

Abstract
Half of estrogen receptor-positive breast cancers contain a subpopulation of cytokeratin 5 (CK5)-expressing cells that are therapy resistant and exhibit increased cancer stem cell (CSC) properties. We and others have demonstrated that progesterone (P4) increases CK5+ breast cancer cells. We previously discovered that retinoids block P4 induction of CK5+ cells. Here we investigated the mechanisms by which progesterone receptors (PR) and retinoic acid receptors (RAR) regulate CK5 expression and breast CSC activity. After P4 treatment, sorted CK5+ compared to CK5 − cells were more tumorigenic in vivo. In vitro, P4-treated breast cancer cells formed larger mammospheres and silencing of CK5 using small hairpin RNA abolished this P4-dependent increase in mammosphere size. Retinoic acid (RA) treatment blocked the P4 increase in CK5+ cells and prevented the P4 increase in mammosphere size. Dual small interfering RNA (siRNA) silencing of RARα and RARγ reversed RA blockade of P4-induced CK5. Using promoter deletion analysis, we identified a region 1.1 kb upstream of the CK5 transcriptional start site that is necessary for P4 activation and contains a putative progesterone response element (PRE). We confirmed by chromatin immunoprecipitation that P4 recruits PR to the CK5 promoter near the − 1.1 kb essential PRE, and also to a proximal region near − 130 bp that contains PRE half-sites and a RA response element (RARE). RA induced loss of PR binding only at the proximal site. Interestingly, RARα was recruited to the − 1.1 kb PRE and the − 130 bp PRE/RARE regions with P4, but not RA alone or RA plus P4. Treatment of breast cancer xenografts in vivo with the retinoid fenretinide reduced the accumulation of CK5+ cells during estrogen depletion. This reduction, together with the inhibition of CK5+ cell expansion through RAR/PR cross talk, may explain the efficacy of retinoids in prevention of some breast cancer recurrences.
INTRODUCTION
Greater than 70% of all breast cancers express estrogen receptor alpha (ER) at diagnosis and display various degrees of dependency on estrogens for proliferation.1 While ER− targeted endocrine therapies have greatly improved survival for patients with ER+ disease, intrinsic or acquired resistance still accounts for half of all breast cancer deaths.2 Furthermore, recurrences can occur after an extended remission (>5 years), suggesting cell populations in ER+ tumors can survive a prolonged dormancy.3,4 One possible explanation for this recurrence is the cancer stem cell (CSC) theory, which posits that tumors contain a small population of cells that exhibit characteristics of normal stem cells including drug resistance, quiescence and replicative immortality, allowing tumors to reform.5 Of note is that breast cancer cells can acquire a CSC phenotype through signaling or therapeutic pressure and thus prevention of the CSC phenotype may be equally as important as targeting existing CSCs.6,7 Understanding how subpopulations of CSCs are regulated in ER+ breast cancers is thus paramount to developing new treatment strategies.
Progesterone receptors (PR) are co-expressed in the majority of ER+ breast cancers and signify initial positive response to endocrine therapy.8 The role of PR itself is complex; it can exert autonomous proliferative signals or oppose the mitogenic effects of estrogens in a context-dependent manner.9–12 In particular, we and others have shown that progesterone (P4) increases a population of ER −, cytokeratin 5 (CK5)+ breast cancer cells.13,14 CK5 is expressed in ER − luminal progenitor cell populations of the normal human breast, which give rise to ER+PR+ luminal cells.15 CK5+ compared with CK5 − breast cancer cells have enhanced mammosphere forming potential, and are chemo- and endocrine therapy resistant.16–18 P4 expansion of CK5+ breast cancer cells involves upregulation of PR target transcription factors such as KLF4, STAT5a and BCL6.19–21 Additionally, endocrine therapy agents such as tamoxifen (Tam), fulvestrant (ICI) or estrogen depletion increase CK5 expression in breast cancer cell lines, and neoadjuvant Tam plus aromatase inhibitor treatment enriches for CK5+ cells in patient biopsy samples.17 Factors that repress CK5+ cells in breast cancer are lesser known.
Via a small molecule screen we previously discovered that several retinoids including all-trans retinoic acid (ATRA) and two synthetic retinoids prevent P4 production of CK5+ breast cancer cells.22 Retinoids (for example, ATRA, 9-cis RA, 13-cis RA) are ligands for nuclear receptors in the retinoid receptor subclass, which includes three retinoic acid (RA) receptors (RARα, −β and −γ) and three retinoid X receptors (RXRα, −β and −γ). These receptors form RAR/RXR heterodimers that can occupy DNA in the absence of ligand and often repress transcription; upon ligand binding they positively or negatively modulate gene transcription to regulate important cellular processes such as differentiation and cell death.23,24 This has led to successful use of ATRA in acute promyelocytic leukemia as a differentiating agent.25 Retinoids are potently antiproliferative in breast cancer cells.26 Treatment studies in breast cancer patients, however, have been mostly disappointing, with use of retinoids in combination treatment with Tam or chemotherapy failing to achieve study end points (reviewed in Garattini et al.27). One exception is the synthetic retinoid fenretinide (Fen, 4-HPR), which has had some efficacy in prevention of premenopausal breast cancer.28 Understanding the interplay between RARs and steroid receptors is important in determining contexts under which they could be therapeutically useful in breast cancer. RARα is positively regulated by estrogens in breast cancer cells,29 and is co-localized at many ER DNA binding sites, where it either acts as a cofactor for ER modulation of gene transcription,30 or competes with ER at promoter binding sites to antagonize gene transcription.31 ATRA reduces PR messenger RNA (mRNA) and protein in breast cancer cells and attenuates activity of a progestin-responsive promoter-reporter.32,33
Here we investigated the molecular mechanisms by which P4 and RA through their cognate receptors regulate expression of CK5 and how this contributes to cancer cell stemness. We demonstrate that P4 induction of large mammospheres is dependent on CK5 expression, and that RA through RARs blocks both P4-mediated CK5 expression and mammosphere size. Furthermore, we identified two regions in the CK5 promoter that are essential for PR/RAR regulation, and describe a mechanism by which RARα selectively controls coactivator recruitment. Finally, we show in vivo that co-treatment with retinoids can prevent the enrichment of CK5+ cells seen during estrogen depletion. Therefore, retooling the use of retinoids to specific cases and timelines may revitalize their usefulness, specifically in conjunction with hormone therapies to abrogate P4 expansion of stem cells, or in some ER − CK5+ breast cancers where retinoids may prevent breast cancer recurrence.
