

Abstract
Background/Aims
Recent findings have demonstrated the occurrence of neutrophil transendothelial migration in the reverse direction (reverse TEM) and that endothelial junctional adhesion molecule C (JAM-C) is a negative regulator of reverse TEM. In this study, we tested the effects of a JAM-C blocking antibody on the resolution of kidney injuries and inflammation in a mouse model of cisplatin-induced acute kidney injury (AKI).
Methods
Cisplatin was administered via intraperitoneal injection. A JAM-C blocking antibody or a control immunoglobulin G was administered intraperitoneal at 1.5 mg/kg, with the injection being delayed until day 4 following cisplatin administration to restrict the effect of antibodies on recovery.
Results
After cisplatin injection, serum creatinine and histologic injuries peaked on day 4. Treatment with a JAM-C blocking antibody on days 4 and 5 promoted the functional and histologic recovery of cisplatin-induced AKI on days 5 and 6. Facilitating recovery with a JAM-C blocking antibody correlated with significantly increased circulating intercellular adhesion molecule 1+ Tamm-Horsfall protein+ neutrophils and significantly decreased renal neutrophil infiltration, indicating that facilitating reverse the TEM of neutrophils from the kidney to the peripheral circulation partially mediated the resolution of inflammation and recovery.
Conclusions
These results demonstrated that reverse TEM is involved in the resolution of neutrophilic inflammation in cisplatin-induced AKI and that JAM-C is an important regulator of this process.
Keywords: Junctional adhesion molecule C, Transendothelial and transepithelial migration, Cisplatin, Acute kidney injury
INTRODUCTION
Cisplatin is one of the most effective chemotherapeutic agents against solid tumors, but renal toxicity occurs in 25% to 30% of patients receiving the drug. Cisplatin- based nephrotoxicity develops in a dose-dependent manner and prevents optimal cisplatin dosing for tumor treatment [1,2]. Cisplatin treatment can lead to tubulointerstitial inflammation, resulting in acute kidney injury (AKI), and its long-term use may cause chronic fibrosis [3]. Inflammation plays an important role in cisplatin nephrotoxicity, and many studies have been conducted in attempt to elucidate the mechanism of this inflammatory response [4,5].
As a sustained immune response may result in severe tissue damage, resolution of inflammation may be an essential step in tissue regeneration and repair. However, the mechanism of recovery following inflammation has not been clearly identified. Inflammatory responses are characterized by the infiltration of neutrophils, macrophages, and natural killer T cells into tissues. Neutrophils in particular are known to penetrate into tissues in the early phase of inflammation, causing an innate immune response [6]. To migrate from the vascular lumen into inflamed tissues, neutrophils must pass through endothelial blood vessel cells, a process known as transendothelial migration (TEM).
The junctional adhesion molecule C (JAM-C) has been reported to block the movement of neutrophils from inflamed tissue into the systemic circulation, a process referred to as reverse TEM. The genetic deletion of JAM-C or the use of a JAM-C-blocking antibody can promote reverse TEM [7]. In a recent study, neutrophils that underwent reverse TEM were reported to express intercellular adhesion molecule 1 (ICAM-1) [8]. When using a JAM-C-blocking antibody in an ischemic/reperfusion injury model, an increase of ICAM-1+ neutrophils in the blood was observed [7]. These results suggest that neutrophils infiltrating into inflamed tissues can be removed through reverse TEM; thereby, mitigating the inflammatory response. However, no reports have documented the effect of a JAM-C-blocking antibody on cisplatin-induced AKI.
We therefore examined the effect of a JAM-C-blocking antibody on the removal of tissue neutrophils in the recovery phase of cisplatin-induced AKI. We also investigated whether these changes could mitigate inflammatory responses in the kidney and lead to functional and histological improvement.
