

Abstract
EuII-containing complexes were studied with respect to properties relevant to their use as contrast agents for magnetic resonance imaging. The influences of molecular parameters and field strength on relaxivity were studied for a series of EuII-containing cryptates and their adducts with β-cyclodextrins, poly-β-cyclodextrins, and human serum albumin. Solid- and solution-phase characterization of EuII-containing complexes is presented that demonstrates the presence of inner-sphere molecules of water. Additionally, relaxivity, water-exchange rate, rotational correlation time, and electronic relaxation time were determined using variable-temperature 17O-NMR, nuclear magnetic relaxation dispersion, and electron paramagnetic resonance spectroscopic techniques. We expect that our results will be instrumental in the design of future EuII-based contrast agents.
Keywords: cryptands, cyclodextrins, europium, lanthanides, magnetic resonance imaging
Graphical abstract
EuII-containing cryptates as contrast agents: Relationship between relaxivity, field strength and molecular parameters for EuII-containing cryptates are discussed using Solomon–Bloembergen–Morgantheory.
Introduction
Magnetic resonance imaging (MRI) is a noninvasive technique used to visualize the interior of objects with high spatial resolution.[1] The contrast observed in most MR images can be enhanced with contrast agents that modulate the attributes, including longitudinal (T1) relaxation times, of protons on proximal water molecules. Contrast agents for MRI are often paramagnetic GdIII-based complexes,[2] but EuII-containing complexes have been studied as redox-active alternatives to GdIII-based complexes.[3] EuII is isoelectronic with GdIII and shortens the T1 relaxation times of nearby water protons to a similar extent as GdIII, leading to enhanced contrast in T1-weighted images. The ability to shorten T1 relaxation times is measured in terms of relaxivity (r1), and the relaxivity of a paramagnetic contrast agent depends on factors such as the applied magnetic field and molecular parameters, including hydration number (q), water-exchange rate (kex), and rotational correlation time (τR). An understanding of the relationships among the structures of paramagnetic complexes, the molecular parameters that influence relaxivity, and magnetic field strength is crucial to the ability to rationally design contrast agents. The effects of paramagnetic metal ions on the relaxation of water molecules are often described using the Solomon–Bloembergen–Morgan equations.[4] Relative to GdIII-based contrast agents, the applicability of these equations to EuII-containing complexes is understudied. To obtain experimental validation of how molecular parameters influence the relaxivity of EuII at different field strengths, we studied three EuII-containing complexes and supramolecular adducts of one of the complexes with cyclodextrins and albumin. These complexes and inclusion complexes that differ in one or more molecular parameters were studied over a range of field strengths. Here, we report the findings from these studies in terms of the relationships among the structures of EuII-containing cryptates; molecular parameters including q, kex, and τR; and field strengths from 0.00021 to 11.7 T.
Results and Discussion
Synthesis and characterization
To understand how molecular parameters influence relaxivity, we studied EuII-containing complexes 1–3 (Figure 1) and supramolecular inclusion complexes of complex 3 with β-cyclodextrin (β-CD), poly-β-cyclodextrin (poly-β-CD), and human serum albumin (HSA). Ligands for EuII-containing complexes 1 and 2 are commercially available, and the synthesis of ligand 7, which was used to form complex 3, is shown in Scheme 1. Briefly, 3-(biphenyl-4-yl)propane-1,2-diol was treated with methyl bromoacetate to obtain diacetate derivative 4 that was saponified then acidified to afford diacid 5. The diacid was converted to diamide 6 via stepwise reactions with thionyl chloride followed by diaza-18-crown-6. The diamide was reduced with borane-tetrahydrofuran (BH3·THF) complex to afford ligand 7.
Figure 1.
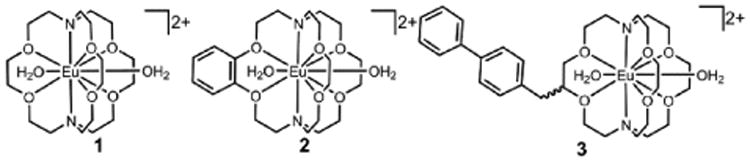
EuII-containing cryptates used in this study. Complexes are drawn with two coordinated molecules of water, and counter anions are not shown for clarity.
Scheme 1.
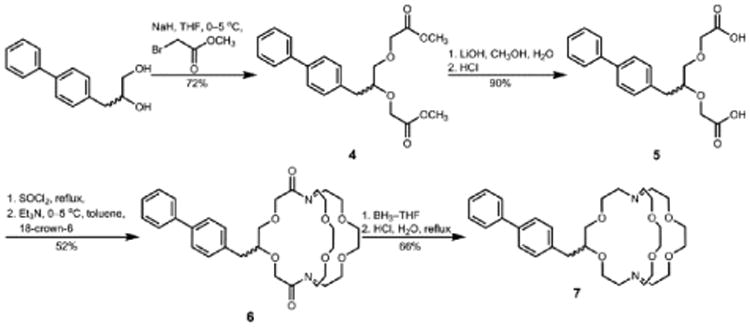
Synthesis of Ligand 7.
EuII-containing complexes 1–3 were prepared by mixing ligands and EuCl2 in water followed by adjustment of pH to 7.4 with phosphate-buffered saline. Formation of complexes 1 and 2 were monitored using water-proton-relaxation-rate enhancement. In this experiment, water-proton-relaxation rate was measured as a function of the amount of metal added to a solution of ligand. Relaxation rate decreased with the addition of ligand up to a 1:1 ligand-to-metal ratio (Figures S1 and S2), and after that point, the rate did not change [analysis of variance (ANOVA single factor analysis, 95% confidence interval)]. Because EuCl2 and complexes 1 and 2 have different relaxivity values, these data suggest that in solution, complexes 1 and 2 exist as 1:1 ligand-to-metal complexes. However, the relaxivity of complex 3 is similar to that of EuCl2, preventing the monitoring of the formation of complex 3 via proton relaxation rate. Also, the UV–visible absorption profiles of complex 3 and EuCl2 overlap, indicating that UV–visible spectroscopy does not provide useful information regarding the formation of EuII-containing complex 3. Therefore, we used inductively coupled plasma mass spectrometry (ICP–MS) to investigate the complexation of EuII by cryptand 7 to form complex 3. We prepared two samples of 7 and EuCl2 with ligand-to-metal ratios of 1:2 and 1:1. Metal not bound to cryptand was precipitated with phosphate-buffered saline,[5] and the concentrations of Eu in the supernatants were measured using ICP–MS. The metal concentrations after precipitation were consistent with the formation of 1:1 ligand-to-metal complexes, even in the presence of excess metal (Table S1). Once complex formation was verified, we characterized the complexes in the solid state and in solution.
To study coordination environments in the solid state, we obtained X-ray crystal structures of 1 and 2. We were unable to grow X-ray quality crystals of complex 3. Complex 1 displays a coordination number of nine in an eclipsed hula-hoop geometry (Figure 2a).[6] Eight coordination sites are occupied by the six oxygen atoms and two nitrogen atoms of the cryptand, and the ninth coordination site is occupied by chloride. Similarly, the crystal structure of complex 2 features a nine-coordinate eclipsed hula-hoop geometry (Figure 2b), where EuII is coordinated by six oxygen atoms and two nitrogen atoms from the cryptand and one chloride ion. These data indicate that, in the solid state, 1 and 2 exist as 1:1 metal-to-ligand complexes, consistent with proton-relaxation-rate-enhancement measurements in solution. Additionally, the crystal structures provide valuable bond distances for modeling relaxometric properties in solution. Accordingly, we turned our attention to further studying the coordination environments of 1–3 in solution. To investigate the coordination of chloride in solution, we measured the molar conductivity of complexes 1–3 in water. The molar conductivities of metal complexes 1, 2, and 3 were 177 ± 3, 183 ± 6, and 199 ± 4 S cm2 mol−1, respectively. These values are consistent with 2:1 dissociation in water,[7] indicating that, on average, both chloride ions are dissociated from EuII in solution. Further, 17O-NMR line-broadening (Tables S2–S4) in the presence of EuII-containing complexes is consistent with the presence of inner-sphere water, suggesting that chloride dissociation enables the coordination of water. Both conductivity and 17O-NMR data are consistent with the presence of EuII-containing complexes that exist in solution as nine-coordinate complexes with one inner-sphere water molecule, ten-coordinate complexes with two inner-sphere water molecules, or a mixture of nine- and ten-coordinate complexes. After studying the coordination environment of EuII-containing cryptates 1–3 in solution, we focused on studying the ability of the complexes to influence relaxation enhancement. Because we expected EuII-containing complexes to behave similarly to GdIII-containing complexes with respect to relaxivity and because aggregation of small GdIII-containing complexes results in increased positive relaxation enhancement,[8] we studied the influence of concentration on relaxation-rate enhancement to determine if, and at what concentration, aggregates form with complexes 1–3.
Figure 2.
