

Abstract
Forkhead box class O family member proteins (FoxOs) of transcription factors are essential regulators of cellular homeostasis, including glucose and lipid metabolism, oxidative stress response and redox signaling, cell cycle progression, and apoptosis. Altered FoxO1 expression and activity have been associated with glucose intolerance, dyslipidemia and complications of diabetes. In the liver, they direct carbons to glucose or lipid utilization, thus providing a unifying mechanism for the two abnormalities of the diabetic liver: excessive glucose production, and increased lipid synthesis and secretion. In the pancreas, FoxO1 is necessary to maintain β‐cell differentiation, and could be promising targets for β‐cell regeneration. In endothelial cells, FoxOs strongly promote atherosclerosis through suppressing nitric oxide production and enhancing inflammatory responses. In the present review, we summarize the basic biology and pathophysiological significance of FoxOs in diabetes.
Keywords: Diabetes, Forkhead box class O family member proteins, Insulin signaling
Introduction
Forkhead box class O family member proteins (FoxOs) of transcription factors control many cellular processes, including cell cycle and survival, and metabolism1, 2. FoxOs consist of four members, namely FoxO1 (also known as FKHR), FoxO3a (also known as FKHRL1), FoxO4 (also known as AFX1) and FoxO6. In addition to the nature and multitude of FoxOs regulatory mechanisms, as well as the variety of gene expression programs regulated by FoxOs, accumulated evidence has led to the integrating hypothesis that FoxOs function to maintain cellular homeostasis in response to internal and external environmental changes. Analyses of FoxOs ablation in animals have provided insight into their physiological significance. Global ablation of FoxO1 is lethal; it results in embryonic cell death because of impaired vascular development3, 4. FoxO3a‐null mice showed age‐dependent infertility, and had abnormal ovarian follicular development5 and a decrease in the neural stem cell pool6. Global ablation of FoxO4 enhances colon inflammation in response to inflammatory stimuli7. Global ablation of FoxO6 has no effects on learning function, but results in impaired memory consolidation8. Therefore, the main role of FoxOs appears to be homeostasis regulation, especially in response to stress.
Here, we focused on recent developments in the biology of FoxOs, including regulatory systems for the activity of FoxOs, and processes that are regulated by these transcription factors. We further described the regulation of FoxOs through post‐translational modifications, including the mechanisms by which FoxOs regulate transcription of target genes. Finally, we described the pathophysiological significance of FoxOs, especially in glucose and lipid metabolism, and diabetic complications.
Regulation of the FoxOs Activity
The activity of FoxOs is mainly regulated by a complex array of post‐translational modifications, which can be activating or inactivating1. Post‐translational modifications change nuclear import and export of FoxOs, modify deoxyribonucleic acid (DNA)‐binding affinity, and alter the pattern of transcriptional activity for target genes. FoxOs share significant sequence homology and possess four distinct functional motifs, namely, forkhead, nuclear localization, nuclear export and transactivation domains (Figure 1). These domains are highly conserved, and FoxO1 and FoxO3a are larger (>650 amino acids) than FoxO4 and FoxO6, which are nearly 500 amino acids in length. FoxOs have nuclear localization and export sequences within the C‐terminal DNA‐binding domain. Two response elements are recognized by FoxOs: Daf‐16 family member‐binding element (5′‐GTAAA[T/C]AA)9, 10 and insulin‐responsive element (5′‐[C/A][A/C]AAA[C/T]AA)11, 12. FoxOs are reported to have higher affinity for the Daf‐16 family member‐binding element13. Kinases and interactions with other proteins modulate the effectiveness of these nuclear localization and export sequences, making the basis of FoxOs shuttling between in and out of the nuclear compartment. The cytoplasmic sequestration of FoxOs is mediated by a combination of binding partners and changes in the properties of FoxOs. The chaperone protein 14‐3‐3 permits the active export by binding to FoxOs in the nucleus14, 15, 16, 17. It is also able to inhibit re‐entry into the nucleus by blocking the nuclear localization signal.
Figure 1.
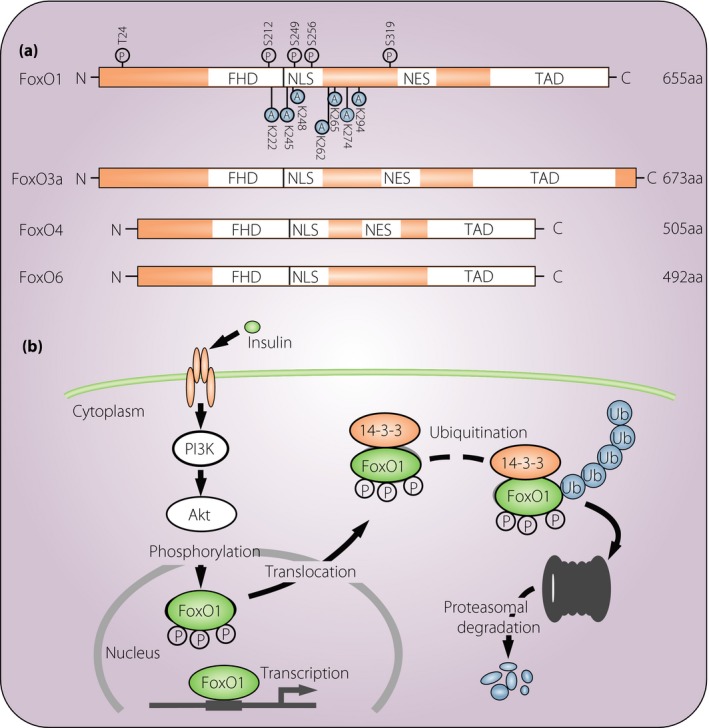
Structure and regulation of forkhead box class O family member proteins (FoxOs). (a) Schematic diagram showing the different domains that characterize human FoxOs. FoxOs share a highly conserved forkhead deoxyribonucleic acid‐binding domain (FHD), which binds to conserved sequences in the target genes. Acetylation and phosphorylation sites are depicted only in FoxO1 as a representative. (b) A model for FoxO1 regulation through insulin‐induced and phosphorylation‐dependent degradation. A, acetylation site; aa, amino acids; NES, nuclear export sequence; NLS, nuclear localization sequence; P, phosphorylation site; TAD, transactivation domain.