METHODS
Animals and drugs
Male C57BL/6 mice (7- to 8-week-old) were purchased from Orient (Charles River Korea, Seoul, Korea) and allowed unrestricted access to food and water before experimentation began. Animal care complied with the rules of the Animal Care Committee of Korea University for the use of laboratory animals in research. Cisplatin (Sigma-Aldrich, St. Louis, MO, USA) was diluted in normal saline to a final concentration of 2 mg/mL. Cisplatin- treated mice were given a single intraperitoneal injection of cisplatin (18 mg/kg), while a control group received the same volume of normal saline. To test the effect of a JAM-C blocking antibody in the recovery phase of cisplatin-induced AKI, either a monoclonal anti-mouse JAM-C antibody (clone H33, Millipore, Billerica, MA, USA) or a control immunoglobulin G (IgG) was administered via intraperitoneal injection at 1.5 mg/kg on day 4 and 5 following cisplatin injection. Mice were sacrificed on days 4, 5, or 6 days after cisplatin administration. Their blood was collected by intracardiac puncture, and both kidneys were processed for histological examination and RNA isolation.
Biochemical analysis and histological examination
Blood urea nitrogen (BUN) and creatinine levels were measured using a Hitachi 747 automatic analyzer (Black Scientific Inc., Bohemia, NY, USA). Renal tissue samples were fixed in 4% buffered paraformaldehyde and embedded in paraffin. After deparaffinization, 5-mm sections were processed and stained with periodic acid-Schiff. Tubular damage was defined by the observations of tubular epithelial swelling, loss of brush border, vacuolar degeneration, necrotic tubules, cast formation, and desquamation. Tubular damage was estimated by examining 8 to 10 high-power fields (HPFs; × 200) persection and using a scoring system based on the percentage of damaged tubules per field (1, < 25%; 2, 25% to 50%; 3, 50% to 75%; 4, > 75%). The mean scores were compared between the groups. For the immunohistochemical detection of neutrophils, kidney tissues were stained with an anti-Ly6G (lymphocyte antigen 6 complex, locus G) antibody (eBioscience, San Diego, CA, USA). Eight to 10 HPFs (× 200) of the outer stripe of the outer medulla were captured, and the mean number of Ly6G-positive cells per HPF was also compared.
Real-time polymerase chain reaction
RNA was extracted from kidney tissues using TRIzol (Invitrogen, Life Technologies, Seoul, Korea) according to the manufacturer’s instructions. RNA was purified using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Then, 1 μg total RNA was reverse transcribed in a reaction volume of 50 μL containing 10 × RT buffer, 5.5 mM MgCl2, 500 μM of each deoxynucleotide, 2.5 μM random hexamers, 0.4 U/μL RNase inhibitor, and 3.125 U/μL MultiScribe Reverse Transcriptase (TaqMan Reverse Transcription Reagents, Applied Biosystems Inc., Foster City, CA, USA) at 25°C for 10 minutes, 48°C for 30 minutes, and 95°C for 5 minutes. Subsequently, real- time polymerase chain reaction (PCR) was performed with the iCycler IQ Real-time PCR Detection system (Bio-Rad, Hercules, CA, USA) under the amplification conditions of 40 cycles of 95°C, 15 seconds and 60°C, 1 minute. We used 18S ribosomal RNA (TaqMan ribosomal control reagent, Applied Biosystems Inc.) as an internal control to normalize the data.
Flow cytometry
To detect ICAM-1+ neutrophils in the blood, peripheral blood cells were stained with antibodies against mouse granulocyte receptor 1 (Gr-1) and mouse ICAM-1 (eBioscience), or mouse ICAM-1 and mouse Tamm-Horsfall protein (BIOSS antibodies, Woburn, MA, USA). Four-color fluorescence analyses were performed (FACSCalibur, BD Bioscience, San Jose, CA, USA) to determine the percentages of ICAM-1+ reverse-transmigrated neutrophils and ICAM-1+ Tamm-Horsfall protein+ neutrophils in the peripheral blood. These data were analyzed using FlowJo software (Tree Star Inc., Ashland, CA, USA).
Statistical analysis
The data were presented as the mean ± SE and analyzed using the Kruskal-Wallis test. A p < 0.05 was considered statistically significant.
