

Abstract
Purpose
Activation of the phosphatidylinositol 3-kinase (PI3K) pathway occurs in over 40% of bladder urothelial cancers. The aim of this study is to determine the therapeutic potential, the underlying action and resistant mechanisms of drugs targeting the PI3K pathway.
Experimental Design
Urothelial cancer cell lines and patient-derived xenografts (PDXs) were analyzed for alterations of the PI3K pathway and for their sensitivity to the small molecule inhibitor pictilisib alone and in combination with cisplatin and/or gemcitabine. Potential predictive biomarkers for pictilisib were evaluated and RNA-sequencing was performed to explore drug resistance mechanisms.
Results
The bladder cancer cell line TCCSUP, which harbors a PIK3CA E545K mutation, was sensitive to pictilisib compared to cell lines with wild type PIK3CA. Pictilisib exhibited stronger anti-tumor activity in bladder cancer PDX models with PI3KCA H1047R mutation or amplification than control PDX model. Pictilisib synergized with cisplatin and/or gemcitabine in vitro, significantly delayed tumor growth and prolonged survival compared with single drug treatment in the PDX models. The phosphorylation of ribosomal protein S6 correlated with response to pictilisib both in vitro and in vivo, and could potentially serve as biomarker to predict response to pictilisib. Pictilisib activated the compensatory MEK/ERK pathways that likely contributed to pictilisib resistance, which was reversed by co-treatment with the RAF inhibitor sorafenib. RNA-sequencing of tumors resistant to treatment suggested that LSP1 down-regulation correlated with drug resistance.
Conclusion
These preclinical results provide new insights into the therapeutic potential of targeting the PI3K pathway for the treatment of bladder cancer.
Keywords: phosphatidylinositol 3-kinase, bladder cancer, targeted therapy, cisplatin, gemcitabine, biomarker, patient-derived xenograft
Introduction
Urothelial carcinoma of the bladder or bladder cancer is among the top ten most common cancers in the United States, leading to approximately 11,820 deaths each year (1). Five to ten percent of patients are already metastatic at diagnosis, and approximately 50% of patients will develop local or distant disease recurrence after radical cystectomy (2). Cisplatin-containing combination chemotherapy has been the standard of care for advanced bladder cancer since the late 1980s, but provides a median survival of only about 14 months for metastatic bladder cancer. Little improvement in survival has been attained during the last few decades (3). Due to the limited efficacy of cisplatin-based chemotherapy in the treatment of advanced bladder cancer, novel therapeutics are currently under investigation, either as single agents or in combination with standard chemotherapy regimens. The Cancer Genome Atlas Project (TCGA) identified multiple new “druggable” targets of bladder cancer, among which 42% were within the phosphatidylinositol 3-kinase (PI3K)/AKT pathway (4). Despite the recurrent genetic aberrations that have been identified to date, there is no targeted therapy approved for bladder cancer by the US Food and Drug Administration (FDA) (5).
PI3K/AKT is a critical signal transduction pathway that regulates apoptosis, survival and proliferation. Activation of the PI3K/AKT pathway frequently occurs due to multiple molecular alterations, including mutations (PIK3CA, AKT1), gene amplification (PIK3CA, AKT1, AKT2), or loss of expression of tumor suppressors (PTEN, TSC2) (6). Activation of the PI3K/AKT pathway in cancer cells may overcome the pro-apoptotic effect of anticancer drugs, mitigate the cytotoxic effect of chemotherapy and cause drug resistance. Therefore, inhibiting this signaling pathway may facilitate chemotherapy treatments (7,8). Preclinical studies and early clinical trials indicate that the treatment of several cancers including breast (9), lung (10), medulloblastoma (11), and pancreatic cancer (6) could benefit from PI3K inhibitors. It is plausible that such targeted therapeutic approaches against the PI3K/AKT pathway can be used in patients with advanced bladder cancer, either as a single agent or in combination, to decrease drug resistance and augment the efficacy of chemotherapy.
According to the structural characteristics and substrate specificity of PI3K, it can be divided into three classes (I, II, and III). Of these, only the class I PI3K comprised of the p110α, β, δ and γ isoforms, is able to activate the downstream effector AKT (12,13). Pictilisib, also known as GDC-0941, is an oral class I pan-PI3K inhibitor with activity in the nanomolar range against all class I PI3K isoforms. Pictilisib binds to the ATP-binding pocket of PI3K p110, prevents activation of PI3K and formation of PIP3, and phosphorylation of downstream targets such as AKT (14,15). Pictilisib is now in advanced stages of clinical trials (16–18). In a phase I, open-label dose-escalation clinical trial of patients with advanced solid tumors, pictilisib was found to have anti-tumor activity, on-target pharmacodynamics activity, and an acceptable safety profile at doses of ≥100 mg. Additionally, pictilisib was well tolerated at doses up to 330 mg with mild to moderate adverse effects and no treatment related deaths (16).
Here we report the activity of pictilisib in selected bladder cancer cell lines and patient-derived xenograft (PDX) models of human bladder cancer. PDXs retain morphological and genetic fidelity, and therefore, more closely mimic the behavior of patient cancers (19,20). Our research group has developed and characterized a panel of 26 bladder cancer PDX models for screening of effective drugs or drug combinations (5). We demonstrated that pictilisib blocked the PI3K/AKT pathway and sensitized bladder cancer cells to cisplatin and gemcitabine treatment in vitro and in vivo. We also demonstrated that the phosphorylation of ribosomal protein S6 (p-S6), a downstream effector of the PI3K/AKT pathway, may serve as a potential biomarker to predict the response to pictilisib.
Materials and Methods
Cell lines and reagents
The bladder cancer cell lines TCCSUP, T24, J82, 5637 and RT4 were purchased from American Type Culture Collection (ATCC, Manassas, VA) in 2007 and stored in liquid N2. Cells were thawed and cultured with the recommended medium and condition by the ATCC. The cell lines have not been tested and authenticated by the authors. Pictilisib, sorafenib and gemcitabine were purchased from LC Laboratories (Woburn, MA); cisplatin was purchased from EMD Biosciences, Inc. (San Diego, CA); antibodies against p-AKT(S473), total AKT (t-AKT), PTEN, p-ERK(Thr202/Tyr204), total ERK (t-ERK), p-S6(S240/244), p-P70S6K(T389) p110α, cleaved-caspase3, Ki-67 and GAPDH were purchased from Cell Signaling Technology (Danvers, MA); β-actin antibody was purchased from Sigma-Aldrich (San Louis, MO). The MTS cell viability assay was purchased from Promega (Madison, WI). Immunohistochemistry kits were purchased from BioGenex (Fremont, CA). Propidium Iodide and Annexin V were purchased from Biolegend (San Diego, CA).
Cell viability and drug sensitivity assays
Pictilisib and sorafenib were dissolved in dimethyl sulfoxide (DMSO) as 10 mM stock solutions. Cisplatin and gemcitabine stock solutions (10 mM) were prepared in PBS. Cells were seeded into 96-well plates (Becton Dickinson, Franklin Lakes, NJ) overnight. Drugs were diluted in culture media and added the next day, and the plates were incubated for an additional 72 hours. The control group contained 0.2% DMSO. Cell viability assays were performed according to the manufacturer’s protocol. The absolute 50% inhibitory concentrations (IC50) were calculated according to Sebaugh (21) and dose-response curves were generated with GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA). The dose range of two drugs for calculating combination indices (CI) was determined based on Chou (22). CI values were calculated with CompuSyn software (http://www.combosyn.com/).
