

Abstract
Repeated presentations of a previously conditioned stimulus lead to a new form of learning known as extinction, which temporarily alters the response to the original stimulus. Previous studies have shown that the consolidation of extinction memory requires de novo protein synthesis. However, the role of specific nodes of translational control in extinction is unknown. Using auditory threat conditioning in mice, we investigated the role of mechanistic target of rapamycin complex 1 (mTORC1) and its effector p70 S6 kinase 1 (S6K1) in the extinction of auditory threat conditioning. We found that rapamycin attenuated the consolidation of extinction memory. In contrast, genetic deletion and pharmacological inhibition of S6K1, a downstream effector of mTORC1, blocked within session extinction, indicating a role for S6K1 independent of protein synthesis. Indeed, the activation of S6K1 during extinction required ERK activation in the basolateral nucleus of the amygdala (BLA) and was necessary for increased phosphorylation of the GluA1 (Thr840) subunit of the AMPA receptor following extinction training. Mice exposed to brief uncontrollable stress showed impaired within session extinction as well as a downregulation of ERK and S6K1 signaling in the amygdala. Finally, using fiber photometry we were able to record calcium signals in vivo, and we found that inhibition of S6K1 reduces extinction-induced changes in neuronal activity of the BLA. These results implicate a novel ERK-S6K1-GluA1 signaling cascade critically involved in extinction.
Introduction
Extinction occurs when a conditioned stimulus (CS) is presented repeatedly in the absence of the unconditioned stimulus (US), leading to the decreased ability of the CS to predict the US. The effects of extinction are context specific and transient, as these memories undergo spontaneous recovery and reinstatement. Extinction is a highly relevant adaptation, as it allows for the updating of old memories based on new information. However, deficits in extinction are present in neuropsychiatric disorders such as post-traumatic stress disorder (PTSD) and are associated with pathological anxiety. Thus, understanding the molecular mechanisms that underlie extinction learning and memory is critical for understanding how this process is disrupted in human disease.
Several studies indicate that the brain circuitry of extinction involves complex reciprocal interactions between various subregions of the prefrontal cortex, hippocampus, and amygdala.1 The current understanding of the molecular mechanisms underlying extinction suggests that there is not only brain region specificity, but temporal specificity as well.2 The basolateral amygdala (BLA), which is comprised of the lateral (LA), basal (BA) and basal medial (BMA) subnuclei of the amygdala, has been implicated in both within session extinction and extinction memory consolidation.2,3 For example, within session extinction is regulated in part by the activation of extracellular signal-regulated kinase (ERK) in the BLA.4 Pharmacological inhibition of L-type voltage-gated calcium channels5 as well as inhibition of N-methyl-D aspartate (NMDA) receptors5 have been shown to impair within session extinction. These studies indicate that the BLA is a critical node for activity-dependent plasticity during extinction training.
The consolidation of extinction also requires molecular mechanisms similar to within session extinction, albeit in different brain regions. For example, inhibition of either ERK signaling or NMDA receptors in the infralimbic subregion (IL) of the prefrontal cortex immediately after extinction training impairs extinction memory.6,7 In addition, de novo protein synthesis in the medial prefrontal cortex (mPFC) is necessary for extinction memory.8 Infusion of anisomycin, a general protein synthesis inhibitor, into the IL before extinction blocks retrieval, but has no effect on within session extinction. Thus, the spatiotemporal requirement of the mechanisms which support extinction memory consolidation are different than those that support within session extinction.
The requirement of new protein synthesis in extinction memory formation is consistent with a general role of protein synthesis in the consolidation of several other types of memory. mTORC1 is a critical regulator of cap-dependent protein synthesis; upon activation, mTORC1 phosphorylates two downstream targets, eIF4E-binding proteins (4E-BPs) and p70 S6 kinase 1 (S6K1).9 Interestingly, the modulation of these downstream targets were shown to differentially regulate the consolidation and reconsolidation of cued threat memory.10,11 Moreover, S6K1 also can be activated by ERK, which can have effects that are unrelated to the regulation of protein synthesis.12 Because a role for new protein synthesis and ERK has been described for extinction memory consolidation and within session extinction, respectively,4,8 the investigation of mTORC1, ERK, and their downstream effectors in extinction learning should provide insight into whether the different phases of extinction are differentially regulated.
As previously mentioned, extinction deficits are a symptom of neuropsychiatric disorders such as PTSD, which can result following a traumatic, stressful life event. Indeed, previous studies indicate that within session extinction is impaired following mild uncontrollable stress in mice.13 This extinction deficit was accompanied by alterations in dendritic morphology within the IL.13 Several other studies indicate an impairment in extinction following chronic stress;14 however, the molecular mechanisms underlying these deficits, particularly within the amygdala, are not well understood.
Herein, we show that mTORC1 is required for cued extinction memory consolidation and that within session extinction requires the activation of a novel ERK-S6K1-GluA1-containing AMPA receptor pathway in the BLA. In addition, inhibition of S6K1 during extinction reduces overall neuronal activity in the BLA. Finally, impairments in the novel S6K1 signaling pathway are associated with extinction deficits that are exhibited following brief uncontrollable stress in mice. These results identify a novel S6K1 signaling cascade that is critically involved with the extinction of auditory threat memory.
Materials and Methods
All procedures involving mice were approved by the New York University Animal Care and Use Committee and followed the National Institutes of Health Guidelines for the use of animals in research.
Housing
Mice were housed on a 12:12 hour regular light/dark cycle (lights on at 7:00 AM) and were provided food and water ad libitum.