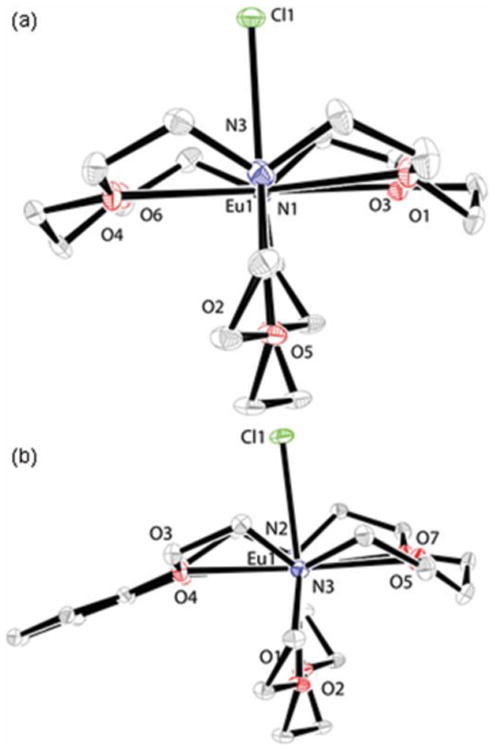
Crystal structures of (a) cryptate 1 (R-factor = 0.0259, resolution = 0.75 Å) and (b) cryptate 2 (R-factor = 0.0203, resolution = 0.55 Å). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms and outer-sphere chloride counter anions are omitted for clarity.
Aggregation Studies
To determine if relaxometric properties allude to the aggregation of complexes 1–3, longitudinal water-proton-relaxation rates were measured as a function of concentration at 37 °C. For complexes 1 and 2, plots of relaxation rate versus concentration afforded linear regression lines (Figure 3). The slopes of the lines afford relaxivities (2.1 and 3.7 mM−1 s−1 for complexes 1 and 2, respectively) that match reported values.[9] These values are also consistent with the relaxivities expected for low molecular weight chelates. Because the data in Figure 3 can be well-described by one line, it is unlikely that complexes 1 and 2 aggregate at concentrations below 10 mM. Unlike complexes 1 and 2, complex 3 showed an inflection point in the relaxation-rate profile around 3.8 mM (Figure 4). Below 3.8 mM, a linear relationship was observed with a relaxivity value of 4.3 mM−1 s−1, expected for complex 3.[9] A change in the slope of the line was observed above 3.8 mM, consistent with a higher relaxivity value of 12.1 mM−1 s−1. This change in slope is likely due to the formation of aggregates occurring through intermolecular hydrophobic interactions between biphenyl moieties. Aggregation increases the molecular tumbling time resulting in larger relaxivities, and these types of hydrophobic intermolecular interactions have been observed with similarly functionalized GdIII-containing complexes.[8a,b] To further study the aggregation of complex 3, dynamic light scattering was performed at two different concentrations (1 and 8 mM). The data (Figure S4) indicate that complex 3 aggregates at the higher concentration (8 mM) but not at the lower concentration (1 mM). This data is consistent with and supports the relaxometric data. It has also been reported that aggregate formation leads to high field bumps in nuclear magnetic relaxation dispersion (NMRD) profiles and affects electronic relaxation parameters.[8b] We used the results of these aggregation studies to avoid aggregation by performing all relaxometric characterizations, including binding-interaction studies of complex 3 with cyclodextrins and HSA, below the self-aggregation concentration of complex 3.
Figure 3.
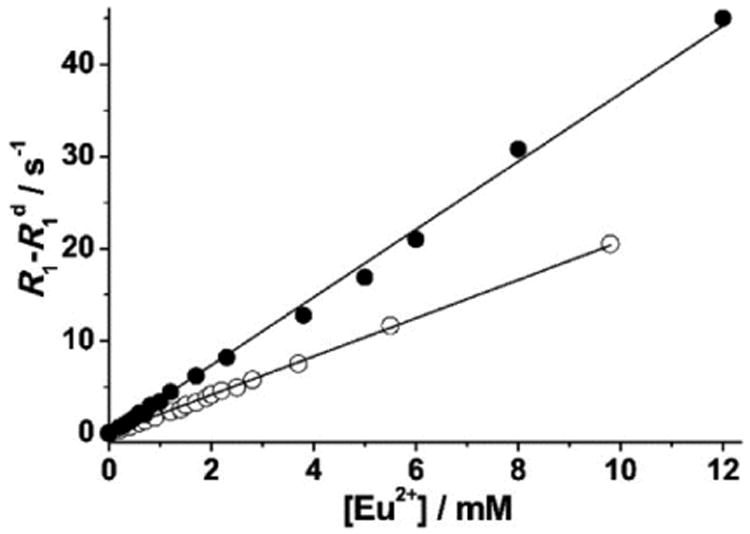
Water-proton-relaxation rates (60 MHz, pH = 7.4, 37 °C) versus concentration of complexes 1 (○) and 2 (●). The solid lines are the linear best fit (R2 = 0.99).
Figure 4.
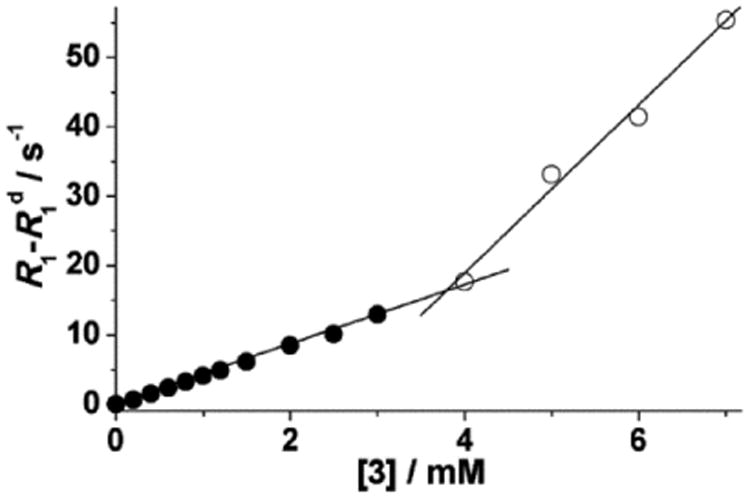
Water-proton-relaxation rates (60 MHz, pH = 7.4, 37 °C) versus concentration of complex 3. The solid lines are linear best fits (R2 = 0.99 for both lines).
Formation of inclusion compounds of complex 3 with β-cyclodextrin (β-CD) and poly-β-cyclodextrin (poly-β-CD)
The formation of host–guest non-covalent interactions between suitably functionalized complexes and slowly tumbling substrates represents a well-established approach to increase relaxivity.[10] The presence of the biphenyl moiety in 3 leads to an interaction of the complex with the hydrophobic cavity of β-CD. The binding interaction was investigated using the well-established proton-relaxation-enhancement technique.[11] This technique consists in measuring the changes of R1 of a solution of a complex as a function of increasing concentrations of the host. With this method, the binding parameters KA, n (the number of equivalent and independent binding sites), and r1b (the relaxivity of the final supramolecular adducts) can be evaluated. A dilute aqueous solution of 3 (0.9 mM) was titrated with β-CD, at 60 MHz and 310 K, and the least-squares fit of R1 versus the binding isotherm of β-CD concentration (Figure 5) provided the value of the affinity constant [KA = (4.0 ± 0.2) × 103 M−1] and the relaxivity of the adduct (r1b = 8.7 ± 0.2 mM−1 s−1). Upon binding of complex 3 with β-CD, a relaxivity enhancement of 2.1× was observed (4.2 ± 0.1 and 8.7 ± 0.2 mM−1 s−1 for 3 and 3/β-CD, respectively). This enhancement of relaxivity (+107%) is likely an effect of the increase in rotational correlation time (τR) associated with the increase in molecular weight.[10] This data led us to use 2.5 equivalents of β-CD for all further relaxometric studies of 3/β-CD to favor (>99% macromolecule-bound complex based on KA) complex 3 being associated with β-CD.
Figure 5.

Water-proton-relaxation rates (60 MHz, pH = 7.4, 37 °C) of solutions of 3 (0.9 mM) as a function of the amount of β-CD. Error bars represent the standard error of the mean of three independently prepared samples.
A similar titration was performed to investigate the larger supramolecular assemblies formed by complex 3 with poly-β-CD. A solution of complex 3 (1 mM) in phosphate-buffered saline was titrated with poly-β-CD, and relaxation-rate enhancement was measured (Figure 6). The poly-β-CD that was used had an average molecular weight of 19 kDa, corresponding to approximately 16 β-CD units, each of which can bind in principle one unit of complex 3. The polymer is characterized by nearly equivalent binding sites and, therefore, is expected to influence the rotational dynamics of each bound complex in an approximately equivalent manner. Data were fit to a simple model that considers the presence of n equivalent and independent binding sites, affording association constants and bound relaxivities. The best-fit of the data was obtained with n = 9 and a KA value of 2.4 (±0.2) × 103 M−1, similar to the value found for the interaction with β-CD. Association of complex 3 with poly-β-CD increased relaxivity from 4.2 to 12.5 mM−1 s−1, which represents an enhancement of +198%, consistent with a pronounced decrease in molecular tumbling.[8a,10a,10b,12] This data led us to use one equivalent (with the assumption of nine binding sites per equivalent) of poly-β-CD for all further relaxometric studies of 3/poly-β-CD to favor (∼95% macromolecule-bound complex based on KA) complex 3 being associated with poly-β-CD.
Figure 6.