Phosphorylation
FoxOs are phosphorylated by various kinds of protein kinases, thereby modifying different sites on FoxOs, and altering their subcellular localization, DNA binding affinity and transcriptional activity1, 2. Major factors for the regulation of FoxOs’ activity as transcriptional factors are insulin and insulin‐like growth factor‐1 (IGF‐1), through the phosphoinositide‐3 kinase (PI3K)/Akt signaling pathway. Both insulin and IGF‐1 induce the PI3K/Akt‐dependent phosphorylation of FoxOs, which promotes its interaction with 14‐3‐3, leading to its nuclear exclusion and eventual ubiquitylation‐dependent proteasomal degradation18. Thus, it is well established that insulin, IGF‐1 and PI3K/Akt play key roles in repressing the transcriptional activity of FoxOs. By contrast, protein kinases including c‐Jun N‐terminal kinases, p38, adenosine monophosphate‐activated protein kinase, cyclin‐dependent kinase 1 and macrophage stimulating 1 facilitate the nuclear localization of FoxOs, and increase their transcriptional activity. Blocking FoxOs binding to 14‐3‐3 also promotes its nuclear localization19. It has been reported that the phosphorylation of FoxOs by Akt also disrupts its interactions with DNA. Phosphorylation of FoxOs at the second of the three Akt sites (Ser256 for FoxO1) introduces a negative charge in the positively charged DNA‐binding domain, thereby inhibiting DNA binding.
Acetylation
Similar to phosphorylation‐dependent regulation, acetylation has been shown to both increase and decrease the transcriptional activity of FoxOs, and alter their biological functions. DNA binding activity of FoxOs is reduced by acetylation and enhanced by deacetylation20, 21. Histone acetyltransferase and histone deacetylases control the effect of acetylation on FoxOs. Acetylation of FoxO1 at K222, K245, K248, K262, K265, K274, and K294 was reported to regulate its DNA binding affinity and sensitivity to Akt phosphorylation22, 23, 24. FoxO3a is also acetylated at K242, K259, K271, K290 and K569 by stress stimuli25. Interestingly, increased acetylation of FoxO3a results in the expression of proapoptotic genes (Bim, tumor necrosis factor‐related apoptosis‐inducing ligand, and FasL), whereas the more deacetylated forms are linked to the expression of anti‐oxidant and cytoprotective genes. The binding of FoxOs to cyclic adenosine monophosphate response element‐binding protein‐binding protein (CBP) and its paralog p300 is essential for the transactivation of target genes. However, acetylation itself attenuates the transcriptional activity of FoxOs20. In FoxO1, cyclic adenosine monophosphate response element‐binding protein‐induced acetylation at two basic residues, K242 and K245, located in the C‐terminal region of the DNA‐binding domain reduces its DNA‐binding affinity and transcriptional activity24.
Other regulation
Peroxisome proliferator‐activated receptor gamma (PPARγ) coactivator 1‐alpha is known as an example of close linkages between FoxOs post‐transcriptional modifications and transcriptional cofactor interactions26. PPARγ coactivator 1‐alpha promotes FoxOs GlcNAcylation27. GlcNAcylation in turn directs FoxOs toward gluconeogenic genes through interactions with additional cofactors or target gene promoter sequences. The interaction can be disrupted by insulin/IGF‐1 signaling. In addition, the amount of active FoxOs is constantly replenished by deacetylation enzymes, such as the sirtuins (SIRTs)28. The presence of multiple acetylation sites (seven lysines in FoxO1) provides the potential for considerable promoter specificity through this mechanism. This system results in the dynamic activation of FoxOs, which is important for quick changes in transcriptional programs.
Pathophysiological Roles of FoxOs in Diabetes
Liver
The roles of FoxOs have been extensively examined in insulin target tissues. The liver adapts to feeding through several insulin‐mediated events including increasing glucose uptake into hepatocytes, suppressing gluconeogenesis and glycogenolysis, and upregulating glycogen synthesis. FoxO1 plays a major role in regulating the insulin response, and the liver is one of its critical sites of action. In fasting, the downregulation of insulin action causes gluconeogenesis through a gene induction of glucose‐6‐phosphatase and phosphoenolpyruvate carboxykinase. This response is largely dependent on the interaction between Akt and FoxO1. Constitutive expression of FoxO1 in liver rises fasting blood glucose29. Conversely, liver‐specific ablation of FoxO1 develops fasting hypoglycemia30, 31. The mechanism behind these phenotypes can be straightforwardly explained; fasting activates FoxO1, in which it is dephosphorylated at the Akt sites and localized to the nucleus. It results in the transcriptional induction of two gluconeogenic enzymes, glucose‐6‐phosphatase catalytic subunit and phosphoenolpyruvate carboxykinase32, leading to increased hepatic glucose production. In the fed state, insulin signaling activates insulin receptor, PI3K, and then subsequently activates Akt. Akt then phosphorylates FoxO1, leading to its nuclear exclusion and inactivation with subsequent suppression of gluconeogenesis.
The activity of FoxO1 as a regulator of blood glucose is also modulated by processes other than Akt phosphorylation; the balance between acetylation and deacetylation is the second order of regulation. Deacetylation of FoxO1 by Sirt1 under cellular stress conditions, including that induced by oxygen free radicals, activates FoxO1, overcoming the nuclear exclusion effect of Akt, and promoting the nuclear translocation/retention and expression of FoxO1 target genes involved in gluconeogenesis33. Other deacetylases, class IIa histone deacetylases, have been identified as positive regulators of hepatic FoxO1 in response to glucagon signaling during fasting. They are phosphorylated by adenosine monophosphate‐activated protein kinase, and they translocate to the nucleus, at which they deacetylate and activate FoxOs, inducing the transcription of gluconeogenic genes34.
