

Abstract
Almost all epidermal growth factor receptor (EGFR)‐mutant lung cancers develop resistance to EGFR‐tyrosine kinase inhibitors. Several mechanisms for this acquired resistance have been identified, including development of an EGFR T790M mutation, MET amplification, hepatocyte growth factor overexpression, loss of phosphatase and tensin homolog expression, epithelial–mesenchymal transition, and transformation to small cell lung cancer. Herein, we report a case of a lung cancer patient with EGFR exon 19 deletion who was resistant to EGFR‐tyrosine kinase inhibitor treatment during disease progression. Using histological and gene sequencing analysis, we observed that the primary adenocarcinoma acquired T790M mutation in EGFR exon 20, and a secondary sarcomatoid carcinoma developed in the vicinity. Assessment of E‐cadherin and Vimentin expression confirmed that the sarcomatoid carcinoma had undergone an epithelial–mesenchymal transition. Therefore, it is important to perform a tissue re‐biopsy after the development of resistance to identify the best treatment options. Surgical resection might be a better “salvage” treatment in cases of oligometastatic progression.
Keywords: Adenocarcinoma, drug resistance, EMT, sarcomatoid carcinoma, T790M
Introduction
Drug resistance is the most challenging problem with epidermal growth factor receptor‐tyrosine kinase inhibitor (EGFR‐TKI) treatment in non‐small cell lung cancer (NSCLC) patients. The major mechanisms giving rise to EGFR‐TKI resistance in NSCLC have been demonstrated, including secondary mutation of EGFR T790M, MET amplification, epithelial–mesenchymal transition, activation of alternative oncogenic pathways, and transformation to small cell lung cancer (SCLC).1 Herein, we present a rare case of EGFR T790M secondary mutation accompanied with the phenotype of sarcomatoid carcinoma after EGFR‐TKI treatment.
Case Report
A non‐smoking 60‐year‐old woman was diagnosed via a left supraclavicular lymph node biopsy in December 2012 with left upper lung adenocarcinoma. A gene mutation test indicated EGFR exon 19 deletion without an exon 20 T790M mutation. After two cycles of gemcitabine plus cisplatin chemotherapy, abdominal computed tomography showed liver metastasis; therefore from January 2013, the patient was administered erlotinib. By October 2015, following erlotinib treatment, the metastatic tumors in the liver and supraclavicular lymph nodes had disappeared and the primary lung tumor had reduced. However, the original lung tumor started to enlarge and bred an adjacent secondary nodule (Fig 1). Although drug resistance had occurred, the patient refused any other treatment and continued to take erlotinib. In July 2016, a left upper lobectomy plus systematic mediastinal lymphadenectomy by video‐assisted thoracoscopic surgery was performed after a comprehensive evaluation showed no sign of extrapulmonary abnormalities and negative T790M mutation from a blood circulating tumor (ct)DNA test. Surgery was approved after multiple disciplinary team‐based evaluation and informed consent was acquired from the patient. The pathological diagnosis of the original lung tumor was confirmed to be adenocarcinoma according to the typical morphology and positivity of thyroid transcription factor 1 and cytokeratin (CK)7. The secondary lung tumor had sarcomatoid histology showing a more spindle‐like mesenchymal morphology with CK and Vimentin positivity (Fig 2). N1 but not N2 lymph nodes were involved. Next generation sequencing confirmed that the original lung adenocarcinoma retained EGFR exon 19 deletion but also detected T790M mutation in EGFR exon 20 (File S1, Table S1). The secondary sarcomatoid carcinoma also displayed EGFR exon 19 deletion and genetic mutations on FANCL and BCL2L2, and amplification on CDK4, MDM2, APFRP1, GNAS, CIC, FANCE, Notch4, and AKT2 (Table 1). Additional histological E‐cadherin and Vimentin staining were performed on the primary biopsy specimen and TKI‐resistant postoperative specimens. In comparison to the adenocarcinoma, the sarcomatoid tumor was strongly positive for Vimentin and almost negative for E‐cadherin, indicating that the sarcomatoid probably underwent EMT (Fig 3). A postoperative ctDNA test did not show any driver gene mutation in the blood, including EGFR exon 20 T790M mutation or exon 19 deletion (Burning Rock Dx, Guangzhou, China). Moreover, postoperative circulating tumor cell analysis showed a value of 4.29 (cut‐off 8.70 FU) (Geno, Shanghai, China) (File S1); therefore, we continued to administer erlotinib. The patient has received thorough imaging evaluations and ct cell tests twice every three months, with no sign of relapse. She continues to take erlotinib and is intensively followed‐up.