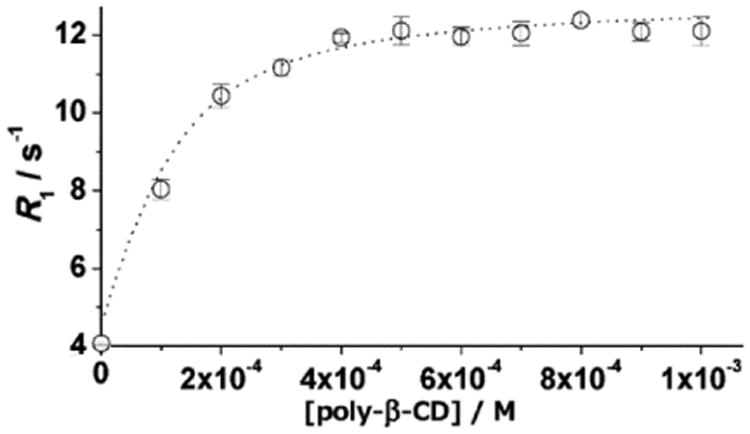
Water-proton-relaxation rates (60 MHz, pH = 7.4, 37 °C) of solutions of 3 (1 mM) as a function of the amount of poly-β-CD. Error bars represent the standard error of the mean of three independently prepared samples.
Interaction of complex 3 with HAS
In addition to forming inclusion complexes with cyclodextrins, the biphenyl group can promote non-covalent interactions with hydrophobic binding sites on HSA. Binding parameters for the interaction of 3 with HSA were determined using the proton-relaxation-enhancement method by measuring R1 of a solution of complex 3 (1 mM) in phosphate-buffered saline as a function of protein concentration (Figure 7). The binding interactions of complex 3 with HSA result in a large relaxivity gain (from 4.2 to 16.6 mM−1 s−1) that we primarily attribute to the decrease of the rotational dynamics of the complex in the adduct with the protein.[13] Analysis of the experimental data was carried out assuming the presence of one class of equivalent and independent binding sites (n = 1), even though the presence of multiple sites with lower affinity on HSA cannot be excluded.[14] In this way, we could estimate the value of the thermodynamic association constant (KA = 2.7 (±0.4) × 103 M−1), which is of the same order of magnitude as found with β-CD and polyβ-CD. Consequently, this data led us to use eight equivalents of HSA for all further relaxometric studies of 3/HSA to favor (∼91% macromolecule-bound complex based on KA) complex 3 being associated with HSA.
Figure 7.
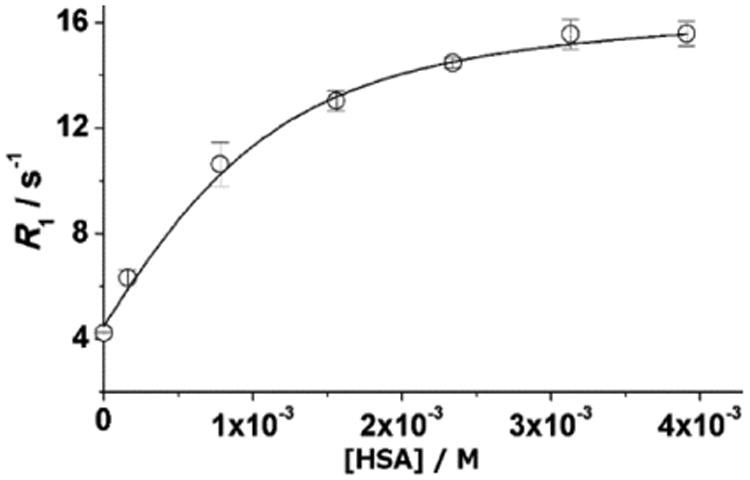
Water-proton-relaxation rates (60 MHz, pH = 7.4, 37 °C) of solutions of 3 (1.0 mM) as a function of the concentration of HSA. Error bars represent the standard error of the mean of three independently prepared samples.
After characterizing EuII-containing complexes 1–3 and the interactions of complex 3 with β-CD, poly-β-CD, and HSA, we studied the relaxometric behavior of those complexes and macromolecular systems at different field strengths.
Relaxometric characterization
To evaluate the molecular parameters that determine the efficiency of the EuII-containing complexes as relaxation agents, variable-field (0.00021 to 11.7 T) longitudinal 1H-NMR relaxation rates (NMRD profiles) were measured via fast-field-cycling relaxometry and variable-temperature 17O-NMR spectroscopy was performed. The NMRD profiles of complexes 1–3 showed that relaxivity was in the order 3>2>1 at every field studied (Figure 8). The relaxivities of complexes 1–3 are highest at low field strengths (<0.02 T), and there is a dispersion between 0.05 and 0.2 T. Around ultra-high fields (≥7 T), the relaxivities of complexes 1–3 increase from 3 to 7 T and then decrease with increasing field strengths above 7 T. When complex 3 forms inclusion complexes with cyclodextrins or HSA, relaxivity changes. The NMRD profiles of inclusion complexes 3/β-CD and 3/poly-β-CD indicate that these complexes possess higher relaxivities than complex 3 at all fields studied (Figure 8). Broad bumps in relaxivity peaking around 1 T were observed, and these relaxivities decreased at higher field strengths. When complex 3 interacted with HSA, relaxivity increased relative to cyclodextrin-bound complexes below 7 T, with a narrow peak around 0.7 T. In the ultra-high field region, the relaxivity of 3/HSA was lower than that of 3/β-CD and 3/poly-β-CD.
Figure 8.

NMRD profiles of solutions of 1 (
 ), 2 (
), 2 (
 ), 3 (◆), 3/β-CD (○), 3/poly-β-CD (Δ), and 3/HSA (◊) at pH 7.4 and 298 K. The solid lines were fitted assuming r = 3.2 Å, q = 2, a = 4.5 Å, and D = 23.0 × 10−10 m2 s−1.
), 3 (◆), 3/β-CD (○), 3/poly-β-CD (Δ), and 3/HSA (◊) at pH 7.4 and 298 K. The solid lines were fitted assuming r = 3.2 Å, q = 2, a = 4.5 Å, and D = 23.0 × 10−10 m2 s−1.
The analysis of these NMRD profiles using the theory of paramagnetic relaxation enables evaluation of the molecular parameters and elucidation of the relationship between these parameters and relaxivity. The field-dependence of r1 arises from modulation of magnetic dipolar interactions between metal ions and solvent nuclei. Modulation occurs either through chemical exchange between bound and bulk water molecules (inner-sphere)[4a] or through long-range interactions with outer-sphere water molecules that diffuse near the paramagnetic ions (outer-sphere).[4a] Experimentally acquired data were analyzed in terms of the Solomon–Bloembergen–Morgan equations and Freed's model for the inner- and outer-sphere contributions, respectively, to proton relaxivity.[4] In the relaxation equations, some parameters were set to known or reasonable values: water-coordination number (q), distance between EuII ions and the nuclei of inner-sphere (r) and outer-sphere water protons (a), and the diffusion coefficient (D). Moreover, as a first approximation, the exchange lifetime τM was set to the value previously determined for 1 by Tóth, i.e. 298τM = 3.2 ns.[15] Water-coordination numbers of complexes 1–3 were assumed to be two (q = 2) based on a ten-coordinate EuII-containing cryptate that was recently reported with two non-adjacent water molecules.[16] In addition, the isomorphic SrII analog of complex 1, provides support of a water-coordination number of two.[15] Another key parameter that is related to coordinated water is the Eu–H distance between the EuII ions and the proton of coordinated water molecules. This value was fixed to 3.2 Å, which was obtained from a crystal structure of a ten-coordinate EuII-containing cryptate with two non-adjacent inner-sphere water molecules.[16] The Eu–H distance of 3.2 Å matches the value reported for another EuII-containing complex (3.2 Å for [EuDTPA(H2O)]3− (DTPA = diethylenetriamine-N,N,N′,N″,N″-pentaacetate).[17] In addition, two other parameters affecting the outer-sphere contributions to relaxivity are the distance between EuII ions and the nuclei of outer-sphere water protons, a, and the relative diffusion coefficient, D. The value of a was set to 4.5 Å based on the distance from EuII to the furthest ligand proton of crystal structure of complex 1. The diffusion coefficient D was fixed to the value for [EuDTPA(H2O)]3−.[17] The parameters q, r, a, and D were fixed to the values mentioned above during the least-squares analyses of the NMRD profiles. In the analyses, the parameters of electron relaxation, Δ2 and τV, and the rotational correlation time, τR, were used as adjustable parameters. The results of the best-fit procedure are reported in Table 1.
Table 1.
