

Abstract
Protein kinase C (PKC) has historically been considered an oncoprotein. This stems in large part from the discovery in the early 1980s that PKC is directly activated by tumor-promoting phorbol esters. Yet three decades of clinical trials using PKC inhibitors in cancer therapies not only failed, but in some cases worsened patient outcome. Why has targeting PKC in cancer eluded successful therapies? Recent studies looking at the disease for insight provide an explanation: cancer-associated mutations in PKC are generally loss-of-function (LOF), supporting an unexpected function as tumor suppressors. And, contrasting with LOF mutations in cancer, germline mutations that enhance the activity of some PKC isozymes are associated with degenerative diseases such as Alzheimer's disease. This review provides a background on the diverse mechanisms that ensure PKC is only active when, where, and for the appropriate duration needed and summarizes recent findings converging on a paradigm reversal: PKC family members generally function by suppressing, rather than promoting, survival signaling.
Keywords: PKC, phorbol esters, tumor suppressor, diacylglycerol, LOF
Introduction
Protein kinase C (PKC) is considered the archetypal transducer of signals that result in phospholipid hydrolysis. The discovery in the late 1970s by Nishizuka and colleagues that PKC is directly activated by diacylglycerol [1] provided the long-sought effector of phosphoinositide hydrolysis, which Hokin and Hokin had shown 25 years previously to result from cholinergic stimulation [2]. All but two members of this family of kinases are allosterically activated by diacylglycerol, a simple lipid whose levels are tightly regulated in the cell [3]. As an essential intermediate in lipid metabolism, diacylglycerol is always present at basal levels, yet small increases resulting from receptor-mediated activation of phospholipase C are readily sensed by PKC. This review describes how the activity of PKC is exquisitely regulated to ensure homeostasis, and discusses recent findings supporting a general role in suppressing survival signaling.
1. PKC Family and Regulation
The PKC Family
PKC isozymes are positioned at the tip of the AGC (protein kinases A, G, and C) branch of the kinome tree, past the branching off points of protein kinase A (PKA), phosphoinositide dependent kinase-1 (PDK-1), S6 kinase, Akt/protein kinase B (PKB), and protein kinase N (PKN) [4]. There are nine mammalian PKC genes that evolved from the single PKC1 in saccharomyces cerevisaie [5, 6]. At the very tip of the branch lie the conventional isozymes (PKC α, β, and γ), preceded on the dendogram by the novel (PKC δ, ε, θ, η) PKC isozymes and, in turn, preceded by the atypical isozymes (PKC ζ and ι) [7]. Splice variants, including the common PKCβI and PKCβII, increase the number of isozymes to considerably more than nine. All PKC isozymes share a common architecture of an amino terminal regulatory moiety and carboxyl terminal kinase domain (Figure 1). The regulatory moiety comprises an autoinhibitory pseudosubstrate segment and modules that control whether this segment is engaged in the substrate-binding cavity of the kinase domain, to maintain the kinase in an ‘off’ conformation, or engaged to binding surfaces, to maintain the kinase in an ‘on’ and open conformation. The nature of the regulatory modules that dictate how the pseudosubstrate is released from the kinase domain determine the subfamilies of PKC.
Figure 1. Domain composition of PKC isozymes grouped by subfamily.
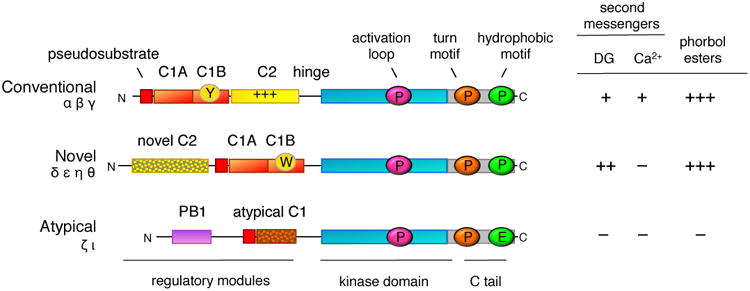
All PKC isozymes comprise an N-terminal regulatory moiety that contains an autoinhibitory pseudosubstrate segment (red) that is immediately followed by a C1A domain (orange) and a C-terminal catalytic moiety. Conventional and novel PKC isozymes have a second C1 domain, the C1B domain (orange), which is the predominant diacylglycerol sensor in the full-length protein; its affinity for diacyglycerol is two orders of magnitude higher in the novel C1B domain compared to the conventional C1B domain because of a Trp (vs Tyr in conventional isozymes) at a site that toggles the affinity of the C1B domain for diacylglycerol (W vs Y indicated on domain). Conventional PKC isozymes have a Ca2+-binding C2 domain (yellow) that contains a basic surface (indicated by +++) that serves as a plasma membrane sensor via its recognition of PIP2. Novel PKC isozymes have a novel C2 domain that does not bind Ca2+ or lipids (mottled). Atypical PKC isozymes have a PB1 domain (purple) that mediates binding to protein scaffolds. The C-terminal kinase moiety contains the catalytic domain that has a priming phosphorylation site by PDK-1 (pink cirle) and a C-terminal tail (C tail; grey) that is phosphorylated at the turn motif (orange circle) and hydrophobic motif (green circle); atypical PKC isozymes have a Glu at the phosphoacceptor site of the hydrophobic motif. The sensitivity to second messengers, diacyglycerol (DG) and Ca2+, and to phorbol esters is shown on the right (+, ++, and +++ indicate relative affinity for C1 domain ligands).
Conventional PKC isozymes are regulated by diacylglycerol, which binds to one of two tandem C1 modules, and Ca2+, which binds to a C2 domain (Figure 1). Novel PKC isozymes are regulated by diacylglycerol, which also binds one of the tandem C1 domains. Although novel PKC isozymes also have a C2 domain, it is not a Ca2+ sensor as key Asp residues required to coordinate Ca2+ are absent. Novel isozymes are able to respond to diacylglycerol alone because a single residue in the C1B domain, present as a Tyr in conventional PKC isozymes and a Trp in novel PKC isozymes, increases the affinity of this module by two orders of magnitude [8]. This enhanced affinity for diacylglycerol allows novel PKC isozymes to respond to elevations in diacylglycerol alone. Atypical PKC isozymes respond to neither diacylglycerol nor Ca2+: they lack a second C1 domain and C2 domain, and although they maintain one C1 domain directly following the pseudosubstrate segment, a ring of basic residues surrounding the binding cleft precludes ligand binding [9, 10]. Instead, these PKC isozymes have a Phox and Bem1p (PB1) protein interaction domain which mediates binding to other PB1 domains on protein scaffolds [11]. This binding tethers the pseudosubstrate out of the substrate-binding cavity to allow substrate phosphorylation. Thus, all PKC isozymes have a pseudosubstrate-C1A module but the mechanisms that regulate how it unmasks the kinase domain differ.
Maturation of PKC
Before PKC isozymes adopt an autoinhibited conformation that is sensitive to second messengers, they undergo a series of ‘processing’ phosphorylations (Figure 2). Studies with the conventional isozyme, PKCβII, reveal that newly synthesized enzyme is in an open conformation in which all its membrane targeting modules and pseudosubstrate are exposed (species i, Figure 2). This species binds the molecular chaperone Hsp90 and its client Cdc37 via a conserved PXXP motif found throughout the AGC family [12], a binding event that is required for PKC to be processed by phosphorylation. By a mechanism that also depends on the kinase complex mTORC2 [13], PKC is first phosphorylated on a segment near the entrance to the active site by the phosphoinositide-dependent kinase-1 (PDK-1) followed by two tightly-coupled phosphorylations on the C-terminal tail, at positions termed the turn motif and hydrophobic motif. Phosphorylation at these latter sites depends on the intrinsic catalytic activity of PKC and, at least in vitro, has been shown to occur by intramolecular autophosphorylation on the hydrophobic motif [14]. The phosphorylated PKC adopts an autoinhibited conformation (species ii in Figure 2) that is stable, with a half-life of approximately 2 days, and resistant to dephosphorylation. In particular, phosphorylation of the hydrophobic motif prevents the degradation of PKC. Mechanisms that prevent any of these steps, including inhibition of mTORC2, result in PKC that cannot become phosphorylated, resulting in the degradation of PKC.
Figure 2. Cartoon showing the multiple inputs that regulate the signaling lifetime of a conventional PKC.