RESULTS
P4-expanded CK5+ breast cancer cells are tumorigenic
We have previously demonstrated that CD44+ breast cancer cells that are enriched in CK5 expression are more tumor-initiating.13 Furthermore, breast cancer cell lines with larger P4-dependent CK5+ populations following suppression of microRNAs (miR)29 and miR141 had increased tumor-initiating ability.19,20 To validate these observations and more directly measure CK5 involvement in tumorigenicity, we used a system in which T47D breast cancer cells are integrated with a CK5 promoter-GFP reporter.16 Cells were treated for 24 h with P4 in vitro to induce a CK5+ cell population, then CK5+ and CK5 − cells were isolated by fluorescence-activated cell sorting (Supplementary Figure 1a). Female nude mice supplemented with estrogen slow release pellets were bilaterally injected with sorted CK5+ and CK5 − cells subcutaneously in opposing fourth mammary fat pads at dilutions ranging from 102 to 105. Tumors were palpated through 6 weeks post injection (Supplementary Figure 1b). Limiting dilution analyses revealed that CK5+ cells initiated tumors more efficiently than CK5− cells (Table 1). These data provide additional confirmation that CK5+ breast cancer cells have enhanced tumor initiation ability.
Table 1.
Tumor-initiating capacity of P4-induced CK5+ compared to CK5 − T47D breast cancer cells
| Number of cells injected per mammary fat pad | Number of tumors per number of injected fat pads | |
|---|---|---|
| Week 4 after implantation | ||
| CK5 − | CK5+ | |
| 1 × 104 | 6/10 | 10/10 |
| 1 × 103 | 3/10 | 8/10 |
| 1 × 102 | 1/10 | 6/10 |
| Tumor-initiating frequency (95% CI) | (1/7275) | (1/360) |
| Tumor-initiating range (95% CI) | (1/14 665–1/3609) | (1/735–1/176) |
| P-value | 1.32 × 10−10 | |
| Week 6 after implantation | ||
| CK5 − | CK5+ | |
| 1 × 104 | 7/10 | 10/10 |
| 1 × 103 | 6/9a | 7/9a |
| 1 × 102 | 5/10 | 9/10 |
| Tumor-initiating frequency (95% CI) | (1/3071) | (1/263) |
| Tumor-initiating range (95% CI) | (1/6425–1/1468) | (1/581–1/119) |
| P-value | 6.05 × 10−9 | |
Abbreviation: CI, confidence interval. Limiting dilution analysis of CK5+ versus CK5 − T47D cells. T47D cells stably harboring a CK5 promoter-GFP reporter were treated for 24 h with 100 nM P4 to induce a population of CK5+ cells for analysis and sorted by fluorescence-activated cell sorting for GFP+ and GFP − cells.
One animal was killed for health reasons prior to week 6, and was excluded from the analysis. Data were analyzed using ELDA software.58
CK5 is necessary for P4-mediated increase in breast cancer cell mammosphere size
P4 increases the mammosphere forming potential of breast cancer cells.20,21 To test whether CK5 is necessary for the P4 effect on mammospheres, we utilized small hairpin RNA (shRNA) inhibition coupled with an adapted in vitro mammosphere assay compatible with automated quantitation. T47D cells constitutively expressing ZsGreen were transduced with three different lentiviral-packaged shRNAs to CK5, resulting in two cell lines with impaired P4 induction of CK5 (Figure 1a). Cell lines transduced with two independent shRNAs (#22 or #78) targeting CK5 or a non-targeting control (shCont) were seeded into mammosphere media with vehicle or P4, and sphere number and size were assessed after 2 weeks (Figure 1b). P4 consistently increased average mammosphere size by 1.6-fold in shCont cells; this increase was attenuated in both shCK5 lines (Figure 1c). A significant difference in the number of mammospheres was also observed in P4 compared to vehicle-treated control cells, albeit only with large mammospheres (>4500 μm2) (Supplementary Figure 2). We thus used mammosphere size as our primary metric for this study. We conclude that CK5 is necessary for P4 to produce large mammospheres.
Figure 1.

The P4-dependent increase in mammosphere size requires expression of CK5. (a) T47D cells with constitutive ZsGreen expression were stably transduced with either a non-targeting shRNA (shCont) or one of three shRNAs targeting CK5 (shCK5). CK5 expression in response to 24 h treatment with 100 nM P4 was analyzed by immunoblot, using α-tubulin as a loading control. CK5 expression is indicated compared to P4-treated control cells. (b) T47D-ZsGreen shCont and shCK5 (#22 or #78) cells were plated in Mammocult media at a density of 100 cells per well in quintuplicate in a 96-well plate and treated with either vehicle (EtOH) or 100 nM P4. After 2 weeks, mammospheres were imaged and analyzed using the IncuCyte Zoom live cell analysis system and software. Experiments were performed three times. Representative images of wells are shown. (c) Mammosphere size depicted for shCont and shCK5 cells treated with vehicle or P4. Data represent mean ± s.e.m. Veh and P4 treatments in each group were compared via Student’s t-test, **P < 0.01.
Retinoids block P4-enhanced CK5 expression and mammosphere size
To validate our previous work that identified retinoids as potent inhibitors of P4-dependent induction of CK5,22,34 we measured CK5 mRNA levels in response to vehicle, P4, 9-cis RA or P4 plus RA in three breast cancer cell lines (T47D, MCF7, BT-474). MCF7 and BT-474 cells were pretreated with 17β-estradiol (E2) for 48 h to induce PR levels. P4 significantly increased CK5 mRNA levels in all three cell lines while RA alone had no effect on CK5 mRNA levels in T47D and BT-474 cells, but significantly decreased CK5 mRNA in MCF7 cells. RA significantly attenuated the P4-mediated increase in CK5 transcripts in all three cell lines (Figure 2a). CK5 protein expression was directly assessed in T47D cells by immunocytochemistry (ICC) and immunoblot. By ICC, P4 increased the population of CK5+ cells to 5.2% compared to vehicle (0%), whereas co-treatment with RA blocked this increase (Figure 2b). Similarly, the P4-mediated increase in CK5 protein was blocked by RA as measured by immunoblot (Figure 2c). We compared a dose–response of RA (1 nM−1 μM) plus or minus P4 and determined via immunoblot that 100 nM was the most effective lowest concentration, and chose that dose for remaining studies (Supplementary Figure 3a). Thus, RA effectively blocks the P4-mediated increase in CK5 expression primarily through a reduction in CK5 mRNA transcripts.
Figure 2.

