

Abstract
Disentangling the interactions between cyanobacteria and associated bacterial community is important for understanding the mechanisms that mediate the formation of cyanobacterial blooms in freshwater ecosystems. Despite the fact that a metagenomic approach enables researchers to profile the structure of microbial communities associated with cyanobacteria, reconstructing genome sequences for all members remains inefficient, due to the inherent enormous microbial diversity. Here, we have established a stable coculture system under high salinity, originally from a mixture of an axenic cyanobacterium Synechococcus sp. PCC 7002 and a non-axenic bloom-forming cyanobacterium Microcystis colony. Metagenomic analysis showed that the coculture consists of S. sp. PCC 7002 and two heterotrophic bacteria, designated as Pseudomonas stutzeri TAIHU and Mesorhizobium sp. TAIHU, respectively. And near-complete genome sequences of both bacteria were reconstructed from the metagenomic dataset with an average completeness of 99.8%. Genome-wide pathway analysis revealed that M. sp. TAIHU carried all the genes involved in the de novo biosynthesis of cobalamin, which is required by S. sp. PCC 7002 for growth. To cope with the high salinity in the coculture, experimental evidence demonstrated that S. sp. PCC 7002 would synthesize the compatible solutes including sucrose and glucosylglycerol, which are supposed to be exploited by both heterotrophic bacteria as potential carbon and/or nitrogen sources. Furthermore, the genes encoding for the biosynthesis of the ectoine, another common osmolyte are found exclusively in P. stutzeri TAIHU, while the genes responsible for the catabolism of ectoine and its derives are present only in M. sp. TAIHU. These genomic evidence indicates beneficial interaction between three members in the coculture. Establishment of the coculture system with relative simplicity provides a useful model system for investigating the interspecies interactions, and genome sequences of both bacteria associated with Microcystis bloom described here will facilitate the researcher to elucidate the role of these heterotrophic bacteria in the formation and maintenance of cyanobacterial bloom in freshwater ecosystem.
Electronic supplementary material
The online version of this article (doi:10.1186/s13568-017-0490-2) contains supplementary material, which is available to authorized users.
Keywords: Biosynthesis of vitamin B12, Compatible solute, Fatty acid, Metagenome, Synechococcus, Mesorhizobium, Microcystis
Introduction
Among the blooms produced by genera of the cyanobacteria, Microcystis blooms might be the most widely spread (Harke et al. 2016). Most Microcystis synthesize and exude toxic secondary metabolites including microcystins, into water environments, causing intoxication of humans and animals (Sivonen and Jones 1999; Bláha et al. 2009). To understand the formation mechanism of Microcystis blooms, various associated biotic and abiotic factors have been studied extensively (Shapiro 1973; Jacoby et al. 2000; Paerl et al. 2001; Oliver and Ganf 2002), especially the roles and diversity of the associated heterotrophic bacteria in the mucilaginous colonies formed by Microcystis. The interactions between Microcystis and bacteria, are also areas of research interest, including the degradation of secondary metabolites, the inhibitory or enhancing effects on the growth of the cyanobacteria, resource competition, and nutrient exchange (Cole 1982; Fuks et al. 2005; Berg et al. 2008; Lemes et al. 2008; Dziallas and Grossart 2011; Shen et al. 2011; Dziallas and Grossart 2012; Briand et al. 2016; Zhu et al. 2016).
Before the advent of the second generation sequencing technology, traditional molecular techniques, including denaturing gradient gel electrophoresis (Muyzer et al. 1993), terminal restriction fragment length polymorphism analysis (Tiedje et al. 1999), and the analysis of conserved marker genes, e.g. 16S rRNA genes (Sogin et al. 2006; McHardy and Rigoutsos 2007), have been applied to profile the diversity of heterotrophic bacteria associated with cyanobacteria (Eiler and Bertilsson 2004; Kolmonen et al. 2004). As a cultivation-independent method, metagenomics provides a way of characterizing the uncultured microbes from their natural habitats in a higher resolution (Mick and Sorek 2014), and also enables the researcher to understand the potential role of these microbes in the community. Consequently, the microbial consortia from various environments, such as ocean water, natural acidophilic biofilms, permafrost, acetate-amended aquifers, and activated sludge bioreactors, have been studied using the metagenomic approach, leading to a better understanding of microbial and functional diversity, the discovery of novel genes, and the reconstruction of genomes from the microbial community (Tyson et al. 2004; Venter et al. 2004; Mackelprang et al. 2011; Wrighton et al. 2012; Albertsen et al. 2013). Metagenomics has also been applied to the study of cyanobacterial blooms (Pope and Patel 2008; Li et al. 2011; Steffen et al. 2012; Mou et al. 2013), showing that Microcystis bloom samples contained an enormous range of heterotrophic bacteria (Li et al. 2011; Steffen et al. 2012). Evidence from the study of long-term laboratory Microcystis colonies revealed that the associated bacteria played an important role in the degradation of benzoate and biosynthesis of vitamin B12 (Xie et al. 2016). These works have provided valuable insight into the molecular mechanisms that underpin the interactions between Microcystis and heterotrophic bacteria in the colony.
Despite its great potential in the analysis of microbial communities, metagenomic approach still has limitations in disentangling the relationships between each organism. The great diversity of bacteria in the sample makes it difficult to obtain all bacterial genomes with high completeness, causing the deficiency of genome information to investigate the interaction between Microcystis and associated bacteria (Li et al. 2011; Steffen et al. 2012). Although the typical approach of isolation followed by cocultivation was often adopted to study the interaction between community members (Shen et al. 2011), isolating associated bacteria and selecting the proper growth media seems time-consuming and tedious (Berg et al. 2008). To study the nature of interactions between bacteria, it is an alternative approach to establish an artificial coculture system with limited number of bacteria species in the laboratory (Amin et al. 2009; Kazamia et al. 2012). Sequencing of a simpler coculture system would lead to a higher completeness of genome sequence from species. It’s also easier to study the potential relationships between community members (Cole et al. 2014; Xie et al. 2016). Compared with samples collected directly from Microcystis bloom in the natural environment, Microcystis colony which maintains in the laboratory for a long term, encompass a limited number of heterotrophic bacteria (Xie et al. 2016; Zhu et al. 2016). Although a coculture system cannot represent the entire microbial diversity in a natural community, the insight gained from the analysis of the coculture could help to develop a fundamental understanding of the interaction between community members in nature.
Synechococcus sp. PCC 7002 is a unicellular cyanobacterium, which was originally isolated from an inshore marine mud sample in Puerto Rico (Baalen 1962). And it can grow over a wide range of NaCl concentration in the laboratory (Batterton and Baalen 1971). In addition, the complete genome sequence of S. sp. PCC 7002 has been determined (see http://www.ncbi.nlm.nih.gov/) and it can be easily genetically transformed (Stevens and Porter 1980; Frigaard et al. 2004), making it one of the most well-studied model cyanobacteria. Here we have established a coculture system consisting of S. sp. PCC 7002 and heterotrophic bacteria associated with Microcystis. The stable coculture system is characterized by high salinity and potential symbiotic interactions between S. sp. PCC 7002 and cobalamin-producing bacteria. The composition of the coculture and the producer of cobalamin, which is required by S. sp. PCC 7002 for growth, were determined through metagenomic analysis. Our results uncovered a potential symbiotic interaction between S. sp. PCC 7002 and Mesorhizobium sp. TAIHU in the coculture. Genes responsible for transporting cobalamin and encoding cobalamin-dependent enzymes were also investigated and compared between individual members in coculture. The approach of establishing a coculture system with symbiosis in our study will facilitate further studies of the relationship between cyanobacteria and its associated bacteria.