Mice
Experiments were performed in adult male (3 months) C57Bl/6 mice (Jackson Laboratories), adult (3–5 months old) S6K1 knockout mice and their wild-type littermates as noted. S6K1 knockout mice were generated as described previously.15 S6K1 knockout mice originally were generated from a 129/SveJ C57BL/6 mixed background and backcrossed at least twice with C57BL/6 mice. All experiments were performed with the examiners blind to drug treatment and genotype.
Cannulation
Cannulas were implanted bilaterally in the basolateral nucleus of the amygdala. Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg), and mounted on a stereotaxic apparatus. Cannulae (26-gauge; Plastics One, Inc) were implanted at the following coordinates: −1.75 mm anterior-posterior, 3.3 medial-lateral and −4.6 mm dorsal-ventral (Supplemental Figure 1). Mice were given one week after surgery to recover.
Drug preparation
Rapamycin (LC Laboratories) was dissolved in a vehicle solution of 1% dimethylsulfoxide (DMSO), 5% Tween-80, and saline and injected at a dose of 40 mg/kg. PF-4708671 (Tocris) was dissolved in a vehicle solution of 17% DMSO, 10% Tween-80, and saline and injected at a dose of 50 mg/kg. For intra-amygdala infusions of PF-4708671, the drug was dissolved in 100% DMSO. SL327 (Tocris) was dissolved in a vehicle solution of 5% DMSO, 5% Tween-80 and saline and injected at a dose of 30 mg/kg. Rapamycin and PF-4708671 were injected intraperitoneally (i.p.) to a volume of 5 ml/kg relative to body weight one hour before the behavioral manipulation began; SL327 was injected 45 minutes before behavior. For intra-amygdala infusions, PF-4708671 (0.2 μl) was infused bilaterally into the basolateral nucleus of the amygdala at a rate of 0.08 μl/min, one hour before behavioral testing began. The injectors remained in the guide cannula for two minutes after the infusion. Control mice received equivalent volumes of vehicle solution. Please see Supplemental Table 1 for a complete list of drugs, vehicle preparations, and injection time points relative to behavior.
Biochemistry
Immediately after extinction training, the amygdala was dissected, homogenized, and analyzed using standard Western blotting procedures as previously described.10,16 For details, see Supplemental Experimental Procedures.
Fear conditioning
Mice were conditioned and extinguished as previously described.10,16 Briefly, mice were habituated, trained, and tested in yoked, sound-insulated chambers (Colbourn Instruments, GraphicState 2.0). On the training day, mice received two conditioned stimuli (CS; 30 sec, 80dB tone) presentations, which co-terminated with an unconditioned stimulus (US; 0.5 mA footshock). Extinction consisted of 15 CS presentations separated by a 40–100 second random intertrial interval, and presented in a novel context. For the experiments using the S6K1 knockout mice, the mice received the same timeline of habituation, training, and extinction, but did not receive any injections. The order of testing between groups was counterbalanced for all behavioral experiments. For details, see the Supplemental Experimental Procedures.
Acute stress
Mice were conditioned with two CS-US pairs. The following day, the mice underwent three days of forced swim stress as previously described.13 Stressed mice underwent 15 minutes of forced swim each day for three consecutive days in a glass cylindrical container (14.5 cm diameter) filled with water (27°C) to a height of 15 cm. Control mice were placed in a dry, glass cylinder (14.5 cm diameter) for an equivalent amount of time. 24 hours after the last day of stress, all mice underwent one day of extinction. The mice were sacrificed immediately afterwards, and the amygdala was dissected for western blotting.
Fiber photometry
Fiber photometry was performed as described previously.17 Animals were anesthetized with 2% isoflurane and placed onto a stereotax. A craniotomy was performed to allow for viral injection at the following coordinates: +1.75 mm anterior-posterior, +3.3 medial-lateral and –4.6 mm dorsal-ventral with respect to Bregma. A 400μm diameter optical fiber was implanted at the following coordinates: +1.75 mm anterior-posterior, +3.3 medial-lateral and –4.4 mm dorsal-ventral (Doric; Quebec, Canada). Viral vectors containing AAV1.Syn.GCaMP6s (UNC vector core) were injected using a 10 μL Nanofil syringe (WPI, Sarasota, FL) with a 33 gauge beveled needle attached to a microsyringe pump and controller (UMP3, Micro4; Sarasota, FL). Virus (300nL) was infused at a rate of 50 nL per minute, and the needle remained at the injection site for 5 minutes post injection and withdrawn slowly. Optical fibers were secured with Metabond.
Two weeks following surgery, all mice were fear conditioned with two CS-US pairs, and GCaMP6s signal was recorded during extinction 24 hours later. To compare calcium signals across animals, fluorescence recordings were normalized to ΔF/F by taking the median signal across the recording period, subtracting this from each data point and then dividing by the median signal. For details, see Supplemental Materials and Methods.
Statistical analysis
Group data are presented as mean ± SEM. Students t-tests were used to analyze the two groups represented in the respective western blotting data. Extinction behavior and fiber photometry data were analyzed using a two-way repeated measure ANOVA and Bonferroni post hoc tests. All groups observed normal distribution.