RA blocks P4-mediated CK5 expression and P4 induction of large mammospheres. (a) Treatment with RA blocks P4-induced CK5 transcription. T47D cells in a were treated with ethanol vehicle (veh), 100 nM P4, 100 nM 9-cis RA or P4 plus RA for 10 h. MCF7 and BT-474 cells were pretreated with 10 nM E2 for 48 h to induce PR expression, then treated in the same manner as T47D cells. Quantitative reverse-transcriptase PCR (qRT-PCR) was used to assess relative CK5 mRNA levels normalized to β-actin. Results are displayed as relative CK5 mRNA expression. Data represent mean ±s.e.m. Within each cell line all groups were compared via analysis of variance (ANOVA)/Tukey, *P<0.05 **P<0.01 ***P<0.001. Experiments were performed three times. (b) CK5+ cells were measured in T47D cells via immunocytochemistry after 24 h of the same treatments as in a. Fluorescent staining shows CK5 (red) and DAPI (blue). Percent CK5+ cells per field are indicated, calculated from five fields taken at × 10 magnification. (c) Immunoblot of CK5, PR (PRA and PRB isoforms indicated) and RARα in T47D cells under the same conditions as in a. α-tubulin was used as a loading control. (d) T47D-ZsGreen cells were plated at a density of 100 cells per well in quintuplicate in mammosphere media in 96-well plates plus indicated treatments. After 2 weeks, mammosphere size was analyzed via scanning on the IncuCyte Zoom. Experiments were repeated three times. Data represent mean ±s.e.m. All groups were compared via ANOVA/Tukey, **P<0.01 ***P<0.001. (e) Merged images of dual ICC for PR (red) and RARα (green) in T47D cells treated as in a for 24 h. Nuclei are counterstained with DAPI (blue). White arrows indicate examples of double positive cells.
We assessed PR and RARα expression and co-localization in T47D cells under four different hormone conditions. By immunoblot PR (both PRA and PRB isoforms) protein decreased with RA treatment, but to a lesser extent than with P4-mediated downregulation (Figure 2c). RARα was present in T47D cells and its levels were unaffected by any treatments (Figure 2c). By dual ICC, PR and RARα were frequently co-localized in the nucleus of T47D cells, with occasional solely PR+ and RARα+, or double-negative cells (Figure 2e). No differences in co-localization were observed under the different hormone conditions. Thus, PR levels decrease slightly with RA treatment but RARα was not affected by P4 treatment.
Since CK5 was required for the P4-mediated increase in mammosphere size, we next assessed if RA inhibition would also impede this process. T47D cells treated with vehicle, P4, RA or P4 plus RA were assessed for mammosphere formation (Figure 2d). Indeed, RA reduced baseline mammosphere size (0.4-fold) and also prevented the P4-mediated increase (1.7-fold, reduced to 0.9-fold) in mammosphere size. Therefore, RA antagonizes the effects of P4 on CK5 transcription and CSC phenotype.
RARs are necessary for retinoid antagonism of P4-dependent increases in CK5 expression and mammosphere size
We next determined if RA was acting through RARs to block P4-mediated induction of CK5. Luminal breast cancer cells mainly express RARα and RARγ.35 Therefore, we used scrambled siRNA (siNT) or small interfering RNA (siRNA) targeting RARα (siRARα), RARγ (siRARγ), alone or simultaneously, in T47D cells stably expressing a CK5 promoter-driven luciferase reporter (Figure 3a). Similar to non-targeting siRNA, when cells were transfected with either siRARα or siRARγ alone, RA still inhibited P4 induction of the CK5 promoter (Figure 3b). When both RARα and RARγ were depleted, however, RA was unable to attenuate P4 activation of the CK5 promoter, indicating that RA can act through either RAR isoform to block CK5 induction. Because 9-cis RA is a ligand for both RARs and RXRs, we next determined if the inhibition of CK5 required RARs using the selective RAR agonist TTNPB. In T47D cells, TTNPB blocked P4 induction of CK5 as efficiently as RA (Figure 3c). TTNPB also attenuated the P4 increase in mammosphere size similar to RA in MCF7 cells (Figure 3d). TTNPB blocked P4 induction of CK5 at doses as low as 1 nM (Supplementary Figure 3b). These data indicate that agonist-bound RARα or RARγ, presumably through dimerization with RXRs, is sufficient to mediate repression of P4-induced CK5 expression and mammosphere size.
Figure 3.

RARs are required for RA inhibition of P4-induced CK5 expression and P4 production of large mammospheres. (a) T47D cells stably expressing a CK5 promoter-driven luciferase reporter were transfected with non-targeting siRNA (siNT), or siRNA to RARα or RARγ for 24 h, then treated with either ethanol vehicle or 100 nM P4 for an additional 24 h. Lysates were collected and analyzed by immunoblot. Relative RARα or RARγ levels are normalized to α-tubulin loading control and indicated relative to the vehicle treated siNT. (b) T47D cells were transfected as above and treated with vehicle, 100 nM P4, 100 nM 9-cis RA or both P4 plus RA. Lysates were collected and luciferase assays performed. Luciferase was graphed as fold change over vehicle control for each group of siRNAs. Experiments were performed three times. Data represent mean ±s.e.m. The four treatments were compared via analysis of variance (ANOVA)/Tukey within each of the siRNA groups, *P<0.05, **P<0.01, ***P<0.001. (c) T47D cells were treated with vehicle or P4 as above with the addition of two groups, 10 nM of the RAR selective agonist TTNPB minus or plus P4. CK5 and PR (PRA and PRB isoforms) expression were measured by immunoblot. (d) MCF7 cells stably expressing ZsGreen were plated at a density of 100 cells per well in quintuplicate in mammosphere media in 96-well plates and treated with 10 nM E2 (to induce PR levels) plus the following hormone combinations: ethanol vehicle, 100 nM P4, 100 nM 9-cis RA, P4 plus RA, 10 nM TTNPB or P4 plus TTNPB. After 2 weeks, mammospheres were analyzed using the IncuCyte Zoom. Data represent mean mammosphere size ±s.e.m. All groups were compared via ANOVA/Tukey, *P<0.05, **P<0.01, ***P<0.001. NS, not significant.
PR regulates CK5 transcription through direct binding to the proximal promoter
We next investigated the mechanism by which PR increases transcription of the CK5 gene. To narrow down the region(s) of the CK5 promoter required for PR responsiveness, we constructed a series of 5′ and internal deletion mutants using a 6 kb region of the human proximal CK5 promoter upstream of luciferase in a lentiviral vector (Figure 4a). Constructs were stably integrated into T47D cells and P4 responsiveness was tested. P4-induced luciferase activity was significantly decreased in four constructs with deletions incorporating a region 1.1 kb upstream of the transcriptional start site (TSS). This region contains a sequence (GGAACAGGGTGGTTC, − 1098 bp from the TSS) with extensive identity to an optimal progesterone response element (PRE) identified by mutagenesis studies36 and coincides with the location of a PR binding site upstream of the CK5 TSS identified by genome-wide analysis of PR DNA binding in T47D cells.37 To confirm that PR was binding at this PRE, we performed chromatin immunoprecipitation (ChIP) for PR followed by quantitative PCR using two independent sets of primers surrounding this region. Indeed, there was significant enrichment of PR bound to the −1.1 kb PRE region in P4 compared to vehicle-treated T47D cells (Figure 4b). quantitative PCR analysis of two negative control regions showed no enrichment compared to IgG control (Supplementary Figure 4). To determine if RA could preclude ligand-activated PR from binding to the − 1.1 kb PRE, we pretreated cells with RA for 30 min followed by P4 treatment. RA did not prevent PR from binding to the − 1.1 kb PRE (Figure 4b), excluding loss of PR DNA binding at this site as a mechanism of RA antagonism of PR-mediated CK5 transcription.