Materials and methods
Strain sources and coculture establishment
Microcystis aeruginosa TAIHU98 strain (FACHB-1752) (Yang et al. 2013, 2015) was obtained from the Freshwater Algae Collection of the Institution of Hydrobiology, Chinese Academy of Sciences (FACHB collection), Wuhan, China. The strain, initially collected from Lake Taihu in 2006, was part of a colony comprising other bacteria. Initially, we cocultured the Microcystis colony with an axenic Synechococcus sp. PCC 7002. At the beginning of the experiment, 500 µl liquid culture of axenic S. sp. PCC 7002 and 500 µl liquid culture of Microcystis colony were transferred together into 100 mL autoclaved BG11 medium (Stanier et al. 1971). The coculture was maintained at 25 ± 1 °C, and illuminated in a 12:12 h light:dark cycle with irradiance of 50 µEm−2 s−1. During subculture, different concentrations of NaCl solution were applied to the BG11 medium in a step gradient (0, 18, 24, 48, 60, 90, and 96 g l−1 NaCl). Previous salt-shock experiments revealed that M. aeruginosa PCC 7806 endured temporary salinity as high as 17.5 g l−1 (Tonk et al. 2007), while the euryhaline cyanobacterium S. sp. PCC 7002 grow over a wide range of NaCl concentration (Batterton and Baalen 1971; Ludwig and Bryant 2012). The final concentration of NaCl in the medium was then adjusted to 60 g l−1, at which the coculture was stable. The mixture of cells was harvested at the exponential phase of algae cell (OD730nm = 1.5) for sequencing (Ludwig and Bryant 2012).
Growth curve and SEM image of the coculture
As a comparison, axenic Synechococcus sp. PCC 7002 was also maintained in BG11 medium with exogenously added vitamin B12 and 60 g l−1 NaCl. The final concentration of vitamin B12 was the same as the concentration in A plus medium (Ludwig and Bryant 2012), which is used to cultivate S. sp. PCC 7002 in the laboratory. The optical density at 730 nm was measured to monitor the cell growth in axenic and coculture conditions. Scanning electron microscopy (SEM) for the two conditions was performed at the Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, China. Samples for the SEM operation were prepared using a published method (Gong et al. 2005).
Sequencing and data cleaning
The algae cells from coculture in the exponential stage were collected for sequencing. Total DNA of the coculture sample was extracted using a previous method (Yang et al. 2013). The sample was sequenced on an Illumina MiSeq sequencer using paired-end technology (301 bp) with the library insert size being 370 bp. To reconstruct the genome sequences of heterotrophic bacteria, the coculture sample was sequenced in another run using MiSeq with library size 420 bp. The library construction and sequencing were conducted in the Institution of Hydrobiology, Chinese Academy of Sciences, according to the manufacturer’s protocol (http://www.illumina.com). Quality control was performed using cutadapt v.1.8.0 (Martin 2011). All the paired reads were merged by FLASH (Magoc and Salzberg 2011) to obtain longer reads. Reads from the coculture sample were pooled together to perform de novo assembly of the coculture sequences.
De novo assembly and contigs binning
De novo assembly was conducted using SPAdes (Bankevich et al. 2012), Velvet (Zerbino and Birney 2008), and IDBA-UD (Peng et al. 2012), respectively. The optimal assembly was selected based on the typical assembly assessment metrics, including N50 [the N50 is defined as the shortest sequence length at 50% of the contigs or genome (Earl et al. 2011)], total size, the largest contigs length and the number of contigs. Different binning strategies were utilized to reconstruct the genome sequences of heterotrophic bacteria from the mini-metagenomic dataset. The binning process was performed using our previous binning method (Xie et al. 2016) combined with the Blobology (Kumar et al. 2013) and MaxBin methods (Wu et al. 2014). The binning result was evaluated further through alignment similarity using BLASTN (Altschul et al. 1997), and some unassigned scaffolds were recruited into individual genome bins according to taxonomy at the family level.
Phylogenetic analysis
Before reconstructing genome sequence of individual from the coculture, 16S rRNA genes were predicted from both the clean reads and assembly results, using ÉMIGRÉ (Miller et al. 2011) and RNAMmer (Lagesen et al. 2007), respectively. To further determine the taxonomic location of strains from the coculture, a comprehensive phylogenetic tree was built based on the alignment of 16S rRNA sequences of all species in the same genus with available draft or complete genome sequences. The genome sequences of 23 Pseudomonas stutzeri strains were downloaded from Pseudomonas database (Winsor et al. 2016), and two strains with partial 16S rRNA were discarded. Genome sequences of 81 strains belonging to the genus Mesorhizobium were downloaded from NCBI in December 2015. The phylogenetic trees were constructed using MEGA6 (Tamura et al. 2013). The genome-to-genome distances, which were used to mimic DNA–DNA hybridization (DDH) (Meier-Kolthoff et al. 2013), and the average nucleotide identity (ANI) (Richter and Rossello-Mora 2009; Chan et al. 2012), were calculated to delineate the bacteria in coculture.
Genome annotation and metabolic pathway analysis
Genome annotation of Pseudomonas stutzeri TAIHU and Mesorhizobium sp. TAIHU in the coculture was performed using the Prokka program (Seemann 2014) and RAST (Aziz et al. 2008), respectively. Open reading frame sequences predicted by Prokka were annotated by BlastKOALA, an online genome annotation service provided by the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2015). Pathway analysis was performed mainly by the “Reconstruct pathway” function of the KEGG mapper, and simultaneously combined with the subsystem function in the RAST server.
Results
Characterization of the coculture system
At the beginning, our coculture was established by mixing the xenic Microcystis colony and pure Synechococcus sp. PCC 7002 strain in the BG 11 medium (Fig. 1). During the step-wise increase of the salinity in the medium (see “Materials and methods” section for the details), Microcystis aeruginosa strain is believed to be absent in the coculture, which is confirmed that the amplification of the conserved marker genes in Microcystis (Additional file 1: Figure S1, Table S1). Our observation is consistent with the previous experimental result that Microcystis aeruginosa PCC 7806 endured temporary salinity as high as 17.5 g l−1 and was unable to maintain the growth under high salinity (Tonk et al. 2007). After the coculture was established stably, the growth of the cells in three condition was compared (Fig. 2a). The strain S. sp. PCC 7002 is unable to grow in the axenic culture in the absence of vitamin B12, but grow in the presence of vitamin B12 as expected, confirming the previous observation that the cyanobacterium required vitamin B12 for growth in the laboratory (Wilhelm and Trick 1995). In the first 2 days after subculture, there is no significant difference in growth rate between coculture and axenic culture supplemented with B12 (Fig. 2a). After that, the cells in the stable coculture grew slower than cells in the axenic culture. On the 15th day after subculture, the cell density in the axenic culture was almost twice that in the coculture. Based on the OD values, the doubling time of two cultures under high salinity is estimated, with ~ 38 h in the axenic culture and ~ 45 h in the coculture. The significant difference in growth rate of cells between two conditions could be explained by several possible factors, including insufficient cobalamin, shortage of nutrients, or high salinity in the coculture. In the coculture where S. sp. PCC 7002 can maintain its growth, there may exists vitamin B12 to support the growth of S. sp. PCC 7002 and the possible cobalamin producer may be some bacteria in the coculture. Furthermore, the scanning electron microscopy (SEM) imaging of the coculture showed that cells with multiple morphological characteristics, including oval and long rod shapes, coexist in the coculture, whereas only an oval morphology was observed in the axenic sample (Fig. 2b). The coccus shape observed in the coculture was consistent with the shape of S. sp. PCC 7002 in the axenic culture, indicating that there was more than one species present in the coculture.
Fig. 1.
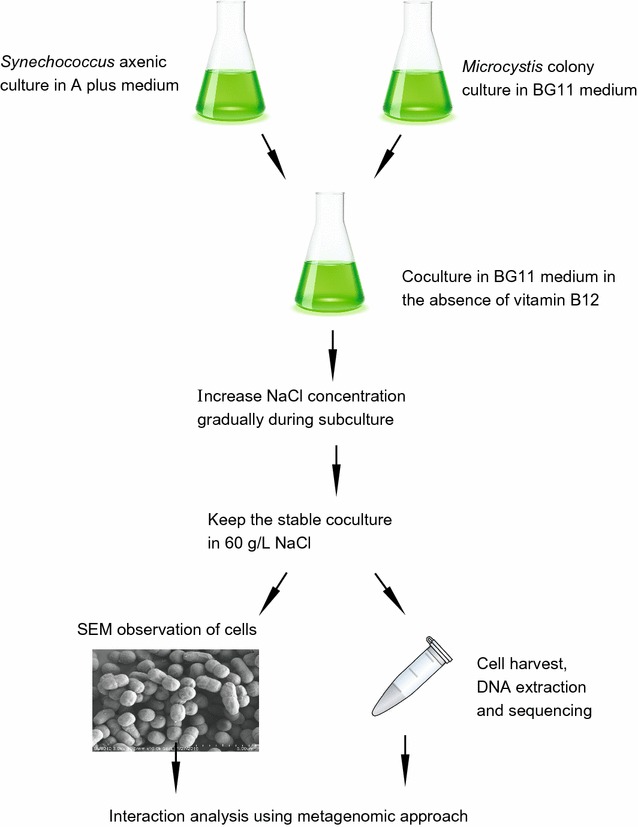
The flow diagram shows the establishment of the coculture system in the study
Fig. 2.