RESULTS
Effects of in vivo blockade of JAM-C on circulating ICAM-1+ neutrophils
Neutrophils that have remigrated from inflamed tissue to the systemic circulation through the endothelial barrier are known to express ICAM-1. Our flow cytometry results showed that, at 5 days after cisplatin administration, the number of ICAM-1+, Gr-1+ neutrophils in the peripheral blood had significantly increased in the JAM-C blocking antibody-treated group, compared with that of the IgG-treated control group (Fig. 1).
Figure 1.
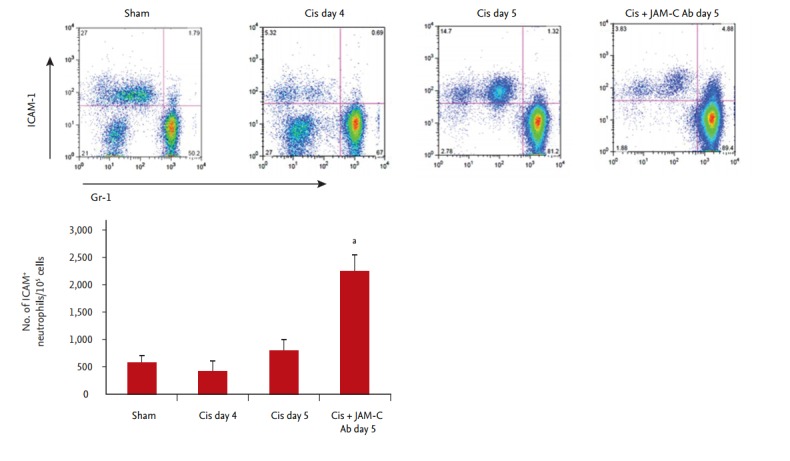
Effect of an in vivo junctional adhesion molecule C (JAM-C) blockade on circulating intercellular adhesion molecule 1 (ICAM-1)+ neutrophils. Flow cytometry results showed the number of ICAM-1+ granulocyte receptor 1+ (Gr-1+) reverse-transmigrated neutrophils in the peripheral blood significantly increased in the JAM-C blocking antibody (Ab)-treated group, compared with that observed in the immunoglobulin G (IgG)-treated control group (n = 6 animals per group). Cis, cisplatin. ap < 0.05, compared with the control IgG group.
Effects of in vivo blockade of JAM-C on circulating ICAM-1+, Tamm-Horsfall protein+ neutrophils
To confirm the origin of the ICAM-1+ reverse-transmigrated neutrophils, we examined their expression of the Tamm-Horsfall protein by flow cytometry. At 5 days after cisplatin injection, more ICAM-1+, Tamm-Horsfall protein+ neutrophils were present in the JAM-C blocking antibody-treated group than in the control IgG group (Fig. 2). These results suggested that the considerable increase in ICAM-1+ neutrophils in the peripheral blood was due to their return to the systemic circulation from previously infiltrated kidney tissue.
Figure 2.

Effects of an in vivo junctional adhesion molecule C (JAM-C) blockade on circulating intercellular adhesion molecule 1+ (ICAM-1+), Tamm-Horsfall protein+ (THP+) neutrophils. After gating neutrophils based on their forward scatter and side scatter properties, we prepared a bivariate histogram showing ICAM-1+ and THP+ cells. ICAM-1+ , THP+ neutrophil levels increased in the JAM-C blocking antibody (Ab)-treated group, compared to those found in control immunoglobulin G (IgG)-treated group on day 5 after cisplatin injection (n = 6 animals per group). Cis, cisplatin. ap < 0.05, compared with control IgG group.