Figure 1.
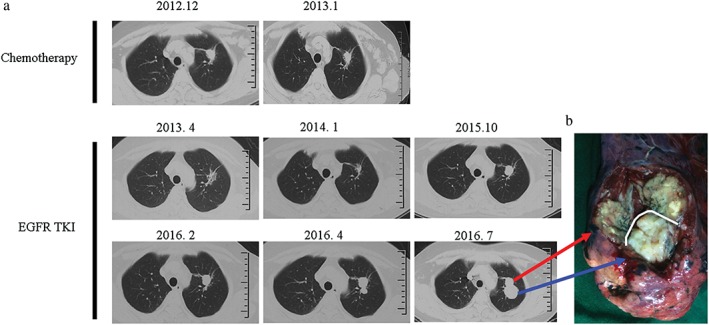
(a) Computed tomography scan and (b) gross specimen. Blue arrow: sarcomatoid carcinoma; red arrow: adenocarcinoma. EGFR‐TKI, epidermal growth factor receptor‐tyrosine kinase inhibitor.
Figure 2.
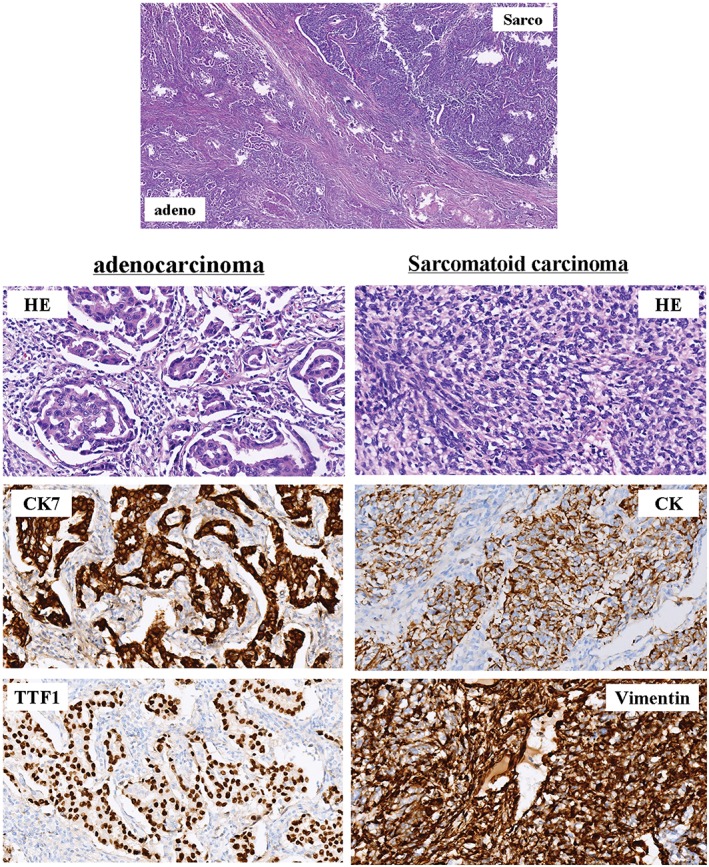
Histology of the two pulmonary nodules after surgical resection. The original lung tumor was confirmed to be adenocarcinoma according to the typical morphology and positivity of thyroid transcription factor 1 (TTF1) and cytokeratin (CK)7. The secondary lung tumor has sarcomatoid histology showing a more spindle‐like mesenchymal morphology with CK and Vimentin positivity (magnification ×100). Adeno, adenocarcinoma; HE, hematoxylin and eosin; Sarco, sarcomatoid carcinoma.
Table 1.
Gene analysis of primary adenocarcinoma and secondary lung sarcomatoid carcinoma from surgical resection (July 2016)
| Gene | Location | Mutation type | Frequency (%) | |
|---|---|---|---|---|
| Adenocarcinoma | Sarcomatoid carcinoma | |||
| EGFR | exon 19 | E746_A750 deletion mut | 27.70 | 28.90 |
| BCL2L2 | exon 3 | A65T missense mut | 16.60 | 49.30 |
| FANCL | exon 1 | S30L missense mut | 15.50 | 51.30 |
| ARFRP1 | 20q13.33 | cn_amplification | 6.19 | 16.99 |
| EGFR | exon 20 | T790M missense mut | 15.20 | 0 |
| TGFBR2 | exon 8 | R553H missense mut | 29.10 | 0 |
| TRRAP | exon 43 | V2098A missense mut | 24.70 | 0 |
| FLT1 | exon 13 | V211 synonymous mut | 17.40 | 0 |
| NF1 | exon 17 | A208 synonymous mut | 8.80 | 0 |
| TBX3 | exon 12 | P63 synonymous mut | 7.00 | 0 |
| SMO | exon 7 | F252 synonymous mut | 0 | 23.10 |
| CDK4 | 12q14.1 | cn_amplification | 0 | 11.38 |
| MDM2 | 12q15 | cn_amplification | 0 | 18.66 |
| AKT2 | 19q13.2 | cn_amplification | 0 | 3.8 |
| CIC | 19q13.2 | cn_amplification | 0 | 4.31 |
| GNAS | 20q13.32 | cn_amplification | 0 | 6.76 |
| FANCE | 6p21.31 | cn_amplification | 0 | 4.12 |
| NOTCH4 | 6p21.32 | cn_amplification | 0 | 3.87 |
Green denotes gene alterations common to adenocarcinoma and sarcomatoid carcinoma; blue denotes gene alterations in adenocarcinoma only; pink: denotes gene alterations in sarcomatoid carcinoma only.