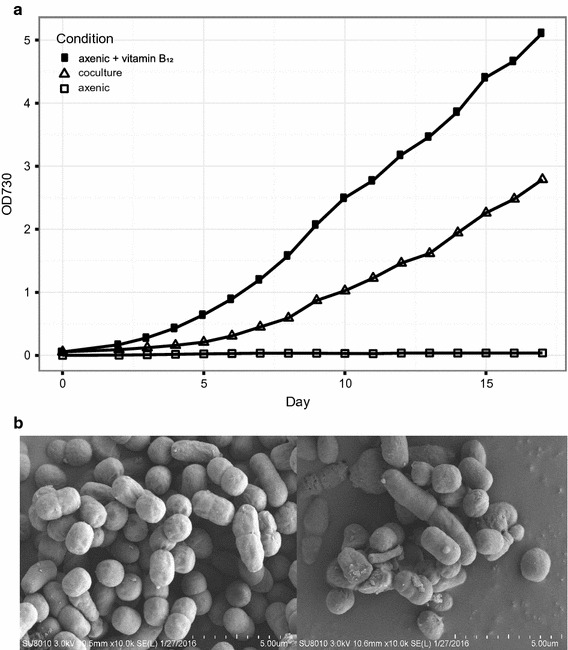
The comparison of algae cell in coculture and axenic culture. a The growth curve of algae cell in coculture (open triangles), axenic Synechococcus sp. PCC 7002 in the presence (filled squares) or absence (open squares) of vitamin B12. The basal medium of the cultures is BG11 medium (see “Materials and methods” section). Obviously, the axenic cyanobacterium is unable to grow in the absence of vitamin B12; b the scanning electron microscopy (SEM) image of cells in coculture (bottom right panel) and axenic culture (bottom left panel)
The composition of the coculture revealed through metagenomics analysis
To investigate the composition and possible cobalamin producer in the coculture, metagenomics approach was applied in our study. The raw sequence was filtered (Additional file 1: Table S2) and then assembled using three metagenome assembly software (Additional file 1: Table S3). Among the assembly, the result produced by SPAdes software (Bankevich et al. 2012) was selected and subject to the following analysis. Three genome bins were reconstructed from the assembly of the coculture sample (Table 1; Additional file 1: Table S4). And the number of genome bin agreed with the number of 16S rRNA genes predicted from the final assembly. Two of three genome bins were 100 and 99% complete, respectively, based on using marker genes specific to a genome’s inferred lineage within the reference genome tree (Parks et al. 2015), indicating that the approach of deep metagenomic sequencing for a simple coculture could improve the quality of assembled genome sequence for the individual members in the coculture.
Table 1.
Statistics of each genome bin reconstructed from coculture
| Group | Total size (bp) | No. scaffold | Longestcontig (bp) | N50 (bp) | GC content (%) | Completeness (%)a | Contamination (%)a | Relative abundance (%)b | Species name |
|---|---|---|---|---|---|---|---|---|---|
| Group1 | 4,830,778 | 38 | 781,449 | 506,457 | 63.63 | 99.8/98.28 | 1.5/0 | 6.68 | Pseudomonas stutzeri TAIHU |
| Group2 | 4,926,263 | 18 | 1,392,957 | 1,200,254 | 63.43 | 99.51/100 | 1.48/0 | 9.19 | Mesorhizobium sp. TAIHU |
| Group3 | 3,401,077 | 39 | 976,673 | 479,380 | 49.2 | 100/100 | 0/0 | 84.14 | Synechococcus sp. PCC7002 |
aThe completeness and contaimnation were determined by the CheckM software, the first value before the slash was the result of lineage-specific workfow and the latter one was the result of taxonomy-specific workflow in the program
bThe relative abundance was the proportion of reads on each genome bin in the total merged read, excluding paired read and the alignment was performed by the BWA software
Based on the binning result, our coculture contained other two bacteria besides Synechococcus sp. PCC 7002. The sequence similarity of the 16S rRNA genes showed that one bacterium was closely related to Pseudomonas stutzeri strains and the other affiliated with the Mesorhizobium genus. In a phylogenetic tree constructed from the complete 16S rRNA sequences of all P. stutzeri strains (Additional file 1: Figure S2), the strain in the coculture was clustered with P. stutzeri T13, an aerobic denitrifying bacterium isolated from activated sludge of a wastewater plant (Li et al. 2012). The estimated DNA–DNA hybridization (DDH) values (Meier-Kolthoff et al. 2013) between eight P. stutzeri strains (including P. stutzeri T13) and the strain in the coculture were greater than 79% (Additional file 1: Table S5), and the corresponding probability that these strains belonged to the same species is higher than 90%. Furthermore, the average nucleotide identity (ANI) values between the eight P. stutzeri strains and the strain identified here ranged from 97.88 to 98.57% (Additional file 1: Table S5), higher than the threshold of 96% proposed by Richter and Rossello-Mora (2009) to circumscribe the prokaryotic species. Taken together, the strain in our coculture was designated Pseudomonas stutzeri TAIHU (Fig. 3). The same analysis strategy was utilized to delineate another bacterium in the coculture (Additional file 1: Table S6), referred to as Mesorhizobium sp. TAIHU (Fig. 3). The bacterium was clustered with M. sp. UASWS1009 in the phylogenetic tree of 16S rRNA in the Mesorhizobium genus (Additional file 1: Figure S3); the latter is a nitrifying bacterium collected from sewage sludge (http://www.ncbi.nlm.nih.gov/nuccore/758551542).
Fig. 3.
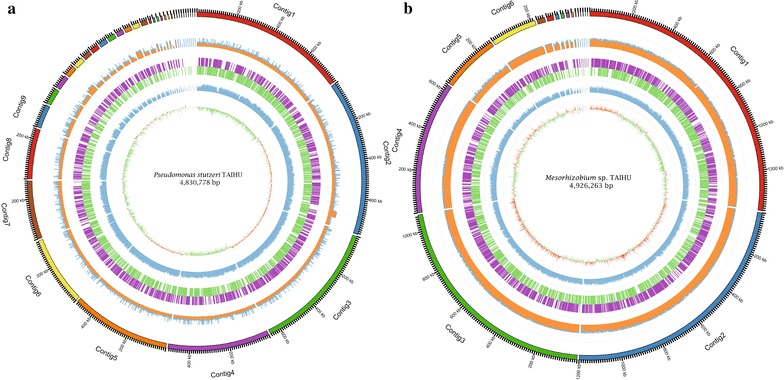
The representation of two bacteria genome sequence recovered from the coculture. The genomic characterization of Mesorhizobium sp. TAIHU (a) and Pseudomonas stutzeri TAIHU (b) is illustrated in the graphic representation generated by Circos program. The rings from outer to inner indicate the contigs, read coverage of each contigs (orange, 1 Kbp window), gene distribution on each strand (positive: purple, negative: green), GC content (blue, 1 Kbp window) and GC skew (orange above and green below zero, 1 Kbp window)
Metabolism of vitamin B12 in the coculture: biosynthesis and transport
Synechococcus sp. PCC 7002 was reported to require vitamin B12 (cobalamin) for in vitro growth (Wilhelm and Trick 1995) and it was confirmed in our study (Fig. 2). Through the genome analysis, most of the genes involved in the cobalamin biosynthesis were found to be absent in the genome of S. sp. PCC 7002 (Fig. 4a), that explains the inability of this organism to synthesize cobalamin de novo. The strain S. sp. PCC 7002 grew well in the coculture in the absence of cobalamin, suggesting that one or both of the other two bacteria in the coculture have the capacity to synthesize vitamin B12. The annotation results for the genomes of two heterotrophic bacteria support this hypothesis. The bacterium Mesorhizobium sp. TAIHU carries all genes involved in the aerobic pathway of cobalamin biosynthesis (Fig. 4a). These genes are distributed into five clusters (Additional file 1: Figure S4), which are located on two contigs. The largest cluster contains 15 adjacent genes. The genes encoding an aerobic cobalt chelatase composed of CobN, CobS and CobT subunits, was present in the genome of M. sp. TAIHU. The ATP-dependent chelatase is responsible for the insertion of cobalt ions in the late stage of the aerobic cobalamin biosynthesis (Rodionov et al. 2003). As in the genome of M. loti MAFF303099 (Kaneko et al. 2000), the genes encoding cobS and cobT subunits are clustered together and separated from other cobalamin genes in the genome of M. sp. TAIHU (Additional file 1: Figure S4). The annotation of the Pseudomonas stutzeri TAIHU genome shows that it lacks most of the genes involved in the synthesis of the corrin ring structure and the gene (cobR) encoding cobalt reductase (Fig. 4a), showing its incapability of synthesizing cobalamin de novo. However, P. stutzeri TAIHU contains the gene encoding cob(I)alamin adenosyltransferase (cobA), which is responsible for the synthesis of adenosylcobalamin (coenzyme vitamin B12) from cobalamin and the synthesis of adenosylcobinamide from cobinamide as a salvage pathway (Escalante-Semerena 2007; Swithers et al. 2012). And this potential enables the bacterium to save the great deal of energy spending on the biosynthesis of the corrin ring structure (Escalante-Semerena 2007).