Parameters relevant to the relaxivity of 1–3 and macromolecular adduct of 3.
| Parameters | 1 | 2 | 3 | 3/β-CD | 3/poly-β-CD | 3/HSA |
|---|---|---|---|---|---|---|
| r1 / mM−1 s−1[a] | 3.85 | 4.61 | 5.64 | 13.1 | 17.8 | 27.9 |
| Δ2 / 1020 s−2[a] | 1.35 ± 0.08 | 1.35 ± 0.11 | 1.23 ± 0.13 | 0.16 ± 0.01 | 0.15 ± 0.02 | 0.09 ± 0.01 |
| τv298 / ps[a] | 3.0 ± 0.3 | 4.2 ± 0.2 | 4.1 ± 0.3 | 23 ± 2 | 30 ± 2 | 64 ± 7 |
| τRL298 / ps[a] | NA | NA | NA | 150 ± 9 | 259 ± 13 | 276 ± 18 |
| τRG298 / ps[a] | 46 ± 2 | 63 ± 3 | 93 ± 5 | 454 ± 25 | 1920 ± 124 | 50,000[c] |
| S2[a] | NA | NA | NA | 0.43 ± 0.02 | 0.17 ± 0.02 | 0.14 ± 0.01 |
| kex298/108 s−1[b] | 2.5 ± 0.2 | 0.5 ± 0.1 | 0.5 ± 0.1 | 0.5[c] | 0.5[c] | 0.5[c] |
| ΔHM / kJ mol−1[b] | 33.4 ± 0.9 | 39.9 ± 1.1 | 31.1 ± 0.6 | NA | NA | NA |
| A0/ħ / 106 rad s−1[b] | −3.9 ± 0.2 | −4.2 ± 0.2 | −4.2 ± 0.2 | NA | NA | NA |
| r / Å[c] | 3.2 | 3.2 | 3.2 | 3.2 | 3.2 | 3.2 |
| q[c] | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 |
| a / Å[c] | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 |
| D298 /10−10 m2 s−1[c] | 23 | 23 | 23 | 23 | 23 | 23 |
| ΔHpp / G[d] | 178.8 | 225.3 | 265.0 | 198.7 | 152.3 | 217.3 |
| 1/T2e / 109 s−1[d] | 2.70[e] | 3.40[e] | 4.00[e] | 3.00 | 2.30 | 3.28[e] |
| τM310 / ns[b] | 2 | 10 | 12 | 12[c] | 12[c] | 12[c] |
Parameters obtained from NMRD fitting.
Parameters obtained from 17O-NMR spectroscopy.
Parameters fixed during the fitting procedure.
Parameters obtained from X-band EPR spectroscopy at 110 K.
Values obtained from reference 18. NA = not applicable
More detailed information on the kinetics of water exchange are obtained by measuring 17O-NMR transverse relaxation rate (R2) as a function of temperature, typically obtained at a relatively high magnetic field strength (>4.7 T). The temperature-dependence of R2 is described by the Swift–Connick equations.[19] These equations involve the bound-water residency lifetime τM, electronic relaxation times T1e and T2e, the hyperfine coupling constant A0/ħ, and q.
Water exchange
Variable-temperature 17O-NMR relaxation rates, R2p, for complexes 1–3 were measured at 9.4 T and pH = 7.4. The data are often reported as reduced transverse relaxation rates, R2r, defined as 1/T2r = R2r = R2p/pM, where pM is the molar fraction of inner-sphere water molecules. The reduced transverse 17O-relaxation rates measured for EuII-containing complexes 1, 2, and 3 are presented in Figure 9. The sign of the temperature dependence of 1/T2r depends on whether the transverse relaxation is dominated by τM, which increases with decreasing temperature, or by the transverse relaxation time of the inner-sphere water molecules, T2m, which increases with increasing temperature. For the complexes 1, 2, and 3, 1/T2r increases with decreasing temperature over the temperature range studied, indicating short residence times for inner-sphere water molecules in these complexes. Thus, all three complexes can be considered to be in the fast-exchange regime for water exchange.
Figure 9.
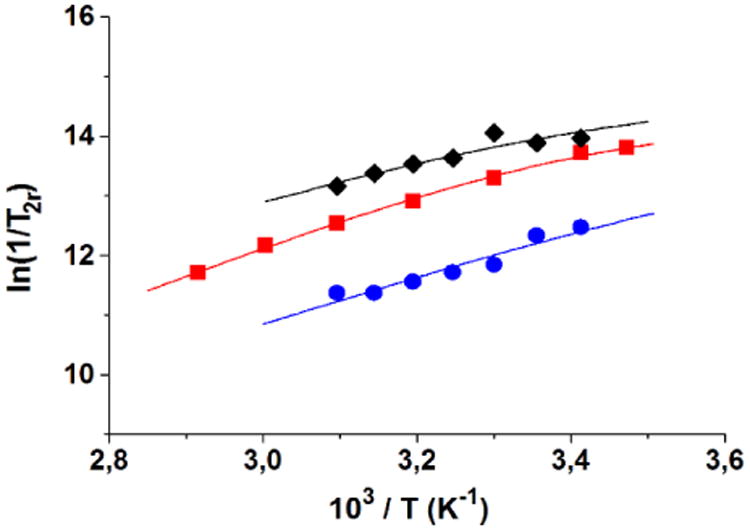
Temperature dependence of reduced transverse 17O-NMR relaxation rates of 1 (
 ), 2 (
), 2 (
 ), and 3 (◆) at 9.4 T and pH 7.4. The solid lines are the best fits of the experimental data points.
), and 3 (◆) at 9.4 T and pH 7.4. The solid lines are the best fits of the experimental data points.
The kex value of 1 is consistent with the previously reported value, obtained from a variable-temperature and pressure 17O-NMR study at 9.4 T.[15] The rates of water exchange of complexes 2 and 3 are quite comparable with each other, with differences of the order of experimental error. Compared to the value of 1, the rates for complexes 2 and 3 are slower by a factor of 5. Although the parameters are affected by a non-negligible uncertainty due to the accuracy of the measurements in the case of low-concentration solutions, the increase in the residency times of the inner-sphere water molecules for complexes 2 (τM298 = 20 ns) and 3 (τM298 = 20 ns) relative to 1 (τM298 = 4 ns) appears to be significant. A well-known complex, investigated in detail, is [Gd(EGTA)(H2O)]− (EGTA = 3,12-bis(carboxymethyl)-6,9-dioxa-3,12-diazatetradecanedioate) for which it has been reported a τM of 30 ns (at 298 K), a surprisingly short value for a q = 1 GdIII-containing complex of a poly(aminocarboxylic) ligand.[20] Two derivatives of [Gd(EGTA)(H2O)]− functionalized with benzene or naphthalene moieties fused to the oxyethylene bridges were synthesized and their relaxometric properties studied.[21] The kex values were found to be roughly twice as high as the corresponding value of [Gd(EGTA)(H2O)]−,[22] an opposite trend to that measured for complexes 1–3. In the absence of variable-pressure 17O-NMR data that can provide information on the mechanism of the exchange process, we can only suggest a plausible hypothesis of this trend. For [Gd(EGTA)(H2O)]−, the mechanism of exchange is dissociative: The faster rate of water exchange measured for the functionalized derivatives was rationalized in terms of a higher steric crowding of the aromatic moiety relative to the ethylene group, inducing a destabilization of the bound water molecule and thus a decrease in its mean residency lifetime. On the other hand, water exchange for 1 was found to proceed via an interchange mechanism,[15] which implies that the incoming water molecule also participates in the rate-determining step. Therefore, the introduction of hydrophobic functional groups can perturb the solvation shell of the complex close to the water coordination sites and make the exchange process slower (longer τM).
To explore the influence of charge on water-exchange rate, the water-exchange rates of complexes 1–3 were compared with other EuII-containing complexes (Figure 10). Two complexes are anionic and have in common the presence of one coordinated water molecule. One is neutral (EuODDA; q = 1) (ODDA = 1,14,10,13-tetraoxa-7,16-diazacyclooctadecane-7,16-diacetate), and 1–3 are cationic with q = 2. The number and the type of EuII-containing complexes are limited, making it impossible to draw firm conclusions; however, the impact of the global electric charge appears to have a predominant role on water-exchange rate, as observed with GdIII-containing complexes.[23] Anionic complexes exhibit significantly faster rates of exchange relative to that of the neutral complex, and the cationic chelates have even slower values of kex. On the other hand, the kex values of the EuII-containing complexes cover a much more limited range than that characteristic of GdIII chelates, i.e. two orders of magnitude instead of five. At least partially, the faster rates of exchange of the EuII-containing complexes compared to those of GdIII can be attributed to the lower charge density and longer metal–coordinated water distance. Both parameters are more favorable for fast water exchange with EuII relative to GdIII.[24]
Figure 10.
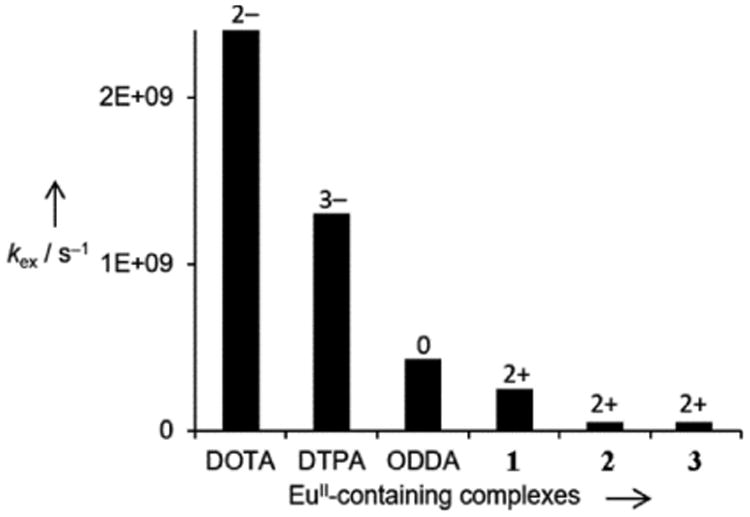
Water-exchange rate of anionic, neutral, and cationic EuII-containing complexes. Data for EuII-containing complexes of DOTA,[3b] DTPA,[25] and ODDA[17] are from published reports.
Because of the short τM values, complexes 1–3 are in the fast-exchange regime, where this parameter does not influence or limit relaxivity. The same condition occurs for the macromolecular adducts of 3. Plots of the temperature-dependence of 1H-relaxivity, determined at 9.4 T in the range of 288 to 318 K (Figure S7), show that r1 decreases with increasing temperature for all the systems. These results indicate that the inclusion complexes of 3 are in the fast water-exchange regime over the entire temperature range studied.
