

Abstract
Aims/Introduction
Overproduction of reactive oxygen species (ROS) in endothelial cells (ECs) plays a pivotal role in endothelial dysfunction. Mitochondrial ROS (mtROS) is one of the key players in the pathogenesis of diabetic vascular complications. Hypoglycemia is linked to increased ROS production and vascular events; however, the underlying mechanisms remain unclear. In the present study, we aimed to determine whether and how low glucose (LG) mediates mtROS generation in ECs, and to examine the impact of LG‐induced mtROS on endothelial dysfunction.
Materials and Methods
Metabolomic profiling, cellular oxygen consumption rate, mtROS, endothelial nitric oxide synthase phosphorylation, and the expression of vascular cell adhesion molecule‐1 or intercellular adhesion molecule‐1 were evaluated in bovine aortic ECs.
Results
We found that LG increased mtROS generation in ECs; which was suppressed by overexpression of manganese superoxide dismutase. Comprehensive metabolic analysis using capillary electrophoresis‐mass spectrometry and oxygen consumption rate assessment showed that the pathway from fatty acid to acetyl‐CoA through fatty acid oxidation was upregulated in ECs under LG conditions. In addition, etomoxir, a specific inhibitor of the free fatty acid transporter, decreased LG‐induced mtROS production. These results suggested that LG increased mtROS generation through activation of fatty acid oxidation. We further revealed that LG inhibited endothelial nitric oxide synthase phosphorylation, and increased the expression of vascular cell adhesion molecule‐1 and intercellular adhesion molecule‐1. These effects were suppressed either by overexpression of manganese superoxide dismutase or by treatment with etomoxir.
Conclusions
The activation of fatty acid oxidation followed by mtROS production could be one of the causes for endothelial dysfunction during hypoglycemia.
Keywords: Fatty acid oxidation, Low glucose, Mitochondrial reactive oxygen species
Introduction
Overproduction of reactive oxygen species (ROS) caused by several factors, such as diabetes, obesity, hypertension, hyperlipidemia and smoking, plays a pivotal role in the pathogenesis of cardiovascular diseases. Excess production of ROS can promote endothelial dysfunction, which is thought to play an important role not only in the initiation of atherosclerosis, but also in the etiology of diabetic vascular complications1, 2, 3, 4. Therefore, to prevent diabetic vascular complications, it is essential to elucidate the mechanisms of ROS generation in the context of diabetes.
The Diabetes Control and Complications Trial5, the United Kingdom Prospective Diabetes Study6, and our Kumamoto Study7 provide strong evidence that hyperglycemia plays an important role in the pathogenesis of microvascular complications in both type 1 and type 2 diabetes. In addition, hyperglycemia‐induced ROS generation has been reported to be involved in vascular endothelial dysfunction in patients with diabetes4. Consistent with these reports, Nishikawa et al. proposed that mitochondrial ROS (mtROS) is the major cause of diabetes‐induced oxidative stress in endothelial cells (ECs)8.
Recent large randomized clinical trials have shown that intensive glycemic control might fail to reduce macrovascular events9. Along with improvements in diabetes management with insulin or insulin secretagogues, there has been an increase in the prevalence of hypoglycemia with efforts to achieve better glycemic control. Severe hypoglycemia is strongly associated with an increased risk for vascular events and death. However, it is not clear whether there is a causal relationship between hypoglycemia and these outcomes10. Hypoglycemic events can trigger inflammation, blood coagulation abnormalities, the sympathoadrenal response and endothelial dysfunction, and these responses are interdependent. As a result, endothelial dysfunction mediated by hypoglycemic abnormalities could contribute to cardiovascular risk11. Previous studies have reported that hypoglycemia induces oxidative stress in type 1 diabetes12, and that recurrent hypoglycemia potentiates long‐term hyperglycemia‐mediated impairment of hippocampal mitochondria and increases the oxidative stress13. Therefore, oxidative stress in ECs might be a common factor linking hyperglycemia, hypoglycemia and the vascular complications of diabetes.