Results
We first sought to determine the role of cap-dependent protein synthesis in threat extinction by using rapamycin, an inhibitor of mTORC1, a well known regulator of the cap-binding translation factor eIF4E. We trained C57BL/6 mice using two pairs of CS (80dB tone) followed by a US (0.5mA footshock).13 All mice behaved similarly in the acquisition phase of threat learning (data not shown). 24 hours later we examined the role of mTORC1 in threat extinction by injecting mice intraperitoneally (i.p.) with either vehicle or rapamycin (40 mg/kg) one hour before the extinction trial. Following the first day of extinction training, all mice underwent an additional extinction trial, drug-free. Consistent with previous studies demonstrating a role for general protein synthesis in threat extinction, we found that mTORC1 was required for extinction memory, but not within session extinction (Supplemental Figure 2).14 On day one of extinction, vehicle- and rapamycin-treated mice showed a similar decrease in their freezing response to the tone (F4,36 = 5.307, p = 0.0018). Both vehicle- and rapamycin-treated mice displayed similar locomotor behavior prior to the initial tone presentation (data not shown). However, on day two of extinction, a mixed-factor repeated measures ANOVA revealed a main effect of time (F4, 36 = 79.44, p < 0.0001) and drug treatment (F1, 36 = 5.366, p = 0.0457; Supplemental Figure 2). These results indicate that with time, all mice showed a decrement in freezing in response to the tone, and that vehicle-treated mice showed lower levels of freezing compared to rapamycin-treated mice. Overall, this study indicates that disruption of mTORC1 with rapamycin impairs the consolidation of extinction memory.
Because the downstream effectors of mTORC1 have been shown to differentially regulate various forms of long-lasting plasticity and long-term memory, we investigated the role S6K1, an mTORC1 downstream effector, in threat extinction using a global, constitutive S6K1 knockout mouse (S6K1 KO).10 Previously, these mice have been shown to be unimpaired in the acquisition and consolidation of auditory threat.18 Indeed, all mice behaved similarly in the acquisition phase of threat learning (data not shown). Surprisingly, the S6K1 KO mice differed in their behavior in response to repeated tone presentations compared to wild-type mice (F4, 40 = 2.644, p = 0.0475; Figure 1A). Bonferroni post hoc analyses revealed a significant difference in freezing levels between wild-type and S6K1 KO mice during the last bin of CS presentations on day one, whereby wild-type mice displayed significantly lower levels of freezing compared to S6K1 KO mice. On day two of extinction, the S6K1 KO mice persisted in their inability to extinguish their response to the tone (F1, 10 = 13.43, p = 0.0044). We then used a pharmacological inhibitor of S6K1, PF-4708671, in wild-type mice to ensure that the phenotype we observed in the S6K1 KO mice was not due to a neurodevelopmental disruption of S6K1 function. Inhibition of S6K1 via PF-4708671 blocked within session extinction compared to vehicle-treated mice on day one (F4, 40 = 3.131, p = 0.0248; Figure 1B); moreover, on day two of extinction, vehicle-treated mice displayed lower levels of freezing behavior compared to the mice that were treated with PF-4708671 (F1,10 = 5.237, p = 0.0451). Overall, these data indicate that inhibition of S6K1, both genetic and pharmacological, impairs within session extinction.
Figure 1. S6K1 is required for the acquisition of extinction memory.
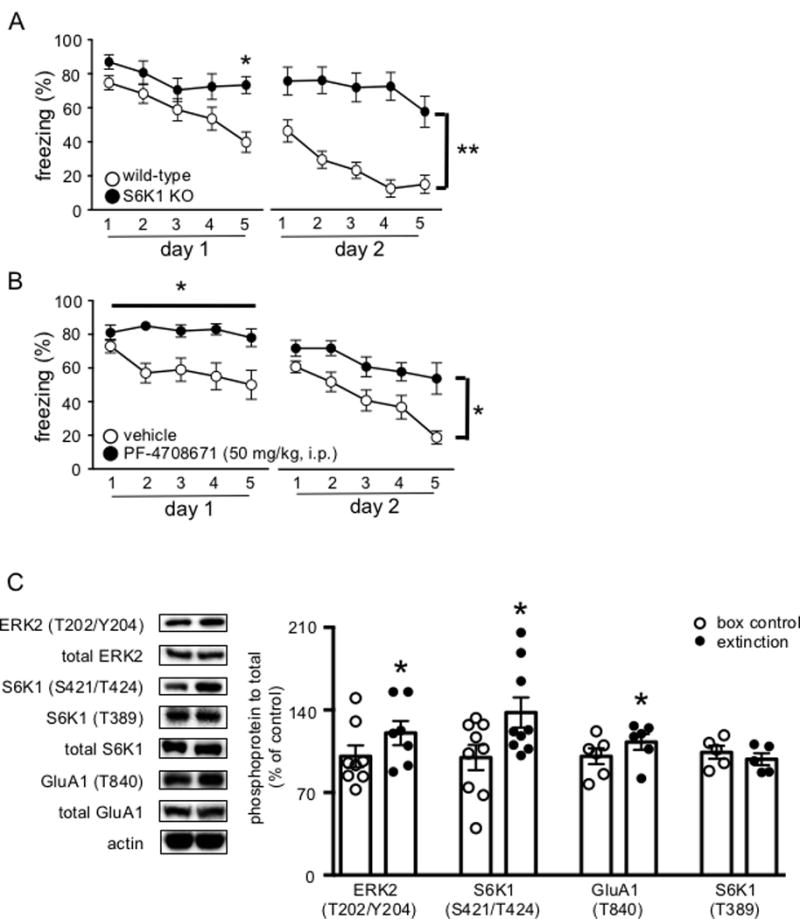
(A) S6K1 knockout mice display impairments in the acquisition of extinction compared to wild-type mice (n = 6 WT, n = 6 KO) (*p < 0.05, **p < 0.01). (B) PF-4708671 (50 mg/kg, i.p.) blocks the acquisition of extinction (*p < 0.05). (C) Left, representative blots for phosphorylated levels of ERK2, S6K1 (Ser421/Thr424), GluA1 (Thr840), S6K1 (Thr389), total protein levels and actin. (n= 6–9 box control, n = 7–9 extinction) Right, cumulative data for blots shown in (C). Data are represented as the mean ± SEM (*p< 0.05).