Fig. 4.
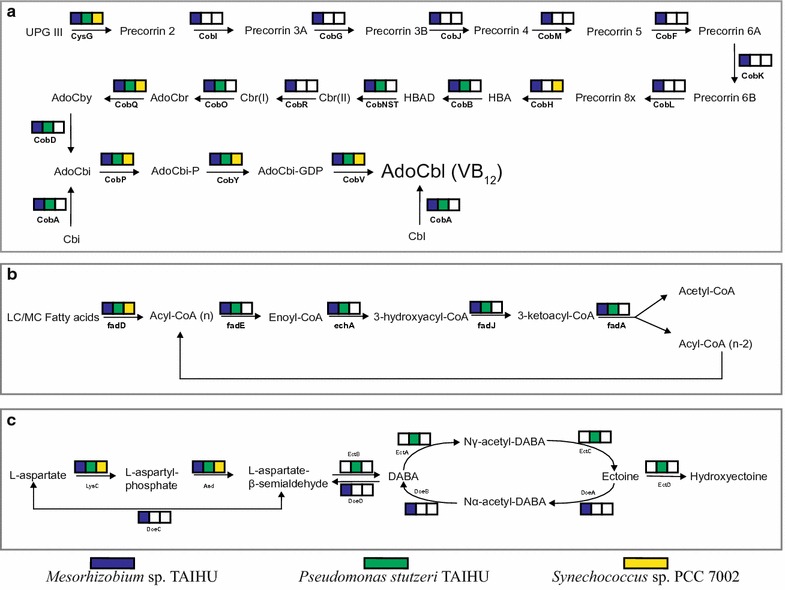
The metabolic pathway analysis for three members in the coculture. a The aerobic cobalamin biosynthesis pathway; b the β-oxidation pathway; c the ectoine biosynthesis and degration pathway. The color box indicates that the individual in the coculture carries the gene encoding the enzyme involved in the pathway, whereas an empty box (white) indicates the absence of the corresponding gene. The genes in Synechococcus sp. PCC 7002, Mesorhizobium sp. TAIHU and Pseudomonas stutzeri TAIHU are shown in yellow, blue and green, respectively. The abbreviations of the metabolites and enzymes used in the figure refer to the reference pathway (map00860, map00071, map00260) in KEGG database and the paper by Swither et al. (2012)
Genes responsible for the transport of cobalt and cobalamin were also investigated in members of the coculture. The gene btuB was identified in the genome of Mesorhizobium sp. TAIHU (Table 2). The outer membrane receptor BtuB is responsible for the transport of vitamin B12 and other corrinoids in prokaryotic cells (Rodionov et al. 2003). The genes for the complete vitamin B12 ABC transporter, consisting of ATPase component BtuD, permease component BtuC and B12-binding component BtuF, were found adjacent to the BtuB receptor in M. sp. TAIHU. The arrangement was consistent with previous reports that the B12-specific components of transporters in Gram-negative bacteria are BtuBFCD (Rodionov et al. 2003; Zhang et al. 2009). The BtuB receptor was also identified in Pseudomonas stutzeri TAIHU, and was encoded next to the gene for the B12-binding component BtuF of the ABC transporter (Table 2). Although the cobalamin transport genes were not identified in the genome of S. sp. PCC 7002 based on the annotation from RAST (Aziz et al. 2008) and Prokka (Seemann 2014), a recent study by Bryant and colleagues discovered the genes responsible for the cobalamin transport (btuBCF) in the cyanobacterium, which were previously annotated as siderophore uptake genes (Pérez et al. 2016a, b). In addition, the role of these genes in the cobalamin transport was demonstrated experimentally in their study (Pérez et al. 2016a, b).
Table 2.
Statistics of functional genes of the three species
| Function pathway or enzymes | Synechococcus sp. PCC 7002a | Mesorhizobium sp. TAIHU | Pseudomonas stutzeri TAIHU |
|---|---|---|---|
| Energy source and carbon source | |||
| Photosynthesis and CO2 fixation | √ | ||
| Beta-oxidation of fatty acid | √ | √ | |
| Cobalamin synthesis and transport | |||
| Cobalamin de novo synthesis | √ | ||
| Cobalamin salvage | MSTH_00675 | PSTH_00549/01543 | |
| Cobalamin and corrinoids transport (btuBFCD) | SYNPCC7002_A0634/0635/0637 | MSTH_03067-03070 | |
| Cobalamin-associated enzymes | |||
| Cobalamin-dependent methionine synthase (metH) | SYNPCC7002_A2466 | MSTH_00354/00815 | PSTH_03850 |
| Cobalamin-independent methionine synthase (metE) | MSTH_01286 | PSTH_01866 | |
| Methylmalonyl-CoA mutase | MSTH_01813/02120 | ||
| Cobalamin-dependent ribonucleotide reductase (class II) | MSTH_00664 | PSTH_00904 PSTH_00905 |
|
| Cobalamin-independent ribonucleotide reductase (class I) | SYNPCC7002_A1350 SYNPCC7002_A0382 |
PSTH_02138 PSTH_02139 |
|
| Anaerobic ribonucleoside-triphosphate reductase (RNR class III) | PSTH_03057 PSTH_03058 |
||
| Compatible solutes | |||
| Ectoine synthesis (ectABCD) | PSTH_01019-01022 | ||
| Glycine betaine (betAB) | MSTH_02104/02105/02862 | PSTH_02582/02583 | |
| Trehalose synthesis (ostAB) | MSTH_01326/01325 | PSTH_04112/04113 | |
| Trehalose (treY, treZ) | PSTH_03770/03772 | ||
| Trehalose (treS) | PSTH_03785 | ||
| Sucrose (sppA, spsA) | SYNPCC7002_A0887/0888 | ||
| Glucosylglycer-(ol/ate) (ggpS, ggpP) | SYNPCC7002_A2841/2851/2852 | ||
| Transport of glycine betaine/proline (proVWX) | MSTH_01321-01323 MSTH_03779-03781 |
PSTH_04547-04549 | |
| Ectoine degradation (doeABCD) | MSTH_03098/03099 MSTH_03109/03110 |
||
aThe check mark indicates that the organism contains the genes responsible for the metabolic ability dictated in the column of the table. Among these, beta-oxidation of fatty acid and biosynthesis of cobalamin are shown in Fig. 4
Biosynthesis of compatible solutes in the coculture
Besides the absence of cobalamin, the medium where the coculture grows is supplemented with 1 M NaCl. Obviously, the high salinity of the medium would impact the growth of the cells in the coculture (Fig. 2a), although the coculture can maintain its growth stably in the laboratory. To maintain normal metabolic activities under hyperosmotic stress, the production or uptake of compatible solutes (e.g., glycine betaine, sucrose, trehalose and ectoine) is one of strategies that the prokaryotes used (Empadinhas and da Costa 2008; Hagemann 2011; Klahn and Hagemann 2011). On the basis of pathway analysis and comparison, genes involved in the biosynthesis of different compatible solutes in each member were identified to mediate the high osmotic stress in the extracellular environment.