Therefore, the aims of the present study were to evaluate whether low glucose (LG) conditions exacerbated mtROS in ECs, similarly to high glucose conditions, and to evaluate whether the suppression of LG‐induced mtROS could ameliorate endothelial dysfunction. Elucidation of the mechanisms by which hypoglycemia promotes endothelial dysfunction might help to prevent diabetic complications.
Methods
Cell culture conditions and materials
Bovine aortic endothelial cells were purchased from TOYOBO (Osaka, Japan). Cells were cultured in Dulbecco's modified Eagle's medium (Wako, Osaka, Japan) with 10% fetal bovine serum and 1.0, 2.5, 4.0, 5.5 or 25 mmol/L glucose in an atmosphere containing 5% CO2 and 95% air at 37°C. Etomoxir, 1‐(2,3,4‐trimethoxybenzyl) piperazine dihydrochloride (trimetazidine), bis‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl) ethyl sulfide and carbonyl cyanide 3‐chlorophenylhydrazone were from Sigma‐Aldrich (St. Louis, Missouri, USA).
Adenoviral vectors
Human manganese superoxide dismutase (MnSOD) adenoviral vectors were provided by Dr M Brownlee (Albert Einstein College of Medicine, Bronx, New York, USA)8. Cells were infected with MnSOD adenovirus or lacZ control adenovirus 48 h before experiments. MnSOD overexpression was confirmed using western blot analysis, as previously described14.
Measurement of mtROS generation
To evaluate the direct generation of mtROS, we used the reduced MitoTracker Red CM‐H2XRos probe (ThermoFisher Scientific, Waltham, Massachusetts, USA), which specifically detects the ROS generation in mitochondria15. Cells were incubated with 300 nmol/L MitoTracker Red CM‐H2XRos at 37°C for 30 min before the end of the experiment.
Measurement of MnSOD activity
MnSOD activity was measured using a SOD assay kit‐WST (Dojindo, Kumamoto, Japan), according to the manufacturer's instructions.
Measurement of metabolites
Measurement of metabolites was carried out according to the manufacturer's instructions (Human Metabolome Technologies, Inc., Tsuruoka, Japan) as follows. Culture cells (8 × 106 cells/sample) were used for the extraction of intracellular metabolites. Culture medium was aspirated from the dish, and cells were washed twice with 5% mannitol solution (10 mL first and then 2 mL). Cells were then treated with 800 μL methanol, and left to rest for 30 s to inactivate enzymes. Next, the cell extract was treated with 550 μL Milli‐Q water containing internal standards (H3304‐1002; Human Metabolome Technologies, Inc.), and left to rest for another 30 s. The extract was obtained and centrifuged at 2,300 g and 4°C for 5 min, and 700 μL of the upper aqueous layer was then filtered by centrifugation through a Millipore 5‐kDa cut‐off filter at 9,100 g and 4°C for 120 min to remove proteins. The filtrate was concentrated by centrifugation and resuspended in 50 μL Milli‐Q water for capillary electrophoresis‐mass spectrometry.
Respirometry (Extracellular Flux Analysis)
The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using an intact cell respirometer designed for adherent cells (XF‐24 Extracellular Flux Analyzer; Seahorse Bioscience, North Billerica, Massachusetts, USA). Bovine aortic endothelial cells were seeded at a density of 10,000 cells/well in 24‐well plates designed for respirometer analysis. Cells reached confluency and were subjected to respirometry at 2 days after seeding.
Mitophagy detection
Mitophagy was evaluated using a Mitophagy Detection Kit (Dojindo), according to the manufacturer's instructions.
Western blot analysis
Western blot analysis was carried out as previously described14. Polyclonal antibodies for phosphorylated acetyl CoA carboxylase (ACC; Ser79), ACC, phosphorylated endothelial nitric oxide synthase (eNOS; Ser1177) and carnitine palmitoyltransferase I (CPT1) A were from Cell Signaling Technologies (Beverly, Massachusetts, USA). Polyclonal antibodies for eNOS and CPT1B were from Abcam (Cambridge, UK).