The requirement for S6K1 in within session extinction was a particularly interesting finding because it affects a different phase of extinction compared to mTORC1, despite the fact that S6K1 is a downstream effector of mTORC1. We hypothesized that S6K1 functions independent of mTORC1 in the context of extinction learning. In fact, previous work has shown that inhibition of extracellular regulated kinase (ERK), which is another upstream activator of S6K1, also impairs within session extinction.8 Therefore, we took advantage of the presence of the two phosphorylation sites, Ser421/Thr424 and Thr389, on S6K1 that are regulated by ERK and mTORC1, respectively, to determine whether there was a differential activation of either site during extinction.9,10 We trained wild-type mice using two CS-US pairs and 24 hours later one group of mice underwent threat extinction (15 CS presentations), whereas the extinction naïve control group was placed into the extinction context, but did not receive any CS presentations. All mice behaved similarly in the acquisition phase of threat learning (data not shown). Consistent with previous findings, mice that underwent extinction exhibited an increase in ERK2 phosphorylation (t5 = 3.187, p = 0.0333; Figure 1C).4 Using immunocytochemistry, we confirmed that ERK2 activation was localized to the basolateral amygdala during extinction (Supplemental Figure 3). We then examined S6K1 phosphorylation on the putative ERK- and mTORC1-dependent phosphorylation sites. Compared to the extinction naïve mice, mice that underwent extinction exhibited an increase in phosphorylation on the ERK-sensitive S6K1 phosphorylation site (Ser421/Thr424) (t7 = 3.364, p = 0.0152; Figure 1C). In contrast, there was no difference of S6K1 phosphorylation on the mTORC1-sensitive site (Thr389) in mice that underwent extinction (t- test, n.s.; Figure 1C). These results indicate, surprisingly, that the activation of S6K1 during extinction is correlated with an increase in ERK activation, and not mTORC1 activation.
We proceeded to investigate downstream targets of S6K1 that might be phosphorylated during extinction. Because extinction has been found to alter synaptic transmission in the basolateral amygdala, we examined the phosphorylation state of the GluA1 subunit at the Thr840 residue that has been reported to be a substrate of S6K1.19 We found that compared to extinction naïve mice, there was an increase in GluA1 (Thr840) phosphorylation in mice that underwent extinction (t5 = 3.082, p = 0.0369, Figure 1C). Thus, our results indicate that there is an increase in ERK2, S6K1 (Ser421/Thr424), and GluA1 (Thr840) phosphorylation during extinction, whereas S6K1 (Thr389), which is the mTORC1-dependent S6K1 phosphorylation site, is unchanged.
Consistent with previous results, our data indicate that during extinction, ERK is activated in the amygdala4 and may be responsible for phosphorylating S6K1. Thus, we tested whether pharmacological inhibition of ERK was capable of blocking within session extinction, as well as the extinction-induced downstream changes in phosphorylation of S6K1 and GluA1 that we observed. All mice behaved similarly in the acquisition phase of threat learning (data not shown). Consistent with previous findings, administration of SL327 (30 mg/kg, i.p.), an inhibitor of MEK-ERK blocked within session extinction (F4,40 = 13.49, p < 0.0001; Supplemental Figure 4A)4. Compared to vehicle-treated mice, mice that were treated with SL327 were unable to extinguish the freezing response to repeated presentations of the tone. Biochemical analysis of the amygdala of mice after extinction training revealed a decrease in ERK (t11 = 2.397, p = 0.0375), S6K1 (Ser421/Thr424) (t9 = 2.336, p = 0.0477) and GluA1 (Thr840) phosphorylation (t8 = 3.293, p = 0.0132; Supplemental Figure 4B). These results indicate that pharmacological inhibition of ERK signaling is sufficient to block within session extinction, as well as the downstream ERK-dependent increases in phosphorylation of S6K1 (Ser421/Thr424) and GluA1 (Thr840).
To confirm that S6K1 activation is required for within session extinction in the BLA, we infused the S6K1 inhibitor PF-4708671 (100 μM) or an equivalent volume of vehicle directly into the BLA of wild-type mice via cannulation. All mice behaved similarly in the acquisition phase of threat learning (data not shown). 24 hours after conditioning, mice received bilateral infusions of PF-4708671 or vehicle and underwent extinction one hour later. We found that pharmacological inhibition of S6K1 in the BLA was sufficient to impair the acquisition of extinction compared to vehicle-infused mice (F4,40 = 2.821, p = 0.0375, Figure 2A). Wild-type mice that were bilaterally infused with vehicle exhibited a decrement in their freezing response to the tone, whereas mice that received bilateral infusions of PF-4708671 mice did not. These results indicate that S6K1 activity in the BLA is required for the acquisition of extinction. We then performed immunocytochemistry in coronal sections of cannulated mice to confirm both the placement of the cannula and that PF-4708671 was indeed inhibiting S6K1 activity by examining S6 ribosomal protein (S6rp) phosphorylation. Compared to vehicle-treated mice, qualitative analysis of S6rp phosphorylation indicated that mice that were infused with PF-4708671 showed decreased levels of S6rp phosphorylation and were comparable to levels observed in the box control mice (Figure 2B). Overall, our results suggest that ERK is activated in the BLA during the acquisition of extinction, which leads to the phosphorylation of S6K1 (Ser421/Thr424), which then results in the phosphorylation of the GluA1 subunit of the AMPA receptor.
Figure 2. Inhibition of S6K1 in the basolateral nucleus of the amygdala blocks the acquisition of extinction.