In S. sp. PCC 7002, experiment evidence showed that sucrose and glucosylglycerol (GG) were detected as the major compatible solutes (Klaehn et al. 2010). Moreover, glucosylglycerate (GGA) as a minor compatible solute was also observed to accumulate in the cyanobacterium. The genes responsible for the biosynthesis of these compounds was identified in the genome. The biosynthesis of GG and GGA was catalyzed by multiple enzymes, including glucosylglycerol-phosphate synthase (GgpS), glucosylglycerol 3-phosphatase (GgpP), and glycerol-3-phosphate dehydrogenase (Ludwig and Bryant 2012). The sucrose synthesis was encoded by two genes in S. sp. PCC 7002 (sppA and spsA) (Cumino et al. 2010). In contrast, the genes for the synthesis of sucrose, GG and GGA was found absent in the two heterotrophs (Table 2). In the case of P. stutzeri TAIHU, the operon (ectABCD) consisting of four ORFs for the biosynthesis of ectoine was identified. Ectoine, also known as tetrahydropyrimidine ectoine, and its derivative 5-hydroxyectoine, are widely used as compatible solutes among microbial communities (Galinski et al. 1985; Cánovas et al. 1998; Pastor et al. 2010). In addition, the other two common compatible solutes glycine betaine (GB) and trehalose were possibly produced by both heterotrophic bacteria, since the genes for biosynthesis pathway (betAB and ostAB) were found in their genomes. Among five pathways for trehalose synthesis (Paul et al. 2008; Moussaid et al. 2015), the ostAB pathway shared in two heterotrophs is most widespread in eubacteria, archaea and plants. The P. stutzeri TAIHU also harbor the genes involved in the two pathway for synthesizing trehalose, that is TreY-TreZ pathway and TreS pathway (Paul et al. 2008). Although the accumulation of trehalose and GB was demonstrated in many cyanobacteria (Hagemann 2011), the genes associated with these osmolytes synthesis was not identified in S. sp. PCC7002 in our analysis. The previous evidence that GB was not detected in the axenic culture of S. sp. PCC 7002 with comparable salinity further corroborated our observation (Cumino et al. 2010).
Potential interaction in the coculture
In the coculture established here, the vitamin-supplying heterotrophs must have an obligate dependency on the cyanobacterium S. sp. PCC 7002 for the fixed carbon. As essential components of membranes, fatty acids are important sources of carbon and energy in all living organisms. Once uptake by bacteria, fatty acids can be degraded via the β-oxidation pathway which include the conversion of acyl-CoA to enoyl-CoA, hydration, oxidation and thiolysis, resulting in the acyl-CoA minus two carbons and acetyl-CoA (Fujita et al. 2007). Through the metabolism analysis for three members in coculture, the genes encoding for the enzymes involved in the degradation pathway were present in the two heterotrophic bacteria whereas absent in the cyanobacterium (Fig. 4b). The gene presence/absence pattern in the fatty acid degradation pathway are suggestive of a possibility that the fatty acid generated by the cyanobacterium might supply the carbon and energy source for the heterotrophies in the coculture.
It is reported that the compatible solutes not only play an important role in protecting against the hyperosmotic stress, but also represent a significant proportion of total cell carbon and/or nitrogen (Welsh 2000; Vargas et al. 2006). As described above, carbohydrate compatible solutes generated by the cyanobacterium S. sp. PCC 7002, such as sucrose and GG can be exploited as potential carbon and energy source by the heterotrophies in coculture. And we have found that two heterotrophic bacteria harbor the genes involved in the phosphoenolpyruvate phosphotransferase system (PTS), with fructose-specific component in the case of P. stutzeri TAIHU and mannose-specific component in the case of M. sp. TAIHU. The PTS is reported to participates the transport and phosphorylation of specific carbohydrates in bacteria, including saccharides, amino sugars and other sugar derivatives (Deutscher et al. 2006). Another interesting observation is that, although the genes for ectoine biosynthesis were identified in P. stutzeri TAIHU as described earlier, the operon responsible for the degradation of ectoines (doeABCD) was found absent in P. stutzeri TAIHU, but present exclusively in M. sp. TAIHU (Fig. 4c). The ectoine degradation is catabolized through four-step reactions (Fig. 4c), supplying aspartate as final product for the cell (Table 2). Moreover, M. sp. TAIHU carries two copies of the operons for one high-affinity transporter system for glycine betaine (proXWV). Early evidences showed that the uptake of ectoine depend on the glycine betaine transport system in Escherichia coli (Jebbar et al. 1992) and a halophilic bacterium, Chromohalobacter salexigens (Vargas et al. 2006). These observations allow us to infer that M. sp. TAIHU might utilize the ectoine secreted by P. stutzeri TAIHU, indicating a potential interaction between both heterotrophic bacteria in the community.
Discussion
Due to the inability to tolerate the high salinity (Tonk et al. 2007), the freshwater cyanobacterium Microcystis species in the colony was replaced by the euryhaline cyanobacterium S. sp. PCC 7002, resulting in the establishment of the stable coculture system, which is characterized by the high salinity (~ 1 M NaCl) and maintained for 5 years in the laboratory. The replacement of photoautotroph in the micro-community has the following advantages: (i) S. sp. PCC 7002 is naturally transformable, making easier molecular genetic operation (Stevens and Porter 1980; Frigaard et al. 2004); (ii) The doubling time for the species is comparably short under optimal conditions (Ludwig and Bryant 2012); Based on these superior characteristics, the coculture system established here provides a useful model to study the microbial interaction between cyanobacteria and bacteria, and the mechanisms for mediating the high salinity in a cooperated way. In addition, further understanding of the nature of interaction will benefit from the genetic transformation of S. sp. PCC 7002, the only photoautotroph in the coculture system.
In our coculture system, both bacteria have the metabolic potential to refresh the cobalamin through biosynthetic or salvage pathway, and that is required by the B12 auxotroph S. sp. PCC 7002. Vitamin B12 are essential cofactor for several important enzymes and methionine synthase is one of most prominent one (Rodionov et al. 2003). The biosynthesis of methionine in organisms can be catalyzed by two isozymes of methionine synthase, namely the vitamin B12-dependent one (MetH) and B12-independent one (MetE). The MetH, also known as 5-methyltetrahydrofolate-homocysteine methyltransferase (N5-MeTHF), catalyzes methyl group transfer from methyltetrahydrofolate to homocysteine to create methionine using B12 as a cofactor (Zydowsky et al. 1986). The cyanobacterium S. sp. PCC 7002 only contains the B12-dependent methionine synthase gene (metH) yet cannot synthesize B12 (Fig. 4a), giving a possible explanation that the strain requires exogenous vitamin B12 for growth in the laboratory (Fig. 2a). And a recent molecular study on S. sp. PCC 7002 have demonstrated the hypothesis and proposed that the enzyme MetH is probably the only enzyme in the cyanobacterium that requires cobalamin as cofactor (Pérez et al. 2016a). In their study, S. sp. PCC 7002 was shown to maintain growth in the absence of cobalamin with added methionine, although less than the growth rate under normal condition (with cobalamin). Based on these evidences, we cannot rule out another possible scenario that the heterotrophic bacteria in the coculture supply the methionine and its derivatives, not just cobalamin, for the cyanobacterium (Xie et al. 2016). In either case, S. sp. PCC 7002 always benefit from the bacteria in the coculture. It should be pointed out that the biosynthesis of cobalamin in the coculture system is not only important for the cyanobacterium, but also for the cobalamin-producer M. sp. TAIHU. Genes encoding the B12-requiring enzymes, including the methylmalonyl-CoA mutase (MCM) and ribonucleotide reductase (RNR class II) were identified in the genome of M. sp. TAIHU (Table 2).
Based on the genomic analysis, each member in the coculture system is found to harbor the genes involved in the biosynthesis of specific compatible solute to cope with the hyperosmotic stress. In addition to stress protection, these diverse compatible solutes also represent a significant proportion of cell carbon and/or nitrogen source, which may be exploited by the community as carbon and energy source, once released to the environment (Welsh 2000; Vargas et al. 2006). Since the medium used for the coculture isn’t supplemented with any organic carbon source, the photosynthates generated from the cyanobacterium S. sp. PCC 7002, including compatible solutes, is considered to be the final source of fixed carbon and energy for the whole community. For example, the carbohydrate compatible solutes (sucrose, GG and GGA) produced by S. sp. PCC 7002 may be utilized by the heterotrophic bacteria. Moreover, both heterotrophic bacteria harbor the genes involved in the phosphoenolpyruvate phosphotransferase system (PTS), which is associated with the transport and phosphorylation of specific carbohydrates in bacteria (Deutscher et al. 2006). Alternatively, the bacteria may obtain the carbon and energy source through beta-oxidation of fatty acid since the genes involved in the catabolism of fatty acid are exclusively present in the heterotrophic bacteria but absent in the cyanobacterium (Fig. 4b).