Western blot and Immunohistochemistry
The procedures for performing western blot analysis were described previously (23), and immunohistochemistry staining was conducted according to the manufacturer’s standard protocol (BioGenex, Fremont, CA).
Flow cytometry analysis
Cells were plated in 6-well plates and incubated overnight. Drugs were added the next day and cells were incubated for a further 48 hours. Cell apoptosis was measured by Annexin-V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) staining according to the manufacturer’s protocol. Cell cycle analysis was performed as described previously (24). Flow cytometry was performed at the UC Davis Comprehensive Cancer Center Flow Cytometer Shared Resource and data was analyzed using FlowJo software (FlowJo, Ashland, OR).
PDX bladder cancer mouse models for in vivo study
All animal studies were performed according to protocols approved by the Jackson Laboratory (JAX) and UC Davis Institutional Animal Care and Use Committees (Protocols No. 12027 and 17794). PDX models (BL0269, JAX#TM00015; BL0293, JAX#TM00016; BL0440, JAX#TM00024) were developed as previously described, fresh PDX specimens (3–5mm3) were implanted subcutaneously into the flanks of 4–5 week old NOD scid gamma (NSG) mice(5). Mice were randomized into each group (8 mice per group) when tumor volumes reached 100 to 300 mm3, and followed by initiation of drug treatment. Pictilisib was dissolved in 30% PEG-300 with 1% Tween80 and 1% DMSO. Mice were dosed daily with 100 mg/kg by oral gavage during the course of study. Cisplatin was given at 2 mg/kg intravenously at days 1, 2, 3, and days 15, 16, 17. Gemcitabine was given by intraperitoneal injection with a dose of 150 mg/kg once a week for four weeks. Drugs were given simultaneously in the combination treatment group. In the sequential treatment group, drugs were administered similarly to the combination treatment group, but the animals were not treated with pictilisib during days1–3 and days 15–17. The tumor volume was calculated using the formula (length × width2) × 0.5. Tumor volumes and body weight were recorded twice weekly. Mice with tumor volume of ≥ 2 cm3 or with losses in body weight of ≥20% were promptly euthanized per IACUC policy.
Gene expression profiling by RNA-sequencing
Total cellular RNA was isolated from two randomly selected tumors from control, pictilisib, cisplatin and combination groups. Tumors were regarded as resistant to treatment when the tumor growth curve reached logarithmic growth phase. RNA samples were submitted to the UC Davis Comprehensive Cancer Center’s Genomics Shared Resource for RNA-sequencing analysis using a “total” RNA-Sequencing approach. The protocol for RNA-sequencing and data analysis were described previously (5). The raw sequence and processed data files are publicly available through the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo) with accession number GSE101419.
Statistics
At least three independent experiments were performed for each analysis described in this article. Student’s t test or one-way ANOVA was used to compare continuous parametric data between two groups and multiple groups, respectively. Overall survival of mice was analyzed using Kaplan-Meier survival curve and log-rank test. Statistical analysis was performed by GraphPad Prism 5 software.
Results
The landscape of genetic alterations along the PI3K/AKT pathway in selected bladder cancer cell lines
We analyzed the genetic alterations of the PI3K/AKT signaling pathway in five common bladder cancer cell lines by searching the Catalogue of Somatic Mutations in Cancer (COSMIC, http://cancer.sanger.ac.uk/cell_lines) and literature review (25–27). As shown in Supplementary Table S1, TCCSUP harbors a hotspot PIK3CA mutation E545K; T24 has a rare PTEN mutation N48I; J82 contains both a rare PIK3CA mutation P124L and homozygous deletion of PTEN. The expression and activation status of key proteins related to the PI3K/AKT signaling pathway were determined (Figure 1A). Consistent with the genetic mutation status, basal levels of AKT phosphorylation were higher in TCCSUP and lack of PTEN protein were observed in J82. Interestingly, AKT phosphorylation was not detected in RT4, which was reported in previous studies (28,29).
Figure 1. Effect of pictilisib on bladder cancer cell lines.
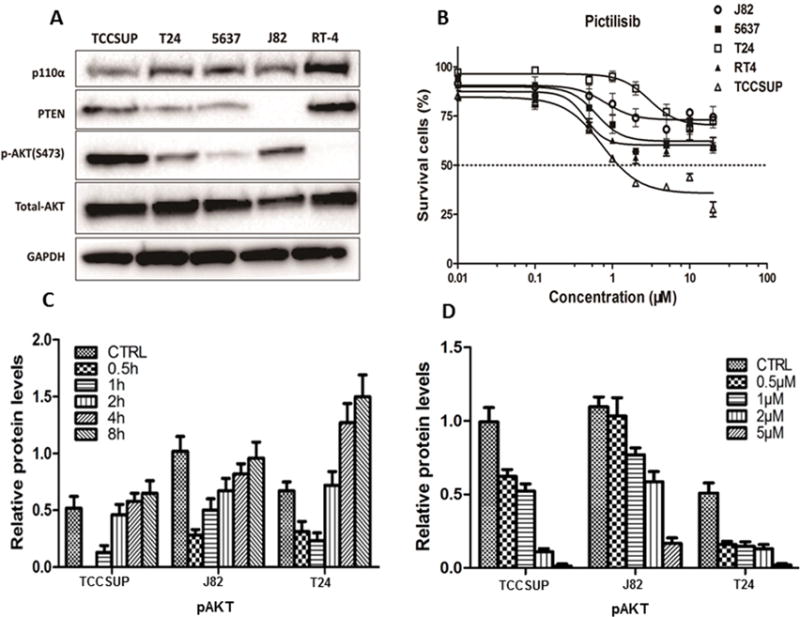
A, Western blot analysis of proteins along the PI3K/AKT pathway in bladder cancer cell lines. p-AKT was higher in TCCSUP and no PTEN protein expression was observed in J82. p-AKT was not detected in RT4. B, Dose response curve of bladder cancer cell lines treated with pictilisib at different concentrations as determined in a 72-hour MTS cell viability assay. The TCCSUP cell line was the most sensitive to pictilisib treatment with an IC50 value of 1.1 μM, while the other four cell lines were less sensitive with IC50 all greater than 20 μM. C, p-AKT(S473) was inhibited by pictilisib treatment (1 μM) in a time-dependent manner. At 0.5h, p-AKT was completely undetectable in sensitive TCCSUP while it persisted in the resistant J82 and T24 cell lines. D, p-AKT(S473) was inhibited in a dose-dependent way after 1hr of pictilisib treatment.
The cytotoxicity of targeting the PI3K pathway in bladder cancer cell lines
The small molecule pan-PI3K inhibitor pictilisib was applied to test the cytotoxicity in bladder cancer cell lines (Figure 1B, Supplementary Table S1). Consistent with the finding that TCCSUP contained an activating mutation, this cell line was the most sensitive to pictilisib treatment with an IC50 of 1.1 μM. The other four cell lines were less sensitive with IC50 all greater than 20 μM.
In order to further characterize how pictilisib affected downstream effectors of PI3K, we examined AKT phosphorylation in pictilisib-sensitive (TCCSUP) and resistant (J82 and T24) cell lines. As shown in Figure 1C and 1D, AKT phosphorylation was decreased by pictilisib in a time- and dose-dependent manner, not only in sensitive TCCSUP cells, but also in resistant T24 and J82 cells. When these cells were treated with pictilisib at a concentration of 1μM, AKT phosphorylation reached a nadir after 30 minutes of treatment with complete disappearance in the most sensitive TCCSUP cells, while measurable levels of AKT phosphorylation persisted in resistant J82 and T24 cells. These data suggested that the PI3K/AKT pathway was not completely suppressed in the two resistant cell lines. AKT phosphorylation gradually rebounded to the baseline level around 4 hours (Figure 1C), and the rebound was not due to inactivation of pictilisib in culture medium (Supplementary Figure S1C). The level of AKT phosphorylation also decreased with increasing concentrations of pictilisib (Figure 1D).