Other mechanisms have also been reported to play a role in FoxO1 regulation and hepatic glucose metabolism. X‐Box binding protein 1, a transcription factor involved in the unfolded protein response that induces the expression of genes involved in endoplasmic reticulum membrane folding, has been shown to increase insulin sensitivity. This activity is independent of its transcriptional effects, and it can be explained by its direct binding to FoxO1 as a chaperone to promote toward proteosomal degradation35. It has also been reported that O‐linked N‐acetylglucosamine modification plays a specific role in regulating the gluconeogenic function of FoxO136, 37. This glycosylation event increases the transcriptional activity of FoxOs independent of nuclear translocation, and results in the upregulation of gluconeogenic genes including glucose‐6‐phosphatase and phosphoenolpyruvate carboxykinase. Paradoxically, hyperglycemia also induces O‐linked N‐acetylglucosamine modification. It results from PPARγ coactivator 1‐alpha binding to O‐linked N‐acetylglucosamine transferase, and targeting it toward nuclear FoxO127.
The second area of liver metabolic function regulated by FoxOs is lipid metabolism. FoxO1 has an important role in the insulin‐dependent regulation of hepatic very‐low‐density lipoprotein (VLDL) production and persistence of VLDL in the circulation. It is mainly mediated by the transcriptional upregulation of two important proteins, apolipoprotein C‐III and microsomal triglyceride transfer protein38. They play a major role in the regulation of circulating triglycerides during fasting. As discussed previously, in the absence of insulin, the activity of Akt is suppressed, and FoxO1 is transcriptionally active. It causes the upregulation of microsomal triglyceride transfer protein, the rate‐limiting enzyme in hepatic VLDL production, thereby increasing VLDL secretion. In addition, FoxO1 activation also results in increased transcriptional activity and the hepatic secretion of apolipoprotein C‐III. In the circulation, this apolipoprotein inhibits the activity of lipoprotein lipase, which is responsible for the hydrolysis and uptake of the triglyceride component of VLDL and chylomicrons, thus prolonging the persistence of VLDL39. In contrast, feeding inactivated FoxO1, shutting down both these mechanisms and inhibiting post‐prandial hyperglycemia. Under the condition of insulin resistance, this suppression of the activity of FoxO1 does not occur, resulting in both hyperglycemia and hypertriglyceridemia.
Studies using various constitutive active mutations of FoxO1 protein suggested both positive and negative effects of FoxOs on lipid production and accumulation. One mouse model carrying constitutively active FoxO1 using a single Ser253‐mutated phosphorylation site led to increased hepatic triglyceride levels, but lower levels in the circulation40. Another mouse model of the expression of constitutively active FoxO1 using alanine substitution at all three Akt phosphorylation sites was associated with normal hepatic triglyceride levels32, but increased the activity of FoxO1 and led to the suppression of a number of proteins required for lipid synthesis, including sterol regulatory element binding protein‐1c, acetyl‐CoA carboxylase‐α and fatty acid synthase32. These inconsistent data are difficult to explain unambiguously; however, the mutated forms of FoxOs might behave in unexpected ways.
Perhaps the best systems in which to study the net effects of FoxOs on hepatic and serum lipid homeostasis are liver‐specific multiple FoxO knockouts. It has been reported that ablation of FoxO1 caused a decrease in plasma glucose content without a significant effect on lipid metabolism, but simultaneous knockout of FoxO1 and FoxO3a resulted in hepatic steatosis, and increased hepatic lipid secretion and serum triglyceride levels41. Although the precise mechanism of these effects could be undetermined, these authors showed a negative transcriptional effect of FoxO3a, particularly in the FoxO1 and FoxO3a combination, on two important genes of lipid synthesis, namely fatty acid synthase and 5‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase.
β‐Cells
β‐Cell function is regulated by FoxO1 through a dual mode of action. FoxO1 inhibits β‐cell proliferation during hyperglycemia, insulin resistance or differentiation in the developing fetal pancreas. In contrast, FoxO1 protects β‐cells against oxidative stress‐induced damage by glucose or lipid loading. Among the many regulators of β‐cell development and mass, pancreatic and duodenal homeobox factor‐1 (Pdx1) expresses in all pancreatic cells during embryonic development, and is restricted to β‐cells in the adult pancreas42. FoxO1 negatively regulates the expression of Pdx1 by competing with the transcription factor FoxA2 for binding to the Pdx1 promoter43. This effect, along with downregulation of neurogenin 3 and Nkx61, which are required for the development of the four endocrine cell lineages of the pancreas and β‐cell development, respectively, mediates the inhibitory effect of FoxO1 on β‐cell differentiation44. At the same time, the nuclear translocation of FoxO1 accompanies nuclear exclusion of Pdx1, and it is considered as an alternative mechanism to inhibit β‐cell proliferation43. Interestingly, FoxO1 ablation at different time‐points during β‐cell development results in vastly different phenotypes, suggesting that FoxO1 appears to have different physiological functions at different stages of pancreas development45. Loss‐of‐function animal models of FoxO1 clearly showed that it suppresses β‐cell proliferation and function46, 47. Removal of one FoxO1 allele in mice rescues the defects in β‐cell development caused by β‐cell‐specific inactivation of Pdx148. Furthermore, FoxO1 haploinsufficiency partially restores the decrease of β‐cell proliferation in insulin receptor substrate‐2‐knockout mice43, and β‐cell‐specific Pdk1‐knocuout mice48. The role of FoxO1 as a negative regulator of insulin signaling in β‐cells in vivo is supported by the fact that selective overexpression of Akt in β‐cells increases β‐cell survival and size49. A report has shown that overexpression of FoxO1 promotes the proliferation of pancreatic cells through the induction of the expression of cyclin D1, when they are cultured under low‐nutrition conditions50. In terms of failure of β‐cell function observed in type 2 diabetes, although multiple factors are likely to underlie the metabolic abnormalities, a widely held theory is that β‐cell chronically exposed to hyperglycemia causes deterioration of their function, a phenomenon known as ‘glucose toxicity’51. Chronic oxidative stress has been proposed to induce glucose toxicity, under which intracellular glucose concentrations exceed the glycolytic capacity of β‐cells. Under these conditions, glucose is shunted to the enolization pathway, resulting in the generation of superoxide anions and induction of β‐cells’ apoptosis52. In contrast to its inhibitory effects on β‐cell proliferation, FoxO1 prevents β‐cell dysfunction from oxidative stress‐induced damage47. Oxidative stresses overcome the effect of insulin/PI3K/Akt signaling on FoxO1 nuclear exclusion by two potential mechanisms through c‐Jun N‐terminal kinases activation. C‐Jun N‐terminal kinases activation can either directly inhibit insulin‐induced Akt activation or promote the nuclear translocation of FoxO1 in β‐cells32. Nuclear FoxO1 is targeted to promyelotic leukemia protein‐containing subdomains, at which it is deacetylated by Sirt1, resulting in increased expression of the Ins2 gene transcription factors, NeuroD and MafA53. In conclusion, under physiological conditions, FoxO1 regulates β‐cell formation and function through a dual mode of action that requires balanced activity of FoxO1. Activated FoxO1 suppresses β‐cell proliferation, but promotes survival by increasing stress resistance. Thus, FoxO1 hyperactivation or hypoactivation could result in β‐cell failure.