Figure 4.

PR is recruited to the CK5 promoter near a PRE that is necessary for P4 transcriptional activation. (a) P4 action requires a region containing a putative PRE 1098 bp upstream of the TSS. Deletion constructs were engineered from a 6 kb fragment of the CK5 promoter upstream of luciferase in a lentiviral vector using existing restriction sites. The left side of the graph indicates relative size of the promoter construct with 5′ or internal deletions, and the location of putative PRE half-sites, or full majority consensus sequence based on that reported by Lieberman et al.36 and Graham et al.37 Constructs were transduced into T47D cells and stable puromycin resistant pools selected. Cells were then seeded at 5000 cells per well in 96-well plates and treated with vehicle or 100 nM P4 for 24 h. Lysates were collected and luciferase activity analyzed using the Luciferase Assay Kit (Promega). Relative fold changes are indicated for each construct over vehicle control. Experiments were repeated three times. Data represent mean ±s.e.m. Data were analyzed by analysis of variance (ANOVA)/Dunnett using the full-length 6 kb promoter construct as the control, **P<0.01, ***P<0.001. (b) ChIP using an antibody for PR or IgG control in T47D cells that were treated with vehicle, 100 nM P4, 100 nM 9-cis-RA or P4 plus RA. Experiments were performed three times. Data represent mean (percent input) ±s.e.m. Primer set one values were compared via Student’s t-test; for primer set two, each group (IgG or PR) was compared via ANOVA/Tukey post hoc, **P<0.01, ***P<0.001. (c) Diagram showing location of primer sets used in b. NS, not significant.
RARα and coactivators are recruited to response elements in the CK5 promoter with P4 but not P4 plus RA treatment
Previous studies in keratinocytes identified that RARα binds to the CK5 promoter ~ 130 bp upstream of the TSS at what was termed a negative RA response element.38,39 We speculated a similar mechanism could occur in breast cancer cells. We therefore treated breast cancer cells with vehicle, P4, RA or RA plus P4 and performed ChIP for RARα followed by quantitative PCR with primers surrounding the negative RARE. Interestingly, RARα was only present at the negative RARE with P4 treatment, and was absent with RA alone or RA plus P4 (Figure 5b). We therefore tested RARα recruitment to the − 1.1 kb PRE under the same conditions, and similarly found that RARα was associated with the PRE region only under P4 conditions, but not with either RA alone or RA plus P4 (Figure 5c). Furthermore, P4 treatment induced PR recruitment at the − 130 bp region, which also contains PRE half-sites,40 but RA blocked this recruitment (Figure 5d). To investigate the activation state of the CK5 promoter, we performed ChIP for coactivators p300 and CBP. p300 was present at both the proximal and distal promoter sites in a P4-dependent manner, but was absent with co-treatment of P4 plus RA. CBP was also recruited in a P4-dependent manner to both promoter sites, but with less efficiency with co-treatment with P4 plus RA. These data suggest that the proximal binding region, while not sufficient for transcription itself, acts as a sensor for positive or negative enhancement through RARs. Figure 5f depicts a schematic of how P4 produces a functional coactivator bridge between PR occupied distal and RARα occupied proximal enhancers, whereas RA treatment disrupts this bridge by removal of RARα/p300.
Figure 5.

P4 recruits RARα and essential coactivators to the CK5 promoter while RA reduces RARα and coactivator occupancy. (a) Diagram showing location of primer sets used in b–e. (b) and (c) ChIP for RARα was performed on T47D cells treated with vehicle, 100 nM P4, P4 plus 100 nM 9-cis RA or P4 plus RA as indicated for 30 min pretreatment with RA or vehicle, followed by 1 h treatment with P4 or vehicle. quantitative PCR (qPCR) was performed for (b) a 200 bp region spanning the − 130 bp RARE, and (c) a 200 bp region spanning the − 1.1 kb PRE. Data represent mean ±s.e.m. IP conditions were compared via ANOVA/Tukey post hoc, ***P<0.001. (d) and (e) ChIP for PR, p300 and CBP was performed in T47D cells using primers for the two promoter regions as described in Figure 4b. nd, not detectable by qPCR. Experiments were repeated three times. Data represent mean (percent input) ±s.e.m. The four treatments within each group (IgG, RARα, PR, p300, CBP) were compared via analysis of variance/Tukey post hoc *P<0.05, **P<0.01, ***P<0.001. (f) Diagram of proposed coactivator bridging between enhancer elements. Under P4 conditions, PR and RARα occupy their respective response elements and coactivators form a functional bridge. RA removes RARα/p300 and reduces CPB occupancy, disrupting the bridge.
Co-treatment with retinoids reduces accumulation of therapy resistant CK5+ cells during endocrine therapy
Breast cancer cells treated with ER-targeted endocrine therapies show a gradual increase in the number of CK5+ cells.17 We reasoned that co-treatment with retinoids may reduce the number of CK5+ cells that accumulate, and thus could prevent recurrences by lessening residual tumor-initiating cells. To test this, we injected female NSG mice with T47D cells supplemented with E2 and allowed tumors to establish to 75 mm3. Tumors were then stratified into four groups: continued on E2 plus vehicle or the synthetic retinoid fenretinide (Fen; 100 mg/kg) or estrogen withdrawal (EWD) plus vehicle or Fen; treatments lasted for 3 weeks. The EWD groups trended toward a decrease in tumor growth compared to E2 alone, achieving significance at the study end point for the EWD only (Figure 6a). EWD tumors showed a robust increase in CK5+ cells compared to E2-treated tumors, while Fen prevented this increase during EWD (Figure 6b and c). Therefore, retinoids can decrease the accumulation of CK5+ cells associated with prolonged endocrine treatment.
Figure 6.