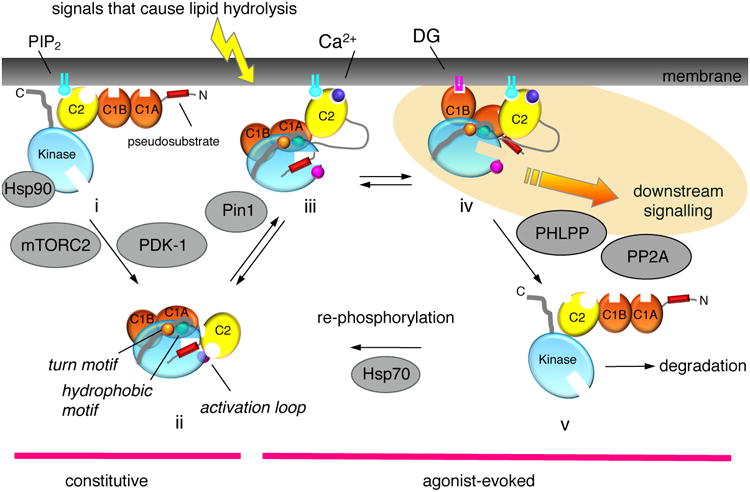
Following its biosynthesis, PKC is in an open, and degradation-sensitive, conformation in which all its regulatory modules are unmasked (species i). It is processed by a series of ordered phosphorylations that depend on the binding of Hsp90, with Cdc37, to a conserved PXXP motif in the kinase domain, the kinase complex mTORC2, and PDK-1. Phosphorylation at three priming sites, the activation loop, and two sites on the C-terminal tail, the turn motif and the hydrophobic motif, promote PKC to adopt an autoinhibited conformation in which the Ca2+-sensing C2 domain (yellow) clamps the autoinhibitory pseudosubstrate segment (red) in the substrate-binding cavity of the kinase domain (cyan), and the diacyglycerol-sensing C1 domains (orange) become masked (species ii). Hydrolysis of PIP2 results in Ca2+-dependent recruitment of PKC to the plasma membrane via engagement of the C2 domain (species iii), where PKC binds its membrane-embedded ligand, diacylglycerol, via primarily the C1B domain (species iv). This active PKC phosphorylates downstream substrates, such as Ras, to suppress oncogenic signaling. The membrane-bound conformation of PKC is sensitive to dephosphorylation, with the first event being dephosphorylation of the hydrophoboic motif catalyzed by PHLPP; subsequent dephosphorylation by PP2A produces a fully dephosphorylated PKC that is shunted for degradation by a proteosomal pathway (species v). However, binding of Hsp70 to the dephosphorylated turn motif allows PKC to become rephosphorylated to sustain the signaling lifetime of the enzyme. Phorbol esters (not shown) bind the C1B domain with two-orders of magnitude higher affinity than diacylglycerol (highlighted in yellow) and are not readily metabolized, trapping PKC in the open, phosphatase-sensitive conformation and resulting in chronic loss, or down-regulation, of PKC. Novel PKC isozymes are regulated by similar mechanisms except their C2 domain does not function as a Ca2+ or plasma membrane sensor, resulting in tht localization of novel PKC isozymes primarily to the more abundant and diacylglycerol-rich Golgi membranes. Atypical PKC isozymes are activated upon binding to specific protein scaffolds that tether the pseudosubstrate out of the substrate-binding cavity. Proteins indicated in grey are key regulators of the steady-state levels of PKC: Hsp70, Hsp90, mTORC2, and PDK-1 function to increase the steady-state levels of PKC by permitting/catalyzing processing phosphorylations; Pin1 and the phosphatases PHLPP and PP2A function to decrease the steady-state levels of PKC by permitting/catalyzing the dephosphorylation of PKC. Targeting any of these proteins will disrupt the balance of PKC signaling.
Atypical PKC isozymes are also processed by phosphorylation, resulting in constitutive phosphorylation, but the mechanism of this regulation differs in one key aspect from the processing phosphorylations of the diacyglycerol-regulated PKC isozymes: the nascent atypical PKC polypeptide is co-translationally phosphorylated on its turn motif by ribosome-associated mTORC2 [15], similar to the co-translational processing of Akt at this site [16]. In addition, Glu occupies the phospho-acceptor position of the hydrophobic motif phosphorylation site and functions sufficiently like a phosphomimetic that unphosphorylated constructs are resistant to degradation.
Activation of PKC
PKC is maintained in an autoinhibited conformation by binding of the pseudosubstrate in the substrate-binding cavity; structural studies of PKC βII [17] have revealed that this interaction is locked in place by the C2 domain [18]. This ensures minimal signaling in the absence of agonist stimulation. In the case of conventional PKC isozymes, hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) results in the activation of PKC by a two step mechanism: first, Ca2+ binds the C2 domain, allowing engagement of PKC on the plasma membrane via bridging with anionic phospholipids, at the Ca2+-binding site, and via binding to PIP2 [19, 20], a lipid restricted to the plasma membrane, via a basic PIP2-binding surface on the C2 domain. Thus, in the presence of Ca2+, engagement of the C2 domain on the plasma membrane tethers the C2 domain away from the kinase domain, a conformational change that results in unmasking of the proteolytically-labile hinge separating the regulatory and catalytic moieties (species iii, Figure 2). Second, membrane-localized PKC binds its membrane-embedded ligand diacylglycerol, primarily via the C1B domain, an event that results in release of the pseudosubstrate from the substrate-binding cavity and accompanying activation (species iv, Figure 2).
As noted above, novel PKC isozymes bind diacylglycerol with two orders of magnitude higher affinity than conventional PKC isozymes and thus do not require pre-targeting to membranes by Ca2+ in order to effectively bind diacylglycerol. Nor do they have a PIP2 sensor on their C2 domain, which restricts conventional PKC isozymes to the plasma membrane. Thus, novel PKC isozymes are primarily recruited to, a signal from, the more abundant and diacylglycerol-rich Golgi membrane.
Atypical PKC isozymes are regulated not by second messengers, but by protein:protein interactions that not only tether the pseudosubstrate out of the substrate-binding cavity, but position atypical PKC isozymes near their substrates. The latter is particularly important for atypical PKC isozymes because their intrinsic catalytic activity is an order of magnitude lower than that of the diacyglycerol-regulated PKC isozymes [15, 21]. Binding to protein scaffolds via their PBI domains results in release of the pseudosubstrate to allow catalysis. For example, binding to Par6 via a PB1:PB1 domain interaction relieves autoinhibition by the pseudosubstrate-C1 module [22]. Similarly, binding to the PB1 domain of the scaffold p62 relieves autoinhibition. In this case the pseudosubstrate is tethered to an acidic surface on the PB1 domain of p62 [23]. The differential affinity of scaffolds for pseudosubstrate fine tunes the activity of atypical PKC isozymes on the relevant scaffold [24]. Additionally, protein interactions can also inhibit atypical PKC isozymes: structural studies have revealed that a segment of Par3 binds the active site of atypical PKC isozymes to block substrate access [25].
Inactivation of PKC
The inactivation kinetics of conventional and novel PKC isozymes mirror the decay of diacylglycerol [26]. Thus, metabolism of diacyglycerol results in the redistribution of PKC to the cytosol, where it regains the autoinhibited conformation (species ii, Figure 2). The spatiotemporal dynamics of this reversible activation of PKC has been well documented with activity reporters of PKC (reviewed in [27]). However, prolonged activation as occurs with ligands that are not readily metabolized, such as phorbol esters, results in the loss of PKC, a process referred to as down-regulation [28]. In its open, membrane-bound conformation (species iv, Figure 2), the phosphatase sensitivity of PKC is two orders of magnitude greater than that of the autoinhibited PKC [29]. Thus, prolonged residency on the membrane results in its dephosphorylation (species v, Figure 2). The first step in this dephosphorylation is catalyzed by the okadaic acid-insensitive PH domain Leucine-rich repeat Protein Phosphatase (PHLPP) [30], which removes the hydrophobic motif phosphate present on conventional and novel PKC isozymes [31]. Its loss results in the subsequent dephosphorylation of the turn motif and activation loop sites by okadaic acid-sensitive PP2A phosphatases [32]. Dephosphorylation is followed by ubiquitination and proteosome-mediated degradation of PKC, although under some conditions the phosphorylated protein can be ubiquitinated and degraded [33]. Interestingly, the down-regulation of PKCε has been reported to require the activity of other PKC isozymes [34], suggesting interplay between the steady-state levels of PKC isozymes. Note that conventional isozymes depend on the peptidyl-prolyl isomerase Pin1 to be converted into a species that can be efficiently down-regulated following activation, providing a ‘timer’ for the lifetime of conventional PKC isozymes [35]: Pin1 catalyzes a post-maturation cis-trans isomerization of the turn motif phospho-Thr-Pro peptidyl bond that then allows the agonist-evoked dephosphorylation, ubiquitination, and degradation of conventional PKC isozymes (Figure 2).