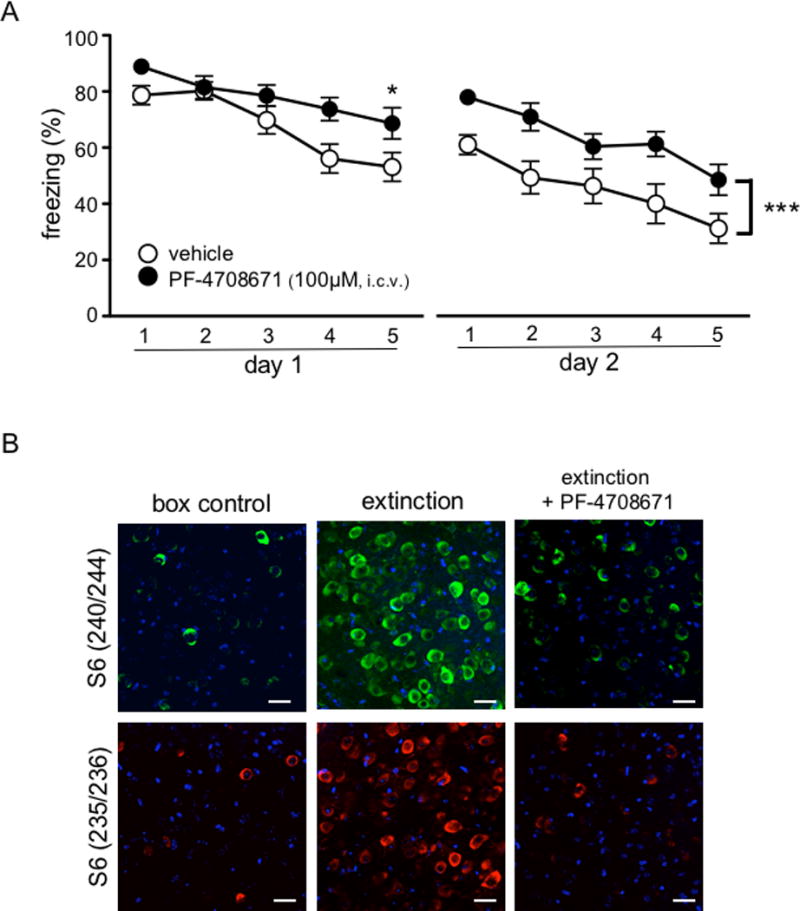
(A) Mice received infusions of either vehicle or PF-4708671 (100μM) (n = 6 per treatment) 45 minutes before the first extinction session. Mice that received PF-4708671 infusions were impaired in within session extinction (*p< 0.05). (B) PF-4708671 (100 μM) blocks S6 phosphorylation (Ser240/244 in green and Ser235/236 in red; DAPI in blue). Data are represented as mean ± SEM in all graphs. Scale bars represent 20μm.
We then turned to a behavioral model in which within session extinction is impaired to determine whether the ERK-S6K1 signaling described above also was impaired. Chronic stress has been shown to enhance the consolidation of threat memory, and impair within session extinction.13,14 Therefore, we used forced swim stress to induce a mild uncontrollable stress in C57/BL6 mice. In order to eliminate the confound of stress induced enhancement on threat memory consolidation, we conditioned the mice before the stress manipulation began 20 24 hours after threat conditioning, we began the first of three consecutive days of a 15-minute, forced swim session. 24 hours after the last day of stress, the mice underwent extinction training. All mice behaved similarly in the acquisition phase of threat learning (data not shown). Consistent with previous findings, chronically stressed mice were impaired in within session extinction compared to control mice (F4, 88 = 2.751, p = 0.0330; Figure 3A).13 Compared to control mice, mice that underwent forced swim stress were unable to extinguish the freezing response to repeated presentations of the tone. The behavioral changes in the stressed mice were accompanied by decreases in p-ERK2 (t10 = 2.433, p = 0.0378; Figure 3B), p-S6K1 (S421/T424) (t10 = 2.263, p = 0.0499, Figure 3C) and p-GluA1 (T840) (t10 = 2.296, p = 0.0446; Figure 3B) compared to control mice. Importantly, forced swim stress did not alter basal phosphorylation levels of ERK2, p-S6K1 (S421/T424) or p-GluA1 (T840) (Supplemental Figure 5). These data suggest that deficits in within session extinction following chronic stress are correlated with a reduction in the ERK - S6K1 - GluA1-containing AMPA receptor signaling pathway.
Figure 3. Mild unpredictable stress impairs within session extinction and is correlated with decreased ERK2, S6K1 (Ser421/Thr424) and GluA1 (Thr840) phosphorylation.