Recent reports have shed light on clinical potential in the field of regenerative medicine; manipulation of FoxOs could convert non‐β‐cells to insulin producing cells. Genetic inactivation of FoxO1 in intestinal endocrine cells results in the expansion of the enteroendocrine neurogenin‐3‐positive progenitor cell pool, and the appearance of functional insulin‐producing cells that express all markers of mature pancreatic β‐cells, and secrete insulin in response to physiological and pharmacological cues. The insulin‐producing cells generated by FoxO1 inactivation in intestinal endocrine cells are able to alleviate diabetes caused by the β‐cell toxin streptozotocin54. Furthermore, FoxO1 inhibition in gut organoid generated from inducible pluripotent stem cells using a dominant‐negative mutant or lentivirus‐encoded small hairpin ribonucleic acid promotes generation of insulin‐positive cells that express all markers of mature pancreatic β‐cells with releasing C‐peptide55. Thus, gut‐targeted FoxO1 inhibition might be a promising strategy to treat human diabetes.
Muscle
FoxO1 promotes the proliferation of myoblasts, the fusion of mononucleated monocytes into myotubes in myogenic lineage specification, and the breakdown of muscle fibers. FoxO1 remains inactive during myoblast proliferation, perhaps through a PI3K/Akt‐independent mechanism of nuclear exclusion; Rho‐associated protein kinase ROCK, a downstream effector of the small GTPase Rho, directly phosphorylates FoxO1 during myoblast proliferation, at the same time it suppresses myoblast differentiation56. In addition to the regulation of myocyte proliferation, mice overexpressing FoxO1 show downregulation of slow‐twitch muscle genes, suggesting that FoxO1 directs myogenic lineage specification57. Consistently, muscle‐specific FoxO1 ablation switches fiber type to MyoD‐containing fast‐twitch myofibers, accompanied with decrease of myogenin‐containing slow‐twitch myofibers58. Additionally, FoxO1 suppresses MyoD‐dependent myogenesis in cultured C2C12 myoblasts. These effects are mediated by a functional and physical interaction of FoxO1 with Notch1 independent of FoxO1's transcriptional function, which leads to co‐repressor clearance from the Notch effector Csl, resulting in the activation of Notch target genes. It involves a direct interaction with Csl, and subsequent stabilization of the FoxO1/Notch1 complex. In vivo studies of FoxO1 inactivation or overexpression showed that it greatly affects skeletal muscle mass. Mice overexpressing FoxO1 lose glycemic control as a result of a decrease in skeletal muscle mass57. In addition to suppression of the myogenic program, this effect is associated with systemic muscle atrophy, a condition that results from the breakdown of muscle fibers. Indeed, transgenic overexpression of FoxO1 in skeletal muscle results in severe muscular atrophy associated with an upregulation of MAFbx/atrogin‐1 and muscle ring‐finger protein 1 expression59. Conversely, muscle‐specific deletion of FoxO members protects from muscle loss as a result of the role of FoxO in the induction of autophagy–lysosome and ubiquitin–proteasome systems60. In the setting of low‐nutrient signaling, the report has shown that FoxOs are required for Akt activity, but not for mTOR signaling. FoxOs control several stress‐response pathways, such as the unfolded protein response, ROS detoxification, DNA repair and translation, suggesting FoxOs in coordinating a variety of stress‐response genes during catabolic conditions. Consistently, muscle‐specific deletion of FoxO1, FoxO3a and FoxO4 completely rescued the reduction of muscle mass in muscle‐specific insulin and IGF‐1 receptors knockout mice61.
FoxO1 also affects glucose and lipid metabolism in skeletal muscle through the expression of three enzymes that switch from the oxidation of carbohydrates during fasting as a major energy source to fatty acids. Under energy deprivation, FoxO1 promotes the expression of pyruvate dehydrogenase kinase‐4, the enzyme that represses glucose oxidation by blocking the activity of pyruvate dehydrogenase62. At the same time, FoxO1 overexpression increases the expression of lipoprotein lipase in C2C12 myoblasts, and increases the plasma levels of the fatty acid translocase CD36, which facilitates fatty acid uptake into skeletal muscle63. In summary, the expression or activity of FoxO1 is increased during fasting to maintain energy homeostasis through the utilization of lipids rather than carbohydrates as the energy source. In starvation, FoxO1 plays a role to supply energy through the breakdown of muscle protein, which consequently causes muscle loss and atrophy, and underlies glucose intolerance in insulin resistance.