2. Phorbol Esters and Tumor Promotion
Phorbol esters are found in the milky sap exuded by plants of the Euphorbiaccae family; the strong irritant property of this sap resulted in its use over the millennia in poison arrows, and croton oil, derived from the shrub Croton tiglium, was used medicinally as a cathartic [36]. Classic studies starting in the 1940s established that croton oil is a tumor promoter [37]: painting a sub threshold amount of a carcinogen on the skin of mice did not result in tumor formation, however, papilomas developed if the carcinogen treatment was followed by repetitive application of croton oil (reviewed in [38]). Indeed in humans, the common use of Croton flavens, one species of Euphorbia, for ‘bush tea’ in Curacao was proposed to be causally related with the high incidence of esophageal cancer on this Caribbean island [39]. In the late 1960s, the active ingredient was identified as a family of diesters of the tetracyclic diterpene phorbol, with varying acyl chains at the C-12 and C-13 positions [40, 41]. The most potent compound was phorbol 12-myristate 13-acetate (PMA, also referred to as TPA for 12-0-tetradecanoyl phorbol 13-acetate). The highly lipophilic properties of these molecules resulted in their nonspecific intercalation into cell membranes, impeding the identification of their ‘receptor’. A breakthrough was made by Blumberg and colleagues, who reasoned that a more water-soluble molecule that still retained the pharmacophore would facilitate identification of the receptor [42, 43]. Their synthesis of phorbol 12,13-dibutyrate (PDBu) not only allowed the identification of PKC as a major receptor for phorbol esters [44], but to this day remains one of the most commonly used tools to modulate signaling pathways in cells.
The identification of PKC as the major receptor for tumor promoting phorbol esters marked the genesis of the concept that PKC would function as an oncoprotein. Yet 30+ years of clinical trials using inhibitors of PKC for cancer not only were ineffective, but in some cases worsened patient outcome. Notably, a meta analysis of 5 clinical trials for non small cell lung carcinomas revealed worsened patient outcome when PKC inhibitors (enzastaurin, an ATP competitive inhibitor, or aprinocarsen, a PKCα antisense oligonucleotide) were combined with chemotherapy compared with chemotherapy alone [45]. Thus, clinical trials unveiled a disconnect between the biology of phorbol esters and the biology of PKC.
Phorbol esters bind the C1 domain of PKC in a competitive manner with respect to the physiological ligand, diacylglycerol. However, unlike diacylglycerol, phorbol esters are not readily metabolized and thus result in constitutive activation of PKC. As noted above, activated PKC is in an open conformation that is sensitive to dephosphorylation and degradation. Thus, while phorbol esters result in acute activation of PKC, this is followed by the chronic loss, or down-regulation, of PKC [28]. As a result, overnight treatment with phorbol esters was a common and effective method to deplete cells of conventional and novel PKC isozymes in the era preceding genetic knockdown. In the paradigm described above for carcinogen-induced tumor promotion, the repetitive application of phorbol esters would be expected to cause a loss of PKC. Indeed, prolonged infusion with bryostatins, marine natural products that, like phorbol esters, also bind the C1 domain with exceptionally high affinity and down-regulate PKC [46], resulted in a significant reduction in the levels of PKCα, PKCε, and PKCη in peripheral blood monocytes of patients with advanced metastatic cancers [47]. Could the tumor-promoting properties of phorbol esters result from the long-term loss of PKC rather than its acute activation?
3. PKC Mutations in Cancer are Loss-of-Function
The first reported cancer-associated mutation in PKC was one in PKCα that was found in human pituitary tumors [48, 49]. Joubert and colleagues reported that this mutation, D294G in the hinge region separating the regulatory and catalytic moieties, abolished the targeting of the enzyme to cell-cell contacts, effectively reducing its function [50]. It took the advent of massive tumor sequencing efforts to provide the resources for a more wide-spread analysis of cancer-associated mutations. There are now over 1,000 cancer-associated somatic mutations in PKC isozymes that are documented in cBioPortal ([51]). They occur in all PKC isozymes and throughout their domain structure. Analysis of approximately 50 of these revealed that two thirds were LOF, with no gain-of function (GOF) mutations identified [52]. Inactivation occurred by diverse mechanisms, from preventing processing phosphorylations (thus decreasing steady-state levels of the mutant PKC), impeding second messenger binding (by mutations in the C1 or C2 domain), or impairing catalysis (by mutations in the kinase domain). Many additional mutations in PKC family members are predicted with high confidence to be LOF as they occur in highly conserved motifs required for catalytic activity. The recently developed software KinView which annotates cancer-associated protein kinase mutations, identifies many additional LOF mutations in PKC, one of which was validated experimentally [53]. Lastly, neomorphic mutations that disrupt normal signaling by redirecting PKC away from physiological substrates, thus potentially engaging novel signaling pathways, may also contribute to cancer. For example, a mutation in PKCγ that alters the substrate specificity of the kinase has been identified in lung cancer [54]. There are also numerous truncating mutations that have been identified in all the PKC isozymes. Lastly, a number of fusion proteins in PKC have also been identified in human cancers [55] and analysis of one such fusion in PKCε in a thyroid cancer cell line reveals that its impaired function protects thyroid cells from apoptosis [56]. Taken together, these results reveal an abundance of cancer-associated mutations in PKC that are LOF and are found in a multitude of cancers.
Cellular analysis of LOF mutations in PKC has revealed that many are dominant negative with respect to the global PKC signaling output of cells. That is, a mutation in one isozyme suppresses the activity of other isozymes. One possible mechanism for this dominant negative effect is that the mutant PKC impairs the processing phosphorylations of other PKC isozymes, thus reducing their steady-state levels. Parker and coworkers showed this to be the case in overexpression studies and suggested that common titratable elements required in the processing of all PKC isozymes may be sequestered by the mutant PKC [57]. In this regard, the concentration of one common regulator of all PKC isozymes, PDK-1, has been estimated to be 10 nM in HeLa cells, considerably below the sum concentration of all the PKC isozymes (>100 nM), not to mention its other kinase substrates [58]. LOF mutations in PKC are generally heterozygous; their ability to function in a dominant negative manner with respect to their wild-type counterpart, and additionally to other PKC isozymes, amplifies their detrimental effects.
Studies in a colon cancer cell line in which a heterozygous cancer-associated mutation in PKCβ was either corrected to wild-type or deleted provide insight into the functional consequences of a mutant PKC. This mutation, found in a colon tumor and in the DLD1 colon cancer cell line, is in the highly conserved APE motif, a key regulatory segment of protein kinases [59]; this motif is a warm spot for LOF mutations in PKC isozymes and impairs processing phosphorylations and the catalytic activity of PKC [52]. Correction of this mutation (A509T) in the endogenous PKCβ allele (PRKCB) using genome editing revealed that the mutant protein is dominant negative with respect to other PKC isozymes: the basal signaling output of PKC was considerably higher in clonal cell lines with corrected PKCβ, and levels of another PKC, PKCα, were elevated more than 2-fold compared to levels in the parental cell line harboring one mutant allele of PKCβ. Cells in which the mutant allele was corrected had significantly reduced anchorage-independent growth compared to the parental cells. And deletion of the the mutant allele by genome editing resulted in an intermediate reduction in anchorage-independent growth, revealing that PKCβ is haploinisufficient with respect to suppressing anchorage-independent growth. Importantly, there was a striking decrease in tumor volume when corrected cell lines were subcutaneously injected in the flanks of nude mice compared to the parental cell lines. This cancer cell line harbours an oncogenic mutation in K-Ras (G13D), underscoring the dominance of PKC in suppressing oncogenic signaling: two alleles of functional PKCβ effectively suppress anchorage-independent growth and tumor growth in a xenograft model, with mutation of one allele unmasking the oncogenic potential of K-Ras.
4. Downstream Substrates of PKC
Considerable evidence supports a role of PKC in serving as ‘brakes’ to oncogenic signaling via its inhibitory phosphorylation of proteins that promote growth and survival signaling, with transmembrane receptors being some of the best characterized substrates of PKC. One of its earliest identified substrates was the EGF receptor: Hunter and colleagues identified an inhibitory phosphorylation on Thr654 [60]. This phosphorylation reduces EGFR tyrosine kinase activity, decreases ligand binding affinity, and promotes internalization of the receptor [61-64]. PKC also phosphorylates and promotes the internalization of the proto-oncogene HER2 [65]. Amplified signaling by growth factor receptors, not only from GOF mutations but also from gene amplification or epigenetic alterations, is associated with a multitude of cancers; functional PKC would keep aberrant signaling in check, whereas LOF mutations in PKC would be predicted to sustain growth factor signaling, even in the context of oncogenic mutations. Inhibitory phosphorylations on the cytoplasmic tails of receptors is a theme in PKC signaling: it phosphorylates and desensitizes not only receptor tyrosine kinases, but also an abundance of G-protein coupled receptors such as the β-adrenergic [66], muscarinic [67], dopamine [68], and histamine [69] receptors, among many others. Thus, one mechanism for the dominant effects of LOF PKC mutations could be through the release of the ‘braking’ function of PKC towards signaling at the level of the cell surface receptor.