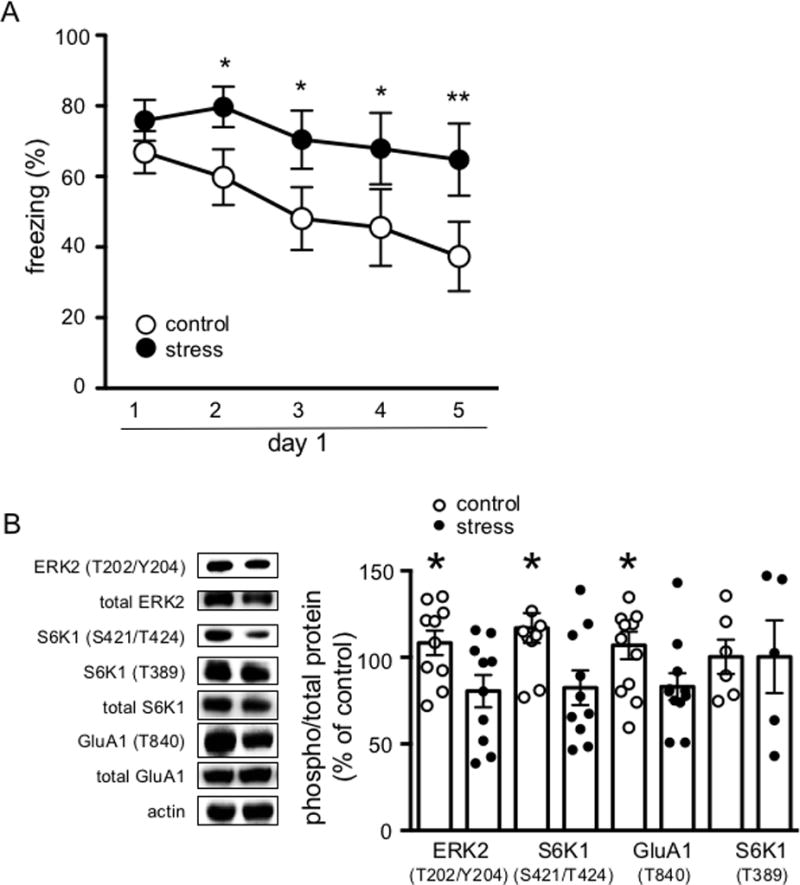
(A) Acute uncontrollable stress impairs within session extinction. (*p<0.05; **p<0.001) and (B) Left, representative blots showing decreased ERK2, S6K1 (Ser421/Thr424) and GluA1 (Thr840) phosphorylation. Right, cumulative data for blots shown on left. Data are represented as the mean ± SEM (n = 12 per group; *p< 0.05).
Finally, to test whether S6K1 is required for activity-dependent changes in calcium signals within the BLA during extinction, we used in vivo fiber photometry to record neuronal activity during extinction training.17 We achieved targeted expression of the genetically encoded calcium indicator, GCaMP6s,21 in the BLA of twenty untrained C57Bl/6 mice by unilaterally injecting an adeno-associated viral vector carrying the GCaMP6S gene on a pan-neuronal promoter (AAV1.hSyn.GCaMP6s) and then placed a chronically implanted fiber optic above the injection site. GCaMP6s viral expression was largely restricted to the BLA (Figure 4A). Two weeks after viral injection, the mice were trained in the standard threat conditioning paradigm described above. All mice behaved similarly in the acquisition phase of threat learning (data not shown). 24 hours after training, mice received a systemic injection of either vehicle or PF-4708671, and then extinction training one hour later. All mice received an additional day of extinction drug-free 24 hours later. We recorded GCaMP6s signals in the BLA during extinction in vehicle- and drug-treated mice on days 1 and 2 of extinction. Consistent with our previous results, inhibition of S6K1 during extinction led to a deficit in within session extinction (F4, 72 = 2.708; p = 0.0368; Figure 4B). Mice that received the S6K1 inhibitor did not display a decrement in freezing behavior compared to vehicle-treated mice. Moreover, in vehicle-treated mice, extinction learning was associated with an increase of neuronal activity in the BLA on day 2 relative to day 1, but this did not occur in mice receiving the S6K1 inhibitor. A two-way repeated measures ANOVA revealed a significant main effect of treatment on the mean amplitude of calcium transients occurring during blocks of 3 CS presentations (F1, 1329 = 11.71, p = 0.0006) (Figure 4D). On day 2 of extinction, vehicle- and PF-4708671-treated mice both displayed within session extinction (F4, 72 = 1.207, p < 0.0001) and vehicle-treated mice showed overall lower levels of freezing compared to PF-4708671-treated mice (F1, 18 = 4.801, p = 0.0418; Figure 4B). These behavioral results were accompanied by a significant interaction between drug treatment and time for the mean amplitude of transients recorded from the GCaMP6s signal (F4, 1599 = 3.828, p = 0.0042) (Figure 4D). Overall, our results indicate that extinction leads to an increase in neuronal signals in the BLA, which is attenuated by inhibiting S6K1.
Figure 4. Inhibition of S6K1 attenuates neuronal activity in the basolateral nucleus of the amygdala during extinction.
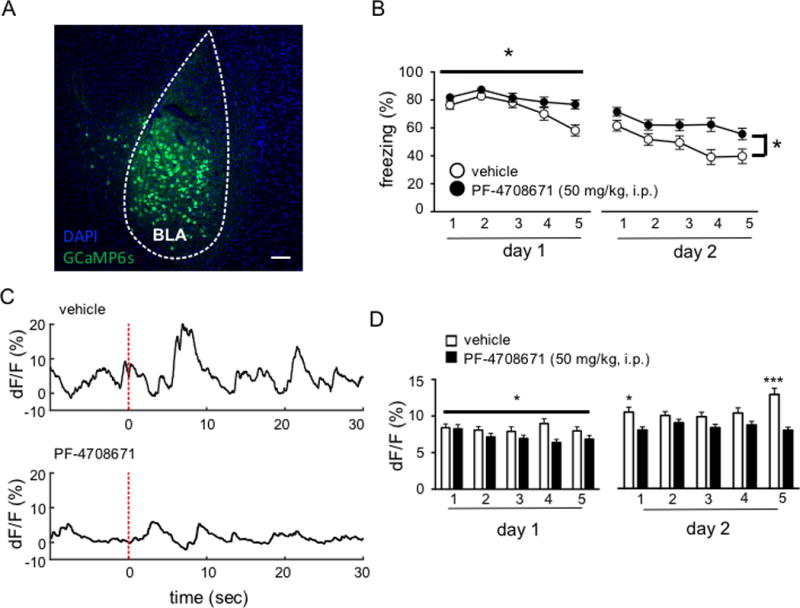
(A) Representative GCaMP6s expression in the BLA. B) Mice that received PF-4708671 infusions were impaired in the acquisition of extinction (*p< 0.05) (n= 8 vehicle, n = 12 PF-4708671) C) Representative traces depicting tone-locked emitted fluorescence. Dashed red line depicts tone onset. (D) PF-4708671 blocks the increase in mean amplitude of calcium events during extinction (*p<0.05; ***p<0.001). Scale bars represent 50μm.