The approach of establishing an artificial system combined with deep sequencing metagenomics, represents a novel attempt to investigate in greater depth the interesting microbial components in communities. The similar mutualism based on cobalamin and nutrient exchange have been described in Microcystis colony (Xie et al. 2016), green algae (Croft et al. 2005; Kazamia et al. 2012), microbial mat consortia (Cole et al. 2014; Romine et al. 2017), and diatom (Durham et al. 2015). Although the coculture system cannot represent the entire microbial diversity in a natural community, the insight gained from the analysis of the simpler coculture could help to develop a fundamental understanding of the interaction between complex community in nature. Given the advantage of natural genetic transformation for S. sp. PCC 7002 (Frigaard et al. 2004), the stable coculture system established here provides a useful model to decipher the nature of interaction between cyanobacteria and heterotrophic bacteria.
Authors’ contributions
TL conceived the study and designed the experiments. GZ and ZY performed the experiments. ZQ, MX and YL were responsible for the library preparation and sample sequencing. MR carried out the data analysis and prepared the first draft of the manuscript. All authors revised the manuscript, participated in discussion of the manuscript and agreed to the final content. All authors read and approved the final manuscript.
Acknowledgements
We thank Dr. Chen Yang for providing the Microcystis colony sample, which is used to establish the coculture in this study. We also thank two anonymous reviewers for valuable comments.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The raw Illumina sequence of coculture described in this manuscript has been deposited in the Sequence Read Archive of the National Center for Biotechnology Information (Accession Number: SRP075385). The coculture assembly has also been deposited at DDBJ/ENA/GenBank under the accession number LYXM00000000. The version described in this paper is version LYXM01000000. The genome sequences of Pseudomonas stutzeri TAIHU and Mesorhizobium sp. TAIHU, can be retrieved from the assembly based on the scaffold list belonging to each species in Additional file 1: Table S7.
Consent for publication
The authors give their consent for publication.
Ethics approval and consent to participate
The article does not contain experiment with human participant or animal.
Funding
This study was funded by the State Key Laboratory of Freshwater Ecology and Biotechnology (2016FBZ08), the National Major Basic Research Program (2015ZX07503-005-003), the Chinese Academy of Sciences Grant (QYZDY-SSW-SMC004), and the National Natural Science Foundation of China (31400215).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ANI
average nucleotide identity
- DDH
DNA–DNA hybridization
- SEM
scanning electron microscopy
- PTS
phosphoenolpyruvate phosphotransferase system
- OD
optical density
Additional file
Additional file 1. Additional figures and tables.
Footnotes
Minglei Ren and Guiying Zhang contributed equally to this work
Electronic supplementary material
The online version of this article (doi:10.1186/s13568-017-0490-2) contains supplementary material, which is available to authorized users.
Contributor Information
Minglei Ren, Email: minglei650@gmail.com.
Guiying Zhang, Email: zhang_guiying@sina.com.
Zi Ye, Email: yezi@ihb.ac.cn.
Zhixian Qiao, Email: qzhxian@ihb.ac.cn.
Meili Xie, Email: xiemeili0101@163.com.
Yan Lin, Email: linyan@ihb.ac.cn.
Tao Li, Phone: +86 136 6722 2098, Email: litao@ihb.ac.cn.
Jindong Zhao, Email: jzhao@ihb.ac.cn.
References
- Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol. 2013;31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin SA, Green DH, Hart MC, Küpper FC, Sunda WG, Carrano CJ. Photolysis of iron–siderophore chelates promotes bacterial–algal mutualism. Proc Natl Acad Sci USA. 2009;106(40):17071–17076. doi: 10.1073/pnas.0905512106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST server: rapid annotations using subsystems technology. BMC Genom. 2008;9(1):75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baalen CV. Studies on marine blue-green algae. Bot Mar. 1962;4(12):129–139. [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterton JC, Baalen CV. Growth responses of blue-green algae to sodium chloride concentration. Arch Mikrobiol. 1971;76(2):151–165. doi: 10.1007/BF00411789. [DOI] [PubMed] [Google Scholar]
- Berg KA, Lyra C, Sivonen K, Paulin L, Suomalainen S, Tuomi P, Rapala J. High diversity of cultivable heterotrophic bacteria in association with cyanobacterial water blooms. ISME J. 2008;3(3):314–325. doi: 10.1038/ismej.2008.110. [DOI] [PubMed] [Google Scholar]
- Bláha L, Babica P, Maršálek B. Toxins produced in cyanobacterial water blooms—toxicity and risks. Interdiscip Toxicol. 2009;2(2):36–41. doi: 10.2478/v10102-009-0006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand E, Humbert J-F, Tambosco K, Bormans M, Gerwick WH. Role of bacteria in the production and degradation of Microcystis cyanopeptides. MicrobiologyOpen. 2016;5(3):469–478. doi: 10.1002/mbo3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cánovas D, Vargas C, Calderón MI, Ventosa A, Nieto JJ. Characterization of the genes for the biosynthesis of the compatible solute ectoine in the moderately halophilic bacterium Halomonas elongata DSM 3043. Syst Appl Microbiol. 1998;21(4):487–497. doi: 10.1016/S0723-2020(98)80060-X. [DOI] [PubMed] [Google Scholar]
- Chan JZM, Halachev MR, Loman NJ, Constantinidou C, Pallen MJ. Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 2012;12(1):302. doi: 10.1186/1471-2180-12-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JJ. Interactions between bacteria and algae in aquatic ecosystems. Annu Rev Ecol Evol Syst. 1982;13:291–314. doi: 10.1146/annurev.es.13.110182.001451. [DOI] [Google Scholar]
- Cole J, Hutchison J, Renslow R, Kim Y-M, Chrisler W, Engelmann H, Dohnalkova A, Hu D, Metz T, Fredrickson J, Lindemann S. Phototrophic biofilm assembly in microbial-mat-derived unicyanobacterial consortia: model systems for the study of autotroph–heterotroph interactions. Front Microbiol. 2014;5:109. doi: 10.3389/fmicb.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature. 2005;438(7064):90. doi: 10.1038/nature04056. [DOI] [PubMed] [Google Scholar]
- Cumino AC, Perez-Cenci M, Giarrocco LE, Salerno GL. The proteins involved in sucrose synthesis in the marine cyanobacterium Synechococcus sp. PCC 7002 are encoded by two genes transcribed from a gene cluster. FEBS Lett. 2010;584(22):4655–4660. doi: 10.1016/j.febslet.2010.10.040. [DOI] [PubMed] [Google Scholar]
- Deutscher J, Francke C, Postma PW. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev. 2006;70(4):939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham BP, Sharma S, Luo H, Smith CB, Amin SA, Bender SJ, Dearth SP, Van Mooy BAS, Campagna SR, Kujawinski EB, Armbrust EV, Moran MA. Cryptic carbon and sulfur cycling between surface ocean plankton. Proc Natl Acad Sci USA. 2015;112(2):453–457. doi: 10.1073/pnas.1413137112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziallas C, Grossart H-P. Temperature and biotic factors influence bacterial communities associated with the cyanobacterium Microcystis sp. Environ Microbiol. 2011;13(6):1632–1641. doi: 10.1111/j.1462-2920.2011.02479.x. [DOI] [PubMed] [Google Scholar]
- Dziallas C, Grossart H-P. Microbial interactions with the cyanobacterium Microcystis aeruginosa and their dependence on temperature. Mar Biol. 2012;159(11):2389–2398. doi: 10.1007/s00227-012-1927-4. [DOI] [Google Scholar]