PKC also phosphorylates and inactivates non receptor oncogenes. PKCα suppresses signaling downstream of the phosphoinositide 3-kinase (PI3K)/Akt cell survival leg of growth factor signaling by catalyzing an inhibitory phosphorylation of the catalytic subunit of PI3K [70]. It also inactivates the proto-oncogene Akt by inducing its PP2A-mediated dephosphorylation [71]. K-Ras is also likely to be a major target for the tumor suppressive function of PKC. Its phosphorylation on Ser181 was reported to inhibit K-Ras function by relocalizing it from the plasma membrane [72]. Although the role of this phosphorylation site in tumors is unclear [73], a recent report by McCormick and colleagues supports a role of PKC in suppressing K-Ras signaling in cancer [74]. These authors showed that the interaction of K-Ras with calmodulin modulates tumor formation in mouse models through inhibition of CaM kinase, an interaction that is prevented by PKC-mediated phosphorylation of K-Ras [74]. Strikingly, oral administration to mice of a weak PKC activator (prostratin [75], see concluding remarks) promoted K-Ras phosphorylation and repressed growth in orthotopic models of human pancreatic cancer. Supporting K-Ras as a target for PKC in cancer, bioinformatics analysis revealed that K-Ras is one of the top ten genes most frequently co-mutated in cancers harbouring LOF mutations in PKC [52]. Taken together, these data point to PKC playing a key role in keeping oncogenes in check, such that LOF mutations in PKC unmask the full signaling potential of the relevant oncogene.
4. PKC Germline Mutations
Whole genome sequencing has resulted in the identification of germline mutations in conventional and novel PKC isozymes that are either causative in, or shown to associate with, disease (Figure 3). For one PKC isozyme, PKCδ, mutations that result in loss of protein expression or activity are causal in juvenile systemic lupus erythematosus (JSLE) and autoimmune lymphoproliferative syndrome [76-79]. These LOF mutations result in increased proliferation and resistance to apoptosis in immune cells. As such, JSLE patients often develop B cell lymphomas [80]. Four LOF mutations have been identified in the disease [81] (Figure 3): an invariant Gly (G248) on one of the ligand binding loops of the C1B domain is mutated to Ser in one patient with SLE-like disorder, a biallelic splice-site mutation causing the absence of protein product was identified in a patient with severe autoimmunity [78], an invariant Gly (G510) in the highly conserved activation loop of AGC kinases is mutated to Ser in three siblings with JSLE, and an Arg in a segment preceding the conserved PXXP motif of the C-terminal tail is mutated to Trp in a patient with autoimmune lymphoproliferative syndrome [77]. Somatic mutations in the latter residue (including to Trp) have also been identified in 3 different colorectal tumors (cBioPortal; [51]).
Figure 3. Germline mutations in PKC associated with disease.
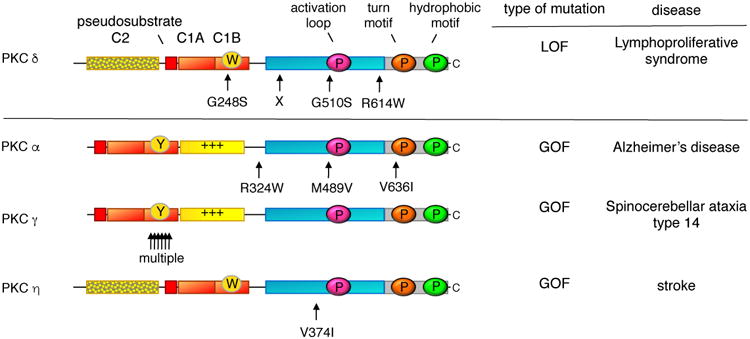
LOF mutations that are causative in a proliferative disease are a hallmark of a bonafide tumor suppressor: indicated are the positions of such germline mutations that have been identified in several families with lymphoproliferative syndrome; X indicates position of biallelic splice mutation that results in no expression of protein. In contrast to LOF mutations, germline mutations that enhance the activity of PKC are associated with degenerative diseases: indicated are rare variants in PKCα that segregated with affected family members with late onset Alzheimer's Disease, the multiple mutations in PKCγ that are causative in spinocerebellar ataxia type 14, and a variant in PKCη that is associated with increased risk to stroke.
The presence of germline mutations that cause human proliferative disorders is a hallmark of a bona fide tumor suppressor [82]. LOF mutations in these tumor suppressors are associated with proliferative phenotypes and increased risk of malignancy. Germline mutations in the classic tumor suppressor, the lipid phosphatase PTEN, are associated with numerous syndromes that predispose to cancer [83, 84]. For example, Cowden Syndrome is caused by germline mutations in PTEN and is associated with increased risk of breast, thyroid, and endometrial cancers. Similarly, LOF mutations in the kinase LKB1 (also known as STK11) lead to the development of Peutz-Jeghers syndrome [85], a disease associated with the development of colonic hamartomas. Thus, the identification of germline mutations in PKCδ that are causal in a proliferative disorder unequivocally establish at least this PKC isozyme as a bona fide tumor suppressor.
In contrast to the LOF mutations in PKCδ that are causal in a proliferative disease, GOF mutations in PKC are associated with degenerative disease (Figure 3). These mutations do not cause constitutive activation of PKC, which would have the paradoxical effect of down-regulating the enzyme, rather they facilitate or enhance the activation of the enzyme. Such enhancing mutations in PKCγ are causal in spinocerebellar ataxia type 14 (SCA14): over 20 mutations have been identified and most occur in the C1B domain. Analysis of some of the C1B mutations reveals that they loosen autoinhibitory constraints to facilitate the ligand-induced ‘open’ conformation of PKCγ. Similarly, mutations that enhance the activation of PKCα are associated with affected individuals in families with Alzheimer's disease [86]. This isozyme of PKC is required for the synaptic depression caused by amyloid-β (Aβ), a cytotoxic peptide associated with Alzheimer's disease. Four Alzheimer's disease-associated mutations have been identified, and they increase the agonist-evoked activity of PKC by a relatively small amount (approximately 10%), too little to promote the down-regulation of the enzyme, however a lifetime of slightly enhanced signaling may sensitize individuals to the detrimental effects of Aβ. A role of enhanced PKC activity in Alzheimer's disease is supported by a recent phosphoproteomics analysis of postmortem brains showing that elevation of PKC signaling is one of the earliest events in Alzheimer's disease [87]. Lastly, a polymorphism in the kinase domain of PKCη (V374I) is associated with increased risk for cerebral infarction (stroke) [88], increased risk of arthritis [89], and severe gastric atrophy [90]; this mutation enhances the kinase activity in vitro [88, 91] and its position on the upper lobe of the kinase domain suggests it may also reduce autoinhibitory constraints. The association of germline mutations that result in enhanced activity of PKC with degenerative diseases supports a general role of PKC isozymes in suppressing survival signaling.
5. Reduced PKC Levels in Human Tumors
Clinical data reveal reduced protein levels of PKC isozymes in tumor tissue compared with cognate normal tissue for a variety of cancers. For example, decreased levels of PKCα, β, δ, ε, and η have been reported in colon cancer [92-94]. In this cancer, low levels of PKCβ in normal tissue correlate with poor survival outcome for patients [95]. Low levels of PKCη in hepatocellular carcinomas have also been shown to correlate with poor survival in liver cancer [96]. Additionally, decreasing PKCδ expression tracks with increasing tumor grade in endometrial cancer [97] and in malignant glioma versus low-grade astrocytoma [98]. While many PKC isozymes have reduced levels in various cancers, one exception may be PKCι: as noted below, PKCι is part of the 3q26 amplicon, but it is unclear how this affects protein expression. Indeed, for PKCα, DNA copy number levels correlate inversely with protein levels in breast cancer [99], the cancer in which PKCα copy number is most amplified [51]. In summary, clinical data reveal that protein levels of PKC have the potential to serve as diagnostic markers for disease prognosis.
6. Could PKC function as an oncoprotein in certain contexts?
Somatic and germline mutations of PKC isozymes are consistent with PKC family members generally suppressing survival signaling: cancer-associated somatic mutations across the PKC family are either inert or LOF, and a germline LOF mutation in PKCδ causes a proliferative disorder. In contrast, germline mutations that enhance activity in several PKC isozymes are associated with degenerative diseases. Thus, reduced function of PKC is associated with cancer and enhanced activity with degeneration. Nonetheless, there may be cancer-specific and isozyme-specific contexts where PKC may function as an oncoprotein.