Discussion
The results of the experiments described herein indicate that the consolidation of extinction memory relies on mTORC1-dependent protein synthesis, and that within-session extinction is regulated in part by a novel signaling cascade involving ERK, S6K1, and GluA1-containing AMPA receptors. Moreover, the involvement of ERK and S6K1 during within session extinction is localized to the basolateral nucleus of the amygdala (BLA). Our results are consistent with a role for de novo protein synthesis in the consolidation of threat extinction.5 Moreover, the signaling cascade described is consistent with previous reports indicating a critical role of the BLA in within session extinction.22 Our results also indicate that inhibition of S6K1 during training attenuates the extinction-induced increase of calcium signals in the BLA. Finally, mild uncontrollable stress leads to an impairment in within session extinction, as well as a downregulation of the ERK-S6K1-GluA1 pathway, suggesting that this signaling cascade is dysregulated following chronic stress. Overall, our results identify a novel intracellular cascade that is critically involved in the extinction of auditory threat conditioning.
Although the exact mechanism(s) underlying extinction are not well understood, several reports have described manipulations that affect within session extinction.2 The BLA has been implicated in the extinction of threat memories and plasticity in this region was linked to within session extinction. For example, inhibition of L-type voltage-gated calcium channels, as well as inhibition of NMDA receptors in the BLA were shown to block within session extinction.4 These reports suggest that calcium-triggered signaling events are critically involved in within session extinction. Our results suggest that within the BLA, GluA1-containing AMPA receptors are one of the final effectors in the cascade; however, the exact function of GluA1 phosphorylation at the Thr840 site by S6K1 has not been studied in vivo. Phosphorylation of GluA1 at Thr840 by protein kinase C has been shown to increase mean channel conductance and AMPA subunit receptor phosphorylation has been shown to have a variety of effects at other phosphorylation sites that mediate long-term plasticity.22,23 Thus, the data suggest that GluA1 (Thr840) phosphorylation during extinction training may be intrinsically involved in the plasticity associated with changes in response to the CS during extinction; however it may not be the only contributing factor. Our in vivo calcium imaging data indicate that indeed, calcium dependent-activity does occur during extinction, and is blocked by inhibition of S6K1. Calcium transients in vehicle-treated mice during both day 1 and day 2 of extinction was greater than that of mice treated with a S6K1 inhibitor. (Figure 4). Although extinction is commonly thought to involve inhibitory processes, previous studies have shown that extinction training can lead to increased firing of certain populations of cells. For example, it was shown that there is increased firing of neurons in the auditory cortex across an extinction trial.24 Moreover, although “fear neurons” in the BA exhibit diminished firing during an extinction trial, “extinction neurons” increase in firing rate during an extinction trial.24,25 Thus, it will be of interest to determine the identity of the cells that underlie the increase in calcium transients observed herein.
Our results indicate that ERK2, S6K1, and GluA1-containing AMPA receptors are proteins that are critically involved in within session extinction and that inhibition of either MEK-ERK or S6K1 is sufficient to block within session extinction. This observation is of clinical relevance because deficits in extinction learning are present in PTSD and in specific phobias. At least one of the molecules, S6K1, has been shown to be linked to major depressive disorder (MDD), which has high comorbidity with PTSD.26 Moreover, in a rat model of depression, decreased S6K1 activity was shown to induce pro-depressive behavior.10 Our experiments indicate that S6K1 activity is also diminished during extinction following acute uncontrollable stress (Figure 3). Interestingly, manipulating S6K1 activity was previously shown to rescue symptoms of various neuropsychiatric disorders including fragile X syndrome and major depressive disorder.10 The downregulation of S6K1 in MDD, and its critical role in the acquisition of extinction may be one node that can lead to comorbidities between MDD and PTSD, which is known to be high in clinical populations.10 These data suggest that targeting S6K1 activity may be a viable therapeutic strategy in developing treatments for major affective disorders in humans, consistent with a well-established role of mTORC1 signaling in a variety of neuropsychiatric diseases.27
The results of these studies indicate there is at least one signaling pathway in place that construes the ability to extinguish aversive memories in the basolateral nucleus of the amygdala. Although extinction memory consolidation requires mTORC1-dependent translation, within session extinction relies on ERK-dependent signaling, through S6K1 (Figure 1C). Interestingly, the role of de novo protein synthesis has been shown to be required in the mPFC during extinction memory consolidation,8 whereas our results indicate that ERK-S6K1-dependent signaling occurs in the basolateral nucleus of the amygdala during the acquisition of extinction. Future studies investigating whether the protein synthesis in the mPFC during the consolidation of extinction also requires mTORC1-dependent translation will further support the notion that the signaling pathways underlying acquisition and consolidation of extinction are spatiotemporally segregated.
Overall, this study indicates that ERK-S6K1-GluA1 signaling is critically involved with the acquisition of extinction. This signaling cascade may be a useful tool in understanding how properties in the BLA change in response to multiple CS presentations during extinction and how the switch between different populations within the BLA to support defensive reactions versus extinction occurs.
Supplementary Material
Acknowledgments
We would like to thank Chris Cain and the LeDoux lab for their input throughout the development of this project, Francis S. Lee for his advice regarding the manuscript text, and Shawn Singh for his assistance with Nissl staining.
Footnotes
Author Contributions
TNH, CL, JL, EK designed the research. TNH, ES, EM, AEF, BSH, RNF performed the experiments. LG, KD, CL contributed to the development of the fiber photometry experiments. TNH & EK analyzed the data. TNH & EK wrote the manuscript, which was edited by all authors. This work was funded by grants from the National Institutes of Health grants to E.K. (NS034007, NS047384, and HD082013) and E.S. (NS087112).