In vivo blockade of JAM-C resulted in faster resolution of neutrophilic inflammation
To determine whether the increase in reverse-transmigrated neutrophils (ICAM-1+, Gr-1+ neutrophils) in the peripheral blood led to a decrease in kidney-infiltrated neutrophils, we also estimated kidney-infiltrated neutrophils at 5 days after cisplatin injection. Ly6G staining revealed that, at 5 days after cisplatin injection, the number of kidney-infiltrated neutrophils significantly decreased in the JAM-C blocking antibody-treated group, compared with that determined for the control IgG-treated group (Fig. 3A). We investigated the effects of the JAM-C blocking antibody on the expression of kidney cytokines (interleukin 6 [IL-6] and tumor necrosis factor α [TNF-α]), using real-time PCR. By day 5, expression of the proinflammatory cytokine IL-6 decreased significantly in the group treated with JAM-C blocking antibody (Fig. 3B). TNF-α levels showed no statistically significant difference between groups.
Figure 3.

In vivo blockade of junctional adhesion molecule C (JAM-C) resulted in faster resolution of neutrophilic inflammation. (A) The number of infiltrated neutrophils in the kidney significantly decreased in the JAM-C blocking antibody (Ab)-treated group, compared to that found in the control immunoglobulin G (IgG)-treated group on day 5 after cisplatin injection (lymphocyte antigen 6 complex, locus G [Ly6G] stain, ×200; n = 6 animals per group). (B) The expression of interleukin 6 (IL-6) significantly decreased in the JAM-C blocking Ab-treated group (n = 6 animals per group). Tumor necrosis factor α (TNF-α) levels showed no statistically significant differences between groups. Cis, cisplatin; HPF, high-power field. ap < 0.05, compared with the sham or control IgG group.
In vivo blockade of JAM-C resulted in faster functional and histological recovery
BUN and creatinine levels peaked on the 4th day of cisplatin administration and subsequently decreased. On the 5th and 6th days, declines in the BUN and creatinine levels were significantly higher in the JAM-C blocking antibody-treated group, compared with the corresponding levels measured in the control IgG-treated group (Fig. 4A). In addition, the tubular injury score measured on the 5th day of cisplatin administration was also less in the JAM-C blocking antibody-treated group than in the control IgG-treated group (Fig. 4B).
Figure 4.
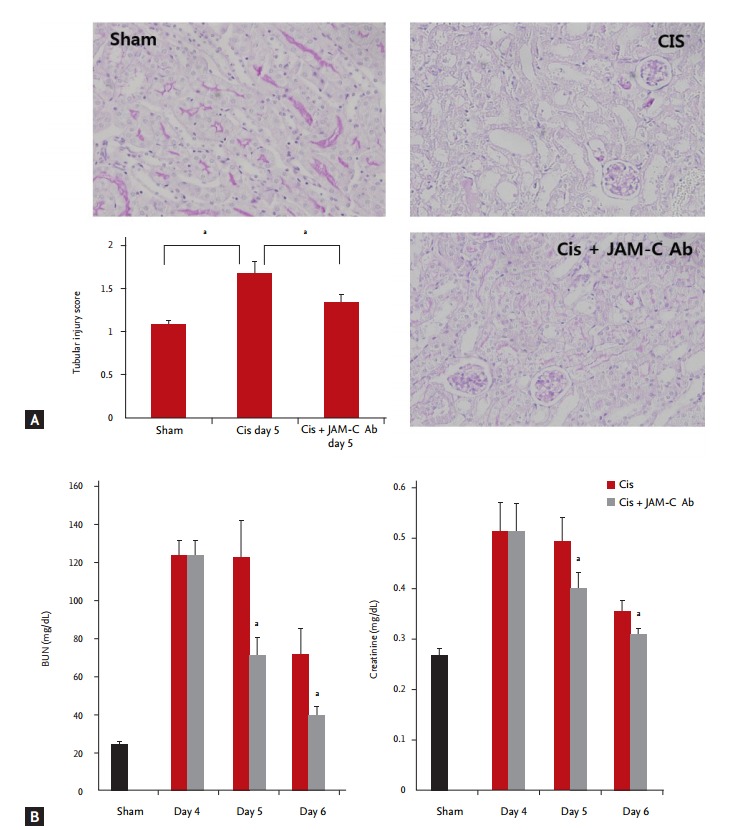
In vivo blockade of junctional adhesion molecule C (JAM-C) resulted in faster functional and histologic recovery. (A) Declines in blood urea nitrogen (BUN) and creatinine levels significantly increased in the JAM-C blocking antibody (Ab)-treated group, compared to those measured in the control immunoglobulin G (IgG)-treated group (n =18 animals per group). (B) Tubular injury scores measured on the 5th day of cisplatin administration decreased in the JAM-C blocking Ab-treated group (lymphocyte antigen 6 complex, locus G [Ly6G] stain, ×200; n = 6 animals per group). Cis, cisplatin. ap < 0.05, compared with the sham or control IgG group.