Figure 3.
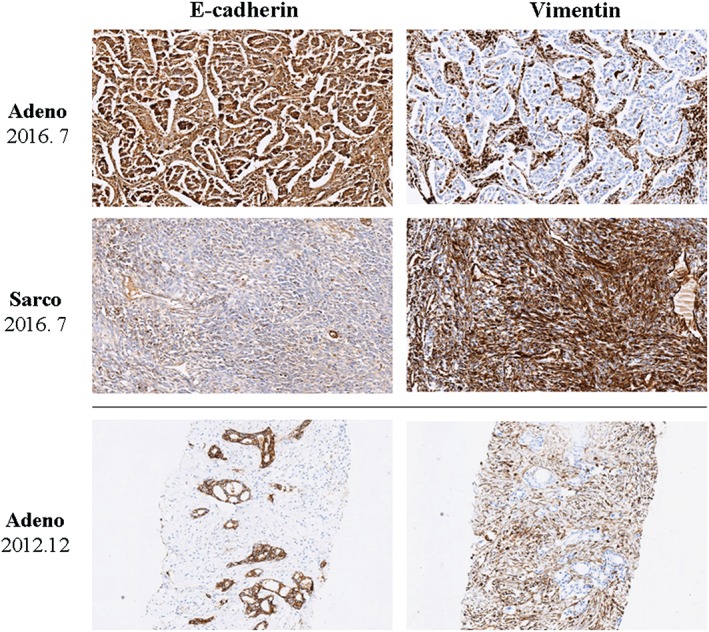
Histological staining of E‐cadherin and Vimentin on the primary biopsy specimen and tyrosine kinase inhibitor‐resistant postoperative specimen (adenocarcinoma and sarcomatoid carcinoma, July 2016). In the adenocarcinoma biopsy (December 2012) and surgical resection (July 2016) specimens, adenocarcinoma cells were strongly positive for E‐cadherin and almost negative for Vimentin, while the surrounding stromal tissues were positive for Vimentin. By contrast, the sarcomatoid histology showed a more spindle‐like mesenchymal morphology with diffuse positivity of Vimentin but lacked E‐cadherin expression (magnification ×100). Adeno, adenocarcinoma; Sarco, sarcomatoid carcinoma.
Discussion
In this case, two pulmonary tumors that acquired resistance to EGFR inhibitors had very distinct phenotypes after EGFR‐TKI treatment. Using in‐depth genetic and histological analyses, two major mechanisms were identified in this patient. The adenocarcinoma exhibited a secondary EGFR exon 20 T790M mutation and the newly developed pulmonary nodule was confirmed to be a sarcomatoid carcinoma, which explains why the primary pulmonary tumor grew more aggressively after effective erlotinib treatment. Secondary EGFR T790M mutation is a well‐known mechanism of EGFR‐TKI resistance, while the development of sarcomatoid carcinoma after EGFR‐TKI treatment is not frequently reported.