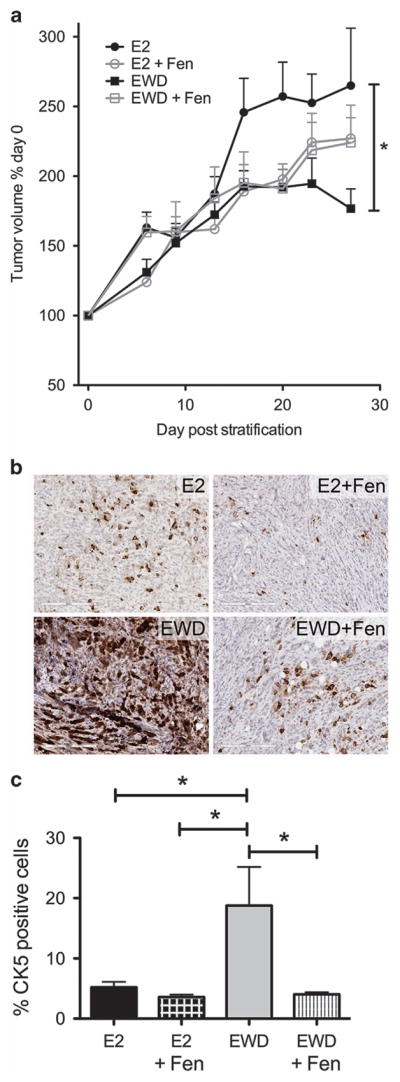
Co-treatment with retinoids during estrogen depletion reduces accumulation of CK5+ breast cancer cells. (a) A total of 1 × 106 T47D cells were implanted into the left and right mammary fat pads of female NOD/SCID mice. Mice were given E2 pellets at time of cell injection. When tumors reached 75 mm3 average volume they were stratified into four treatment groups: continued on E2 (n =10 tumors), E2 plus fenretinide (Fen) (n =10 tumors), EWD (n =10 tumors) or EWD plus Fen (n =10 tumors). Change in tumor volumes relative to treatment start is plotted versus the number of days post treatment. Data represent mean ±s.e.m. Tumor volumes at the last time point were compared via analysis of variance (ANOVA)/Tukey, *P<0.05 (EWD versus E2). (b) Representative immunohistochemistry for CK5 in tumor sections from all treatment groups. (c) The percent of CK5+ cells was analyzed using an Aperio digital pathology microscope for whole sections of tumors in each group (n =3) and plotted as percent positive cells ±s.e.m. All groups were compared via ANOVA/Tukey, *P<0.05.
DISCUSSION
CKs are intermediate filament proteins that form tetrameric complexes of two Type I and two Type II peptides important for cell structure and motility.41 However, CKs can influence other cellular processes such as proliferation, migration, invasion and stress-related signaling.42–45 For example, CK17 interaction with the scaffolding protein 14-3-3σ in keratinocytes facilitates its translocation to the cytoplasm to activate Akt signaling and cell cycle progression.43 Similarly, 14-3-3σ interacts with CK5, CK17 and actin in basal-like breast cancer cells to facilitate cell migration and invasion.46 CK14 and CK5 are found in cells at the leading edge of breast cancer invadopodia,46,47 suggesting they are important for cancer cell invasion. Thus, CKs can influence a wide variety of processes important for cancer cells. CK5 is a poor prognostic marker in both basal-like and luminal-like breast cancer.14,48 Here we show that CK5+ cells (induced by P4 treatment) have high tumorigenic potential and that CK5 is necessary for the P4-mediated increase in mammosphere size. P4 is a key hormone promoting expansion of murine mammary stem cells and human breast progenitor cells,49–51 an action that is maintained in some breast cancers.13,14,16 On the contrary, RA promotes stem cell differentiation and inhibits expression of CK5 in the skin.38,52 Therefore, a balance of PR-RAR regulation of CK5 may be pivotal in dictating a breast CSC phenotype.
In this paper we investigated the mechanism by which P4 and RA modulate expression of CK5 through their cognate receptors. Both PR and RARs are members of the nuclear receptor family of ligand-activated transcription factors; PRs belong to the steroid hormone receptor subclass, whereas RARs belong to the RXR heterodimer subclass.53 Previous studies demonstrated progestins and RA cross regulate expression of each other’s receptors in breast cancer cells. RA decreased PR mRNA and protein levels, and conversely progestins decreased RARα and RARγ at the transcript level.32,33,54 We confirmed that RA decreases PR protein levels but did not observe the reverse within a 24 h window (Figure 2c). Therefore, while RA could partially reduce progestin potency by lowering PR levels, we speculated other mechanisms likely contribute to this cross talk and investigated functional interactions between PR and RARs at the gene level.
Here we define unique convergence between RAR and PR signaling at the CK5 locus in breast cancer cells (Figure 5f). We describe two core promoter regions that affect transcription: a more distal (−1.1 kb from the TSS) region containing a PRE and a proximal region centered at − 130 bp that contains a previously described RARE38,39 and glucocorticoid receptor half-sites.40 The distal PRE region is essential for robust P4 activation of the gene (Figure 4a), while the proximal region alone is not sufficient itself for activation. P4 recruited PR and essential coactivators (p300 and CBP) to the promoter region when using primers for either the proximal or distal sites. However, we were surprised that P4 alone was sufficient to recruit RARα, and that RA alone did not induce RARα binding at the proximal − 130 bp/RARE region. In keratinocytes, RA induces RARα binding to negative RAREs of multiple basal CKs promoters including CK5 (−130 bp), CK6, CK14, and CK17, and prevents their expression, likely through recruitment of co-repressors, to maintain differentiation.38,39 We conclude that RAR regulates CK5 in breast cancer cells through a different mechanism. We propose that coactivators form a bridge between two nuclear receptor-occupied CK5 promoter elements in breast cancer cells, and that RARα acts as a sensor for RA, which redirects RARα and p300 away to disrupt the bridge (Figure 5f). Thus a reduction in RARs was not sufficient to decrease P4 transcription of CK5 suggests that RARα is not an essential positive cofactor, but is required for negative regulation through the proximal site. This proposal is intriguing in light of reports that describe widespread co-localization of RARα near ER binding sites in breast cancer cells; RARα therein acts as either as a positive cofactor or a negative regulator of ER gene transcription.30,31 We have found no evidence that either estrogens or ER directly affect CK5 transcription in breast cancer cells. The gradual increase in CK5+ cells in response to antiestrogen treatment17 suggests that this occurs through indirect transcriptional mechanisms. Collectively, our data support that CK5 is an important target for hormonal and nuclear receptor regulation that affects the downstream phenotype of breast cancer cells.
Retinoids have been extensively explored clinically against breast cancer based on promising preclinical studies.26 However, several trials using retinoids as single agents or in combination with Tam or chemotherapy for breast cancer failed to meet study objectives (reviewed in Garattini et al.27). One exception is the synthetic retinoid-like compound fenretinide, which is effective in prevention of secondary breast cancer in young women.28 Our unbiased screening results revealed that retinoids prevent the P4 expansion of CK5+ breast cancer cells.22 RAs may therefore be more efficacious in preventing initial transforming events, or conversion to a CSC phenotype, but may be less effective in reverting existing CSCs. On the basis of our data, we suggest that under the right contexts retinoids may reduce the acquisition of cells prone to tumor recurrence, an issue of particular relevance to luminal breast cancer, which can have long dormancy periods prior to relapse.4 In addition, the progestin-associated increase in breast cancer incidence during hormone replacement therapy is hypothesized to occur through expansion of stem cells;55 RA co-treatment could prospectively prevent this.