Electronic relaxation time
Electronic relaxation is characterized by two parameters: the trace of the square of the transient zero field splitting (ZFS) tensor, Δ2, and the correlation time that describes the modulation of the ZFS, τV. In the least-squares fitting procedure, these parameters are essentially determined from NMRD profiles. The values of Δ2 and τV of complexes 1–3 are similar to those reported for [EuII(H2O)8]2+ and to each other.[26] As noted by Tóth and coworkers, this is a different behavior from GdIII-containing complexes for which, typically, electron spin relaxation is slower after the aqua ion is chelated.[27]
Unlike GdIII-containing chelates, 1/T1e cannot be considered negligible compared to 1/τR. Therefore, electronic relaxation can limit relaxivity at typical imaging fields, even for low-molecular-weight EuII-containing complexes. For example, in the case of complex 3, 1/T1e contributes to the value of 1/τC about 40% at 0.5 T and 4.5% at 3 T (Figure S8). The contribution becomes negligible in the ultra-high field range (<1% at 7 T). This influence, albeit limited, of the electronic relaxation time on relaxivity might explain the small bump near 7 T, observable in NMRD profiles of 1–3. The X-band EPR spectra of 1–3 were recorded at 110 K.[18] The X-band (0.34 T) line width (ΔHpp, G) values show a slight increase from 1 to 3, reflecting small difference in their electronic relaxation times. From the bandwidth, the T2e values were calculated (Table 1), using simplifications introduced by Reuben and adapted by Merbach.[28] The T2e values compare favorably to the values estimated from fitting the NMRD profiles using the Morgan equation.[4]
In the analysis of 3/β-CD, 3/poly-β-CD, and 3/HSA, the parameters for electronic relaxation were used as empirical fitting parameters and do not have precise physical meanings for these slowly tumbling systems.[29]
Rotational correlation time
In the clinically relevant region of proton Larmor frequencies (>10 MHz), rotational correlation represents the limiting factor of relaxivity. This relationship implies that the overall correlation time, τC, is largely dominated by the rotational correlation time and that the contribution of both the exchange lifetime and the electronic relaxation can be safely neglected. Because τR is roughly proportional to the molecular weight of EuII-containing chelates, a linear dependence of τR on the molecular masses of the complexes should exist. A plot of the τR values of 1–3 (Table 1) as a function of molecular mass yields a straight line (R2 = 0.987). This linear behavior implies that the outer-sphere contribution to r1 is nearly identical for the three complexes. In comparison to low-molecular-weight GdIII-containing complexes, the τR values are slightly shorter, even accounting for the larger size of EuII relative to GdIII.[4a,17] This discrepancy might arise, in part, from the choice of the distance value r. For example, if the value of r is fixed to 3.3 instead of 3.2, then the τR value of 2, which has the same molecular mass as [Gd(DOTA)(H2O)]− (τR = 66 ps),[30] increases from 63 to 75 ps. Additionally, a different solvation sphere due to the different charge densities of the two ions might contribute to the differences in rotational correlation times.
The presence of a biphenyl group in complex 3 facilitates the formation of adducts with cyclodextrins and HSA, and the rotational correlation time of those adducts was also studied. To obtain information regarding the rotational dynamics of the inclusion complexes of 3, the Lipari–Szabo model was used.[31] This model-free approach considers the occurrence of an internal rotation, characterized by a correlation time τRL, superimposed on a global motion described in terms of the correlation time τRG. The degree of correlation between the two types of motion is described by the parameter S2 that assumes the value of zero when the two motions are completely independent; in the absence of local fluctuations, the complex is immobilized, the motion is isotropic, and S2 = 1. In complex 3, the biphenyl moiety is connected to the cryptand through a methylene group that can freely rotate causing local rotation superimposed to global tumbling motion of the complex. Therefore, a five-parameter (Δ2, τV, τRL, τRG, and S2) least-squares fit of the data was performed (Table 1).
The global rotational correlation times of complexes 1–3 and of cyclodextrins and HSA bound to complex 3 (Table 1) correlate with the molecular weights of the molecules, where increasing molecular weight roughly correlates with increasing rotational correlation time.
The marked decrease in molecular tumbling following the formation of the supramolecular adducts is signaled by the formation of broad relaxivity peaks in the high field region of the NMRD profiles, distinctive of slowly rotating systems. However, an increase of the local rotational flexibility of the metal complex at the binding site is highlighted by the increasing difference between the local and global values of the rotational correlation times, which limits the relaxivity. This local rotational flexibility is also shown by the decrease of S2 from 0.43 for 3/β-CD to 0.14 for 3/HSA. The S2 obtained for complex 3/β-CD indicates that the inclusion complex prevents internal rotation to some extent. This observation is further evidenced by global rotational correlation time of 3/β-CD being only 3.0× longer than the local rotational correlation time. The relaxivity enhancement of 2.1× (at 1.5 T) observed upon formation of inclusion complexes between complex 3 and β-CD further supports the presence of a rigid adduct with restricted internal rotation in 3/β-CD. As expected, an even larger increase in rotational correlation time was observed when complex 3 was mixed with poly-β-CD. The larger difference between local and global rotations of the macrocyclic complex 3/poly-β-CD and the lower value of the order parameter S2 indicate a higher degree of rotational flexibility than 3/β-CD. Because of internal rotation, the relaxivity enhancement observed in 3/poly-β-CD at 1.4 T is only 3.5 despite the global rotational correlation time of 3/poly-β-CD being 20× longer than that of complex 3.
The biphenyl moiety in complex 3 also interacts with HSA to yield an increase in global rotational correlation time. However, this slowdown in molecular motion is partly offset by the high degree of local motion: The relaxivity enhancement of complex 3 obtained upon binding with HSA (20.9 mM−1 s−1, 1.4 T) is only 3.9× that of complex 3. Therefore, the relaxivity of the adduct is primarily limited by the poor correlation between global and local motions. A more restricted local rotation of the complex at the binding site of the protein would favor a larger enhancement of relaxivity. It has to be noted that the fitting (continuous lines in Figure 8) was restricted to frequencies above 3 MHz because of the inadequacy of Solomon–Bloembergen–Morgan theory for the description of the rotational dynamics of slow-rotating systems at low magnetic field strengths.
Conclusions
The present study describes a detailed analysis of the contribution of molecular parameters (water-exchange rate, electronic relaxation time, and rotational correlation time) to the relaxivity of EuII-containing cryptates over a wide range of field strengths using 1H-NMR, 17O-NMR, and EPR spectroscopies. The results reported here demonstrate for the first time that it is possible to achieve a high relaxivity enhancement through non-covalent interactions between suitably functionalized EuII-containing complexes and slowly tumbling substrates like β-CD, poly-β-CD, and HSA. Similarly to the case of well-known GdIII-based complexes, the relaxivities of rapidly rotating EuII chelates are essentially limited by the value of the rotational correlation time. A lengthening of τR causes a considerable increase in the longitudinal relaxation rate of water protons. However, unlike what is often observed in the case of GdIII-based complexes, the slow exchange process of the bound water molecules does not hamper the increase in relaxivity at the imaging fields of the supramolecular adducts. This is because the EuII complexes investigated to date have solvent exchange rates values in the range necessary to achieve high relaxivity. This property, in the case of macromolecular systems, largely compensates for the negative effect on r1 of a greater distance between the water protons and the paramagnetic ion, thus enabling relaxivity values comparable to those of the analogous GdIII-based systems.
Despite these interesting and encouraging results, the number and type of complexes studied is limited and there is a need to expand the available library of EuII-based systems to obtain more in-depth information on the correlation between solution structure and molecular relaxation parameters. However, these preliminary results clearly indicate that in the case of EuII-containing complexes, it is possible to design and develop highly sensitive macromolecular or nano-sized probes for advanced MRI applications.
Experimental Section
Commercially available chemicals were of reagent-grade purity or better and were used without further purification unless otherwise stated. Water was purified using a PURELAB Ultra Mk2 water purification system (ELGA) and degassed under reduced pressure prior to use. 3-(Biphenyl-4-yl)propane-1,2-diol was synthesized according to a reported procedure.[9] Analytical thin-layer chromatography (TLC) was carried out on ASTM TLC plates precoated with silica gel 60 F254 (250 μm layer thickness). TLC visualization was accomplished using a UV lamp followed by charring with potassium permanganate stain (3 g KMnO4, 20 g K2CO3, 5 mL 5% w/v aqueous NaOH, 300 mL H2O). Flash chromatography was performed using silica gel 60, 230–400 mesh (EMD chemicals). 1H-NMR, correlation spectroscopy (COSY), 13C-NMR, distortionless enhancement by polarization transfer (DEPT), and heteronuclear multiple quantum coherence (HMQC) spectra were obtained using an MR400 (9.4 T) or V500 (11.7 T) spectrometers. Peaks were assigned using DEPT, COSY, and HMQC spectra or by comparison with reported spectra.[9] Chemical shifts are reported relative to residual solvent signals (CD3CN: 1H: δ 1.94;13C: δ 1.32). 1H-NMR data are assumed to be first order, and the apparent multiplicity is reported as “s” = singlet, “d” = doublet, “dd” = doublet of doublet, “ddd” = doublet of doublet of doublets, “m” = multiplet, and “brs” = broad singlet. Italicized elements are those that are responsible for the shifts. High-resolution electrospray ionization mass spectra (HRESIMS) were obtained using an electrospray time-of-flight high-resolution Waters Micromass LCT Premier XE mass spectrometer.