Three possible pathophysiological mechanisms underlying the development of sarcomatoid carcinoma might be involved: (i) intra‐patient tumor heterogeneity, in which the sarcomatoid carcinoma may already be present and gradually becomes dominant after EGFR‐TKI therapy; (ii) metachronous development of the secondary cancer; or (iii) direct transformation from adenocarcinoma to sarcomatoid carcinoma. It is difficult to explain the reasons by pathological diagnosis alone. Thus, next generation sequencing was performed with a broad gene panel. The genetic sequencing data explicitly shows that the sarcomatoid carcinoma shared the same genetic phenotype as the primary adenocarcinoma, including EGFR 19 deletion (with comparable mutation frequency) and three other genes (Table 1). In view of the computed tomography imaging and sequencing results, it seems logical that the sarcomatoid carcinoma retaining the original EGFR mutation may have been directly derived from the primary adenocarcinoma. Sequist et al. reported sarcomatoid morphology with an EMT phenotype at the time of EGFR‐TKI resistance.2 In our case, an assessment of E‐cadherin and Vimentin expression confirmed that the sarcomatoid carcinoma had also undergone EMT. EMT indicates that a cancer cell has lost its epithelial morphology and thus develops a more spindle‐like mesenchymal morphology, and is often associated with a more invasive phenotype. Previous studies have shown that EMT is a well‐established mechanism for acquired resistance in EGFR‐mutant NSCLC.3, 4, 5 However, concurrent EGFR T790M secondary mutation and EMT in a lung adenocarcinoma patient with EGFR‐TKI drug resistance has rarely been reported in the literature.
Although blood biopsy techniques have developed significantly, tissue re‐biopsy is still very important after EGFR‐TKI resistance and should not be replaced by liquid biopsy in these cases. Before the surgery, a blood ctDNA test was performed and only EGFR 19 deletion was detected, with a frequency of 0.08%; T790M was not found. If tissue re‐biopsy and surgery had not been performed, we would not have information on the underlying mechanisms of acquired resistance and this patient might have been treated with chemotherapy or radiotherapy. As sarcomatoid carcinomas are less sensitive to chemotherapy or radiotherapy, we can presume that the treatment response would have been poor. This case also indicates the necessity of multiple disciplinary team‐based evaluation and treatment, especially for patients with EGFR‐TKI acquired resistance. If performance status and pulmonary function are good, a complete surgical resection may be the best alternative for a patient with local disease progression or oligometastasis. Because of the patient's refusal, we did not perform a biopsy of the lung mass and only tested for EGFR mutation on the left supraclavicular lymph nodes. Although the patient has responded well to TKI treatment, a secondary lung biopsy may be necessary because of the heterogeneity between the primary lung tumor and metastatic lymph nodes.
The clinical implications of this case provide significant insight that two or more mechanisms may be simultaneously involved in EGFR‐TKI resistance. Therefore, if possible, tumor biopsies should be performed after the development of resistance to identify the best treatment option for patients.
Disclosure
No authors report any conflict of interest.
Supporting information
File S1. Next generation sequencing (NGS) and circulating tumor cell (ctC) analysis.
Table S1. The OncoScreen panel captures and sequences all exons of a panel of 287 cancer‐related genes simultaneously, along with intronic regions from 22 genes from which we want to catch all fusion/translocation events. All of the tested genes cover the most important solid tumor related signaling pathways and targeted therapeutics.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81301812, 81172233); the Specialized Research Fund for the Doctoral Program of Higher Education (20131202120004); the Science & Technology Foundation for Selected Overseas Chinese Scholar, Ministry of Personnel of China; the Science & Technology Foundation for Selected Overseas Chinese Scholar, Bureau of Personnel of China Tianjin; the Tianjin Key Project of Natural Science Foundation (17JCZDJC36200); the Tianjin Educational Committee Foundation (20120117); and the Tianjin Medical University General Hospital Young Incubation Foundation (ZYYFY2015015).
Contributor Information
Yan Wang, Email: wangyan5701@sohu.com.
Jun Chen, Email: huntercj2004@yahoo.com.
References
- 1. Suda K, Bunn PA Jr, Rivard CJ, Mitsudomi T, Hirsch FR. Primary double‐strike therapy for cancers to overcome EGFR kinase inhibitor resistance: Proposal from the bench. J Thorac Oncol 2017; 12: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sequist LV, Waltman BA, Dias‐Santagata D et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011; 3: 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chung JH, Rho JK, Xu X et al. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR‐TKIs. Lung Cancer 2011; 73: 176–82. [DOI] [PubMed] [Google Scholar]
- 4. Suda K, Tomizawa K, Fujii M et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor‐mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol 2011; 6: 1152–61. [DOI] [PubMed] [Google Scholar]
- 5. Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, Oyama T. Epithelial‐mesenchymal transition in EGFR‐TKI acquired resistant lung adenocarcinoma. Anticancer Res 2010; 30: 2513–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1. Next generation sequencing (NGS) and circulating tumor cell (ctC) analysis.
Table S1. The OncoScreen panel captures and sequences all exons of a panel of 287 cancer‐related genes simultaneously, along with intronic regions from 22 genes from which we want to catch all fusion/translocation events. All of the tested genes cover the most important solid tumor related signaling pathways and targeted therapeutics.