DISCUSSION
The mechanisms of cisplatin-induced nephrotoxicity include DNA damage, alteration of the cellular transport system, mitochondrial dysfunction, oxidative stress, inflammatory responses, activation of mitogen-activated protein kinases, activation of apoptotic pathways, and endothelial dysfunction [3]. Inflammatory responses are particularly well known to mediate cisplatin-induced nephrotoxicity. Increased inflammatory cells and proinflammatory cytokines were observed in cisplatin-induced AKI in previous studies [5,9,10]. In this study, increases in infiltrated neutrophils and IL-6 levels were observed in kidney tissue at 4 days after cisplatin administration. These findings suggested that immune cells and inflammatory responses were involved in cisplatin- induced AKI. In addition, previous data showed that infiltrated inflammatory cells from the blood could amplify inflammatory cytokine production and chemokine secretion; thereby, promoting tissue damage and ultimately a reduction in renal function and fibrosis [11]. For the induction of inflammation, neutrophils in the blood must infiltrate inflamed tissue by crossing the vascular endothelial barrier. This process involves a variety of factors, such as PECAM-1 (platelet endothelial cell adhesion molecule 1), ICAM-2, CD99, ESAM (endothelial cell-selective adhesion molecule), and junctional adhesion molecules. To end inflammation, infiltrated inflammatory cells such as neutrophils, macrophages, and natural killer T-cells must be removed. Until recently, the resolution of inflammation was thought to be a passive process related to the disappearance of proinflammatory stimuli. However, recent findings have demonstrated the involvement of an active, tightly regulated process that induces the reduction of proinflammatory mediators and the removal of inflammatory cells [12,13]. Although the mechanisms of infiltrated neutrophil removal are known to include apoptosis and phagocytosis, recent data have shown that infiltrated neutrophils can be removed through their reentry into systemic circulation by reverse TEM [7,14,15].
JAM-C is located in the endothelial cell junction and regulates the unidirectional TEM of neutrophils from the blood to tissues [16,17]. Woodfin et al. [7] presented real-time confocal images showing that neutrophil TEM was delayed or even reversed following ischemic/reperfusion injury. These phenomena are related to the disappearance of JAM-C at the endothelial cell junction. The genetic deletion of JAM-C or employing a JAM-C-blocking antibody promoted the reverse TEM of neutrophils [7].
Because this study was aimed at investigating the effects of a JAM-C-blocking antibody on the recovery phase of cisplatin-induced AKI, the JAM-C-blocking antibody was administered beginning at day 4 postcisplatin administration, when maximum renal injury is normally observed. On day 5, the number of infiltrated neutrophils in the kidneys of animals treated with JAM-C-blocking antibody had decreased. TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining was performed to detect apoptotic neutrophils in the kidney. No significant differences were observed in the number of apoptotic neutrophils detected between the JAM-C-blocking antibody-treated group and the control IgG-treated group (data not shown). In addition, on day 5, in contrast to tissue neutrophils, ICAM-1+ reverse-transmigrated neutrophils in the peripheral blood increased in the JAM-C-blocking antibody-treated group. To detect the origin of these ICAM-1+ neutrophils in the blood, we stained for the Tamm-Horsfall protein, which is released from the apical domain of the thick ascending limb of the loop of Henle into the interstitium and circulation during the recovery phase of kidney injury [18]. In this study, we observed that ICAM-1+, Tamm-Horsfall protein+ neutrophils increased in the blood of the JAM-C-blocking antibody-treated group. These results suggested that blocking JAM-C facilitates the reverse TEM of neutrophils in inflamed kidney tissue.