One context in which a PKC isozyme may serve an oncogenic function is in Adult T Cell Leukemia (ATTL). Whole genome and whole exome sequencing has revealed frequent (33% of patients) mutations in PKCβ, with a hotspot at Asp427 [100]. Overexpression studies indicate that this mutation increases the activity of PKCβ as assessed by several cellular readouts, including accelerated phorbol ester-dependent membrane translocation and enhanced NF-κB transcription. These activating effects are, however, so great that it begs the question as to whether this enhanced open conformation of PKC may promote the degradation of the mutants. Analysis of the steady-state levels of the mutant PKCβ in the patients will be important.
Although no GOF mutations in cancer have been identified to date in novel PKC isozymes, numerous reports suggest they function as oncoproteins in certain contexts. PKCδ, which has roles both in survival and apoptotic pathways [38, 101], has been reported to promote tumor progression in pancreatic cancer [102], and mice deficient in this isozyme have an increased incidence of lung tumors [103]. Similarly, genetic ablation of PKCε in a transgenic mouse model of prostate adenocacinoma inhibits prostate cancer development and metastasis [104]. Conversely, Kaznaietz and colleagues have shown that transgenic mice overexpressing PKCε in the prostate develop preneoplastic lesions [105]. PKCε expression is frequently elevated not only in prostate tumors, but also those of breast and other cancers [105].
Whether a specific PKC could function as a tumor suppressor in some cancers and an oncoprotein in others is also a possibility. For example, very few mutations in PKC are observed in breast cancer relative to other cancers [52]. Reyland and colleagues have shown that elevated PKCδ mRNA levels negatively correlate with prognosis in Erb2-positive breast cancer, with mouse models suggesting that it is required for ErbB2-driven mammary gland tumorigenesis [106]. Elevated PKCδ mRNA has also been reported to correlate with poor survival outcome in estrogen receptor-positive breast cancer [107, 108]. Nonetheless, evaluation of the mutational status of conventional and novel PKC isozymes in breast cancer suggests that PKC isozymes will be tumor suppressors in this cancer as well. Notably, there are several truncation and frameshift mutations observed in the genes of the conventional and novel PKC isozymes, including PKCδ, in human primary breast tumors. In addition, a previously characterized LOF mutation in PKCβ, A509V [52], is also observed in an invasive breast carcinoma. Higher expression of PKCβ mRNA also correlates with improved survival in hormone-insensitive, but not hormone-sensitive, breast cancers [108]. Establishing whether PKC isozymes may play oncogenic roles in specific contexts in specific cancers, such as breast, awaits functional characterization of mutations in PKC isozymes in these cancers.
What about atypical PKC isozymes? LOF mutations have been identified in PKCζ and mutations of the highly conserved APE motif have been identified in both PKCζ (E421K in a breast cancer) and PKCι (E423D in lung cancer) [52, 109]. Additionally, a frequently observed mutation in PKCι is neomorphic: mutation of R471C in PKCι changes its substrate specificity [110]. Low levels of PKCζ correlate with poor patient outcome in colon cancer, and functional studies in intestinal cells reveal that loss of PKCζ promotes metabolic reprogramming by two mechanisms – regulating the activity of a key metabolic enzyme, 3-phosphoglycerate dehydrogenase, and regulating the nuclear translocation of the transcription factors YAP and β-catenin [111, 112]. PKCι has also been proposed to have a tumor suppressive function in the intestine: this isozyme is lost in the intestinal epithelium of patients with Crohn's disease, a pathology associated with high risk of cancer, and mice lacking PKCι in their intestinal epithelium have increased inflammation and tumorigenesis [113]. However, the PRKCI gene is part of the 3q amplicon and considerable evidence supports a role for PKCι as an oncoprotein [114]. Notably, Fields and coworkers have identified an unambiguous oncogenic role for PKCι in lung cancer: in lung squamous cell carcinomas, PKCι was shown to phosphorylate SOX2, a master transcriptional regulator of stemness, thus allowing the expression of hedgehog acetyl transferase to permit growth in soft agar [115, 116], and in K-Ras-mediated lung adenocarcinomas, PKCι was shown to promote a tumor initiating phenotype by phosphorylating ELF-3 to control Notch expression [115]. Glioblastoma may also be a cancer in which atypical PKC isozymes function as oncoproteins: Ghosh and coworkers showed that high atypical PKC immunoreactivity, primarily PKCι, correlated with poor disease prognosis in patients with glioblastoma and that an atypical PKC inhibitor reduced tumor growth in a mouse model of glioblastoma [117]. This suggests that atypical PKC isozymes have oncogenic functions in certain contexts.
Hints that PKC isozymes function generally as tumor suppressors populate the literature. As early as the 1990s, Black and workers established a role for PKCα in suppressing cell growth [118, 119]. Furthermore, mice deleted in PKCα spontaneously develop colon tumors and, on an ApcMin/+ background, loss of PKCα facilitates the formation of more aggressive tumors and decreases survival [120]. PKCα deletion also increases lung tumor formation in mouse models with an activated K-Ras background [121]. Mouse models also supported a tumor suppressive role for atypical PKC isozymes: deletion of PKCζ in mice that are phosphatase and tensin homolog (PTEN) haploinsufficient resulted in larger and more invasive prostate tumors [122]. Similarly, deletion of PKCz in ApcMin/- mice hus, while PKC function is generally lost in cancer, there are clearly specific contexts in which PKC isozymes, particularly atypical ones, have oncogenic functions.
8. Concluding Remarks
The finding that PKC function is often lost in cancer sheds new light on the two stage carcinogen paradigm that established phorbol esters as potent tumor promoters. In the first step, the sub-threshold treatment with a carcinogen causes an activating mutation in an oncogene, such as K-Ras. This single hit is insufficient for carcinogenesis because PKC is suppressing the function of the oncogene. In the second step, the repeated treatments with phorbol esters would cause the loss of PKC, allowing unchecked signaling of the oncogene. It should be noted that phorbol esters have many other targets in addition to PKC [123] and also induce local inflammatory responses, functions that likely also contribute to their tumor-promoting properties. Nonetheless, it is their ability to cause the chronic loss of PKC, rather than their ability to cause its short-term activation, that likely contributes to their tumor-fpromoting properties vis à vis PKC.
The unexpected finding that PKC isozymes generally function as tumor suppressors suggests that cancer therapies should henceforth focus on restoring, rather than inhibiting, activity. Furthermore, inhibitors that failed in clinical trials for cancer may have potential for degenerative diseases in which PKC activity is enhanced. Developing novel therapies to restore the function of PKC presents its own challenges. As discussed above, forcing the activation of PKC locks the protein in an open conformation that eventually results in its degradation, epitomized by the effects of phorbol esters in inducing down-regulation. Thus, strategies to enhance PKC activity would need to be accompanied by strategies to prevent the down-regulation of the PKC. One possibility would be through the use of molecules that only slightly enhance the activity of the relevant PKC isozyme, an approach that would recapitulate the PKC mutations in Alzheimer's disease that only subtly enhance activity and thus do not significantly affect PKC stability. Prostratin, a very weakly-activating phorbol ester (12-deoxyphorbol 13-acetate; [75]), may hold promise: its oral administration has been reported to reduce tumor volume in mouse xenograft studies of pancreatic cancer [74]. Another possibility is to reduce the ability of faulty PKC to act in a dominant-negative manner towards other PKC isozymes, for example by targeting the mutant PKC for degradation or masking binding surfaces that may sequester elements required for the processing of other PKC isozymes. Modulating the activity of enzymes that control the steady-state levels of PKC in the cell might be another mechanism to promote PKC signaling. In terms of repurposing inhibitors for degenerative disease, it is noteworthy that bryostatin, which failed in cancer trials and which down-regulate PKC, is showing promise in mouse models for Alzheimer's disease and is currently in clinical trials for the disease [124].
The dominant role of PKC isozymes in suppressing survival signaling lends caution to targeting proteins that control the steady-state levels of PKC. Notably, mTOR inhibitors [125] and Hsp90 inhibitors [126], which are currently in use in the clinic, prevent processing of PKC [12, 13]. As a result, these drugs will have the unintended and detrimental consequence of suppressing PKC levels, removing its protective and tumor suppressive function. If this is the case, one might predict that rapamycin, which targets mTORC1, might be more effective in cancer treatment than general mTOR kinase inhibitors, which target both mTORC1 and mTORC2.
The paradigm of kinases as oncoproteins has been challenged by recent findings that many kinases, like PKC, are tumor suppressive. These findings suggest that, in cases where PKC function is impaired, innovative approaches to restore PKC activity should be coupled to chemotherapies targeting primary oncogenic drivers. Patients with low levels of PKC expression or with LOF mutations would benefit from such combined therapies.
Acknowledgments
I thank members of my lab for thoughtful comments. This work was supported by NIH GM 43154 and Cure Alzheimer's Fund.