Apoptosis and cell cycle analysis were performed to evaluate the mechanism for the anti-proliferative effect of pictilisib on drug-sensitive TCCSUP and drug-resistant T24 cell lines. TCCSUP showed a modest (<3%) but significantly dose-dependent increase in apoptosis (Supplementary Figure S1B), and more dramatic (>13%) S phase arrest (Supplementary Figure S1A) after pictilisib treatment. In contrast, no significant effect was observed in T24 cells (Supplementary Figure S1).
Pictilisib exhibited anti-tumor activity in bladder cancer PDXs
Next, we determined whether the cytotoxic effect of pictilisib could be translated into in vivo anti-tumor activity in bladder cancer PDXs. If so, pictilisib can potentially be developed for treatment of advanced bladder cancers that have alterations in the PI3K pathway. For this experiment, we compared the efficacy of pictilisib between PDX BL0269 and BL0293. BL0269 harbors a PIK3CA hotspot mutation (H1047R) and BL0293 harbors a rare and non-hotspot mutation (D549Y) (5). Pictilisib significantly inhibited tumor growth in BL0269 (Figure 2A). The median time to achieve a tumor volume of ten times the baseline increased from 15.5 days in control to 27 days with pictilisib (p<0.01). Compared to the median survival of 18.5 days for the control, pictilisib treatment prolonged overall survival to 33.5 days (p<0.01) (Figure 2B). Tumors obtained at day 3 revealed that pictilisib treatment substantially reduced the level of p-AKT (Figure 2C) with diminished Ki-67 (Supplementary Figure S2A), and increased levels of cleaved caspase 3 (Supplementary Figure S2B). In contrast, tumor growth was only slightly retarded by pictilisib in BL0293 (Supplementary Figure S3G) with the median time to achieve a tumor volume of 7.5 times the baseline was 19 days and 24 days in control and pictilisib treatment groups, respectively (p=0.630).
Figure 2. Pictilisib exhibited anti-tumor activity and synergized with cisplatin in vitro and in vivo.
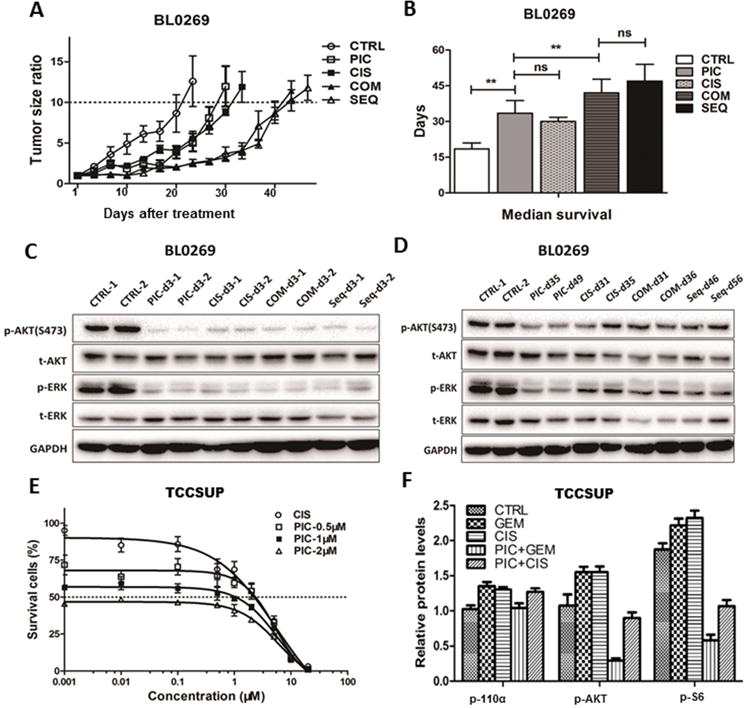
A, Tumor growth curve of PDX BL0269 (JAX#TM00015). Tumors grew more slowly in all treatment groups than the control group (CTRL). The median time of the tumor growth to 10 times the baseline was 15.5, 27, 28, 41.5 and 42 days for control (CTRL), pictilisib (PIC), cisplatin (CIS), concurrent combination (COM) and sequential (SEQ) treatment groups, respectively. B, Median survival of treatment groups and control. Median survival of CTRL, PIC, CIS, COM and SEQ groups were 18.5, 33.5, 30, 42 and 47 days, respectively. All treatment groups had longer lifespan than the control group (p<0.01). No significant difference was found between CIS and PIC (p=0.1864), or between COM and SEQ groups (p=0.1562). There was longer survival in the COM and SEQ groups than in the PIC and CIS groups (p<0.01). C and D, Western blot analysis of tumor samples obtained 3 days after treatment. The expression of p-AKT and p-ERK were suppressed among all treatment groups (C). In contrast, the expression of p-AKT and p-ERK rebounded when tumor became resistant (D). E, Dose response curve of TCCSUP bladder cancer cells treated with different concentrations of pictilisib and cisplatin as determined in a 72-hour cell viability assay. Compared to treatment with cisplatin alone, pictilisib enhanced the cytotoxicity of cisplatin, especially at a lower dose of cisplatin (<1μM). F, Quantitative analysis of western blot of p-AKT and p-S6 in TCCSUP cells after treatment with cisplatin (2 μM) or gemcitabine (0.5 μM) for 24 hours, or in combination with pictilisib (1 μM) for 1 hour. Pictilisib reduced the up-regulation of p-AKT induced by cisplatin treatment at 24 hours and decreased the expression of the downstream effector p-S6 in TCCSUP cells. ns=non-significant, * p<0.05; ** p<0.01.
Pictilisib potentiated the effect of cisplatin in vitro and in vivo
We evaluated whether pictilisib could potentiate the cytotoxicity and anti-tumor activity of cisplatin. First, we used the CI method to determine the drug-drug interaction between pictilisib and cisplatin in vitro. Cells were treated with various concentrations of cisplatin or with a fixed concentration of pictilisib (0.5, 1, or 2 μM), which corresponded to known patient plasma levels (maximum plasma concentration = 2 μM) (16). Compared to treatment with cisplatin alone, pictilisib enhanced the cytotoxicity of cisplatin, especially at a lower dose of cisplatin (<1μM, Figure 2E). The CI values of the two drugs suggested a synergistic effect (CI<0.9, Supplementary Table S2). Furthermore, we found that pictilisib could reduce the up-regulation of p-AKT induced by cisplatin treatment at 24 hours and decrease the expression of the downstream effector p-S6 in TCCSUP cells (Figure 2F).
Next, we determined whether the synergistic effect of pictilisib and cisplatin could be translated to in vivo anti-tumor activity with PDXs. We compared the anti-tumor efficacy of single agent pictilisib, cisplatin, concurrent combination and sequential treatment with pictilisib and cisplatin in NSG mice bearing the PDX BL0269. Combination and sequential treatment groups significantly delayed the tumor growth and prolonged lifespan compared with single drug treatment with pictilisib or cisplatin (p<0.01, Figure 2A and 2B, Supplementary Figure S3A and 3B), while there was no significant difference in overall survival between the combination and sequential treatment groups (p=0.156, Figure 2B, Supplementary Figure S3B). Furthermore, we revealed that pictilisib combined with gemcitabine and cisplatin (GC) was also more effective than the gemcitabine and cisplatin regimen in inhibiting the tumor growth of BL0269 (Supplementary Figure S3I). Body weight was slightly decreased in all treatment groups (p<0.01 at day 20), while there was no significant difference between single drug treatment groups and combination drug treatment groups (p=0.053 at day 20 Supplementary Figure S3). When tumors became resistant to any treatment, we observed up-regulation of p-AKT, p-ERK (Figure 2D), and Ki-67 (Supplementary Figure S2A) and down-regulation of cleaved caspase 3 (Supplementary Figure S2B). These data suggested that alternative oncogenic pathways were activated to compensate for the PI3K/AKT pathway.