IL-6, a proinflammatory cytokine, is known to increase in cisplatin-induced AKI [9,10]. In this study, IL-6 levels were also increased on day 4 after cisplatin injection, after which a tendency towards decreased production was observed. Significant declines in IL-6 production were observed in JAM-C-blocking antibody-treated animals, compared to the control IgG-treated group on day 5. This finding suggested that the removal of tissue neutrophils by reverse TEM might facilitate the reduction of inflammation. TNF-α also plays a central role in mounting inflammatory responses by stimulating the expression of other inflammatory mediators, such as transforming growth factor β, RANTES (regulated on activation, normal T cell expressed and secreted), MIP-2 (macrophage inflammatory protein 2), monocyte chemoattractant protein 1 (MCP-1), IL-1b, and ICAM-1 during cisplatin-induced nephrotoxicity [19]. However, in this study we did not observe a statistically significant difference in TNF-α levels between the JAM-C-blocking antibody-treated group and the control IgG-treated group. This outcome may have occurred because the TNF-α level was already reduced on day 5, regardless of JAM-C antibody treatment.
Finally, we observed that declines in BUN and creatinine levels were significantly enhanced in the JAM-C-blocking antibody-treated animals during the recovery phase of cisplatin-induced AKI. In histologic examinations, tubular injury scores measured on the 5th day of cisplatin administration were also less in the JAM-C-blocking antibody-treated group. These results showed that the removal of infiltrated tissue neutrophils and the reduction of neutrophilic inflammation through the promotion of reverse TEM facilitated functional and histological kidney recovery. During this process, JAM-C is an important regulator of reverse TEM in cisplatin-induced AKI. However, it is possible that the reduction of local inflammatory responses through the promotion of reverse TEM is associated with systemic inflammation. ICAM-1+ reverse-transmigrated neutrophils have a greater ability to generate reactive oxygen species [8]. Woodfin et al. [7] demonstrated that the number of ICAM-1+ neutrophils in the lung vasculature was significantly higher in the JAM-C-blocking antibody-treated group than the control IgG-treated group in a cremaster muscle ischemic/reperfusion injury model. A more recent study showed evidence that severe lung injury was induced in JAM-C knock out pancreatitis model mice [20]. In this study, we use a JAM-C-blocking antibody instead of JAM-C knockout mice. The use of low-dose JAM-C-blocking antibody or injection of JAM-C during the recovery phase may facilitate recovery from kidney injury, without significant damage to other organs. Thus, further evaluation of the dose or time dependence of JAM-C blocking may be required.
In conclusion, the use of a JAM-C-blocking antibody during cisplatin-induced AKI facilitated reverse TEM. This reduced the number of infiltrated tissue neutrophils and the inflammatory response, resulting in the promotion of functional and histological kidney recovery. These findings are expected to be useful for establishing new therapeutic strategies that may assist in the recovery from cisplatin-induced AKI.
KEY MESSAGE
1. Junctional adhesion molecule C (JAM-C) is an important regulator of reverse transendothelial migration (TEM) in cisplatin-induced acute kidney injury.
2. In vivo blockade of JAM-C was associated with significantly increased circulating intercellular adhesion molecule 1+ Tamm-Horsfall protein+ neutrophils and significantly decreased renal neutrophil infiltration.
3. The removal of infiltrating tissue neutrophils through the promotion of reverse TEM facilitated functional and histological kidney recovery.