Adipocytes
Adipose tissue has a pivotal role in the regulation of energy homeostasis as energy reservoirs that store triglycerides and mobilize them by oxidization during energy deprivation64. In adipocytes, FoxO1 suppresses adipogenesis; expression of a constitutively active form of FoxO1 prevents the differentiation of a preadipocytic cell line65. FoxO1 regulates the expression and activity of two master adipogenic transcription factors, PPARγ and C/EBPα. It suppresses expression of PPARγ through direct binding to its promoter, and activity of PPARγ by competitively inhibiting the formation of the PPARγ/RXR functional complex66. On the contrary, FoxO1 physically interacts with C/EBPα to promote expression of adiponectin65. A dominant‐negative form of FoxO1, with a truncated C‐terminal transactivation domain, promotes adipogenesis in vitro 66, and restores the adipocyte differentiation of embryonic fibroblasts from insulin receptor‐knockout mice. FoxO1 haploinsufficiency also restores adipocyte counts and size in mice fed a high‐fat diet67. Adipose tissue‐specific FoxO1 transgenic mice that express dominant‐negative FoxO1 in white adipose tissue show glucose tolerance and insulin sensitivity, and increases in energy expenditure under normal as well as high‐fat diet‐feeding68. FoxO1 inhibition in brown adipose tissue increases oxygen consumption and the expression of genes promoting mitochondrial metabolism, PPARγ coactivator 1 and uncoupling protein 1. The anti‐adipogenic actions of FoxO1 appear to be under the control of insulin signaling, because cells lacking the insulin receptor, insulin receptor substrate or Akt show increased FoxO1 activation and impaired differentiation69, 70, 71.
Acetylation‐dependent alteration of FoxO1 also affects the biology of adipocytes. Sirt1‐ and Sirt2‐mediated FoxO1 deacetylation inhibits adipocyte differentiation. They simultaneously prevent FoxO1 nuclear exclusion, showing that FoxO1 might also be involved in these suppressive effects in adipocytes72. FoxO1 suppresses adipogenesis at the early stages of adipocyte differentiation, and at the end‐stage of clonal expansion and terminal differentiation by inducing cell cycle arrest through upregulation of the cell cycle inhibitor, p2167. These observations suggest that FoxO1 has a dual function in white and brown adipose tissue. It regulates energy and nutrient homeostasis through energy storage in white adipose tissue, whereas it regulates energy expenditure in brown adipose tissue.
Endothelial cells
In endothelial cells, it has been found that FoxO1 inhibits and increases transactivity of endothelial nitric oxide (NO) synthase (NOS)73 and inducible NOS74, respectively, supporting the expected proatherogenic role of FoxO under insulin‐resistant states in type 2 diabetes. Indeed, endothelial cells from endothelium‐specific FoxO‐1/3a/4‐deficient mice show marked increases of endothelial NOS (eNOS)‐dependent NO production and inhibition of nuclear factor kappa‐B activation. In a model of advanced atherosclerosis and metabolic dysfunction (endothelium‐specific FoxO‐1/3a/4‐deficient mice with low‐density lipoprotein‐receptor‐null background), the animals showed strong protection against the development of vascular dysfunction and atherosclerosis induced by a Western diet75. The mice showed increased arterial relaxation in response to acetylcholine, indicative of increased bioavailability of NO. Consistent with these findings, aortae from these mice showed reduced endothelial inflammation, oxidative stress and atherosclerosis. In this model of metabolic and vascular dysfunction, no effect on metabolic parameters or glucose homeostasis was observed. These data suggest a relatively straightforward mechanism whereby increasing insulin sensitivity in the endothelium has a favorable effect on NO bioavailability and atherosclerosis.
Intriguingly, under standard diet feeding, the endothelium‐specific FoxO‐1/3a/4‐deficient mice were glucose‐intolerant accompanied with hepatic insulin resistance76. In liver sinusoidal endothelial cells of the mice, as expected, eNOS‐derived NO levels increased. Surprisingly, the excess of NO blunted hepatic insulin sensitivity through tyrosine nitration of the insulin receptor in liver. The report suggests that the eNOS‐derived NO has been shown to have a pathophysiological role in obesity‐related insulin resistance at an early stage76. Complementary studies of hyperinsulinemia produced similar results, and pharmacological eNOS inhibition in a model of early insulin resistance partially restored glucose intolerance. It will be interesting to confirm whether, despite insulin resistance and glucose intolerance, FoxOs deletion in the endothelium has a favorable effect on endothelial cell function in atherosclerosis‐prone vessels.
Macrophages
Myeloid ablation of all three FoxO isoforms increased the proliferation rate of granulocyte‐macrophage progenitors in bone marrow, resulting in elevated peripheral numbers of monocytes and granulocytes associated with decreased gene expression related to cell cycle inhibitors, such as cyclin‐dependent kinase inhibitors, to enhance atherosclerotic lesion formation in mice with low‐density lipoprotein‐receptor‐null background77. The FoxO‐1/3a/4‐deficient macrophages showed reduced sensitivity to cholesterol‐induced apoptosis and increased levels of markers of oxidative stress. Furthermore, macrophages from the myeloid‐specific FoxO‐1/3a/4‐deficient mice showed impaired insulin signaling, a hallmark of the insulin‐resistant state. Similarly, reduced hepatic insulin signaling was associated with inducible NOS‐dependent NO production accompanied with increases of protein cysteine nitrosylation and tyrosine nitration in the liver. Inhibition of reactive oxygen species in these mice with the anti‐oxidant N‐acetyl‐l‐cysteine reversed the increase in peripheral white blood cell and monocytes counts, atherosclerotic lesion formation, and the insulin‐resistant phenotype. These findings are not always consistent with the two other reports showing that FoxO1 increases inflammation in macrophages by enhancing the expression of C‐C chemokine receptor type 278, interleukin‐1β79 or components of toll‐like receptor‐4 signaling80. However, these findings provide a significant insight in the pathophysiological significance of myeloid FoxOs in the context of atherosclerotic lesion formation.