Ribonucleic acid extraction and quantitative reverse transcription polymerase chain reaction analysis
After incubation for 1 h under each experimental condition, total cellular ribonucleic acid (RNA) was extracted with an RNeasy Mini Kit (Qiagen, Hilden, Germany). All polymerase chain reaction (PCR) analyses were carried out with a LightCycler System (Roche Molecular Biochemicals, Indianapolis, Indiana, USA) using SYBR Green I master mix. Specific primers for PCR were as follows: vascular cell adhesion molecule‐1 (VCAM‐1) forward, 5′‐TTGCGCAGATTGGTGACTCT‐3′; VCAM‐1 reverse, 5′‐CCACTCGGATTGCTTTCTCC‐3′; intercellular adhesion molecule‐1 (ICAM‐1) forward, 5′‐GGACCATGGCACCAATTTCT‐3′; ICAM‐1 reverse, 5′‐GAGGCTGGGAACAGTCCATC‐3′; CPT1B forward, 5′‐ GGTCGACTTCCAGCTCAGTC‐3′; CPT1B reverse, 5′‐ AGTAGGAGGAACCCGCTGTT‐3′; 18S forward, 5′‐CTCAACACGGGAAACCTCAC‐3′; and 18S reverse, 5′‐AGACAAATCGCTCCACCAAC‐3′. Expression of 18S was used for normalization in gene expression analysis.
Statistical analysis
Data are presented as mean ± SD. Statistical analysis was carried out using unpaired Student's t‐tests for two‐group comparisons and one‐way analysis of variance (anova) followed by Tukey's multiple comparison tests or Games–Howell multiple comparison tests for more than two groups using SPSS software (ver. 21; IBM, Armonk, New York, USA). A P‐value of <0.05 was considered statistically significant.
Results
LG induced mtROS production in ECs
To determine whether LG could induce mtROS production, we used the reduced MitoTracker Red CM‐H2XRos probe in ECs. The fluorescence intensity of the MitoTracker Red CM‐H2XRos probe was significantly increased in ECs cultured with 2.5 mmol/L glucose (149.4 ± 11.5% that of 5.5 mmol/L glucose) compared with that of 5.5 mmol/L glucose. MitoTracker Red CM‐H2XRos fluorescence was suppressed with overexpression of MnSOD, a specific SOD targeting mtROS (Figure 1a,b). Additional experiments confirmed that lower glucose levels induced greater MitoTracker Red CM‐H2XRos fluorescence in ECs independent of osmotic pressure (Figure 1c,d). In contrast, MnSOD activity and the glutathione‐to‐glutathione disulfide ratio in ECs did not change under LG conditions (Figure 1e; Table 1). We also investigated the dose‐dependent effects of glucose on mtROS generation. The fluorescence intensity of the MitoTracker Red CM‐H2XRos probe was significantly increased in ECs cultured with 2.5 mmol/L glucose compared with that of 4.0 mmol/L glucose, and was comparable with that of 1.0 mmol/L glucose. In contrast, the MitoTracker Red CM‐H2XRos fluorescence of 25 mmol/L glucose was significantly higher than that of 2.5 mmol/L glucose (Figure 1f,g). In addition, we investigated whether fluctuations in glucose levels influenced mtROS generation. The fluorescence intensity of the MitoTracker Red CM‐H2XRos probe was significantly increased in ECs cultured with 25 mmol/L glucose for 1 h and 2.5 mmol/L glucose for 1 h compared with that in ECs cultured with 5.5 mmol/L glucose for 1 h and 2.5 mmol/L glucose for 1 h (Figure 1h,i).
Figure 1.