Taken together, our work describes cross talk among nuclear receptors PR and RARs at a single gene that is tied to a breast CSC phenotype. However, we speculate this cross talk occurs at a broader genome level in breast cancer cells. An emerging paradigm in nuclear receptor research is co-occupancy of multiple nuclear receptors at regulatory sites and co-dependency for transcriptional activation, or conversely transrepression.11,12,30 Further understanding of these complex relationships at the genome-wide level and in whole-tumor models may allow for better use of multiple-hormone treatments that target specific nuclear receptor relationships and co-dependencies to prevent or treat breast cancers.
MATERIALS AND METHODS
Cell culture and shRNA
Breast cancer cell lines (T47D, MCF7, BT-474) were obtained from the University of Colorado Cancer Center Tissue Culture core. Cells were maintained in minimal Eagle’s medium, 5% fetal bovine serum, 1 × NEAA, 1 × 10−9M insulin, 0.1 mg/ml sodium pyruvate and 2 mM L-glutamine. Cell lines were authenticated using short tandem repeat analysis, and tested negative for mycoplasma using the MycoAlert mycoplasma detection kit (Lonza, Basel, Switzerland). shRNAs targeting CK5 (TRCN0000425222, TRCN0000433559, TRCN0000083878) and a non-targeting clone (SHC0002) were purchased from Sigma Mission shRNA library (Functional Genomics facility, University of Colorado, Boulder, CO, USA). Cells were transduced with virus containing the shRNAs and stable pools selected with puromycin.
CK5 promoter deletion constructs
Constructs were created from the previously described 6 kb fragment of the human CK5 promoter cloned into a lentiviral vector upstream of firefly luciferase16 using the restriction enzymes listed in Supplementary Table 1. All constructs were analyzed via gel electrophoresis to verify size deletions and sequenced to confirm appropriate deletion. Cells were transduced with virus containing each construct and stable pools selected with puromycin.
Luciferase reporter assay and siRNA
T47D cells stably expressing CK5 promoter-luciferase reporter constructs were grown in media containing charcoal stripped serum for 24 h. Cells were then transfected with DharmaFECT 1 (T-2001-02, Dharmacon, Lafayette, CO, USA) non-targeting siRNA (D-0081810-10), or 10 nM siRNA targeting RARα (L-003437-00, Dharmacon), RARγ (L-003439-00, Dharmacon) or both together for 24 h, followed by treatment with vehicle (EtOH) or P4 (100 nM) for an additional 24 h. Lysates were harvested and assayed using the Luciferase Assay System (Promega, Madison, WI, USA).
Chromatin immunoprecipitation assay
For ChIP experiments, T47D cells were grown to 70–80% confluency in 15 cm2 dishes in phenol red-free media containing charcoal stripped serum. The next day, cells were pretreated for 30 min with 100 nM RA or vehicle then treated with 100 nM P4 or vehicle for 1 h. Immunoprecipitation for PR was performed using antibody PR Ab-8 (recognizes both PRA and PRB, MS-298-P, Thermo Fisher, Grand Island, NY, USA), RARα, p300, (ab41934, ab54984, Abcam, Cambridge, MA, USA) or CBP (7389, Cell Signaling, Danvers, MA, USA). Cells were processed using the ChIP-IT Express kit (Active Motif, Carlsbad, CA, USA). Chromatin was sheared using an S220 Focused Ultrasonicator (Covaris, Woburn, MA, USA).
Mammosphere formation assay
ZsGreen-labeled cells were plated at a density of 100 cells per well in 96-well ultra-low attachment plates in quintuplicate in 100 μl MammoCult Media (Stemcell Technologies, Vancouver, BC, Canada) containing 1% methylcellulose plus indicated treatments. Cells were grown for 2 weeks, fed with additional media without methylcellulose containing treatment once per week, then imaged and analyzed for sphere number and size using parameters on the FITC channel using the IncuCyte ZOOM Live Cell Analysis System (Essen Bioscience, Ann Arbor, MI, USA).
Immunoblotting
Whole-cell lysates were collected in RIPA buffer following indicated treatments. Proteins were immunoblotted with primary antibodies to CK5 (mouse NCL-L-CK5, Leica Biosystems, Buffalo Grove, IL, USA), RARα (sc551, Santa Cruz, Dallas, TX, USA; ab76074, Abcam), RARγ (sc550, Santa Cruz), alpha-tubulin (ST1568, Sigma, St Louis, MO, USA) or PR (PgR 1294, recognizes both PRA and PRB, Dako, Carpinteria, CA, USA) followed by IRDye 800CW Goat-Anti-Mouse IgG (926-32210, Li-Cor Biosciences, Lincoln, NE, USA) and IRDye 680LT Goat-Anti-Rabbit IgG (926-68021, Li-Cor Biosciences). The Odyssey Infrared Imaging System (Li-Cor Biosciences) was used to image immunoblots, and Image Studio Lite (Li-Cor Biosciences) was used for blot analysis.
Immunohistochemistry and Immunocytochemistry
Immunohistochemistry and ICC were performed essentially as previously described.20,56 Primary antibodies to CK5 (NCL-L-CK5, Leica Biosystems, 1:200), RARα (ab28767, Abcam, 1:100) and PR (PgR 1294, Dako, 1:500) were applied, followed by secondary antibodies, and developed using ImmPRESS Peroxidase detection kit (Vector Laboratories, Burlingame, CA, USA) for immunohistochemistry and secondary fluorescent antibodies (A11029, A11037, Invitrogen, Grand Island, NY, USA) for ICC. For immunohistochemistry, slides were scanned into the Aperio digital pathology system (Leica Biosystems) and whole sections analyzed for percent of positive cells using an algorithm tuned for CK5, therefore blinding was not necessary.
Quantitative reverse transcription PCR
RNA was harvested using QIAzol lysis reagent (Qiagen, Venlo, the Netherlands), qRT-PCR was performed using Absolute Blue Sybr Green (Thermo Fisher). Analysis was performed using the Pfaffl method for quantitative PCR.57 Primers targeting genomic DNA are as follows:
−1.1 kb primer set 1 (Fwd 5′-GAGTGGGTGTGGTTTAGAACAG-3′, Rev 5′-GTCTATGGATTGTCCTGCCAG-3′),
−1.1 kb primer set 2 (Fwd 5′-CTGGCAGGACAATCCATAGAC-3′, Rev 5′-CCAGCAAGCTCTATTCCACTAG-3′),
−130 bp primer set (Fwd 5′-CCAAGAGATCAGTGCTGCAAGG-3′, Rev 5′-GTTACCCAGGAACGGTGATGC-3′).