Pictilisib potentiated the effect of gemcitabine in vitro and in vivo
Gemcitabine is another drug used in first-line chemotherapy for bladder cancer, specifically used in combination with platinum agents (gemcitabine plus cisplatin/carboplatin; GC) (3). We thus investigated whether gemcitabine and pictilisib had a similar synergistic drug-drug interaction. First, we found a significant left shift of the dose-response curve with combination of pictilisib and gemcitabine in TCCSUP cells compared to gemcitabine alone (Figure 3A). The CI values of gemcitabine and pictilisib in all treatment groups were 0.20 or less, suggesting a very strong synergistic cytotoxic effect between pictilisib and gemcitabine (Supplementary Table S2). Furthermore, we revealed that pictilisib could inhibit the up-regulation of p-AKT induced by gemcitabine treatment at 24 hours and decrease the levels of p-S6 in TCCSUP cells (Figure 2F).
Figure 3. Pictilisib synergized with gemcitabine in vitro and in vivo.
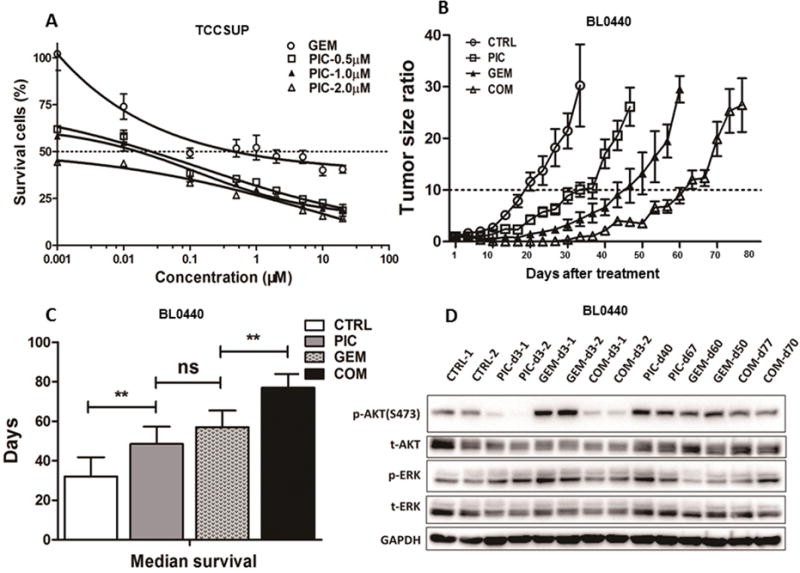
A, Dose response curve of bladder cancer cell lines treated with different concentrations of pictilisib (PIC) and gemcitabine (GEM) as determined in a 72-hour cell viability assay. A significant left shift of the dose-response curve with combination of pictilisib and gemcitabine compared to gemcitabine alone in TCCSUP cells. B, Tumor growth curve of PDX BL0440 (JAX#TM00024). In all treatment groups, tumors grew more slowly than the control group (CTRL), and the combination (COM) treatment was the most effective. The median time of the tumor growth to 10 times the baseline was 18.5, 33.5, 47 and 61.5 days for CTRL, PIC, GEM and COM treatment groups, respectively. C, Median survival of treatment groups and control. Median survival of CTRL, PIC, GEM and COM groups was 30, 48.5, 57 and 77 days, respectively. All treatment groups had longer lifespan than the control group (p<0.01). No significant difference was found between GEM and PIC (p=0.2766). There was longer survival in the COM groups than in the PIC and GEM groups (p<0.01). D, Western blot analysis of tumor samples obtained 3 days after treatment and at resistance. PIC and COM treatment reduced p-AKT, while GEM slightly increased p-AKT and p-ERK. p-AKT returned to the basal level in tumors that became resistant to treatment. ns=non-significant, *p<0.05; **p<0.01.
Next, we determined whether the synergistic cytotoxic effect of pictilisib and gemcitabine could be translated to in vivo anti-tumor effect. For this experiment, we used PDX BL0440, which had PIK3CA copy number amplification and was partially resistant to gemcitabine chemotherapy. We found the in vivo results correlated well with in vitro efficacy of pictilisib and gemcitabine. As shown in Figure 3B and Supplementary Figure S3D, the median time of the tumor growth to ten times the baseline increased from 18.5 days for the control to 33.5 days (p<0.01) with pictilisib, 47 days (p<0.01) with gemcitabine, and 66.5 days (p<0.01) with the combination group. The median survival of the combination group (77 days) was significantly longer than pictilisib (48.5 days) or gemcitabine (57 days) (p<0.01, Figure 3C and Supplementary Figure S3E). No significant difference (p=0.277) was found between pictilisib (median=48.5 days) and gemcitabine (median=57 days) single drug treatment groups. Body weight was slightly reduced in all treatment groups (p<0.01 at day 30), while no significant difference was observed between single drug groups and the combination treatment groups (p=0.327 at day 30, Supplementary Figure S3). Western blot analysis showed that p-AKT was significantly suppressed in the pictilisib and combination groups, but not in the gemcitabine group (Figure 3D). As pictilisib has no direct effect on the MEK/ERK pathway, no significant decrease of p-ERK was observed. In fact, there might be slight increase of p-ERK after pictilisib treatment, possibly secondary to positive feedback of the MEK/ERK pathway after inhibition of the PI3K/AKT pathway. Immunohistochemistry staining of tumors collected at day 3 revealed that Ki-67 (Supplementary Figure S4A) was significantly reduced, and cleaved caspase 3 was substantially increased in treatment groups (Supplementary Figure S4B). When tumors became resistant to different treatment drugs, the levels of p-AKT, Ki-67 and cleaved caspase 3 returned to baseline levels (Figure 3D and Supplementary Figure S4). These results suggested that pictilisib was not only effective in bladder cancer with PIK3CA activation mutation, but also with PIK3CA amplification, and it could potentiate the antitumor efficacy of gemcitabine.
p-S6 level correlated with response to pictilisib both in vitro and in vivo
Signaling transduction is a complicated network associated with positive and negative feedback loops, and each component of the pathways is regulated by many factors (Figure 4A). For example, the MEK/ERK pathway was suggested to be activated by PI3K/AKT pathway inhibition through PI3K-independent or PI3K-dependent feedback loop, causing drug resistance to the PI3K inhibitor (30,31). In the present study, we evaluated several key components of the PI3K/AKT pathway (p110α, p-P70S6K, p-S6) and MEK/ERK pathway (p-ERK) to determine which component correlates with drug response and may be useful as biomarkers for guiding pictilisib treatment. After exposure to 1 μM pictilisib for 1 hour, p-ERK expression increased in TCCSUP cells; p-P70S6K was down-regulated in both TCCSUP and J82 cells; and only p-S6 down-regulation was observed in the pictilisib-sensitive TCCSUP cells (Figure 4B). These results prompted us to determine whether p-S6 could potentially serve as biomarker to predict the response of pictilisib. As shown in Supplementary Figure S5, p-P70S6K levels in TCCSUP and J82 cells were modulated in a time-dependent fashion after pictilisib treatment, as was AKT phosphorylation (Figure 1 C). The phosphorylation of S6 was reduced by pictilisib in a time- and dose-dependent manner in TCCSUP cells, which was consistent with its response to pictilisib. Although the level of p-P70S6K started to rebound after approximately 2 hours in TCCSUP cells, its downstream effector p-S6 could be suppressed for 8 hours without rebound. These data strongly suggested that p-S6 might be a potential biomarker of pictilisib efficacy. Next, we investigated the expression of these key regulators of the PI3K/AKT pathway in the BL0269 and BL0440 PDX tumors. In line with the in vitro findings, we found a rebound of p-S6 in pictilisib resistant tumors of both PDX models, which further demonstrated that p-S6 levels correlated well, and could potentially serve as a biomarker to predict response to pictilisib (Figure 4C and 4D).