Abbreviations
- Aβ
amyloid-β
- GOF
gain-of-function
- JSLE
juvenile systemic lupus erythematosus
- LOF
loss-of-function
- mTORC
mammalian target of rapamycin complex
- PB1
Phox and Bem1p
- PDBu
phorbol 12,13-dibutyrate
- PDK-1
phosphoinositide-dependent kinase-1
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PKA
protein kinase A
- PKB
protein kinase B
- PKC
protein kinase C
- PKN
protein kinase N
- PMA
phorbol 12-myristate 13-acetate
Footnotes
The author has no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takai Y, et al. Unsaturated Diacylglycerol as a Possible Messenger for the Activation of Calcium-activated, Phospholipid-dependent Protein Kinase System. Biochem Biophys Res Comm. 1979;91:1218–1224. doi: 10.1016/0006-291x(79)91197-5. [DOI] [PubMed] [Google Scholar]
- 2.Hokin MR, Hokin LE. Enzyme Secretion and the Incorporation of P32 into Phospholipids of Pancreas Slices. J Biol Chem. 1953;203:967–977. [PubMed] [Google Scholar]
- 3.Carrasco S, Merida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. 2007;32(1):27–36. doi: 10.1016/j.tibs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Manning G, et al. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 5.Levin DE, et al. A Candidate Protein Kinase C Gene, PKC1, Is Required for the S. cerevisiae Cell Cycle. Cell. 1990;62:213–224. doi: 10.1016/0092-8674(90)90360-q. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe M, Chen CY, Levin DE. Saccharomyces cerivisiae PKC1 encodes a protein kinase C (PKC) homolog with a substrate specificity similar to that of mammalian PKC. J Biol Chem. 1994;269:16829–16836. [PubMed] [Google Scholar]
- 7.Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298(3):E395–402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282(2):826–30. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 9.Kazanietz MG, et al. Zinc Finger Domains and Phorbol Ester Pharmacophore: Analysis of binding to mutated form of protein kinase C z and the vav and c-raf proto-oncogene products. J Biol Chem. 1994;269:11590–11594. [PubMed] [Google Scholar]
- 10.Pu Y, et al. Effects on ligand interaction and membrane translocation of the positively charged arginine residues situated along the C1 domain binding cleft in the atypical protein kinase C isoforms. J Biol Chem. 2006;281(44):33773–88. doi: 10.1074/jbc.M606560200. [DOI] [PubMed] [Google Scholar]
- 11.Lamark T, et al. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278(36):34568–81. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- 12.Gould CM, et al. The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J Biol Chem. 2009;284(8):4921–35. doi: 10.1074/jbc.M808436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guertin DA, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11(6):859–71. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Behn-Krappa A, Newton AC. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr Biol. 1999;9(14):728–737. doi: 10.1016/s0960-9822(99)80332-7. [DOI] [PubMed] [Google Scholar]
- 15.Tobias IS, et al. Protein kinase Czeta exhibits constitutive phosphorylation and phosphatidylinositol-3,4,5-triphosphate-independent regulation. Biochem J. 2016;473(4):509–23. doi: 10.1042/BJ20151013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Facchinetti V, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. Embo J. 2008;27(14):1932–43. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonard TA, et al. Crystal structure and allosteric activation of protein kinase C betaII. Cell. 2011;144(1):55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antal CE, et al. Intramolecular C2 Domain-Mediated Autoinhibition of Protein Kinase C betaII. Cell Rep. 2015;12(8):1252–60. doi: 10.1016/j.celrep.2015.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corbalan-Garcia S, et al. A new phosphatidylinositol 4,5-bisphosphate-binding site located in the C2 domain of protein kinase Calpha. J Biol Chem. 2003;278(7):4972–80. doi: 10.1074/jbc.M209385200. [DOI] [PubMed] [Google Scholar]
- 20.Evans JH, et al. Specific translocation of protein kinase Calpha to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Mol Biol Cell. 2006;17(1):56–66. doi: 10.1091/mbc.E05-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakanishi H, Exton JH. Purification and characterization of the zeta isoform of protein kinase C from bovine kidney. J Biol Chem. 1992;267(23):16347–54. [PubMed] [Google Scholar]
- 22.Graybill C, et al. Partitioning-defective protein 6 (Par-6) activates atypical protein kinase C (aPKC) by pseudosubstrate displacement. J Biol Chem. 2012;287(25):21003–11. doi: 10.1074/jbc.M112.360495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai LC, et al. Zeta Inhibitory Peptide Disrupts Electrostatic Interactions That Maintain Atypical Protein Kinase C in Its Active Conformation on the Scaffold p62. J Biol Chem. 2015;290(36):21845–56. doi: 10.1074/jbc.M115.676221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tobias IS, Newton AC. Protein Scaffolds Control Localized Protein Kinase Czeta Activity. J Biol Chem. 2016;291(26):13809–22. doi: 10.1074/jbc.M116.729483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soriano EV, et al. aPKC Inhibition by Par3 CR3 Flanking Regions Controls Substrate Access and Underpins Apical-Junctional Polarization. Dev Cell. 2016;38(4):384–98. doi: 10.1016/j.devcel.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallegos LL, Kunkel MT, Newton AC. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J Biol Chem. 2006;281:30947–30956. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- 27.Antal CE, Newton AC. Spatiotemporal dynamics of phosphorylation in lipid second messenger signaling. Mol Cell Proteomics. 2013;12(12):3498–508. doi: 10.1074/mcp.R113.029819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaken S, Tashjian AH, Jr, Blumberg PM. Characterization of phorbol ester receptors and their down-modulation in GH4C1 rat pituitary cells. Cancer Res. 1981;41(6):2175–81. [PubMed] [Google Scholar]
- 29.Dutil EM, et al. In vivo regulation of protein kinase C by trans-phosphorylation followed by autophosphorylation. J Biol Chem. 1994;269(47):29359–29362. [PubMed] [Google Scholar]
- 30.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18(1):13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Gao T, Brognard J, Newton AC. The phosphatase PHLPP controls the cellular levels of protein kinase C. J Biol Chem. 2008;283(10):6300–11. doi: 10.1074/jbc.M707319200. [DOI] [PubMed] [Google Scholar]
- 32.Hansra G, et al. Multisite dephosphorylation and desensitization of conventional protein kinase C isotypes [In Process Citation] Biochem J. 1999;342(Pt 2):337–44. [PMC free article] [PubMed] [Google Scholar]
- 33.Leontieva OV, Black JD. Identification of two distinct pathways of protein kinase Calpha down-regulation in intestinal epithelial cells. J Biol Chem. 2004;279(7):5788–801. doi: 10.1074/jbc.M308375200. [DOI] [PubMed] [Google Scholar]
- 34.Goode NT, Hajibagheri MAN, Parker PJ. Protein Kinase C (PKC)-induced PKC Down-regulation. J Biol Chem. 1995;270:2669–2673. doi: 10.1074/jbc.270.6.2669. [DOI] [PubMed] [Google Scholar]
- 35.Abrahamsen H, et al. Peptidyl-prolyl Isomerase Pin1 Controls Down-regulation of Conventional Protein Kinase C Isozymes. J Biol Chem. 2012;287(16):13262–78. doi: 10.1074/jbc.M112.349753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hecker E. Cocarcinogenic principles from the seed oil of Croton tiglium and from other Euphorbiaceae. Cancer Res. 1968;28(11):2338–49. [PubMed] [Google Scholar]
- 37.Berenblum I, Shubik P. The role of croton oil applications, associated with a single painting of a carcinogen, in tumour induction of the mouse's skin. Br J Cancer. 1947;1(4):379–82. doi: 10.1038/bjc.1947.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7(4):281–94. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 39.Weber J, Hecker E. Cocarcinogens of the diterpene ester type from Croton flavens L. and esophageal cancer in Curacao. Experientia. 1978;34(6):679–82. doi: 10.1007/BF01947253. [DOI] [PubMed] [Google Scholar]
- 40.Hecker E, et al. The chemical structure of a cocarcinogen and of phorbol isolated from croton oil. J Med Chem. 1966;9(2):246–7. doi: 10.1021/jm00320a024. [DOI] [PubMed] [Google Scholar]
- 41.Hecker E. Co-carcinogens or modulators of carcinogenesis. New aspects of the etiology of human tumors and of the molecular mechanisms of carcinogenesis. Naturwissenschaften. 1978;65(12):640–8. doi: 10.1007/BF00401906. [DOI] [PubMed] [Google Scholar]