Limiting dilution analysis and tumor growth
Experiments involving animals were performed under an approved University of Colorado Institutional Animal Care and Use Committee protocol. For limiting dilution analysis, T47D cells harboring CK5 promoter-GFP were treated with 100 nM P4 for 24 h, GFP+ and GFP − cells collected by fluorescence-activated cell sorting, serially diluted into Cultrex (Trevigen, Gaithersburg, MD, USA) and an equal number of GFP+ and GFP − cells as indicated were injected bilaterally into opposing fourth mammary fat pads of 8-week-old female nu/nu mice (Jackson Labs, Bar Harbor, ME, USA). All animals were supplemented with silastic pellets containing 17β-estradiol (1 mg). Tumors were palpated twice weekly for 6 weeks. For treatment experiments, T47D xenografts were developed by injecting 1 × 106 T47D cells in Cultrex (Trevigen) bilaterally into the fourth mammary fat pads of 8-week-old female NOD/SCID mice (Jackson Labs) supplemented with silastic pellets containing 17β-estradiol (1 mg). When tumors reached an average of 75 mm2, mice were stratified into four treatment groups with equal average tumor volume (n = 5 animals each); continued on E2 or EWD, both plus/minus fenretinide. Tumors were measured 2× per week and volumes estimated by the formula l(w2)/2. Fenretinide (2.5 mg/mouse) or peanut oil vehicle was administered subcutaneously 2× per week; EWD was performed by surgical removal of the silastic estrogen pellet. After 3 weeks of treatment, mice were killed, and tumors collected and analyzed by immunohistochemistry. Sample number was calculated at 80% power and α = 0.05.
Statistical methods
Data are represented as mean ± s.e.m. unless otherwise noted, and analyzed using a two-tailed Student’s t-test or one-way analysis of variance followed by either a Tukey or Dunnett multiple comparison post hoc test as indicated. Prism 6.0 (GraphPad Software, La Jolla, CA, USA) was used for statistical analyses when samples met variance and normality tests. P<0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank the University of Colorado Cancer Center Flow Cytometry, Biorepository Core, and Tissue Culture Cores supported by P30CA046934 and the University of Colorado Department of Pathology Sequencing Core for their technical assistance and services. We thank Andrea Osypuk and Story Wilson for their assistance with Aperio imaging and analysis. This work was supported by grants from the Colorado Clinical and Translational Sciences Institute NIH TL1 TR001081 (LMF), National Institutes of Health grants NIH F31 CA210519 (LMF), NIH 2R01 CA140985 (CAS) and Breast Cancer Research Foundation (16-072, CAS, co-PI).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
LMF performed most of the studies. OM performed experiments in Figure 2a and b, and Figure 5b and c. JF-S performed experiments in Table 1 and Supplementary Figure 1. SKN engineered and provided reagents for Figure 4. LMF, JF-S, DVL, SKN and CAS contributed intellectual design and interpretation of results. LMF wrote the manuscript. JF-S, SKN and CAS provided editorial assistance. All authors read and approved the final manuscript.
References
- 1.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2006;26:87–98. doi: 10.3233/bd-2007-26108. [DOI] [PubMed] [Google Scholar]
- 4.Guedj M, Marisa L, de Reynies A, Orsetti B, Schiappa R, Bibeau F, et al. A refined molecular taxonomy of breast cancer. Oncogene. 2012;31:1196–1206. doi: 10.1038/onc.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 6.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osborne CK, Schiff R, Arpino G, Lee AS, Hilsenbeck VG. Endocrine responsiveness: understanding how progesterone receptor can be used to select endocrine therapy. Breast. 2005;14:458–465. doi: 10.1016/j.breast.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 9.Daniel AR, Gaviglio AL, Knutson TP, Ostrander JH, D’Assoro AB, Ravindranathan P, et al. Progesterone receptor-B enhances estrogen responsiveness of breast cancer cells via scaffolding PELP1- and estrogen receptor-containing transcription complexes. Oncogene. 2015;34:506–515. doi: 10.1038/onc.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knutson TP, Daniel AR, Fan D, Silverstein KA, Covington KR, Fuqua SA, et al. Phosphorylated and sumoylation-deficient progesterone receptors drive proliferative gene signatures during breast cancer progression. Breast Cancer Res. 2012;14:R95. doi: 10.1186/bcr3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature. 2015;523:313–317. doi: 10.1038/nature14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singhal H, Greene ME, Tarulli G, Zarnke AL, Bourgo RJ, Laine M, et al. Genomic agonism and phenotypic antagonism between estrogen and progesterone receptors in breast cancer. Sci Adv. 2016;2:e1501924. doi: 10.1126/sciadv.1501924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci USA. 2008;105:5774–5779. doi: 10.1073/pnas.0706216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato T, Tran TH, Peck AR, Girondo MA, Liu C, Goodman CR, et al. Prolactin suppresses a progestin-induced CK5-positive cell population in luminal breast cancer through inhibition of progestin-driven BCL6 expression. Oncogene. 2014;33:2215–2224. doi: 10.1038/onc.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 16.Axlund SD, Yoo BH, Rosen RB, Schaack J, Kabos P, Labarbera DV, et al. Progesterone-inducible cytokeratin 5-positive cells in luminal breast cancer exhibit progenitor properties. Horm Cancer. 2013;4:36–49. doi: 10.1007/s12672-012-0127-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabos P, Haughian JM, Wang X, Dye WW, Finlayson C, Elias A, et al. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res Treat. 2011;128:45–55. doi: 10.1007/s10549-010-1078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knox AJ, Scaling AL, Pinto MP, Bliesner BS, Haughian JM, Abdel-Hafiz HA, et al. Modeling luminal breast cancer heterogeneity: combination therapy to suppress a hormone receptor-negative, cytokeratin 5-positive subpopulation in luminal disease. Breast Cancer Res. 2014;16:418. doi: 10.1186/s13058-014-0418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P, et al. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2013;32:2555–2564. doi: 10.1038/onc.2012.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finlay-Schultz J, Cittelly DM, Hendricks P, Patel P, Kabos P, Jacobsen BM, et al. Progesterone downregulation of miR-141 contributes to expansion of stem-like breast cancer cells through maintenance of progesterone receptor and Stat5a. Oncogene. 2015;34:3676–3687. doi: 10.1038/onc.2014.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman CR, Sato T, Peck AR, Girondo MA, Yang N, Liu C, et al. Steroid induction of therapy-resistant cytokeratin-5-positive cells in estrogen receptor-positive breast cancer through a BCL6-dependent mechanism. Oncogene. 2016;35:1373–1385. doi: 10.1038/onc.2015.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoo BH, Axlund SD, Kabos P, Reid BG, Schaack J, Sartorius CA, et al. A high-content assay to identify small-molecule modulators of a cancer stem cell population in luminal breast cancer. J Biomol Screen. 2012;17:1211–1220. doi: 10.1177/1087057112452138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.le Maire A, Alvarez S, Shankaranarayanan P, Lera AR, Bourguet W, Gronemeyer H. Retinoid receptors and therapeutic applications of RAR/RXR modulators. Curr Top Med Chem. 2012;12:505–527. doi: 10.2174/156802612799436687. [DOI] [PubMed] [Google Scholar]