Figure 4. The impact of pictilisib on downstream effectors of the PI3K/AKT pathway in bladder cancer.
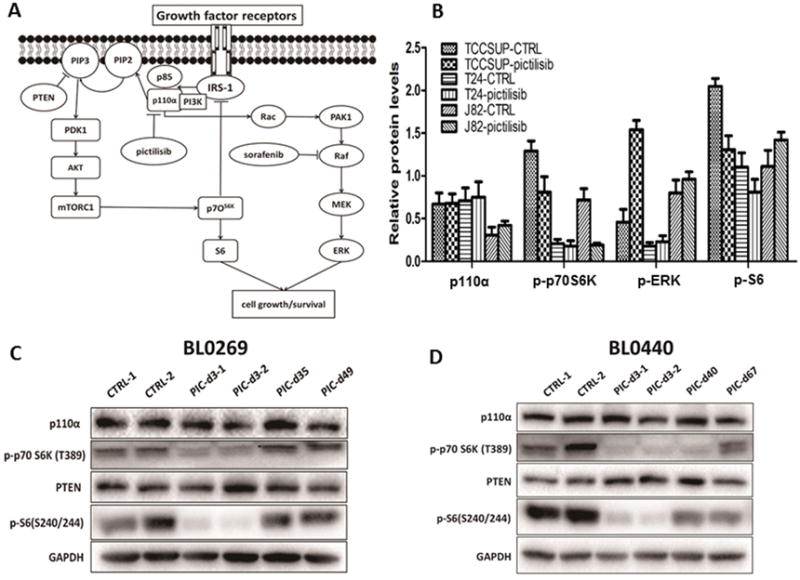
A, Pathway diagram showing the PI3K/AKT pathway and the PI3K-dependent feedback loop with the MEK/ERK pathway. B, Quantitative analysis of western blot of bladder cancer cells treated with 1 μM pictilisib for one hour. Alteration of pS6 (down-regulation) and p-ERK (up-regulation) was only observed in TCCSUP cells. C, Comparison of the PI3K/AKT pathway proteins in pictilisib-sensitive and -resistant tumors of the PDX BL0269 model. D, Comparison of the PI3K/AKT pathway proteins in pictilisib-sensitive and -resistant tumors of the PDX BL0440 model. A rebound of p-S6 in pictilisib-resistant tumors was found in both PDX models.
Compensatory activation of the MEK/ERK pathway by pictilisib treatment
It was previously reported that the anticancer activity of PI3K/AKT inhibitors could be countered by MEK/ERK pathway activation (30,31). Here we showed that in pictilisib-sensitive TCCSUP, p-ERK was upregulated in a time-dependent, but not dose-dependent manner (Figure 5A, 5B), suggesting that even at a concentration below the IC50 (1.10 μM), the MEK/ERK pathway was already activated. We did not observe any significant change of p-ERK in the resistant J82 and T24 cells (Supplementary Figure S5C and S5D). Since both pathways are important for many cellular functions, we hypothesized that simultaneous blockade of the PI3K/AKT and MEK/ERK pathways by pictilisib and sorafenib, respectively, might achieve improved or even synergistic anti-tumor activity. Sorafenib is effective in suppressing Raf/MEK/ERK pathway activity, and has already been used for the treatment of advanced hepatocellular cancer and renal cell cancer (32). Sorafenib alone was moderately effective with an IC50 of 9.4 μM in TCCSUP (Figure 5C). A synergistic to additive effect of pictilisib and sorafenib was observed at a variety of concentrations in TCCSUP (Supplementary table S2), and the activation of p-ERK induced by pictilisib could be inhibited when cells were treated with the combination of pictilisib and sorafenib (Figure 5D).
Figure 5. Sorafenib inhibited the ERK pathway and synergized with pictilisib.
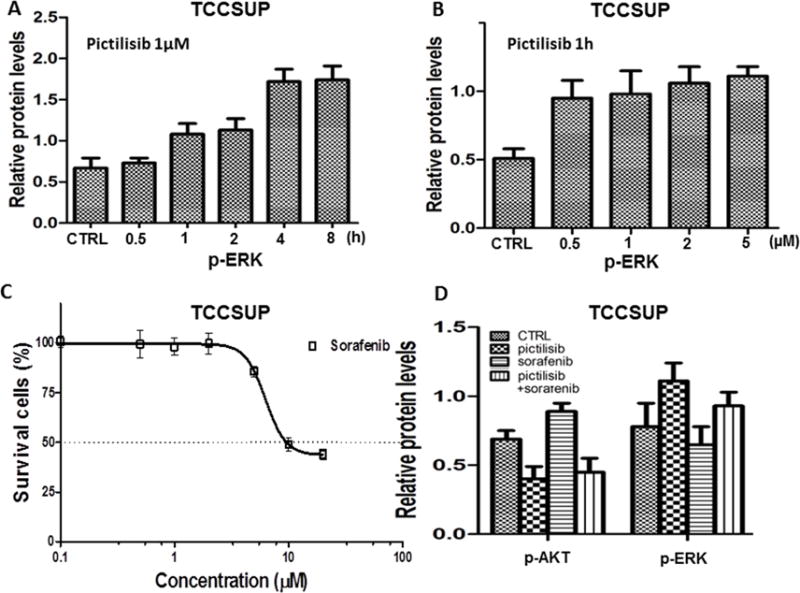
A and B, Expression of p-ERK was up-regulated in a time-dependent (A), but not dose-dependent (B) manner in TCCSUP cells treated with pictilisib. C, The dose response curve of TCCSUP cells treated with different concentrations of sorafenib, showing the absolute IC50 was 9.4 μM. D, Quantitative analysis of western blot of p-AKT and p-ERK in TCCSUP cells after treatment with pictilisib (1 μM) or sorafenib (5 μM) or the combination of pictilisib (1 μM) and sorafenib (5 μM) for 1 hour. Sorafenib mitigated the activation of p-ERK induced by pictilisib.