- 42.Driedger PE, Blumberg PM. Specific Binding of Phorbol Ester Tumor Promoters. Proc Natl Acad Sci U S A. 1980;77:567–571. doi: 10.1073/pnas.77.1.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blumberg PM, et al. Mechanism of action of the phorbol ester tumor promoters: specific receptors for lipophilic ligands. Biochem Pharmacol. 1984;33(6):933–40. doi: 10.1016/0006-2952(84)90448-9. [DOI] [PubMed] [Google Scholar]
- 44.Castagna M, et al. Direct Activation of Calcium-activated, Phospholipid-dependent Protein Kinase by Tumor-promoting Phorbol Esters. J Biol Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 45.Zhang LL, et al. The protein kinase C (PKC) inhibitors combined with chemotherapy in the treatment of advanced non-small cell lung cancer: meta-analysis of randomized controlled trials. Clin Transl Oncol. 2015;17(5):371–7. doi: 10.1007/s12094-014-1241-3. [DOI] [PubMed] [Google Scholar]
- 46.de Vries DJ, et al. Demonstration of sub-nanomolar affinity of bryostatin 1 for the phorbol ester receptor in rat brain. Biochem Pharmacol. 1988;37(21):4069–73. doi: 10.1016/0006-2952(88)90097-4. [DOI] [PubMed] [Google Scholar]
- 47.Marshall JL, et al. Phase I study of prolonged infusion Bryostatin-1 in patients with advanced malignancies. Cancer Biol Ther. 2002;1(4):409–16. doi: 10.4161/cbt.1.4.17. [DOI] [PubMed] [Google Scholar]
- 48.Prevostel C, et al. The natural protein kinase C alpha mutant is present in human thyroid neoplasms. Oncogene. 1995;11(4):669–74. [PubMed] [Google Scholar]
- 49.Prevostel C, et al. Protein kinase C(alpha) actively downregulates through caveolae- dependent traffic to an endosomal compartment. J Cell Sci. 2000;113(Pt 14):2575–84. doi: 10.1242/jcs.113.14.2575. [DOI] [PubMed] [Google Scholar]
- 50.Vallentin A, Lo TC, Joubert D. A single point mutation in the V3 region affects protein kinase Calpha targeting and accumulation at cell-cell contacts. Mol Cell Biol. 2001;21(10):3351–63. doi: 10.1128/MCB.21.10.3351-3363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antal CE, et al. Cancer-Associated Protein Kinase C Mutations Reveal Kinase's Role as Tumor Suppressor. Cell. 2015;160(3):489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McSkimming DI, et al. KinView: a visual comparative sequence analysis tool for integrated kinome research. Mol Biosyst. 2016;12(12):3651–3665. doi: 10.1039/c6mb00466k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Creixell P, et al. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell. 2015;163(1):202–17. doi: 10.1016/j.cell.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stransky N, et al. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Knauf JA, et al. Involvement of protein kinase Cepsilon (PKCepsilon) in thyroid cell death. A truncated chimeric PKCepsilon cloned from a thyroid cancer cell line protects thyroid cells from apoptosis. J Biol Chem. 1999;274(33):23414–25. doi: 10.1074/jbc.274.33.23414. [DOI] [PubMed] [Google Scholar]
- 57.Garcia-Paramio P, et al. The broad specificity of dominant inhibitory protein kinase C mutants infers a common step in phosphorylation. Biochem J. 1998;333(Pt 3):631–6. doi: 10.1042/bj3330631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hein MY, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163(3):712–23. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 59.Meharena HS, et al. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 2013;11(10):e1001680. doi: 10.1371/journal.pbio.1001680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hunter T, Ling N, Cooper JA. Protein Kinase C Phosphorylation of the EGF Receptor at a Threonine Residue Close to the Cytoplasmic Face of the Plasma Membrane. Nature. 1984;311:480–483. doi: 10.1038/311480a0. [DOI] [PubMed] [Google Scholar]
- 61.Hunter T, Ling N, Cooper JA. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature. 1984;311(5985):480–3. doi: 10.1038/311480a0. [DOI] [PubMed] [Google Scholar]
- 62.Livneh E, et al. Release of a phorbol ester-induced mitogenic block by mutation at Thr-654 of the epidermal growth factor receptor. Mol Cell Biol. 1988;8(6):2302–8. doi: 10.1128/mcb.8.6.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Livneh E, et al. An insertional mutant of epidermal growth factor receptor allows dissection of diverse receptor functions. EMBO J. 1987;6(9):2669–76. doi: 10.1002/j.1460-2075.1987.tb02558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santiskulvong C, Rozengurt E. Protein kinase Calpha mediates feedback inhibition of EGF receptor transactivation induced by Gq-coupled receptor agonists. Cell Signal. 2007;19(6):1348–57. doi: 10.1016/j.cellsig.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 65.Ouyang X, Gulliford T, Epstein RJ. The duration of phorbol-inducible ErbB2 tyrosine dephosphorylation parallels that of receptor endocytosis rather than threonine-686 phosphorylation: implications for the physiological role of protein kinase C in growth factor receptor signalling. Carcinogenesis. 1998;19(11):2013–9. doi: 10.1093/carcin/19.11.2013. [DOI] [PubMed] [Google Scholar]
- 66.Pitcher J, et al. Desensitization of the isolated beta 2-adrenergic receptor by beta-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry. 1992;31(12):3193–7. doi: 10.1021/bi00127a021. [DOI] [PubMed] [Google Scholar]
- 67.Hosey MM, et al. Multiple mechanisms involving protein phosphorylation are linked to desensitization of muscarinic receptors. Life Sci. 1995;56(11-12):951–5. doi: 10.1016/0024-3205(95)00033-3. [DOI] [PubMed] [Google Scholar]
- 68.Namkung Y, Sibley DR. Protein kinase C mediates phosphorylation, desensitization, and trafficking of the D2 dopamine receptor. J Biol Chem. 2004;279(47):49533–41. doi: 10.1074/jbc.M408319200. [DOI] [PubMed] [Google Scholar]
- 69.Fujimoto K, et al. Identification of protein kinase C phosphorylation sites involved in phorbol ester-induced desensitization of the histamine H1 receptor. Mol Pharmacol. 1999;55(4):735–42. [PubMed] [Google Scholar]
- 70.Sipeki S, et al. PKCalpha reduces the lipid kinase activity of the p110alpha/p85alpha PI3K through the phosphorylation of the catalytic subunit. Biochem Biophys Res Commun. 2006;339(1):122–5. doi: 10.1016/j.bbrc.2005.10.194. [DOI] [PubMed] [Google Scholar]
- 71.Tanaka Y, et al. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J Biol Chem. 2003;278(36):33753–62. doi: 10.1074/jbc.M303313200. [DOI] [PubMed] [Google Scholar]
- 72.Bivona TG, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21(4):481–93. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 73.Barcelo C, et al. Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014;74(4):1190–9. doi: 10.1158/0008-5472.CAN-13-1750. [DOI] [PubMed] [Google Scholar]
- 74.Wang MT, et al. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell. 2015;163(5):1237–51. doi: 10.1016/j.cell.2015.10.041. [DOI] [PubMed] [Google Scholar]
- 75.Szallasi Z, Blumberg PM. Prostratin, a nonpromoting phorbol ester, inhibits induction by phorbol 12-myristate 13-acetate of ornithine decarboxylase, edema, and hyperplasia in CD-1 mouse skin. Cancer Res. 1991;51(19):5355–60. [PubMed] [Google Scholar]
- 76.Belot A, et al. Protein kinase cdelta deficiency causes mendelian systemic lupus erythematosus with B cell-defective apoptosis and hyperproliferation. Arthritis Rheum. 2013;65(8):2161–71. doi: 10.1002/art.38008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuehn HS, et al. Loss-of-function of the protein kinase C delta (PKCdelta) causes a B-cell lymphoproliferative syndrome in humans. Blood. 2013;121(16):3117–25. doi: 10.1182/blood-2012-12-469544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Salzer E, et al. B-cell deficiency and severe autoimmunity caused by deficiency of protein kinase C delta. Blood. 2013;121(16):3112–6. doi: 10.1182/blood-2012-10-460741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kiykim A, et al. Potentially Beneficial Effect of Hydroxychloroquine in a Patient with a Novel Mutation in Protein Kinase Cdelta Deficiency. J Clin Immunol. 2015;35(6):523–6. doi: 10.1007/s10875-015-0178-9. [DOI] [PubMed] [Google Scholar]
- 80.Bernatsky S, et al. An international cohort study of cancer in systemic lupus erythematosus. Arthritis and rheumatism. 2005;52(5):1481–90. doi: 10.1002/art.21029. [DOI] [PubMed] [Google Scholar]
- 81.Salzer E, et al. Protein Kinase C delta: a Gatekeeper of Immune Homeostasis. J Clin Immunol. 2016;36(7):631–40. doi: 10.1007/s10875-016-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Payne SR, Kemp CJ. Tumor suppressor genetics. Carcinogenesis. 2005;26(12):2031–45. doi: 10.1093/carcin/bgi223. [DOI] [PubMed] [Google Scholar]
- 83.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183–98. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 84.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11(4):289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007;26(57):7825–32. doi: 10.1038/sj.onc.1210594. [DOI] [PubMed] [Google Scholar]