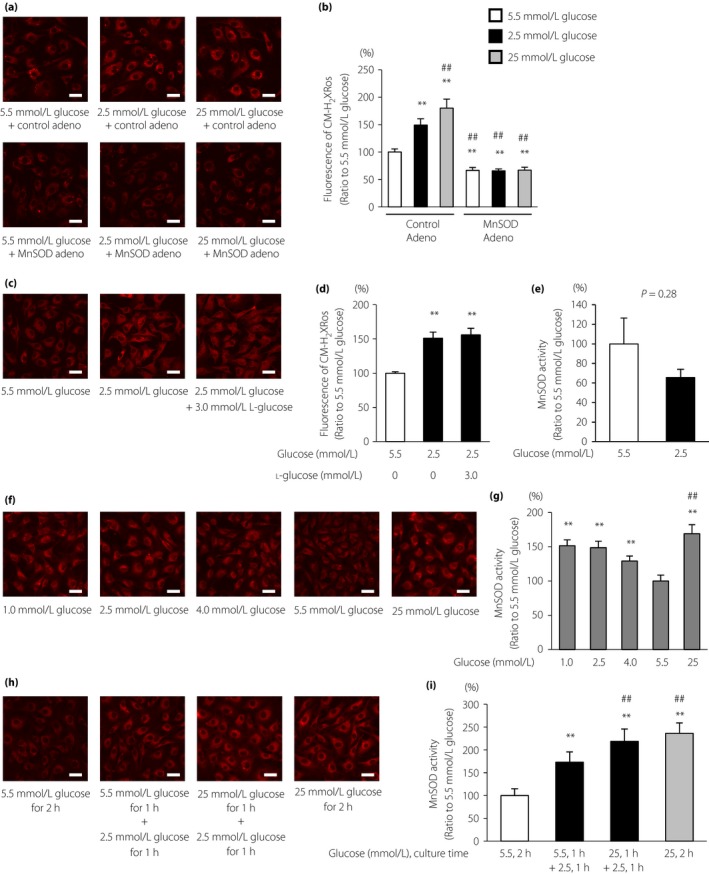
Low glucose induced mitochondrial reactive oxygen species (mtROS) production. (a) mtROS production in bovine aortic endothelial cells (BAECs) and the effects of manganese superoxide dismutase (MnSOD) overexpression on mtROS production. BAECs overexpressing MnSOD (lower panel) or control adenovirus (upper panel) were incubated under the indicated conditions for 1 h and treated with 300 nmol/L MitoTracker Red CM‐H2XRos (red) for 30 min. Scale bars, 30 μm. (b) The relative fluorescence intensity of MitoTracker Red CM‐H2XRos was measured. Control Adeno, control adenovirus; MnSOD Adeno, MnSOD adenovirus. Data are four independent experiments carried out in duplicate (mean ± SD). **P < 0.01 compared with 5.5 mmol/L glucose and control adenovirus; ## P < 0.01 compared with 2.5 mmol/L glucose and control adenovirus. White bars, 5.5 mmol/L glucose; black bars, 2.5 mmol/L glucose; gray bars, 25 mmol/L glucose. (c) Effects of osmotic pressure on mtROS generation under low glucose conditions. BAECs were incubated under the indicated conditions for 1 h and treated with 300 nmol/L MitoTracker Red CM‐H2XRos (red) for 30 min. Scale bars, 30 μm. (d) The relative fluorescence intensity of the MitoTracker Red CM‐H2XRos probe was measured. Data are four independent experiments carried out in duplicate (mean ± SD). **P < 0.01 compared with 5.5 mmol/L glucose. (e) MnSOD activity in BAECs. BAECs were incubated under the indicated conditions for 1 h. MnSOD activity was measured using a SOD assay kit‐WST. Data are mean ± SD (n = 5/group). (f) The mtROS production in BAECs and the effects of dose‐dependent glucose levels or (h) the fluctuation of glucose levels on mtROS production. BAECs were incubated under the indicated conditions and treated with 300 nmol/L MitoTracker Red CM‐H2XRos (red) for 30 min. Scale bars, 30 μm. (g,i) The relative fluorescence intensity of MitoTracker Red CM‐H2XRos was measured. Data are four independent experiments carried out in duplicate (mean ± SD). **P < 0.01 compared with 5.5 mmol/L glucose; ## P < 0.01 compared with 2.5 mmol/L glucose.
