dankort D, Mcmahon M, Mooi WJ, Peeper DS. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012;26:1055–69. doi: 10.1101/gad.187252.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Phenotypic characteristics. (A) PCR showing mouse genotypes before and after 4-hydroxytamoxifen treatment. These mice were induced at birth with 3 consecutive applications of 4-hydroxytamoxifen within the first three days of life. (B) Skin coat color, gross morphologic differences, and mouse size differences are depicted in the representative examples (Tyr::CreERT2; BrafCA/CA and Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox). (C) H&E sections of the epidermis and blue nevus-like melanocytic hyperplasia of the Tyr::CreERT2; BrafCA/CA mice, as well as epidermis and underlying blue nevus-like melanocytic hyperplasia and/or spindle cell melanoma cell proliferation of the Tyr::CreERT2; BrafCA/CA; Fbxw7flox/flox mice are shown (upper panels). Immunohistochemistry analysis of p16INK4A of the nevus and tumor sections is depicted (lower panels).