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.dos Santos NA, Carvalho Rodrigues MA, Martins NM, dos Santos AC. Cisplatin-induced nephrotoxicity and targets of nephroprotection: an update. Arch Toxicol. 2012;86:1233–1250. doi: 10.1007/s00204-012-0821-7. [DOI] [PubMed] [Google Scholar]
- 2.Peres LA, da Cunha AD., Jr Acute nephrotoxicity of cisplatin: molecular mechanisms. J Bras Nefrol. 2013;35:332–340. doi: 10.5935/0101-2800.20130052. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez-Gonzalez PD, Lopez-Hernandez FJ, Lopez-Novoa JM, Morales AI. An integrative view of the pathophysiological events leading to cisplatin nephrotoxicity. Crit Rev Toxicol. 2011;41:803–821. doi: 10.3109/10408444.2011.602662. [DOI] [PubMed] [Google Scholar]
- 4.Choi DE, Jeong JY, Lim BJ, Lee KW, Shin YT, Na KR. Pretreatment with darbepoetin attenuates renal injury in a rat model of cisplatin-induced nephrotoxicity. Korean J Intern Med. 2009;24:238–246. doi: 10.3904/kjim.2009.24.3.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang KP, Kim DH, Jung YJ, et al. Alpha-lipoic acid attenuates cisplatin-induced acute kidney injury in mice by suppressing renal inflammation. Nephrol Dial Transplant. 2009;24:3012–3020. doi: 10.1093/ndt/gfp242. [DOI] [PubMed] [Google Scholar]
- 6.Kelly KJ, Williams WW, Jr, Colvin RB, et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodfin A, Voisin MB, Beyrau M, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12:761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckley CD, Ross EA, McGettrick HM, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol. 2006;79:303–311. doi: 10.1189/jlb.0905496. [DOI] [PubMed] [Google Scholar]
- 9.Faubel S, Lewis EC, Reznikov L, et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther. 2007;322:8–15. doi: 10.1124/jpet.107.119792. [DOI] [PubMed] [Google Scholar]
- 10.Kim MG, Yang HN, Kim HW, Jo SK, Cho WY, Kim HK. IL-10 mediates rosiglitazone-induced kidney protection in cisplatin nephrotoxicity. J Korean Med Sci. 2010;25:557–563. doi: 10.3346/jkms.2010.25.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramesh G, Reeves WB. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol. 2003;285:F610–F618. doi: 10.1152/ajprenal.00101.2003. [DOI] [PubMed] [Google Scholar]
- 12.Henson PM. Dampening inflammation. Nat Immunol. 2005;6:1179–1181. doi: 10.1038/ni1205-1179. [DOI] [PubMed] [Google Scholar]
- 13.Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 14.Saverymuttu SH, Peters AM, Keshavarzian A, Reavy HJ, Lavender JP. The kinetics of 111indium distribution following injection of 111indium labelled autologous granulocytes in man. Br J Haematol. 1985;61:675–685. doi: 10.1111/j.1365-2141.1985.tb02882.x. [DOI] [PubMed] [Google Scholar]
- 15.Wei Q, Dong G, Chen JK, Ramesh G, Dong Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013;84:138–148. doi: 10.1038/ki.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aurrand-Lions M, Lamagna C, Dangerfield JP, et al. Junctional adhesion molecule-C regulates the early influx of leukocytes into tissues during inflammation. J Immunol. 2005;174:6406–6415. doi: 10.4049/jimmunol.174.10.6406. [DOI] [PubMed] [Google Scholar]
- 17.Bradfield PF, Scheiermann C, Nourshargh S, et al. JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood. 2007;110:2545–2555. doi: 10.1182/blood-2007-03-078733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Achkar TM, McCracken R, Liu Y, et al. Tamm-Horsfall protein translocates to the basolateral domain of thick ascending limbs, interstitium, and circulation during recovery from acute kidney injury. Am J Physiol Renal Physiol. 2013;304:F1066–F1075. doi: 10.1152/ajprenal.00543.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835–842. doi: 10.1172/JCI15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu D, Zeng Y, Fan Y, et al. Reverse-migrated neutrophils regulated by JAM-C are involved in acute pancreatitis-associated lung injury. Sci Rep. 2016;6:20545. doi: 10.1038/srep20545. [DOI] [PMC free article] [PubMed] [Google Scholar]