Table 1.
Metabolites examined during metabolome analysis
| 5.5 mmol/L glucose | 2.5 mmol/L glucose | P‐value | |
|---|---|---|---|
| Glucose 6‐phosphate | 39.3 ± 8.0 | 31.1 ± 20.9 | 0.58 |
| Fructose 6‐phosphate | 10.7 ± 1.4 | 10.3 ± 5.4 | 0.92 |
| Fructose 1,6‐diphosphate | 158.2 ± 18.5 | 120.8 ± 15.8 | 0.06 |
| 3‐Phosphoglyceric acid | 70.4 ± 4.8 | 46.1 ± 9.3 | 0.03 |
| 2‐Phosphoglyceric acid | 7.0 ± 0.6 | 4.9 ± 0.8 | 0.03 |
| Phosphoenolpyruvic acid | 20.6 ± 3.6 | 9.5 ± 4.7 | 0.03 |
| Pyruvic acid | 286.8 ± 23.0 | 242.3 ± 18.4 | 0.06 |
| Lactic acid | 2344.3 ± 223.9 | 1263.7 ± 144.6 | <0.01 |
| Acetyl CoA | 0.24 ± 0.03 | 0.30 ± 0.07 | 0.23 |
| Citric acid | 753.5 ± 50.7 | 809.1 ± 42.5 | 0.22 |
| cis‐Aconitic acid | 12.0 ± 1.1 | 13.2 ± 1.3 | 0.28 |
| Isocitric acid | 14.2 ± 3.4 | 15.6 ± 2.2 | 0.58 |
| 2‐Oxoglutaric acid | 715.9 ± 25.3 | 786.5 ± 87.2 | 0.29 |
| Succinic acid | 144.9 ± 4.4 | 198.3 ± 38.3 | 0.14 |
| Fumaric acid | 462.6 ± 35.0 | 447.3 ± 14.5 | 0.54 |
| Malic acid | 1967.1 ± 59.6 | 1838.9 ± 84.9 | 0.11 |
| ATP | 5386.2 ± 140.3 | 5635.0 ± 266.4 | 0.25 |
| GTP | 1185.1 ± 24.2 | 1204.8 ± 59.9 | 0.64 |
| Glutamate | 48806.0 ± 680.1 | 51153.4 ± 1815.9 | 0.14 |
| Glutathione (GSSG) | 2257.7 ± 217.2 | 2493.4 ± 170.6 | 0.22 |
| Glutathione (GSH) | 1860.5 ± 248.0 | 1644.0 ± 184.2 | 0.30 |
Data are mean ± standard deviation (n = 3/group). ATP, adenosine triphosphate; GSH, glutathione; GSSG, glutathione‐SS‐glutathione; GTP, guanosine 5'‐triphosphate.
LG stimulation increased fatty acid oxidation in ECs
To investigate the mechanism through which LG induces mtROS, metabolomic profiling was carried out using capillary electrophoresis‐time‐of‐flight mass spectrometry in ECs treated with either 2.5 or 5.5 mmol/L glucose (Figure 2a). Metabolome analysis showed that levels of intermediate metabolites in glycolysis, such as fructose 1,6‐diphosphate, 3‐phosphoglyceric acid, 2‐phosphoglyceric acid, phosphoenolpyruvic acid, pyruvic acid and lactic acid, decreased with 2.5 mmol/L glucose treatment for 1 h compared with that of normal glucose treatment (Table 1). However, treatment of ECs with 2.5 mmol/L glucose did not alter the levels of adenosine triphosphate, guanosine 5′‐triphosphate or tricarboxylic acid cycle metabolites, such as citric acid, isocitric acid and succinic acid. Interestingly, acetyl‐CoA levels were not altered with LG stimulation. Acetyl‐CoA can be synthesized through fatty acid oxidation (FAO), as well as glycolysis (Figure 2a; Table 1). The levels of metabolic intermediates of glutamine did not change in ECs under LG conditions (Table 1). These results show that the pathway from fatty acid to acetyl‐CoA through FAO was upregulated in ECs during LG stimulation.