The concentrations of Eu were determined using ICP–MS or energy-dispersive X-ray fluorescence (EDXF) spectroscopy. Samples for ICP–MS were diluted using aqueous nitric acid (2%). Standard solutions were prepared by serial dilution of a commercially available standard (Eu2O3 in aqueous nitric acid (5%) 1000 ppm, Alfa Aesar). Calibration curves were generated using the 153Eu isotope ion counts for a 10–100 ppb concentration range. ICP–MS analyses were performed using an Agilent Technologies 7700× ICP–MS instrument in the Lumigen Instrument Center of Wayne State University. EDXF measurements were performed with a Shimadzu EDX-7000 spectrometer. Calibration curves were generated using fluorescence intensity at 5.845 keV for a 250–1000 ppm concentration range.
Longitudinal proton relaxation times were measured using six different instruments. A Bruker minispec was used at 1.4 T and 37 °C using an inversion recovery technique. NMRD profiles were measured at 25 °C over a continuum of magnetic fields from 0.0002 to 0.2 T on a fast field-cycling Stelar SmartTracer relaxometer, and data points at high field strengths were measured on a Stelar Spinmaster relaxometer (0.5–1.6 T) equipped with a BrukerWP80 magnet. Susceptibility weighted imaging (SWI) was performed to acquire relaxivity data at 3 (Siemens TRIO) and 7 T (Clinscan). Multiple flip angles (5, 10, 15, 20, 25, and 30°) were used in the SWI experiment to allow for the determination of T1 following a reported procedure.[32] MR images were processed using SPIN (SVN Revision 1757), and Matlab (T.12.0.635 R2011a) was used to calculate effective transverse relaxation times, T2*, and corrected T1 maps. Data points at 9.4 and 11.7 T were measured using inverse recovery methods with Varian unity 400 and Varian 500S instruments.
Variable-temperature 17O-NMR measurements were performed using a Varian-500S (9.4 T) spectrometer for samples of 1 (5 mM), 2 (5 mM), and 3 (1.2 mM) in phosphate-buffered saline, with a blank of phosphate-buffered saline (pH = 7.4) with 1% 17O-enriched water (diluted from 20% enriched H217O). Transverse relaxation rates were calculated from line widths at half height following a reported procedure.[9]
EPR spectroscopy was performed on a Bruker EMX X-band spectrometer equipped with an Oxford variable-temperature cryostat. EPR samples were prepared and sealed under an atmosphere of N2 in Norell SEPR250S EPR tubes and were frozen in liquid N2 before insertion into the sample cavity. EPR spectra of 3/β-CD and 3/poly-β-CD were acquired at 110 K (Figures S5 and S6). Acquisition parameters included a microwave frequency of 9.38 GHz, a microwave power of 1.99 mW, a modulation frequency of 100 kHz, a receiver gain of 30 dB, and a modulation amplitude of 4.0 G.
Molar conductivity measurements were carried out using an Omega CDH 280 portable conductivity meter that was calibrated with aqueous KCl (0.01 M, 1.413 mS cm−1). Conductivity values were calculated from three independently prepared solutions of metal complexes (1.00 mM, 4.00 mL) measured in water under an atmosphere of N2 at ambient temperature. Results are reported as mean ± standard error of three independently prepared samples.
Dynamic light scattering data were obtained using a Malvern Zetasizer Nano-ZS instrument (ZEN3600) operating with a 633 nm wavelength laser. Dust was removed from samples by filtering through 0.2 μm hydrophilic filters. Samples of complex 3 (1 and 8 mM in phosphate-buffered saline) were prepared in air-tight cuvettes inside a wet glove box under an atmosphere of N2.
Dimethyl-2,2′-([1,1′-biphenyl]-4-yl)propane-1,2-dily)bis(oxy) diacetate (4): To a solution of 3-(biphenyl-4-yl)propane-1,2-diol (2.00 g, 8.76 mmol, 1 equiv) in anhydrous tetrahydrofuran (15 mL) under an atmosphere of Ar at ambient temperature was added NaH (58% in oil, 1.45 g, 35.0 mmol, 4 equiv) followed by the dropwise addition of methyl bromoacetate (6.60 mL, 70.1 mmol, 8 equiv). The resulting reaction mixture was stirred at ambient temperature for 22 h, and excess NaH was quenched with methanol (3 mL). Solvents were removed under reduced pressure, and the resulting residue was extracted with diethyl ether (5 × 30 mL). Purification was performed using silica gel chromatography (5:1 hexanes/ethyl acetate) to yield 2.31 g (71%) of 4 as a colorless oil. TLC: Rf = 0.20 (3:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CD3CN): δ = 2.90 (d, 3J (H,H) = 4.0 Hz, 2H;CH2), 3.56 (ddd, 3J (H,H) = 4.0, 12.0, 40.0 Hz, 2H; CH2CH), 3.66 (s, 3H; CH3), 3.68 (s, 3H; CH3), 3.80–3.87 (m, 1H; CHCH2), 4.09 (s, 2H; CH2C=O), 4.18 (s, 2H; CH2C=O), 7.31–7.66 ppm (m, 9H; CHCH); 13C NMR (101 MHz, CD3CN): δ = 37.8 (CH2), 52.1 (CH3), 68.0 (CH2C=O), 69.1 (CH2C=O), 73.5 (CH2CH), 81.0 (CHCH2), 127.6 (CHCH), 127.7 (CHCH), 128.2 (CHCH), 129.8 (CHCH), 131.0 (CHCH), 131.4 (CHCH), 138.6, 139.7, 141.6, 171.6, 171.8 ppm; HRESIMS (m/z): [M + Na]+ calcd for C21H24O6Na, 395.1471; found, 395.1477.
2,2′-((3-([1,1′-Biphenyl]-4-yl)propane-1,2-dily)bis(oxy) diacetic acid (5): To a solution of 4 (2.00 g, 5.38 mmol, 1 equiv) in methanol (9 mL) and water (3 mL) was added LiOH (1.03 g, 43.0 mmol, 8 equiv), and the reaction mixture was stirred at ambient temperature for 6 h. The resulting yellow solution was concentrated under reduced pressure, and the residue was dissolved in water (8 mL) and acidified with HCl (6 M, 3 mL). The resulting white solid was collected by filtering, washed with water (15 mL), and dried under reduced pressure to yield 1.85 g (quantitative) of 5 as a white solid. 1H NMR (400 MHz, CD3CN): δ = 2.88 (dd, 3J (H,H) = 2.0, 6.4 Hz, 2H; CH2), 3.47–3.68 (m, 2H; CH2), 3.83–3.91 (m, 1H; CH), 4.03–4.19 (m, 4H; CH2), 7.29–7.69 (m, 9H; CHCH), 9.60 ppm (brs, 2H; OH); 13C NMR (101 MHz, CD3CN): δ = 37.7 (CH2), 68.2 (CH2), 68.7 (CH2), 73.9 (CH2), 82.0 (CH), 127.7 (CHCH), 127.8 (CHCH), 128.3 (CHCH), 129.9 (CHCH), 131.1 (CHCH), 138.0, 140.0, 141.5, 171.8, 172.2 ppm; HERESIMS (m/z): [M + Na]+ calcd for C19H20O6Na, 367.1158; found, 367.1148.
5-([1,1′-Biphenyl]-4-ylmethyl)-4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane-2,9-dione (6): Thionyl chloride (3.60 mL, 49.4 mmol, 10 equiv) was added to 5 (1.70 g, 4.94 mmol, 1 equiv), and the resulting solution was heated at reflux for 3 h under an atmosphere of Ar. The reaction mixture was concentrated under reduced pressure to obtain a yellow oil that was dissolved in anhydrous toluene (150 mL) under an atmosphere Ar. To a separate solution of 4,13-diaza-18-crown-6-ether (1.30 g, 4.94 mmol, 1 equiv) in anhydrous toluene (150 mL) was added triethylamine (2.00 mL, 14.8 mmol, 3 equiv), and the resulting solution was stirred for 15 min. The two solutions were added simultaneously over 60 min to anhydrous toluene (300 mL) at 0–5 °C. The resulting mixture was stirred for 24 h while allowing to warm to ambient temperature. Solvent was removed under reduced pressure, and the residue was extracted with CH2Cl2 (5 × 10 mL). Purification was performed using silica gel chromatography (10:1 CH2Cl2/methanol) to yield 1.46 g (52%) of 6 as a yellow oil. TLC: Rf = 0.32 (10:1 CH2Cl2/methanol); 1H-NMR (400 MHz, CD3CN): δ = 2.60–3.10 (m, 5H; CH2), 3.20–4.80 (m, 28H, CH and CH2), 7.20–7.70 ppm (m, 9H; CH); 13C NMR (101 MHz, CD3CN): δ = 37.2 (CH2), 37.4 (CH2), 37.7 (CH2), 38.0 (CH2), 47.3 (CH2), 47.7 (CH2), 48.2 (CH2), 48.3 (CH2), 48.4 (CH2), 49.0 (CH2), 49.1 (CH2), 49.3 (CH2), 49.35 (CH2) 49.4 (CH2), 49.6 (CH2), 49.65 (CH2) 49.7 (CH2), 49.8 (CH2), 49.9 (CH2), 68.7 (CH2), 69.2 (CH2), 69.3 (CH2), 69.7 (CH2), 69.8 (CH2), 69.9 (CH2), 70.0 (CH2), 70.1 (CH2), 70.2 (CH2), 70.3 (CH2), 70.4 (CH2), 70.9 (CH2), 71.0 (CH2), 71.1 (CH2), 71.28 (CH2), 71.3 (CH2), 71.4 (CH2), 71.6 (CH2), 71.7 (CH2), 71.8 (CH2), 71.9 (CH2), 72.0 (CH2), 72.1 (CH2), 72.2 (CH2), 72.4 (CH2), 73.6 (CH2), 73.7 (CH2), 74.9 (CH2), 78.6 (CH), 79.7 (CH), 81.6 (CH), 82.7 (CH), 127.5 (CHCH), 128.0 (CHCH), 129.7 (CHCH), 130.7 (CHCH), 130.8 (CHCH), 138.5, 139.6, 141.4, 169.5, 169.6 ppm; HRESIMS (m/z): [M + H]+ calcd for C31H43N2O8, 571.3019; found 571.3012.