Conclusions
The multiple actions of FoxOs in glucose and lipid‐regulating organs suggest FoxO as master regulators of energy metabolism. In addition, accumulating evidence on other organ cells, such as endothelial cells, macrophages and gut epithelial cells, strongly suggests crucial roles of FoxOs to maintain whole‐body homeostasis and diabetes‐related diseases (Figure 2). In light of the different cellular functions, regulation of FoxOs by antagonists in some disease states or agonists in normal and disease conditions might be useful in treating or preventing a wide variety of disorders. A further understanding of their function will provide essential insight into both basic and clinical processes.
Figure 2.
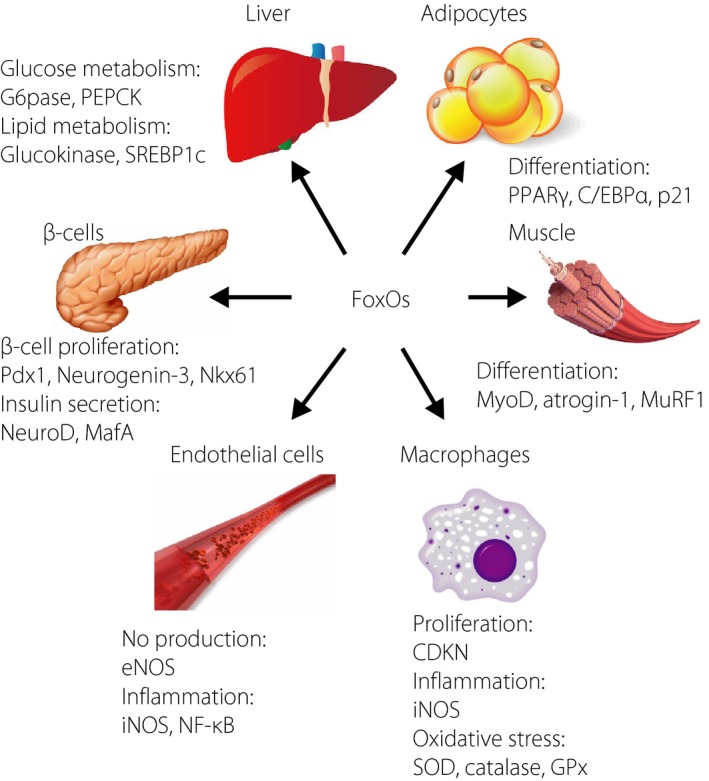
Roles of forkhead box class O family member proteins in various organs. CDKN, cycline‐dependent kinase inhibitor; C/EBPα, CCAAT/enhancer binding protein alpha; eNOS, endothelial nitric oxide synthase; GPx, glutathione peroxidase; iNOS, inducible nitric oxide synthase; MafA; v‐maf musculoaponeurotic fibrosarcoma oncogene family, protein A; MuRF1, muscle ring‐finger protein‐1; MyoD, myogenic differentiation; NeuroD, neurogenic differentiation; NF‐κB, nuclear factor‐kappa B; NOS, nitric oxide synthase; PEPCK, phosphoenolpyruvate carboxykinase; PPAR‐γ, peroxisome proliferator‐activated receptor gamma; SOD, superoxide dismutase; SREBP‐1c, sterol regulatory element‐binding protein 1c.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
Kyoichiro Tsuchiya expresses special appreciation and thanks to Professor Domenico Accili, his mentor at Columbia University.
J Diabetes Investig 2017; 8: 726–734
References
- 1. Lam EW, Brosens JJ, Gomes AR, et al Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer 2013; 13: 482–495. [DOI] [PubMed] [Google Scholar]
- 2. Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 2013; 14: 83–97. [DOI] [PubMed] [Google Scholar]
- 3. Hosaka T, Biggs WH 3rd, Tieu D, et al Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci USA 2004; 101: 2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Furuyama T, Kitayama K, Shimoda Y, et al Abnormal angiogenesis in Foxo1 (Fkhr)‐deficient mice. J Biol Chem 2004; 279: 34741–34749. [DOI] [PubMed] [Google Scholar]
- 5. Lin L, Hron JD, Peng SL. Regulation of NF‐kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity 2004; 21: 203–213. [DOI] [PubMed] [Google Scholar]
- 6. Renault VM, Rafalski VA, Morgan AA, et al FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 2009; 5: 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou W, Cao Q, Peng Y, et al FoxO4 inhibits NF‐kappaB and protects mice against colonic injury and inflammation. Gastroenterology 2009; 137: 1403–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salih DA, Rashid AJ, Colas D, et al FoxO6 regulates memory consolidation and synaptic function. Genes Dev 2012; 26: 2780–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biggs WH 3rd, Cavenee WK, Arden KC. Identification and characterization of members of the FKHR (FOX O) subclass of winged‐helix transcription factors in the mouse. Mamm Genome 2001; 12: 416–425. [DOI] [PubMed] [Google Scholar]
- 10. Furuyama T, Nakazawa T, Nakano I, et al Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF‐16 homologues. Biochem J 2000; 349: 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hall RK, Yamasaki T, Kucera T, et al Regulation of phosphoenolpyruvate carboxykinase and insulin‐like growth factor‐binding protein‐1 gene expression by insulin. The role of winged helix/forkhead proteins. J Biol Chem 2000; 275: 30169–30175. [DOI] [PubMed] [Google Scholar]
- 12. Hribal ML, Nakae J, Kitamura T, et al Regulation of insulin‐like growth factor‐dependent myoblast differentiation by Foxo forkhead transcription factors. J Cell Biol 2003; 162: 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brent MM, Anand R, Marmorstein R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure 2008; 16: 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brunet A, Bonni A, Zigmond MJ, et al Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96: 857–868. [DOI] [PubMed] [Google Scholar]
- 15. Biggs WH 3rd, Meisenhelder J, Hunter T, et al Protein kinase B/Akt‐mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA 1999; 96: 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin‐sensitive pathway. J Biol Chem 1999; 274: 15982–15985. [DOI] [PubMed] [Google Scholar]
- 17. Tang ED, Nunez G, Barr FG, et al Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem 1999; 274: 16741–16746. [DOI] [PubMed] [Google Scholar]
- 18. van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol 2007; 8: 440–450. [DOI] [PubMed] [Google Scholar]
- 19. Obsilova V, Vecer J, Herman P, et al 14‐3‐3 Protein interacts with nuclear localization sequence of forkhead transcription factor FoxO4. Biochemistry 2005; 44: 11608–11617. [DOI] [PubMed] [Google Scholar]
- 20. Lalmansingh AS, Karmakar S, Jin Y, et al Multiple modes of chromatin remodeling by Forkhead box proteins. Biochim Biophys Acta 2012; 1819: 707–715. [DOI] [PubMed] [Google Scholar]
- 21. Calnan DR, Brunet A. The FoxO code. Oncogene 2008; 27: 2276–2288. [DOI] [PubMed] [Google Scholar]