Figure 2.
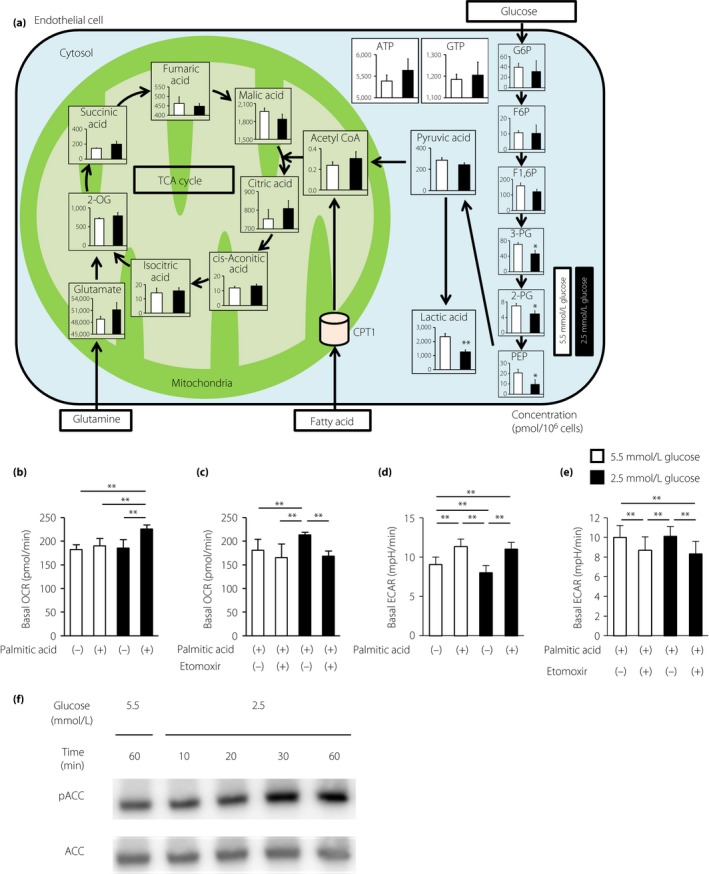
Low glucose increased fatty acid oxidation in endothelial cells. (a) Metabolome analysis. Intracellular concentrations (pmol/million cells) of key metabolites involved in glycolysis are shown. Error bars indicate standard deviation (n = 3/group). Total metabolites were extracted with methanol from bovine aortic endothelial cells (BAECs) with 5.5 mmol/L glucose (white) or 2.5 mmol/L glucose (black) for 1 h. *P < 0.05, **P < 0.01 vs 5.5 mmol/L glucose. 2‐OG, 2‐oxoglutaric acid; 2‐PG, 2‐phosphoglyceric acid; 3‐PG, 3‐phosphoglyceric acid; F1,6P, fructose 1,6‐diphosphate; F6P, fructose 6‐phosphate; G6P, glucose 6‐phosphate; PEP, phosphoenolpyruvic acid. (b) Basal oxygen consumption rates (OCRs) of BAECs treated with palmitic acid (100 μmol/L) or left untreated. BAECs were incubated under the indicated conditions for 1 h. (c) Basal OCRs of BAECs treated with palmitic acid in the presence of DMSO control or etomoxir (100 μmol/L). (d) Basal extracellular acidification rates (ECARs) of BAECs treated with palmitic acid (100 μmol/L) or left untreated. BAECs were incubated under the indicated conditions for 1 h. (e) Basal ECARs of BAECs treated with palmitic acid in the presence of DMSO control or etomoxir (100 μmol/L). White bars, 5.5 mmol/L glucose; black bars, 2.5 μmol/L glucose. Data are mean ± SD (n = 3–4/group). **P < 0.01. (f) Representative images of protein expression determined by western blotting of BAECs cultured under the indicated conditions for 1 h. ACC, acetyl CoA carboxylase; pACC, phosphorylated acetyl CoA carboxylase.