- Earl DA, Bradnam K, St. John J, Darling A, Lin D, Faas J, Yu HOK, Vince B, Zerbino DR, Diekhans M, Nguyen N, Nuwantha P, Sung AW-K, Ning Z, Haimel M, Simpson JT, Fronseca NA, Birol İ, Docking TR, Ho IY, Rokhsar DS, Chikhi R, Lavenier D, Chapuis G, Naquin D, Maillet N, Schatz MC, Kelly DR, Phillippy AM, Koren S, Yang S-P, Wu W, Chou W-C, Srivastava A, Shaw TI, Ruby JG, Skewes-Cox P, Betegon M, Dimon MT, Solovyev V, Kosarev P, Vorobyev D, Ramirez-Gonzalez R, Leggett R, MacLean D, Xia F, Luo R, Xie Y, Liu B, Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Yin S, Sharpe T, Hall G, Kersey PJ, Durbin R, Jackman SD, Chapman JA, Huang X, DeRisi JL, Caccamo M, Li Y, Jaffe DB, Green R, Haussler D, Korf I, Paten B. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res. 2011 doi: 10.1101/gr.126599.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiler A, Bertilsson S. Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environ Microbiol. 2004;6(12):1228–1243. doi: 10.1111/j.1462-2920.2004.00657.x. [DOI] [PubMed] [Google Scholar]
- Empadinhas N, da Costa MS. Osmoadaptation mechanisms in prokaryotes: distribution of compatible solutes. Int Microbiol. 2008;11(3):151–161. [PubMed] [Google Scholar]
- Escalante-Semerena JC. Conversion of cobinamide into adenosylcobamide in bacteria and archaea. J Bacteriol. 2007;189(13):4555–4560. doi: 10.1128/JB.00503-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigaard N-U, Sakuragi Y, Bryant DA. Gene Inactivation in the cyanobacterium Synechococcus sp. PCC 7002 and the green sulfur bacterium Chlorobium tepidum using in vitro-made DNA constructs and natural Transformation. In: Carpentier R, editor. Photosynthesis research protocols. Totowa: Humana Press; 2004. pp. 325–340. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Matsuoka H, Hirooka K. Regulation of fatty acid metabolism in bacteria. Mol Microbiol. 2007;66(4):829–839. doi: 10.1111/j.1365-2958.2007.05947.x. [DOI] [PubMed] [Google Scholar]
- Fuks D, Radić J, Radić T, Najdek M, Blažina M, Degobbis D, Smodlaka N. Relationships between heterotrophic bacteria and cyanobacteria in the northern Adriatic in relation to the mucilage phenomenon. Sci Total Environ. 2005;353(1–3):178–188. doi: 10.1016/j.scitotenv.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Galinski EA, Pfeiffer H-P, TrÜPer HG. 1,4,5,6-Tetrahydro-2-methyl-4-pyrimidinecarboxylic acid. Eur J Biochem. 1985;149(1):135–139. doi: 10.1111/j.1432-1033.1985.tb08903.x. [DOI] [PubMed] [Google Scholar]
- Gong YC, Gu XW, Yu YH, Shen YF, Gu FK, Ni B. Preparation of the flagellate specimens for surface scanning electron microscopy. J Ocean U CHINA. 2005;35(3):496–498. [Google Scholar]
- Hagemann M. Molecular biology of cyanobacterial salt acclimation. FEMS Microbiol Rev. 2011;35(1):87–123. doi: 10.1111/j.1574-6976.2010.00234.x. [DOI] [PubMed] [Google Scholar]
- Harke MJ, Steffen MM, Gobler CJ, Otten TG, Wilhelm SW, Wood SA, Paerl HW. A review of the global ecology, genomics, and biogeography of the toxic cyanobacterium, Microcystis spp. Harmful Algae. 2016;54:4–20. doi: 10.1016/j.hal.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Jacoby JM, Collier DC, Welch EB, Hardy FJ, Crayton M. Environmental factors associated with a toxic bloom of Microcystis aeruginosa. Can J Fish Aquat Sci. 2000;57(1):231–240. doi: 10.1139/f99-234. [DOI] [Google Scholar]
- Jebbar M, Talibart R, Gloux K, Bernard T, Blanco C. Osmoprotection of Escherichia coli by ectoine: uptake and accumulation characteristics. J Bacteriol. 1992;174(15):5027–5035. doi: 10.1128/jb.174.15.5027-5035.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2015 doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, Sasamoto S, Watanabe A, Idesawa K, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Mochizuki Y, Nakayama S, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000;7(6):331–338. doi: 10.1093/dnares/7.6.331. [DOI] [PubMed] [Google Scholar]
- Kazamia E, Czesnick H, Nguyen TTV, Croft MT, Sherwood E, Sasso S, Hodson SJ, Warren MJ, Smith AG. Mutualistic interactions between vitamin B12-dependent algae and heterotrophic bacteria exhibit regulation. Environ Microbiol. 2012;14(6):1466–1476. doi: 10.1111/j.1462-2920.2012.02733.x. [DOI] [PubMed] [Google Scholar]
- Klaehn S, Steglich C, Hess WR, Hagemann M. Glucosylglycerate: a secondary compatible solute common to marine cyanobacteria from nitrogen-poor environments. Environ Microbiol. 2010;12(1):83–94. doi: 10.1111/j.1462-2920.2009.02045.x. [DOI] [PubMed] [Google Scholar]
- Klahn S, Hagemann M. Compatible solute biosynthesis in cyanobacteria. Environ Microbiol. 2011;13(3):551–562. doi: 10.1111/j.1462-2920.2010.02366.x. [DOI] [PubMed] [Google Scholar]
- Kolmonen E, Sivonen K, Rapala J, Haukka K. Diversity of cyanobacteria and heterotrophic bacteria in cyanobacterial blooms in Lake Joutikas, Finland. Aquat Microb Ecol. 2004;36(3):201–211. doi: 10.3354/ame036201. [DOI] [Google Scholar]
- Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet. 2013 doi: 10.3389/fgene.2013.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemes GAF, Kersanach R, Pinto L, Dellagostin OA, Yunes JS, Matthiensen A. Biodegradation of microcystins by aquatic Burkholderia sp. from a South Brazilian coastal lagoon. Ecotoxicol Environ Saf. 2008;69(3):358–365. doi: 10.1016/j.ecoenv.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Li N, Zhang L, Li F, Wang Y, Zhu Y, Kang H, Wang S, Qin S. Metagenome of microorganisms associated with the toxic cyanobacteria Microcystis aeruginosa analyzed using the 454 sequencing platform. Chin J Oceanol Limn. 2011;29(3):505–513. doi: 10.1007/s00343-011-0056-0. [DOI] [Google Scholar]
- Li A, Gai Z, Cui D, Ma F, Yang J, Zhang X, Sun Y, Ren N. Genome sequence of a highly efficient aerobic denitrifying bacterium, Pseudomonas stutzeri T13. J Bacteriol. 2012;194(20):5720. doi: 10.1128/JB.01376-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M, Bryant DA. Synechococcus sp. strain PCC 7002 transcriptome: acclimation to temperature, salinity, oxidative stress, and mixotrophic growth conditions. Front Microbiol. 2012;3:354. doi: 10.3389/fmicb.2012.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackelprang R, Waldrop MP, DeAngelis KM, David MM, Chavarria KL, Blazewicz SJ. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature. 2011 doi: 10.1038/nature10576. [DOI] [PubMed] [Google Scholar]
- Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011 [Google Scholar]
- McHardy AC, Rigoutsos I. What’s in the mix: phylogenetic classification of metagenome sequence samples. Curr Opin Microbiol. 2007;10(5):499–503. doi: 10.1016/j.mib.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Meier-Kolthoff JP, Auch AF, Klenk H-P, Goeker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013;14(1):60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick E, Sorek R. High-resolution metagenomics. Nat Biotech. 2014;32(8):750–751. doi: 10.1038/nbt.2962. [DOI] [PubMed] [Google Scholar]
- Miller CS, Baker BJ, Thomas BC, Singer SW, Banfield JF. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011 doi: 10.1186/gb-2011-12-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou X, Jacob J, Lu X, Robbins S, Sun S, Ortiz JD. Diversity and distribution of free-living and particle-associated bacterioplankton in Sandusky Bay and adjacent waters of Lake Erie Western Basin. J Great Lakes Res. 2013;39(2):352–357. doi: 10.1016/j.jglr.2013.03.014. [DOI] [Google Scholar]
- Moussaid S, Domínguez-Ferreras A, Muñoz S, Aurag J, Berraho EB, Sanjuán J. Increased trehalose biosynthesis improves Mesorhizobium ciceri growth and symbiosis establishment in saline conditions. Symbiosis. 2015;67(1):103–111. doi: 10.1007/s13199-015-0338-y. [DOI] [Google Scholar]
- Muyzer G, de Waal EC, Uitterlinden AG. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59(3):695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver RL, Ganf GG. Freshwater Blooms. In: Whitton BA, Potts M, editors. The ecology of cyanobacteria: their diversity in time and space. Dordrecht: Springer; 2002. pp. 149–194. [Google Scholar]