5-([1,1′-Biphenyl]-4-ylmethyl)-4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (7): To a solution of 6 (0.800 g, 1.40 mmol, 1 equiv) in anhydrous tetrahydrofuran under an atmosphere of Ar at 0°C was added borane–tetrahydrofuran complex (14 mL, 14 mmol, 1 M in tetrahydrofuran, 10 equiv) in a drop-wise manner, and the resulting mixture was heated at reflux for 24 h before the reaction mixture was allowed to cool to ambient temperature. To the reaction mixture was added aqueous HCl (6 M, 14 mL), and the mixture was heated at reflux for 4 h. The pH of the reaction solution was adjusted to 11 by adding concentrated ammonium hydroxide (20 mL). The resulting solution was concentrated under reduced pressure to obtain white solid that was purified using silica gel chromatography (20:1 CH2Cl2 / methanol) to yield 0.5 g (66%) of 7 as a yellow oil. TLC: Rf= 0.23 (10:1 CH2Cl2/methanol); 1H-NMR (400 MHz, CD3CN): δ = 2.55–2.88 (m, 10H; CH2), 3.03–3.11 (m, 1H; CH2), 3.32–3.82 (m, 26H; CH and CH2), 7.30–7.66 ppm (m, 9H; CH); 13C NMR (101 MHz, CD3CN): δ = 38.4 (CH2), 53.4 (CH2), 53.5 (CH2), 53.7 (CH2), 53.8 (CH2), 54.1 (CH2), 54.3 (CH2), 66.1 (CH2), 68.0 (CH2), 68.1 (CH2), 68.57 (CH2), 68.59 (CH2), 68.8 (CH2), 68.9 (CH2), 69.2 (CH2), 69.6 (CH2), 71.4 (CH2), 79.4 (CH), 126.7 (CH), 127.7 (CH), 127.8 (CH), 128.3 (CH), 129.2 (CH), 129.9 (CH), 131.0 (CH), 138.4, 139.6, 141.5 ppm; HRESIMS (m/z): [M + H]+ calcd for C31H47N2O6, 543.3434; found 543.3114.
Metal complexes (1–3) were prepared according to reported procedures,[9] using a wet glove box under an atmosphere of N2 using degassed water. The resulting EuII-containing complexes were in phosphate-buffered saline (11.9 mM phosphates, 2.7 mM potassium chloride, 137 mM sodium chloride, pH 7.4). Samples for NMRD measurements or long-term studies were flame sealed, and samples for other NMR and EPR experiments were sealed using paraffin wax and used immediately. For NMRD measurements, macromolecules (β-CD, poly-β-CD, and HSA) were used in the blanks and the concentration of macromolecule in each sample was kept constant: 2.5, 1.0, and 4.0 mM for β-CD, poly-β-CD, and HSA, respectively.
To study the stability of complex 2 in phosphate-buffered saline, an aliquot (300 μL) of a solution of complex 2 in phosphate-buffered saline was flame sealed in an NMR tube under an atmosphere of N2. The relaxivity of this sample was measured (60 MHz, 37 °C) over the course of one month and was triplicated with independently prepared samples (Figure S3). No change in relaxivity was observed, indicating that any competition between phosphate and the cryptand in 2 was below the detection limit of the technique.
X-ray quality crystals of EuII-containing complexes were obtained by vapor diffusion. A solution of metal complex was prepared by dissolving EuCl2 and ligand in methanol under an inert atmosphere. The resulting clear solution was stirred for 1 h then filtered through a 0.2 μm hydrophilic filter. Tetrahydrofuran was diffused into the filtrate to obtain needle-like crystals. Crystal structure analyses were performed on a Bruker APEX-II Kappa geometry diffractometer with Mo radiation and a graphite monochromator using a Bruker-charge-coupled-device-based diffractometer equipped with an Oxford Cryostream low-temperature apparatus. Data were acquired at 100 K. Structures were solved by the direct method using the SHELXS-97 program that is part of APEX II2 and refined by the least squares method, SHELXL 2012 incorporated into ShelXle.[33] Single crystals of 1 contained one cation of 1 with one chloride, and no water molecules bound, one non-covalent chloride counter ion, and one non-covalent molecule of water in the asymmetric unit. The structure of 1 was solved with a resolution of 0.75 Å in space group P121/c1. Single crystals of 2 contained one cation of 2 with one bound chloride, no water molecules bound, and one non-covalent molecule of water in the asymmetric unit. The structure of 2 was solved with a resolution of 0.55 Å in space group P121/c1. All non-hydrogen atoms were refined anisotropically. CCDC 1549045 and CCDC 1549046 contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01EB013663). M.B. and F.C. are grateful to Università del Piemonte Orientale for financial support (Ricerca locale 2016). The authors thank Dr. Derek J. Averill from Shimadzu Scientific Instruments for use of the EDXF spectrometer, Prof. Jeremy Kodanko for use of the conductivity meter, and Levi Ekanger for assistance acquiring EPR spectra.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Radecki G, Nargeot R, Jelescu IO, Le Bihan D, Ciobanu L. Proc Natl Acad Sci USA. 2014;111:8667–8672. doi: 10.1073/pnas.1403739111. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) van Veluw SJ, Zwanenburg JJM, Engelen-Lee J, Spliet WGM, Hendrikse J, Luijten PR, Biessels GJ. J Cereb Blood Flow Metab. 2013;33:322–329. doi: 10.1038/jcbfm.2012.196. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) van de Bank BL, Voogt IJ, Italiaander M, Stehouwer BL, Boer VO, Luijten PR, Klomp DWJ. NMR Biomed. 2013;26:367–375. doi: 10.1002/nbm.2868. [DOI] [PubMed] [Google Scholar]
- 2.a) Boros E, Polasek M, Zhang Z, Caravan P. J Am Chem Soc. 2012;134:19858–19868. doi: 10.1021/ja309187m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Laus S, Ruloff R, Tóth É, Merbach AE. Chem Eur J. 2003;9:3555–3566. doi: 10.1002/chem.200204612. [DOI] [PubMed] [Google Scholar]; c) Banerjee SR, Ngen EJ, Rotz MW, Kakkad S, Lisok A, Pracitto R, Pullambhatla M, Chen Z, Shah T, Artemov D, Meade TJ, Bhujwalla ZM, Pomper MG. Angew Chem Int Ed. 2015;54:10778–10782. doi: 10.1002/anie.201503417. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2015;127:10928–10932. [Google Scholar]; d) Aime S, Botta M, Crich SG, Giovenzana G, Pagliarin R, Sisti M, Terreno E. Magn Reson Chem. 1998;36:S200–S208. [Google Scholar]; e) Laurent S, Elst LV, Muller RN. Contrast Med Mol Imaging. 2006;1:128–137. doi: 10.1002/cmmi.100. [DOI] [PubMed] [Google Scholar]
- 3.a) Ekanger LA, Polin LA, Shen Y, Haacke EM, Martin PD, Allen MJ. Angew Chem Int Ed. 2015;54:14398–14401. doi: 10.1002/anie.201507227. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2015;127:14606–14609. [Google Scholar]; b) Burai L, Tóth É, Moreau GE, Sour A, Scopelliti R, Merbach AE. Chem Eur J. 2003;9:1394–1404. doi: 10.1002/chem.200390159. [DOI] [PubMed] [Google Scholar]; c) Ekanger LA, Ali MM, Allen MJ. Chem Commun. 2014;50:14835–14838. doi: 10.1039/c4cc07027e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Regueiro-Figueroa M, Barriada JL, Pallier A, Esteban-Gómaz D, Blas AD, Rodríguez-Blas T, Tóth É, Platas-Iglesias C. Inorg Chem. 2015;54:4940–4952. doi: 10.1021/acs.inorgchem.5b00548. [DOI] [PubMed] [Google Scholar]; e) Ekanger LA, Mills DR, Ali MM, Polin LA, Shen Y, Haacke EM, Allen MJ. Inorg Chem. 2016;55:9981–9988. doi: 10.1021/acs.inorgchem.6b00629. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Ekanger LA, Polin LA, Haacke EM, Allen MJ. Contrast Media Mol Imaging. 2016;11:299–303. doi: 10.1002/cmmi.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Gamage NDH, Mei Y, Garcia J, Allen MJ. Angew Chem Int Ed. 2010;49:8923–8925. doi: 10.1002/anie.201002789. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2010;122:9107–9109. [Google Scholar]; h) Basal LA, Yan Y, Shen Y, Haacke EM, Mehrmohammadi M, Allen MJ. ACS Omega. 2017;2:800–805. doi: 10.1021/acsomega.6b00514. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Ekanger LA, Basal LA, Allen MJ. Chem Eur J. 2017;23:1145–1150. doi: 10.1002/chem.201604842. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Botta M, Ravera M, Barge A, Bottaro M, Osella D. Dalton Trans. 2003:1628–1633. [Google Scholar]