Conflict of Interest
The authors declare that there are no competing or conflicting interests.
Supplemental Information
Supplemental information is available at Molecular Psychiatry’s website.
References
- 1.Sotres-Bayon F, Quirk GJ. Prefrontal control of fear: more than just extinction. Current Opinion in Neurobiology. 2010;20:231–235. doi: 10.1016/j.conb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quirk GJ, Mueller D. Neural Mechanisms of Extinction Learning and Retrieval. Neuropsychopharmacology. 2007;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2006;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- 4.Herry C, Trifilieff P, Micheau J, Lüthi A, Mons N. Extinction of auditory fear conditioning requires MAPK/ERK activation in the basolateral amygdala. Eur J Neurosci. 2006;24:261–269. doi: 10.1111/j.1460-9568.2006.04893.x. [DOI] [PubMed] [Google Scholar]
- 5.Sotres-Bayon F, Bush DEA, LeDoux JE. Acquisition of Fear Extinction Requires Activation of NR2B-Containing NMDA Receptors in the Lateral Amygdala. Neuropsychopharmacology. 2007;32:1929–1940. doi: 10.1038/sj.npp.1301316. [DOI] [PubMed] [Google Scholar]
- 6.Hugues S. Postextinction Infusion of a Mitogen-Activated Protein Kinase Inhibitor Into the Medial Prefrontal Cortex Impairs Memory of the Extinction of Conditioned Fear. Learning & Memory. 2004;11:540–543. doi: 10.1101/lm.77704. [DOI] [PubMed] [Google Scholar]
- 7.Sotres-Bayon F, Diaz-Mataix L, Bush DEA, Ledoux JE. Dissociable Roles for the Ventromedial Prefrontal Cortex and Amygdala in Fear Extinction: NR2B Contribution. Cerebral Cortex. 2009;19:474–482. doi: 10.1093/cercor/bhn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santini E. Consolidation of Fear Extinction Requires Protein Synthesis in the Medial Prefrontal Cortex. Journal of Neuroscience. 2004;24:5704–5710. doi: 10.1523/JNEUROSCI.0786-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santini E, Huynh TN, Klann E. Mechanisms of Translation Control Underlying Long-Lasting Synaptic Plasticity and the Consolidation of Long-Term Memory. 1st. Elsevier Inc; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huynh TN, Santini E, Klann E. Requirement of Mammalian Target of Rapamycin Complex 1 Downstream Effectors in Cued Fear Memory Reconsolidation and Its Persistence. Journal of Neuroscience. 2014;34:9034–9039. doi: 10.1523/JNEUROSCI.0878-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoeffer CA, Cowansage KK, Arnold EC, Banko JL, Moerke NJ, Rodriguez R, et al. Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proceedings of the National Academy of Sciences. 2011;108:3383–3388. doi: 10.1073/pnas.1013063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iijima Y. c-Raf/MEK/ERK Pathway Controls Protein Kinase C-mediated p70S6K Activation in Adult Cardiac Muscle Cells. Journal of Biological Chemistry. 2002;277:23065–23075. doi: 10.1074/jbc.M200328200. [DOI] [PubMed] [Google Scholar]
- 13.Izquierdo A. Brief Uncontrollable Stress Causes Dendritic Retraction in Infralimbic Cortex and Resistance to Fear Extinction in Mice. Journal of Neuroscience. 2006;26:5733–5738. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman AN, Lorson NG, Sanabria F, Foster Olive M, Conrad CD. Chronic stress disrupts fear extinction and enhances amygdala and hippocampal Fos expression in an animal model of post-traumatic stress disorder. Neurobiology of Learning and Memory. 2014;112:139–147. doi: 10.1016/j.nlm.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. The EMBO Journal. 1998;17:6649–6659. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, et al. Natural Neural Projection Dynamics Underlying Social Behavior. Cell. 2014;157:1535–1551. doi: 10.1016/j.cell.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antion MD, Merhav M, Hoeffer CA, Reis G, Kozma SC, Thomas G, et al. Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learning & Memory. 2008;15:29–38. doi: 10.1101/lm.661908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delgado JY, Coba M, Anderson CNG, Thompson KR, Gray EE, Heusner CL, et al. NMDA Receptor Activation Dephosphorylates AMPA Receptor Glutamate Receptor 1 Subunits at Threonine 840. Journal of Neuroscience. 2007;27:13210–13221. doi: 10.1523/JNEUROSCI.3056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aubry AV, Serrano PA, Burghardt NS. Molecular Mechanisms of Stress-Induced Increases in Fear Memory Consolidation within the Amygdala. Front Behav Neurosci. 2016;10:191. doi: 10.3389/fnbeh.2016.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen T-W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins MA, Wells G, Bachman J, Snyder JP, Jenkins A, Huganir RL, et al. Regulation of GluA1 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor function by protein kinase C at serine-818 and threonine-840. Mol Pharmacol. 2014;85:618–629. doi: 10.1124/mol.113.091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dwyer JM, Maldonado-Avilés JG, Lepack AE, DiLeone RJ, Duman RS. Ribosomal protein S6 kinase 1 signaling in prefrontal cortex controls depressive behavior. Proceedings of the National Academy of Sciences. 2015;112:6188–6193. doi: 10.1073/pnas.1505289112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic Removal of p70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Neuron. 2012;76:325–337. doi: 10.1016/j.neuron.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1774–1779. doi: 10.1016/j.pnpbp.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santini E, Klann E. Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Science Signaling. 2014;7:re10–re10. doi: 10.1126/scisignal.2005832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.