Down regulation of LSP1 in drug resistant tumors
To explore the mechanisms potentially underlying the development of drug resistance to pictilisib in bladder cancer, we performed RNA-sequencing analysis to comprehensively define the transcriptomic changes associated with the drug-resistant PDX BL0269 tumor model. For this, gene-level expression of 21,597 annotated, protein-coding transcripts was quantified for each sample and differentially-expressed genes (i.e., relative to the control) were subsequently determined for each group of tumors that developed resistance to pictilisib, cisplatin, or the combination treatment. Criteria for filtering of the results included a cut-off for differential expression of an absolute log2 fold change of ≥1 (i.e., ≥2-fold change) and a minimum threshold for an elevated level of expression of per kilobase of transcript per million mapped (FPKM) >1 (equivalent to one transcript per cell) (33,34). Hierarchical clustering was performed on the resulting gene sets for the pictilisib-, cisplatin-, and combination treatment-resistant tumors, and the expression patterns visualized as heat maps (Figure 6A). Intersection analysis was utilized to identify common resistance-associated expression changes, and as shown in the Venn diagram (Figure 6B), the expression of three genes, lymphocyte-specific protein 1 (LSP1), peptidase inhibitor 3 (PI3), small nucleolar RNA H/ACA box 24 (SNORA24) were altered in all three groups, and had moderate to high levels of expression (e.g., 11–519 FPKM) (Figure 6C). Compared with the control group, although the alteration of PI3 was not consistent, down-regulation of LSP1 and up-regulation of SNORA24 were observed in all drug-resistant groups. SNORA24 was reported to participate in the synthesis of pseudouridine residues in the human 18S and 28S rRNAs and the spliceosomal small nuclear RNAs (35). However, whether SNORA24 plays a role in carcinogenesis or drug metabolism is not yet known. LSP1 acts as tumor suppressor gene (36). Mutation or single nucleotide polymorphisms of LSP1 have been found to be associated with increased risk of breast cancer and hepatocellular carcinoma. Loss of LSP1 function may remove suppressing effects on the ERK/MAPK pathway, leading to increased proliferation and migration of tumor cells (36,37). In fact, we observed that p-ERK expression was increased when tumors become resistant to drug treatment in the PDX BL0269 model (Figure 2C). Consistent with the findings for LSP1 transcript levels (i.e., RNA-seq), Western blotting further confirmed the down-regulation of LSP1 protein in resistant tumor tissues (Figure 6D).
Figure 6. Gene expression profiling of control and drug-resistant PDX BL0269 tumors.
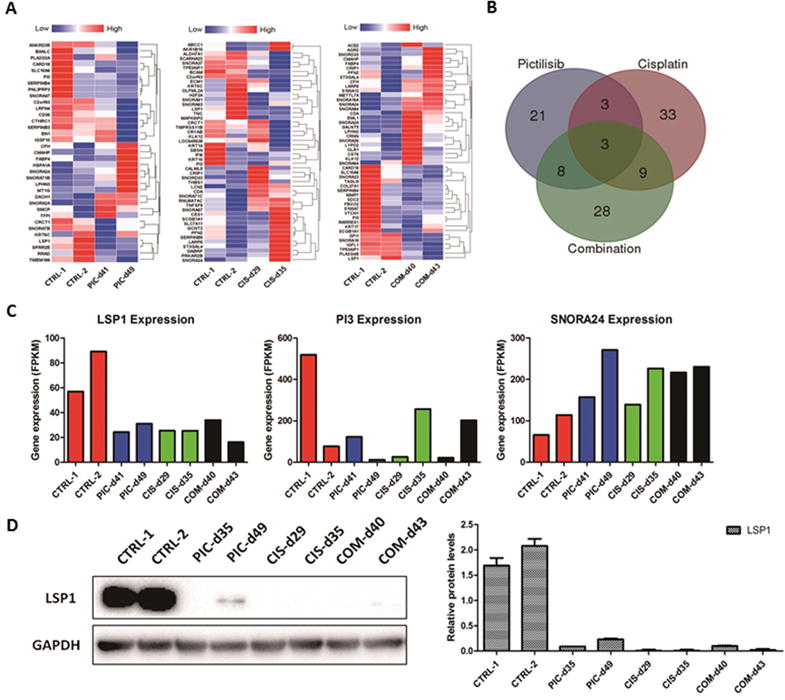
A, Hierarchical clustering of differentially-expressed genes in pictilisib (PIC)-, cisplatin (CIS)- and combination (COM)-resistant tumors of PDX BL0269 model (i.e., relative to the controls) was performed and the results visualized as heat maps. B, The relatedness of the differentially-expressed gene sets identified for the three groups of drug-resistant tumors were depicted as a Venn diagram. C, Gene expression level determined by the fragments per kilobase of transcript per million mapped (FPKM). Three genes (LSP1, PI3, SNORA24) were differentially expressed in all three drug resistant groups. Compared with the control group, although the alteration of PI3 was not consistent, down-regulation of LSP1 and up-regulation of SNORA24 were observed in all three drug resistant groups. D, Western blotting analysis showing down-regulation of LSP1 in drug-resistant tumors compared to the control group.
Discussion
Standard treatment with cisplatin-based chemotherapy for advanced bladder cancer patients has reached an efficacy plateau with very little improvement in survival over the last few decades (2). Gaining insight into the biology of bladder cancer reveals new “druggable” targets, which allow the development of molecularly guided targeted therapy. Whole exome sequencing of 131 high-grade muscle-invasive urothelial bladder carcinomas identified that alterations in the PI3K/AKT pathway occurred in 42% of tumors, and therefore presents a promising therapeutic target (4). Drugs targeting PI3K may avoid the activation of negative feedback loops of PI3K/AKT pathway and suppress this pathway robustly enough to elicit a durable response (31).
The present study was initiated to determine the feasibility of targeting the PI3K pathway to kill bladder cancer cells and to potentiate the antitumor activity of cisplatin and gemcitabine. Pictilisib was chosen to show the proof of principle in this study merely because it has already reached the Phase II clinical trial stage (17,18,38). We believe other PI3K inhibitors could possibly achieve similar effects as we previously showed with another PI3K inhibitor BEZ235 (5). The growth of five bladder cancer cell lines could be inhibited by pictilisib to some extent, but only PIK3CA E545K mutant TCCSUP cells had an IC50 as low as 1 μM. These observations are consistent with previous studies that suggested sensitivity to PI3K inhibitors was dependent on hotspot PIK3CA mutation status (9,10,29). We also revealed that AKT phosphorylation in TCCSUP cells could be inhibited by pictilisib to a nadir after 30 minutes, while remaining measurable in resistant J82 and T24 cells. It was reported that AKT could be activated by both PI3K dependent and by the PI3K independent signaling, such as serine/threonine (TBK1, KBKE) and tyrosine (Src, PTK6) kinases (39,40). We speculated that PI3K might not serve as the major regulator of AKT activity in J82 and T24 cells. Meanwhile the rapid kinetics of AKT phosphorylation suggested that alternate AKT activators also need to be considered as drug targets. We further demonstrated that pictilisib effectively inhibited tumor growth and prolonged lifespan of mice bearing PDX tumors harboring the PIK3CA hotspot mutation (H1047R) or PIK3CA amplification. PIK3CA, which encodes the p110α catalytic isoform of class I PI3K, and is one of the most commonly mutated or amplified kinases in a variety of tumors (7). By collecting data from public databases (http://www.cbioportal.org/), we identified that the frequency of mutation and amplification of PIK3CA in bladder cancer ranged from 15.2% to 26.0%. Of PI3KCA mutations in bladder cancer, E545K (41.0%) and E542K (18.7%) mutations in the helical domain are the most common. In contrast, mutation at H1047R (2.2%) in the kinase domain is more common in other cancers, but is less so in bladder cancer (Supplementary Table S1) (41). Our study provided the first preclinical evidence indicating bladder cancer patients with PIK3CA mutations (E545K and H1047R) or amplification may be appropriate for future clinical trials with a PI3K inhibitor.