- 24.Perissi V, Rosenfeld MG. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol. 2005;6:542–554. doi: 10.1038/nrm1680. [DOI] [PubMed] [Google Scholar]
- 25.Gianni M, Kalac Y, Ponzanelli I, Rambaldi A, Terao M, Garattini E. Tyrosine kinase inhibitor STI571 potentiates the pharmacologic activity of retinoic acid in acute promyelocytic leukemia cells: effects on the degradation of RARalpha and PML-RARalpha. Blood. 2001;97:3234–3243. doi: 10.1182/blood.v97.10.3234. [DOI] [PubMed] [Google Scholar]
- 26.Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- 27.Garattini E, Bolis M, Garattini SK, Fratelli M, Centritto F, Paroni G, et al. Retinoids and breast cancer: from basic studies to the clinic and back again. Cancer Treat Rev. 2014;40:739–749. doi: 10.1016/j.ctrv.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Veronesi U, Mariani L, Decensi A, Formelli F, Camerini T, Miceli R, et al. Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast cancer. Ann Oncol. 2006;17:1065–1071. doi: 10.1093/annonc/mdl047. [DOI] [PubMed] [Google Scholar]
- 29.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 30.Ross-Innes CS, Stark R, Holmes KA, Schmidt D, Spyrou C, Russell R, et al. Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes Dev. 2010;24:171–182. doi: 10.1101/gad.552910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hua S, Kittler R, White KP. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell. 2009;137:1259–1271. doi: 10.1016/j.cell.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarke CL, Graham J, Roman SD, Sutherland RL. Direct transcriptional regulation of the progesterone receptor by retinoic acid diminishes progestin responsiveness in the breast cancer cell line T-47D. J Biol Chem. 1991;266:18969–18975. [PubMed] [Google Scholar]
- 33.Clarke CL, Roman SD, Graham J, Koga M, Sutherland RL. Progesterone receptor regulation by retinoic acid in the human breast cancer cell line T-47D. J Biol Chem. 1990;265:12694–12700. [PubMed] [Google Scholar]
- 34.Reid BG, Jerjian T, Patel P, Zhou Q, Yoo BH, Kabos P, et al. Live multicellular tumor spheroid models for high-content imaging and screening in cancer drug discovery. Curr Chem Genomics Transl Med. 2014;8:27–35. doi: 10.2174/2213988501408010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Widschwendter M, Berger J, Daxenbichler G, Muller-Holzner E, Widschwendter A, Mayr A, et al. Loss of retinoic acid receptor beta expression in breast cancer and morphologically normal adjacent tissue but not in the normal breast tissue distant from the cancer. Cancer Res. 1997;57:4158–4161. [PubMed] [Google Scholar]
- 36.Lieberman BA, Bona BJ, Edwards DP, Nordeen SK. The constitution of a progesterone response element. Mol Endocrinol. 1993;7:515–527. doi: 10.1210/mend.7.4.8388996. [DOI] [PubMed] [Google Scholar]
- 37.Clarke CL, Graham JD. Non-overlapping progesterone receptor cistromes contribute to cell-specific transcriptional outcomes. PLoS One. 2012;7:e35859. doi: 10.1371/journal.pone.0035859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jho SH, Radoja N, Im MJ, Tomic-Canic M. Negative response elements in keratin genes mediate transcriptional repression and the cross-talk among nuclear receptors. J Biol Chem. 2001;276:45914–45920. doi: 10.1074/jbc.M103144200. [DOI] [PubMed] [Google Scholar]
- 39.Radoja N, Diaz DV, Minars TJ, Freedberg IM, Blumenberg M, Tomic-Canic M. Specific organization of the negative response elements for retinoic acid and thyroid hormone receptors in keratin gene family. J Invest Dermatol. 1997;109:566–572. doi: 10.1111/1523-1747.ep12337483. [DOI] [PubMed] [Google Scholar]
- 40.Radoja N, Komine M, Jho SH, Blumenberg M, Tomic-Canic M. Novel mechanism of steroid action in skin through glucocorticoid receptor monomers. Mol Cell Biol. 2000;20:4328–4339. doi: 10.1128/mcb.20.12.4328-4339.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- 42.Chung BM, Rotty JD, Coulombe PA. Networking galore: intermediate filaments and cell migration. Curr Opin Cell Biol. 2013;25:600–612. doi: 10.1016/j.ceb.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441:362–365. doi: 10.1038/nature04659. [DOI] [PubMed] [Google Scholar]
- 44.Seltmann K, Fritsch AW, Kas JA, Magin TM. Keratins significantly contribute to cell stiffness and impact invasive behavior. Proc Natl Acad Sci USA. 2013;110:18507–18512. doi: 10.1073/pnas.1310493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toivola DM, Strnad P, Habtezion A, Omary MB. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010;20:79–91. doi: 10.1016/j.tcb.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boudreau A, Tanner K, Wang D, Geyer FC, Reis-Filho JS, Bissell MJ. 14-3-3sigma stabilizes a complex of soluble actin and intermediate filament to enable breast tumor invasion. Proc Natl Acad Sci USA. 2013;110:E3937–E3944. doi: 10.1073/pnas.1315022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639–1651. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14:1368–1376. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 49.Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010;465:798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 50.Graham JD, Mote PA, Salagame U, van Dijk JH, Balleine RL, Huschtscha LI, et al. DNA replication licensing and progenitor numbers are increased by progesterone in normal human breast. Endocrinology. 2009a;150:3318–3326. doi: 10.1210/en.2008-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465:803–807. doi: 10.1038/nature09091. [DOI] [PubMed] [Google Scholar]
- 52.Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011;226:322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roman SD, Clarke CL, Hall RE, Alexander IE, Sutherland RL. Expression and regulation of retinoic acid receptors in human breast cancer cells. Cancer Res. 1992;52:2236–2242. [PubMed] [Google Scholar]
- 55.Horwitz KB, Sartorius CA. Progestins in hormone replacement therapies reactivate cancer stem cells in women with preexisting breast cancers: a hypothesis. J Clin Endocrinol Metab. 2008;93:3295–3298. doi: 10.1210/jc.2008-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kabos P, Finlay-Schultz J, Li C, Kline E, Finlayson C, Wisell J, et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res Treat. 2012;135:415–432. doi: 10.1007/s10549-012-2164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.