- Paerl HW, Fulton RS, Moisander PH, Dyble J. Harmful freshwater algal blooms, with an emphasis on cyanobacteria. Sci World J. 2001;1:76–113. doi: 10.1100/tsw.2001.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor JM, Salvador M, Argandoña M, Bernal V, Reina-Bueno M, Csonka LN, Iborra JL, Vargas C, Nieto JJ, Cánovas M. Ectoines in cell stress protection: uses and biotechnological production. Biotechnol Adv. 2010;28(6):782–801. doi: 10.1016/j.biotechadv.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Paul MJ, Primavesi LF, Jhurreea D, Zhang YH. Trehalose metabolism and signaling. Annu Rev Plant Biol. 2008;59:417–441. doi: 10.1146/annurev.arplant.59.032607.092945. [DOI] [PubMed] [Google Scholar]
- Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28(11):1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- Pérez AA, Liu Z, Rodionov DA, Li Z, Bryant DA. Complementation of cobalamin auxotrophy in Synechococcus sp. strain PCC 7002 and validation of a putative cobalamin riboswitch in vivo. J Bacteriol. 2016;198(19):2743–2752. doi: 10.1128/JB.00475-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez AA, Rodionov DA, Bryant DA. Identification and regulation of genes for cobalamin transport in the cyanobacterium Synechococcus sp. strain PCC 7002. J Bacteriol. 2016;198(19):2753–2761. doi: 10.1128/JB.00476-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope PB, Patel BKC. Metagenomic analysis of a freshwater toxic cyanobacteria bloom. FEMS Microbiol Ecol. 2008;64(1):9–27. doi: 10.1111/j.1574-6941.2008.00448.x. [DOI] [PubMed] [Google Scholar]
- Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009;106(45):19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J Biol Chem. 2003;278(42):41148–41159. doi: 10.1074/jbc.M305837200. [DOI] [PubMed] [Google Scholar]
- Romine MF, Rodionov DA, Maezato Y, Osterman AL, Nelson WC. Underlying mechanisms for syntrophic metabolism of essential enzyme cofactors in microbial communities. ISME J. 2017 doi: 10.1038/ismej.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- Shapiro J. Blue–green algae: why they become dominant. Science. 1973;179(4071):382–384. doi: 10.1126/science.179.4071.382. [DOI] [PubMed] [Google Scholar]
- Shen H, Niu Y, Xie P, Tao MIN, Yang XI. Morphological and physiological changes in Microcystis aeruginosa as a result of interactions with heterotrophic bacteria. Freshw Biol. 2011;56(6):1065–1080. doi: 10.1111/j.1365-2427.2010.02551.x. [DOI] [Google Scholar]
- Sivonen K, Jones G. Cyanobacterial toxins. In: Chorus I, Bartram J, editors. Toxic cyanobacteria in water: a guide to public health significance. London: Monitoring and Management, Routledge; 1999. [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci USA. 2006;103(32):12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanier RY, Kunisawa R, Mandel M, Cohen-Bazire G. Purification and properties of unicellular blue-green algae (order Chroococcales) Bacteriol Rev. 1971;35(2):171–205. doi: 10.1128/br.35.2.171-205.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen MM, Li Z, Effler TC, Hauser LJ, Boyer GL, Wilhelm SW. Comparative metagenomics of toxic freshwater cyanobacteria bloom communities on two continents. PLoS ONE. 2012 doi: 10.1371/journal.pone.0044002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SE, Porter RD. Transformation in Agmenellum quadruplicatum. Proc Natl Acad Sci USA. 1980;77(10):6052–6056. doi: 10.1073/pnas.77.10.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers KS, Petrus AK, Secinaro MA, Nesbø CL, Gogarten JP, Noll KM, Butzin NC. Vitamin B12 synthesis and salvage pathways were acquired by horizontal gene transfer to the Thermotogales. Genome Biol Evol. 2012;4(8):842–851. doi: 10.1093/gbe/evs057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedje JM, Asuming-Brempong S, Nüsslein K, Marsh TL, Flynn SJ. Opening the black box of soil microbial diversity. Appl Soil Ecol. 1999;13(2):109–122. doi: 10.1016/S0929-1393(99)00026-8. [DOI] [Google Scholar]
- Tonk L, Bosch K, Visser PM, Huisman J. Salt tolerance of the harmful cyanobacterium Microcystis aeruginosa. Aquat Microb Ecol. 2007;46(2):117–123. doi: 10.3354/ame046117. [DOI] [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428(6978):37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Vargas C, Jebbar M, Carrasco R, Blanco C, Calderón MI, Iglesias-Guerra F, Nieto JJ. Ectoines as compatible solutes and carbon and energy sources for the halophilic bacterium Chromohalobacter salexigens. J Appl Microbiol. 2006;100(1):98–107. doi: 10.1111/j.1365-2672.2005.02757.x. [DOI] [PubMed] [Google Scholar]
- Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers Y-H, Smith HO. Environmental genome shotgun sequencing of the sargasso sea. Science. 2004;304(5667):66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Welsh DT. Ecological significance of compatible solute accumulation by micro-organisms: from single cells to global climate. FEMS Microbiol Rev. 2000;24(3):263–290. doi: 10.1111/j.1574-6976.2000.tb00542.x. [DOI] [PubMed] [Google Scholar]
- Wilhelm SW, Trick CG. Effects of vitamin B12 concentration on chemostat cultured Synechococcus sp. strain PCC 7002. Can J Microbiol. 1995;41(2):145–151. doi: 10.1139/m95-019. [DOI] [Google Scholar]
- Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FSL. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016;44(D1):D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science. 2012;337(6102):1661–1665. doi: 10.1126/science.1224041. [DOI] [PubMed] [Google Scholar]
- Wu YW, Tang YH, Tringe SG, Simmons BA, Singer SW. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome. 2014;2(1):26. doi: 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Ren M, Yang C, Yi H, Li Z, Li T, Zhao J. Metagenomic analysis reveals symbiotic relationship among bacteria in Microcystis-dominated community. Front Microbiol. 2016 doi: 10.3389/fmicb.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Zhang W, Ren M, Song L, Li T, Zhao J. Whole-genome sequence of Microcystis aeruginosa TAIHU98, a nontoxic bloom-forming strain isolated from Taihu Lake, China. Genome Announc. 2013;1(3):e00333. doi: 10.1128/genomeA.00333-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Lin F, Li Q, Li T, Zhao J. Comparative genomics reveals diversified CRISPR-Cas systems of globally distributed Microcystis aeruginosa, a freshwater bloom-forming cyanobacterium. Front Microbiol. 2015 doi: 10.3389/fmicb.2015.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Rodionov DA, Gelfand MS, Gladyshev VN. Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genom. 2009;10:78. doi: 10.1186/1471-2164-10-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Zancarini A, Louati I, De Cesare S, Duval C, Tambosco K, Bernard C, Debroas D, Song L, Leloup J, Humbert JF. Bacterial communities associated with four cyanobacterial genera display structural and functional differences: evidence from an experimental approach. Front Microbiol. 2016;7:1662. doi: 10.3389/fmicb.2016.01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zydowsky TM, Courtney LF, Frasca V, Kobayashi K, Shimizu H, Yuen LD, Matthews RG, Benkovic SJ, Floss HG. Stereochemical analysis of the methyl transfer catalyzed by cobalamin-dependent methionine synthase from Escherichia coli B. J Am Chem Soc. 1986;108(11):3152–3153. doi: 10.1021/ja00271a081. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw Illumina sequence of coculture described in this manuscript has been deposited in the Sequence Read Archive of the National Center for Biotechnology Information (Accession Number: SRP075385). The coculture assembly has also been deposited at DDBJ/ENA/GenBank under the accession number LYXM00000000. The version described in this paper is version LYXM01000000. The genome sequences of Pseudomonas stutzeri TAIHU and Mesorhizobium sp. TAIHU, can be retrieved from the assembly based on the scaffold list belonging to each species in Additional file 1: Table S7.