- 22. Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem 2010; 285: 27396–27401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsuzaki H, Daitoku H, Hatta M, et al Acetylation of Foxo1 alters its DNA‐binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA 2005; 102: 11278–11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daitoku H, Hatta M, Matsuzaki H, et al Silent information regulator 2 potentiates Foxo1‐mediated transcription through its deacetylase activity. Proc Natl Acad Sci USA 2004; 101: 10042–10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brunet A, Sweeney LB, Sturgill JF, et al Stress‐dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004; 303: 2011–2015. [DOI] [PubMed] [Google Scholar]
- 26. Puigserver P, Rhee J, Donovan J, et al Insulin‐regulated hepatic gluconeogenesis through FOXO1‐PGC‐1alpha interaction. Nature 2003; 423: 550–555. [DOI] [PubMed] [Google Scholar]
- 27. Housley MP, Udeshi ND, Rodgers JT, et al A PGC‐1alpha‐O‐GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J Biol Chem 2009; 284: 5148–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tanno M, Sakamoto J, Miura T, et al Nucleocytoplasmic shuttling of the NAD+‐dependent histone deacetylase SIRT1. J Biol Chem 2007; 282: 6823–6832. [DOI] [PubMed] [Google Scholar]
- 29. Matsumoto M, Han S, Kitamura T, et al Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest 2006; 116: 2464–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsumoto M, Pocai A, Rossetti L, et al Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 2007; 6: 208–216. [DOI] [PubMed] [Google Scholar]
- 31. Haeusler RA, Kaestner KH, Accili D. FoxOs function synergistically to promote glucose production. J Biol Chem 2010; 285: 35245–35248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang W, Patil S, Chauhan B, et al FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem 2006; 281: 10105–10117. [DOI] [PubMed] [Google Scholar]
- 33. Motta MC, Divecha N, Lemieux M, et al Mammalian SIRT1 represses forkhead transcription factors. Cell 2004; 116: 551–563. [DOI] [PubMed] [Google Scholar]
- 34. Mihaylova MM, Vasquez DS, Ravnskjaer K, et al Class IIa histone deacetylases are hormone‐activated regulators of FOXO and mammalian glucose homeostasis. Cell 2011; 145: 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou Y, Lee J, Reno CM, et al Regulation of glucose homeostasis through a XBP‐1‐FoxO1 interaction. Nat Med 2011; 17: 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Housley MP, Rodgers JT, Udeshi ND, et al O‐GlcNAc regulates FoxO activation in response to glucose. J Biol Chem 2008; 283: 16283–16292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuo M, Zilberfarb V, Gangneux N, et al O‐glycosylation of FoxO1 increases its transcriptional activity towards the glucose 6‐phosphatase gene. FEBS Lett 2008; 582: 829–834. [DOI] [PubMed] [Google Scholar]
- 38. Kamagate A, Qu S, Perdomo G, et al FoxO1 mediates insulin‐dependent regulation of hepatic VLDL production in mice. J Clin Invest 2008; 118: 2347–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Altomonte J, Cong L, Harbaran S, et al Foxo1 mediates insulin action on apoC‐III and triglyceride metabolism. J Clin Invest 2004; 114: 1493–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qu S, Altomonte J, Perdomo G, et al Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology 2006; 147: 5641–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang K, Li L, Qi Y, et al Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 2012; 153: 631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ahlgren U, Jonsson J, Jonsson L, et al beta‐cell‐specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta‐cell phenotype and maturity onset diabetes. Genes Dev 1998; 12: 1763–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kitamura T, Nakae J, Kitamura Y, et al The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 2002; 110: 1839–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Al‐Masri M, Krishnamurthy M, Li J, et al Effect of forkhead box O1 (FOXO1) on beta cell development in the human fetal pancreas. Diabetologia 2010; 53: 699–711. [DOI] [PubMed] [Google Scholar]
- 45. Kitamura T, Kitamura YI, Kobayashi M, et al Regulation of pancreatic juxtaductal endocrine cell formation by FoxO1. Mol Cell Biol 2009; 29: 4417–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakae J, Biggs WH 3rd, Kitamura T, et al Regulation of insulin action and pancreatic beta‐cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 2002; 32: 245–253. [DOI] [PubMed] [Google Scholar]
- 47. Kitamura YI, Kitamura T, Kruse JP, et al FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab 2005; 2: 153–163. [DOI] [PubMed] [Google Scholar]
- 48. Hashimoto N, Kido Y, Uchida T, et al Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat Genet 2006; 38: 589–593. [DOI] [PubMed] [Google Scholar]
- 49. Dickson LM, Rhodes CJ. Pancreatic beta‐cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? Am J Physiol Endocrinol Metab 2004; 287: E192–E198. [DOI] [PubMed] [Google Scholar]
- 50. Ai J, Duan J, Lv X, et al Overexpression of FoxO1 causes proliferation of cultured pancreatic beta cells exposed to low nutrition. Biochemistry 2010; 49: 218–225. [DOI] [PubMed] [Google Scholar]
- 51. Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta‐cell function. Nature 2001; 414: 788–791. [DOI] [PubMed] [Google Scholar]
- 52. Robertson RP, Harmon J, Tran PO, et al Glucose toxicity in beta‐cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003; 52: 581–587. [DOI] [PubMed] [Google Scholar]
- 53. Kawamori D, Kaneto H, Nakatani Y, et al The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX‐1 through its intracellular translocation. J Biol Chem 2006; 281: 1091–1098. [DOI] [PubMed] [Google Scholar]