- 86.Alfonso SI, et al. Gain-of-function mutations in protein kinase Calpha (PKCalpha) may promote synaptic defects in Alzheimer's disease. Sci Signal. 2016;9(427):ra47. doi: 10.1126/scisignal.aaf6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tagawa K, et al. Comprehensive phosphoproteome analysis unravels the core signaling network that initiates the earliest synapse pathology in preclinical Alzheimer's disease brain. Hum Mol Genet. 2015;24(2):540–58. doi: 10.1093/hmg/ddu475. [DOI] [PubMed] [Google Scholar]
- 88.Kubo M, et al. A nonsynonymous SNP in PRKCH (protein kinase C eta) increases the risk of cerebral infarction. Nat Genet. 2007;39(2):212–7. doi: 10.1038/ng1945. [DOI] [PubMed] [Google Scholar]
- 89.Takata Y, et al. Genetic association between the PRKCH gene encoding protein kinase Ceta isozyme and rheumatoid arthritis in the Japanese population. Arthritis Rheum. 2007;56(1):30–42. doi: 10.1002/art.22262. [DOI] [PubMed] [Google Scholar]
- 90.Goto Y, et al. PRKCH gene polymorphism is associated with the risk of severe gastric atrophy. Gastric Cancer. 2010;13(2):90–4. doi: 10.1007/s10120-009-0542-7. [DOI] [PubMed] [Google Scholar]
- 91.Zurgil U, et al. PKCeta promotes senescence induced by oxidative stress and chemotherapy. Cell Death Dis. 2014;5:e1531. doi: 10.1038/cddis.2014.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pongracz J, et al. Expression of protein kinase C isoenzymes in colorectal cancer tissue and their differential activation by different bile acids. Int J Cancer. 1995;61(1):35–9. doi: 10.1002/ijc.2910610107. [DOI] [PubMed] [Google Scholar]
- 93.Craven PA, DeRubertis FR. Loss of protein kinase C delta isozyme immunoreactivity in human adenocarcinomas. Dig Dis Sci. 1994;39(3):481–9. doi: 10.1007/BF02088331. [DOI] [PubMed] [Google Scholar]
- 94.Suga K, et al. Down-regulation of protein kinase C-alpha detected in human colorectal cancer. Biochem Mol Biol Int. 1998;44(3):523–8. doi: 10.1080/15216549800201552. [DOI] [PubMed] [Google Scholar]
- 95.Dowling CM, et al. Protein kinase C beta II suppresses colorectal cancer by regulating IGF-1 mediated cell survival. Oncotarget. 2016 doi: 10.18632/oncotarget.8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu HC, et al. Analysing the expression of protein kinase C eta in human hepatocellular carcinoma. Pathology. 2009;41(7):626–9. doi: 10.3109/00313020903273076. [DOI] [PubMed] [Google Scholar]
- 97.Reno EM, et al. Analysis of protein kinase C delta (PKC delta) expression in endometrial tumors. Hum Pathol. 2008;39(1):21–9. doi: 10.1016/j.humpath.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mandil R, et al. Protein kinase Calpha and protein kinase Cdelta play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 2001;61(11):4612–9. [PubMed] [Google Scholar]
- 99.Myhre S, et al. Influence of DNA copy number and mRNA levels on the expression of breast cancer related proteins. Mol Oncol. 2013;7(3):704–18. doi: 10.1016/j.molonc.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kataoka K, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47(11):1304–15. doi: 10.1038/ng.3415. [DOI] [PubMed] [Google Scholar]
- 101.Basu A, Pal D. Two faces of protein kinase Cdelta: the contrasting roles of PKCdelta in cell survival and cell death. ScientificWorldJournal. 2010;10:2272–84. doi: 10.1100/tsw.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mauro LV, et al. PKC Delta (PKCdelta) promotes tumoral progression of human ductal pancreatic cancer. Pancreas. 2010;39(1):e31–41. doi: 10.1097/MPA.0b013e3181bce796. [DOI] [PubMed] [Google Scholar]
- 103.Symonds JM, et al. Protein kinase C delta is a downstream effector of oncogenic K-ras in lung tumors. Cancer Res. 2011;71(6):2087–97. doi: 10.1158/0008-5472.CAN-10-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hafeez BB, et al. Genetic ablation of PKC epsilon inhibits prostate cancer development and metastasis in transgenic mouse model of prostate adenocarcinoma. Cancer Res. 2011;71(6):2318–27. doi: 10.1158/0008-5472.CAN-10-4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Garg R, et al. Protein kinase C and cancer: what we know and what we do not. Oncogene. 2014;33(45):5225–37. doi: 10.1038/onc.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Allen-Petersen BL, et al. Protein kinase Cdelta is required for ErbB2-driven mammary gland tumorigenesis and negatively correlates with prognosis in human breast cancer. Oncogene. 2014;33(10):1306–15. doi: 10.1038/onc.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McKiernan E, et al. Protein kinase Cdelta expression in breast cancer as measured by real-time PCR, western blotting and ELISA. Br J Cancer. 2008;99(10):1644–50. doi: 10.1038/sj.bjc.6604728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gyorffy B, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–31. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 109.Galvez AS, et al. Protein kinase Czeta represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol Cell Biol. 2009;29(1):104–15. doi: 10.1128/MCB.01294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Linch M, et al. A cancer-associated mutation in atypical protein kinase Ciota occurs in a substrate-specific recruitment motif. Sci Signal. 2013;6(293):ra82. doi: 10.1126/scisignal.2004068. [DOI] [PubMed] [Google Scholar]
- 111.Ma L, et al. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell. 2013;152(3):599–611. doi: 10.1016/j.cell.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Llado V, et al. Repression of Intestinal Stem Cell Function and Tumorigenesis through Direct Phosphorylation of beta-Catenin and Yap by PKCzeta. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nakanishi Y, et al. Control of Paneth Cell Fate, Intestinal Inflammation, and Tumorigenesis by PKClambda/iota. Cell Rep. 2016;16(12):3297–310. doi: 10.1016/j.celrep.2016.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parker PJ, et al. Atypical protein kinase Ciota as a human oncogene and therapeutic target. Biochem Pharmacol. 2014;88(1):1–11. doi: 10.1016/j.bcp.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ali SA, et al. Protein Kinase Ciota Drives a NOTCH3-dependent Stem-like Phenotype in Mutant KRAS Lung Adenocarcinoma. Cancer Cell. 2016;29(3):367–78. doi: 10.1016/j.ccell.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Justilien V, et al. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014;25(2):139–51. doi: 10.1016/j.ccr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kusne Y, et al. Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFalpha in glioblastoma. Sci Signal. 2014;7(338):ra75. doi: 10.1126/scisignal.2005196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Saxon ML, Zhao X, Black JD. Activation of protein kinase C isozymes is associated with post-mitotic events in intestinal epithelial cells in situ. J Cell Biol. 1994;126(3):747–63. doi: 10.1083/jcb.126.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Frey MR, et al. Protein kinase C isozyme-mediated cell cycle arrest involves induction of p21(waf1/cip1) and p27(kip1) and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J Biol Chem. 1997;272(14):9424–35. doi: 10.1074/jbc.272.14.9424. [DOI] [PubMed] [Google Scholar]
- 120.Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 2006;66(14):6955–63. doi: 10.1158/0008-5472.CAN-06-0268. [DOI] [PubMed] [Google Scholar]
- 121.Hill KS, et al. Protein kinase Calpha suppresses Kras-mediated lung tumor formation through activation of a p38 MAPK-TGFbeta signaling axis. Oncogene. 2014;33(16):2134–44. doi: 10.1038/onc.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim JY, et al. c-Myc phosphorylation by PKCzeta represses prostate tumorigenesis. Proc Natl Acad Sci U S A. 2013;110(16):6418–23. doi: 10.1073/pnas.1221799110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kazanietz MG. Eyes wide shut: protein kinase C isozymes are not the only receptors for the phorbol ester tumor promoters. Mol Carcinog. 2000;28(1):5–11. doi: 10.1002/(sici)1098-2744(200005)28:1<5::aid-mc2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 124.Schrott LM, et al. Acute oral Bryostatin-1 administration improves learning deficits in the APP/PS1 transgenic mouse model of Alzheimer's disease. Curr Alzheimer Res. 2015;12(1):22–31. doi: 10.2174/1567205012666141218141904. [DOI] [PubMed] [Google Scholar]
- 125.Don AS, Zheng XF. Recent clinical trials of mTOR-targeted cancer therapies. Rev Recent Clin Trials. 2011;6(1):24–35. doi: 10.2174/157488711793980147. [DOI] [PubMed] [Google Scholar]
- 126.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]