- 4.a) Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]; b) Bloembergen N, Purcell EM, Pound RV. Phys Rev. 1948;73:679–712. [Google Scholar]; c) Solomon I. Phys Rev. 1955;99:559–565. [Google Scholar]; d) Bloembergen N. J Chem Phys. 1957;27:572–573. [Google Scholar]; e) Bloembergen N, Morgan LO. J Chem Phys. 1961;34:842–850. [Google Scholar]
- 5.Garcia J, Kuda-Wedagedara ANW, Allen MJ. Eur J Inorg Chem. 2012;2012:2135–2140. doi: 10.1002/ejic.201101166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Ruiz-Martinez A, Casanova D, Alvarez S. Chem Eur J. 2008;14:1291–1303. doi: 10.1002/chem.200701137. [DOI] [PubMed] [Google Scholar]; b) Kuda-Wedagedara ANW, Wang C, Martin PD, Allen MJ. J Am Chem, Soc. 2015;137:4960–4963. doi: 10.1021/jacs.5b02506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Apelblat A, Esteso MA, Bešter-Rogač M. J Phys Chem B. 2013;117:5241–5248. doi: 10.1021/jp4024074. [DOI] [PubMed] [Google Scholar]
- 8.a) Avedano S, Botta M, Haigh JS, Longo DL, Woods M. Inorg Chem. 2013;52:8436–8450. doi: 10.1021/ic400308a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Webber BC, Cassino C, Botta M, Woods M. Inorg Chem. 2015;54:2085–2087. doi: 10.1021/ic5028724. [DOI] [PubMed] [Google Scholar]; c) Schühle DT, Schatz J, Laurent S, Elst LV, Muller RN, Stuart MCA, Peters JA. Chem Eur J. 2009;15:3290–3296. doi: 10.1002/chem.200802099. [DOI] [PubMed] [Google Scholar]; d) Vanasschen C, Bouslimani N, Thonon D, Desreux JF. Inorg Chem. 2011;50:8946–8958. doi: 10.1021/ic2010997. [DOI] [PubMed] [Google Scholar]
- 9.Garcia J, Neelavalli J, Haacke EM, Allen MJ. Chem Commun. 2011;47:12858–12860. doi: 10.1039/c1cc15219j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Aime S, Botta M, Frullano L, Crich SG, Giovenzana GB, Pagliarin R, Palmisano G, Sisti M. Chem Eur J. 1999;5:1253–1260. [Google Scholar]; b) Aime S, Botta M, Fedeli F, Gianolio E, Terreno E, Anelli P. Chem Eur J. 2001;7:5261–5269. doi: 10.1002/1521-3765(20011217)7:24<5261::aid-chem5261>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]; c) Aime S, Crich SG, Gianolio E, Terreno E, Beltrami A, Uggeri F. Eur J Inorg Chem. 1998:1283–1289. [Google Scholar]; d) Aime S, Botta M, Panero M. Magn Reson Chem. 1991;29:923–927. [Google Scholar]
- 11.a) Dwek RA. Nuclear Magnetic Resonance in Biochemistry: Applications to Enzyme Systems. Oxford University Press; Oxford: 1973. [Google Scholar]; b) Jenkins BG, Armstrong E, Lauffer RB. Magn Reson Med. 1991;17:164–178. doi: 10.1002/mrm.1910170120. [DOI] [PubMed] [Google Scholar]; c) Aime S, Botta M, Fasano M, Terreno E. In: The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging. 1st. Merbach AE, Tóth É, editors. John Wiley & Sons, Ltd; Chichester, UK: 2001. pp. 193–242. [Google Scholar]
- 12.Battistini E, Gianolio E, Gref R, Couvreur P, Fuzerova S, Othman M, Aime S, Badet B, Durand P. Chem Eur J. 2008;14:4551–4561. doi: 10.1002/chem.200701587. [DOI] [PubMed] [Google Scholar]
- 13.a) Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JWM, Amedio JC, Looby RJ, Supkowski RM, Horrocks WD, Jr, McMurry TJ, Lauffer RB. J Am Chem Soc. 2002;124:3152–3162. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]; b) Garcia J, Allen MJ. Inorg Chim Acta. 2012;393:324–327. doi: 10.1016/j.ica.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Henrotte V, Elst LV, Laurent S, Muller RN. J Biol Inorg Chem. 2007;12:929–937. doi: 10.1007/s00775-007-0247-5. [DOI] [PubMed] [Google Scholar]; d) Parac-Vogt TN, Kimpe K, Laurent S, Elst LV, Burtea C, Chen F, Muller RN, Ni Y, Verbruggen A, Binnemans K. Chem Eur J. 2005;11:3077–3086. doi: 10.1002/chem.200401207. [DOI] [PubMed] [Google Scholar]; e) Aime S, Botta M, Crich SG, Giovenzana GB, Pagliarin R, Piccinini M, Sisti M, Terreno E. J Biol Inorg Chem. 1997;2:470–479. [Google Scholar]
- 14.Aime S, Botta M, Fasano M, Crich SG, Terreno E. J Biol Inorg Chem. 1996;1:312–319. [Google Scholar]
- 15.Burai L, Scopelliti R, Tóth É. Chem Commun. 2002:2366–2367. doi: 10.1039/b206709a. [DOI] [PubMed] [Google Scholar]
- 16.Jin GX, Bailey MD, Allen MJ. Inorg Chem. 2016;55:9085–9090. doi: 10.1021/acs.inorgchem.6b01659. [DOI] [PubMed] [Google Scholar]
- 17.Burai L, Tóth É, Seibig S, Scopelliti R, Merbach AE. Chem Eur J. 2000;6:3761–3770. doi: 10.1002/1521-3765(20001016)6:20<3761::aid-chem3761>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 18.Garcia J. Ph D Thesis. Wayne State University; Detroit, MI: 2013. Studies on the Physiochemical Properties of Eu2+ Cryptates: Implications to Contrast Agents for Magnetic Resonance Imaging. [Google Scholar]
- 19.Swift TJ, Connick RE. J Chem Phys. 1962;37:307. [Google Scholar]
- 20.Aime S, Barge A, Borel A, Botta M, Chemerisov S, Merbach AE, Müller U, Pubanz D. Inorg Chem. 1997;36:5104–5112. [Google Scholar]
- 21.Avedano S, Tei L, Lombardi A, Giovenzana GB, Aime S, Longo D, Botta M. Chem Commun. 2007:4726–4728. doi: 10.1039/b714438e. [DOI] [PubMed] [Google Scholar]
- 22.Botta M, Avedano S, Giovenzana GB, Lombardi A, Longo D, Cassino C, Tei L, Aime S. Eur J Inorg Chem. 2011:802–810. [Google Scholar]
- 23.Siriwardena-Mahanama BN, Allen MJ. Molecules. 2013;18:9352–9381. doi: 10.3390/molecules18089352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Powell DH, Dhubhghaill OMN, Pubanz D, Helm L, Lebedev YS, Schlaepfer W, Merbach AE. J Am Chem Soc. 1996;118:9333–9346. [Google Scholar]; b) Tóth É, Dhubhghail OMN, Besson G, Helm L, Merbach AE. Magn Reson Chem. 1999;37:701–708. [Google Scholar]
- 25.Seibig S, Tóth É, Merbach AE. J Am Chem Soc. 2000;122:5822–5830. [Google Scholar]
- 26.Caravan P, Tóth É, Rockenbauer A, Merbach AE. J Am Chem Soc. 1999;121:10403–10409. [Google Scholar]
- 27.Tóth É, Burai L, Merbach AE. Coord Chem Rev. 2001;216-217:363–382. [Google Scholar]
- 28.a) Reuben J. J Phys Chem. 1971;75:3164–3167. [Google Scholar]; b) Powell DH, Merbach AE, González G, Brücher E, Micskei K, Ottaviani MF, Köhler K, Zelewsky AV, Grinberg OY, Lebedev YS. Helv Chim Acta. 1993;76:2129–2146. [Google Scholar]
- 29.Helm L. Prog Nucl Magn Reson Spectrosc. 2006;49:45–64. [Google Scholar]
- 30.Aime S, Botta M, Garda Z, Kucera BE, Tircso G, Young VG, Woods M. Inorg Chem. 2011;50:7955–7965. doi: 10.1021/ic2012827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.a) Lipari G, Szabo A. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]; (b) Lipari G, Szabo A. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 32.Haacke EM, Brown RW, Thompson MR, Venkatesan R. Magnetic Resonance Imaging Physical Principles and Sequence Design. John Wiley & Sons, Inc.; New York: 1999. p. 654. [Google Scholar]
- 33.Hübschle CB, Sheldrick GM, Dittrich B. J Appl Cryst. 2011;44:1281–1284. doi: 10.1107/S0021889811043202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.