- 54. Talchai C, Xuan S, Kitamura T, et al Generation of functional insulin‐producing cells in the gut by Foxo1 ablation. Nat Genet 2012; 44: S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bouchi R, Foo KS, Hua H, et al FOXO1 inhibition yields functional insulin‐producing cells in human gut organoid cultures. Nat Commun 2014; 5: 4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bowker‐Kinley MM, Davis WI, Wu P, et al Evidence for existence of tissue‐specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998; 329: 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Constantin‐Teodosiu D, Constantin D, Stephens F, et al The role of FOXO and PPAR transcription factors in diet‐mediated inhibition of PDC activation and carbohydrate oxidation during exercise in humans and the role of pharmacological activation of PDC in overriding these changes. Diabetes 2012; 61: 1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rauramaa R, Kuusela P, Hietanen E. Adipose, muscle and lung tissue lipoprotein lipase activities in young streptozotocin treated rats. Horm Metab Res 1980; 12: 591–595. [DOI] [PubMed] [Google Scholar]
- 59. Kamei Y, Mizukami J, Miura S, et al A forkhead transcription factor FKHR up‐regulates lipoprotein lipase expression in skeletal muscle. FEBS Lett 2003; 536: 232–236. [DOI] [PubMed] [Google Scholar]
- 60. Milan G, Romanello V, Pescatore F, et al Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015; 6: 6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. O'Neill BT, Lee KY, Klaus K, et al Insulin and IGF‐1 receptors regulate FoxO‐mediated signaling in muscle proteostasis. J Clin Invest 2016; 126: 3433–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Furuyama T, Kitayama K, Yamashita H, et al Forkhead transcription factor FOXO1 (FKHR)‐dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 2003; 375: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bastie CC, Nahle Z, McLoughlin T, et al FoxO1 stimulates fatty acid uptake and oxidation in muscle cells through CD36‐dependent and ‐independent mechanisms. J Biol Chem 2005; 280: 14222–14229. [DOI] [PubMed] [Google Scholar]
- 64. Large V, Peroni O, Letexier D, et al Metabolism of lipids in human white adipocyte. Diabetes Metab 2004; 30: 294–309. [DOI] [PubMed] [Google Scholar]
- 65. Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1‐C/enhancer‐binding protein alpha transcriptional complex. J Biol Chem 2006; 281: 39915–39924. [DOI] [PubMed] [Google Scholar]
- 66. Armoni M, Harel C, Karni S, et al FOXO1 represses peroxisome proliferator‐activated receptor‐gamma1 and ‐gamma2 gene promoters in primary adipocytes. A novel paradigm to increase insulin sensitivity. J Biol Chem 2006; 281: 19881–19891. [DOI] [PubMed] [Google Scholar]
- 67. Nakae J, Kitamura T, Kitamura Y, et al The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell 2003; 4: 119–129. [DOI] [PubMed] [Google Scholar]
- 68. Nakae J, Cao Y, Oki M, et al Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes 2008; 57: 563–576. [DOI] [PubMed] [Google Scholar]
- 69. Accili D, Taylor SI. Targeted inactivation of the insulin receptor gene in mouse 3T3‐L1 fibroblasts via homologous recombination. Proc Natl Acad Sci USA 1991; 88: 4708–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miki H, Yamauchi T, Suzuki R, et al Essential role of insulin receptor substrate 1 (IRS‐1) and IRS‐2 in adipocyte differentiation. Mol Cell Biol 2001; 21: 2521–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tseng YH, Kriauciunas KM, Kokkotou E, et al Differential roles of insulin receptor substrates in brown adipocyte differentiation. Mol Cell Biol 2004; 24: 1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab 2007; 6: 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Potente M, Urbich C, Sasaki K, et al Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest 2005; 115: 2382–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tanaka J, Qiang L, Banks AS, et al Foxo1 links hyperglycemia to LDL oxidation and endothelial nitric oxide synthase dysfunction in vascular endothelial cells. Diabetes 2009; 58: 2344–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tsuchiya K, Tanaka J, Shuiqing Y, et al FoxOs Integrate Pleiotropic Actions of Insulin in Vascular Endothelium to Protect Mice from Atherosclerosis. Cell Metab 2012; 15: 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tsuchiya K, Accili D. Liver sinusoidal endothelial cells link hyperinsulinemia to hepatic insulin resistance. Diabetes 2013; 62: 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tsuchiya K, Westerterp M, Murphy AJ, et al Expanded granulocyte/monocyte compartment in myeloid‐specific triple FoxO knockout increases oxidative stress and accelerates atherosclerosis in mice. Circ Res 2013; 112: 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kawano Y, Nakae J, Watanabe N, et al Loss of Pdk1‐Foxo1 signaling in myeloid cells predisposes to adipose tissue inflammation and insulin resistance. Diabetes 2012; 61: 1935–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Su D, Coudriet GM, Hyun Kim D, et al FoxO1 links insulin resistance to proinflammatory cytokine IL‐1beta production in macrophages. Diabetes 2009; 58: 2624–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fan W, Morinaga H, Kim JJ, et al FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J 2010; 29: 4223–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]