The activation of PI3K/AKT pathway was reported to contribute to cisplatin and gemcitabine resistance, both of which are currently the frontline choice for advanced bladder cancer (15,42,43). Arjumand et al (15) suggested that cervical cancer cells engineered with a PIK3CA E545K mutation were more resistant to cisplatin. Previously, we observed that PDX BL0269 with a PIK3CA-H1047R mutation showed resistance to both cisplatin and gemcitabine (5). To our knowledge, combined pictilisib plus cisplatin and/or gemcitabine as a potential therapeutic approach in bladder cancer has not previously been investigated. We found pictilisib combined with cisplatin or gemcitabine resulted in an enhanced effect in decreasing proliferation of bladder cancer in vitro and in vivo. Pandey et al (44) suggested that sequential administration of a PI3K/AKT inhibitor following treatment with cisplatin resulted in enhanced antitumor efficacy for breast cancer. We found that sequential treatment with pictilisib and cisplatin is as effective as the concurrent combination treatment. Given the fact that as high as 50% of bladder cancer patients are resistant to cisplatin and gemcitabine chemotherapy (45), our concurrent combination experiment of pictilisib and cisplatin or gemcitabine may have high clinically relevance.
The OPPORTUNE clinical trial combining pictilisib and anastrozole showed significantly increased antitumor response in patients with early stage breast cancer than anastrozole alone (38). In contrast, the phase II PEGGY clinical trial, Vuylsteke et al (18) reported that patients with hormone receptor-positive, HER2-negative locally recurrent or metastatic breast cancer did not get significant benefit from pictilisib plus paclitaxel treatment compared with paclitaxel alone. In the phase II clinical trial FERGI, treatment with pictilisib alone also failed to increase progression-free survival in locally advanced or metastatic breast cancer compared with placebo, and PIK3CA mutation status was found to have no effect on predicting the benefit of pictilisib (17). Although preclinical studies have found that mutations or amplification in PIK3CA correlated with pictilisib sensitivity, clinical trials reported little or no association between PIK3CA mutation status and outcome (9,17,18). Currently, a major challenge is to identify biomarkers for predicting pictilisib response in cancer treatment. Prediction of sensitivity may require more complex signatures rather than single mutational event (7). Our results indicated that treatment-induced down-regulation of p-S6 correlated with the response to pictilisib in vitro and in vivo, and rebound of p-S6 was observed when tumors became resistant to pictilisib in PDX models. Yang et al (46) and Brien et al (9) also suggested that p-S6 could be used as a pharmacodynamic biomarker to predict PI3K inhibitor response. As a result, the alteration of p-S6 after pictilisib treatment may be a candidate biomarker for evaluating pictilisib response, but needs to be validated in clinical trials.
We performed RNA-sequencing and western blot, and found that LSP1 was down-regulated in all drug resistant groups. LSP1 was reported to interact with kinase suppressor of Ras, which functions as a scaffold for MAPK and Raf kinases and is a key regulator of cellular growth (37). Loss of LSP1 function removes ERK/MAPK pathway suppression, leading to aberrant proliferation or migration (36). In line with this mechanism, we observed that the expression of p-ERK was increased in all treatment resistant groups of the PDX BL0269 model. Similarly, Carracedo et al (30) reported that patients with solid tumors and treated with an mTOR inhibitor everolimus showed a marked increase in ERK activation. Rozengurt et al (31) suggested that PI3K/mTOR inhibitors relieved a negative feedback on receptor tyrosine kinases that leads to RAF/MEK/ERK activation. We observed that TCCSUP cells with the PIK3CA E545K mutation showed a time-dependent increase of p-ERK after pictilisib treatment, and inhibition of an upstream factor by a RAF inhibitor sorafenib could synergize with pictilisib to inhibit tumor cell and abrogate the up-regulation of p-ERK by pictilisib. These data implicate the importance of rational combinations of therapeutic agents to overcome drug resistance that was induced by compensatory activation of other pathways.
In summary, we demonstrated for the first time that pictilisib effectively enhanced the antitumor effect of cisplatin and gemcitabine in human bladder cancer in vitro and in vivo. Our results also revealed that the expression of p-S6 is a potential candidate biomarker for evaluating pictilisib response. Furthermore, our preclinical data provided good insight for combination of pictilisib and chemotherapeutic agents for bladder cancer treatment in further clinical trials.
Supplementary Material
Statement of translational relevance.
Perturbation of the PI3K/AKT pathway is frequently observed in advanced bladder cancer. The small molecule PI3K inhibitor pictilisib exhibited cytotoxic effects, and synergized with cisplatin and gemcitabine both in vitro with cell cultures and in vivo with patient-derived xenograft models carrying a PI3K mutation or amplification. The phosphorylation level of ribosomal protein S6 correlated well with response to pictilisib both in vitro and in vivo, and could potentially serve as a biomarker to predict pictilisib sensitivity. Down-regulation of LSP1 correlates with resistance to pictilisib. Taken together, our preclinical results provide strong evidence that the PI3K/AKT pathway can serve as a therapeutic target, and that pictilisib can be used as a single agent or in combination with chemotherapeutic agents for the treatment of advanced bladder cancer, and therefore warrants further clinical investigation.
Acknowledgments
We thank Qilai Long and Daniel Zhu for their assistance in conducting experiments. We are also grateful to Stephenie Y. Liu and Ryan R. Davis for their expert technical assistance in performance of the RNA-sequencing analyses.
Financial Support: Work was supported in part by Merit Review (Award # I01 BX001784, C.-X. Pan) from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program (The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government); NCI Cancer Center Support Grant (PI: de Vere White; Grant #: 2 P30 CA 0933730); The Laney Foundation (PI: de Vere White).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Trenta P, Calabro F, Cerbone L, Sternberg CN. Chemotherapy for Muscle-Invasive Bladder Cancer. Curr Treat Options Oncol. 2016;17:6. doi: 10.1007/s11864-015-0376-y. [DOI] [PubMed] [Google Scholar]
- 3.Alfred Witjes J, Lebret T, Comperat EM, Cowan NC, De Santis M, Bruins HM, et al. Updated 2016 EAU Guidelines on Muscle-invasive and Metastatic Bladder Cancer. Eur Urol. 2017;71:462–75. doi: 10.1016/j.eururo.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 4.Network CGAR. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan CX, Zhang H, Tepper CG, Lin TY, Davis RR, Keck J, et al. Development and Characterization of Bladder Cancer Patient-Derived Xenografts for Molecularly Guided Targeted Therapy. PLoS One. 2015;10:e0134346. doi: 10.1371/journal.pone.0134346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soler A, Figueiredo AM, Castel P, Martin L, Monelli E, Angulo-Urarte A, et al. Therapeutic benefit of selective inhibition of p110alpha PI3-kinase in pancreatic neuroendocrine tumors. Clin Cancer Res. 2016;22:5805–17. doi: 10.1158/1078-0432.CCR-15-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Workman P, Clarke PA, Raynaud FI, van Montfort RL. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res. 2010;70:2146–57. doi: 10.1158/0008-5472.CAN-09-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badinloo M, Esmaeili-Mahani S. Phosphatidylinositol 3-kinases inhibitor LY294002 potentiates the cytotoxic effects of doxorubicin, vincristine, and etoposide in a panel of cancer cell lines. Fundam Clin Pharmacol. 2014;28:414–22. doi: 10.1111/fcp.12043. [DOI] [PubMed] [Google Scholar]
- 9.O’Brien C, Wallin JJ, Sampath D, GuhaThakurta D, Savage H, Punnoose EA, et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3′ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16:3670–83. doi: 10.1158/1078-0432.CCR-09-2828. [DOI] [PubMed] [Google Scholar]
- 10.Walls M, Baxi SM, Mehta PP, Liu KK, Zhu J, Estrella H, et al. Targeting small cell lung cancer harboring PIK3CA mutation with a selective oral PI3K inhibitor PF-4989216. Clin Cancer Res. 2014;20:631–43. doi: 10.1158/1078-0432.CCR-13-1663. [DOI] [PubMed] [Google Scholar]
- 11.Ehrhardt M, Craveiro RB, Holst MI, Pietsch T, Dilloo D. The PI3K inhibitor GDC-0941 displays promising in vitro and in vivo efficacy for targeted medulloblastoma therapy. Oncotarget. 2015;6:802–13. doi: 10.18632/oncotarget.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, et al. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441:366–70. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 13.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 14.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–32. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 15.Arjumand W, Merry CD, Wang C, Saba E, McIntyre JB, Fang S, et al. Phosphatidyl inositol-3 kinase (PIK3CA) E545K mutation confers cisplatin resistance and a migratory phenotype in cervical cancer cells. Oncotarget. 2016;7:82424–39. doi: 10.18632/oncotarget.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17:811–21. doi: 10.1016/S1470-2045(16)00106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vuylsteke P, Huizing M, Petrakova K, Roylance R, Laing R, Chan S, et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann Oncol. 2016;27:2059–66. doi: 10.1093/annonc/mdw320. [DOI] [PubMed] [Google Scholar]
- 19.Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res. 2007;13:3989–98. doi: 10.1158/1078-0432.CCR-07-0078. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–97. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sebaugh JL. Guidelines for accurate EC50/IC50 estimation. Pharm Stat. 2011;10:128–34. doi: 10.1002/pst.426. [DOI] [PubMed] [Google Scholar]
- 22.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 23.Vinall RL, Hwa K, Ghosh P, Pan CX, Lara PN, Jr, de Vere White RW. Combination treatment of prostate cancer cell lines with bioactive soy isoflavones and perifosine causes increased growth arrest and/or apoptosis. Clin Cancer Res. 2007;13:6204–16. doi: 10.1158/1078-0432.CCR-07-0600. [DOI] [PubMed] [Google Scholar]
- 24.Wang S, Zhang H, Cheng L, Evans C, Pan CX. Analysis of the cytotoxic activity of carboplatin and gemcitabine combination. Anticancer Res. 2010;30:4573–8. [PMC free article] [PubMed] [Google Scholar]
- 25.Dickstein RJ, Nitti G, Dinney CP, Davies BR, Kamat AM, McConkey DJ. Autophagy limits the cytotoxic effects of the AKT inhibitor AZ7328 in human bladder cancer cells. Cancer Biol Ther. 2012;13:1325–38. doi: 10.4161/cbt.21793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nickerson ML, Witte N, Im KM, Turan S, Owens C, Misner K, et al. Molecular analysis of urothelial cancer cell lines for modeling tumor biology and drug response. Oncogene. 2016;36:35–46. doi: 10.1038/onc.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Platt FM, Hurst CD, Taylor CF, Gregory WM, Harnden P, Knowles MA. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res. 2009;15:6008–17. doi: 10.1158/1078-0432.CCR-09-0898. [DOI] [PubMed] [Google Scholar]
- 28.Sathe A, Guerth F, Cronauer MV, Heck MM, Thalgott M, Gschwend JE, et al. Mutant PIK3CA controls DUSP1-dependent ERK 1/2 activity to confer response to AKT target therapy. Br J Cancer. 2014;111:2103–13. doi: 10.1038/bjc.2014.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross RL, McPherson HR, Kettlewell L, Shnyder SD, Hurst CD, Alder O, et al. PIK3CA dependence and sensitivity to therapeutic targeting in urothelial carcinoma. BMC Cancer. 2016;16:553. doi: 10.1186/s12885-016-2570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther. 2014;13:2477–88. doi: 10.1158/1535-7163.MCT-14-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer R, Fetterly G, Lugade A, Thanavala Y. Sorafenib: a clinical and pharmacologic review. Expert Opin Pharmacother. 2010;11:1943–55. doi: 10.1517/14656566.2010.496453. [DOI] [PubMed] [Google Scholar]
- 33.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 34.Brooks MJ, Rajasimha HK, Roger JE, Swaroop A. Next-generation sequencing facilitates quantitative analysis of wild-type and Nrl(−/−) retinal transcriptomes. Mol Vis. 2011;17:3034–54. [PMC free article] [PubMed] [Google Scholar]
- 35.Kiss AM, Jady BE, Bertrand E, Kiss T. Human box H/ACA pseudouridylation guide RNA machinery. Mol Cell Biol. 2004;24:5797–807. doi: 10.1128/MCB.24.13.5797-5807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koral K, Paranjpe S, Bowen WC, Mars W, Luo J, Michalopoulos GK. Leukocyte-specific protein 1: a novel regulator of hepatocellular proliferation and migration deleted in human hepatocellular carcinoma. Hepatology. 2015;61:537–47. doi: 10.1002/hep.27444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison RE, Sikorski BA, Jongstra J. Leukocyte-specific protein 1 targets the ERK/MAP kinase scaffold protein KSR and MEK1 and ERK2 to the actin cytoskeleton. J Cell Sci. 2004;117:2151–7. doi: 10.1242/jcs.00955. [DOI] [PubMed] [Google Scholar]
- 38.Schmid P, Pinder SE, Wheatley D, Macaskill J, Zammit C, Hu J, et al. Phase II Randomized Preoperative Window-of-Opportunity Study of the PI3K Inhibitor Pictilisib Plus Anastrozole Compared With Anastrozole Alone in Patients With Estrogen Receptor-Positive Breast Cancer. J Clin Oncol. 2016;34:1987–94. doi: 10.1200/JCO.2015.63.9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahajan K, Mahajan NP. PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J Cell Physiol. 2012;227:3178–84. doi: 10.1002/jcp.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radisavljevic Z. AKT as locus of cancer positive feedback loops and extreme robustness. J Cell Physiol. 2013;228:522–4. doi: 10.1002/jcp.24167. [DOI] [PubMed] [Google Scholar]
- 41.Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305–16. doi: 10.1007/s10555-009-9198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang X, Fraser M, Moll UM, Basak A, Tsang BK. Akt-mediated cisplatin resistance in ovarian cancer: modulation of p53 action on caspase-dependent mitochondrial death pathway. Cancer Res. 2006;66:3126–36. doi: 10.1158/0008-5472.CAN-05-0425. [DOI] [PubMed] [Google Scholar]
- 43.Strouch MJ, Milam BM, Melstrom LG, McGill JJ, Salabat MR, Ujiki MB, et al. The flavonoid apigenin potentiates the growth inhibitory effects of gemcitabine and abrogates gemcitabine resistance in human pancreatic cancer cells. Pancreas. 2009;38:409–15. doi: 10.1097/MPA.0b013e318193a074. [DOI] [PubMed] [Google Scholar]
- 44.Pandey A, Kulkarni A, Roy B, Goldman A, Sarangi S, Sengupta P, et al. Sequential application of a cytotoxic nanoparticle and a PI3K inhibitor enhances antitumor efficacy. Cancer Res. 2014;74:675–85. doi: 10.1158/0008-5472.CAN-12-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von der Maase H, Hansen SW, Roberts JT, Dogliotti L, Oliver T, Moore MJ, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol. 2000;18:3068–77. doi: 10.1200/JCO.2000.18.17.3068. [DOI] [PubMed] [Google Scholar]
- 46.Yang W, Hosford SR, Dillon LM, Shee K, Liu SC, Bean JR, et al. Strategically Timing Inhibition of Phosphatidylinositol 3-Kinase to Maximize Therapeutic Index in Estrogen Receptor Alpha-Positive, PIK3CA-Mutant Breast Cancer. Clin Cancer Res. 2016;22:2250–60. doi: 10.1158/1078-0432.CCR-15-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.