To characterize FAO‐associated metabolic changes, an extracellular flux analyzer was used to assess the OCR, which mainly reflects mitochondrial oxidative phosphorylation activity. Without palmitic acid, there was no significant difference between the basal OCR under normal glucose conditions and that under LG conditions. In contrast, compared with that in ECs under normal glucose conditions, treatment with 100 μmol/L palmitic acid increased the basal OCR in ECs under LG conditions (189.8 ± 10.4 vs 225.9 ± 9.2 pmol/min, respectively; P < 0.01), suggesting a high rate of FAO under LG conditions (Figure 2b).
CPT1 is the rate‐limiting enzyme that transfers long‐chain fatty acyl‐CoA to the mitochondria for beta‐oxidation, and is inhibited by unphosphorylated ACC16. When treated with etomoxir, a CPT1 inhibitor, enhancement of the basal OCR in ECs under LG conditions with palmitic acid treatment was reduced to the level observed with normal glucose conditions (213.5 ± 6.1 vs 168.6 ± 11.0 pmol/min, respectively; P < 0.01; Figure 2c).
Furthermore, we evaluated the ECAR, which is used as a measure of anaerobic glycolysis, as it mainly reflects the production of lactate, under LG conditions. Without palmitic acid, the basal ECAR in ECs under LG conditions decreased compared with that in ECs under normal glucose conditions (8.0 ± 0.9 vs 9.0 ± 1.0 mpH/min, respectively; P < 0.01), suggesting a decrease in glycolytic metabolism to lactate (Figure 2d). In contrast, with palmitic acid, there was no significant difference between the basal ECAR under normal glucose conditions and that under LG conditions.
When treated with etomoxir, ECARs in ECs under normal glucose and LG conditions decreased compared with those in ECs that were not treated with etomoxir. However, when treated with palmitic acid and etomoxir, there was no significant difference between the basal ECAR under normal glucose conditions and that under LG conditions (Figure 2e).
We next examined the effects of LG conditions on ACC activity. Treatment with LG increased the phosphorylation of ACC at Ser79 after 30 and 60 min of incubation, which indicated the inactivation of ACC (Figure 2e). Therefore, LG stimulation increased the rate of mitochondrial beta‐oxidation through inhibition of ACC and activation of CPT1 in ECs. Furthermore, we investigated CPT1 expression by western blot analysis and quantitative reverse transcription PCR, and evaluated the mitophagy status of bovine aortic endothelial cells. LG conditions had no significant effect on CPT1 expression or mitophagy status compared with normal glucose conditions (Figure S1).
LG increased mtROS generation through activation of FAO
To confirm the source of mtROS production during LG stimulation, ECs under LG conditions were supplemented with etomoxir or trimetazidine. When ECs were cultured under normal or high glucose conditions, there were no significant differences in the reduced MitoTracker Red CM‐H2XRos fluorescence intensities of cells treated with vehicle or etomoxir (Figure 3a,c,d,f,g). In contrast, treatment of cells with LG conditions increased the fluorescence of MitoTracker Red CM‐H2XRos, which was completely suppressed by etomoxir (Figure 3b,e,g). In addition, we evaluated the effects of trimetazidine, another pharmacological FAO inhibitor, on LG‐induced mtROS generation. Trimetazidine is a competitive inhibitor of 3‐ketoacyl CoA thiolase, a key enzyme in beta‐oxidation. Trimetazidine suppressed LG‐induced mtROS generation as with etomoxir (Figure 3h–n).
Figure 3.
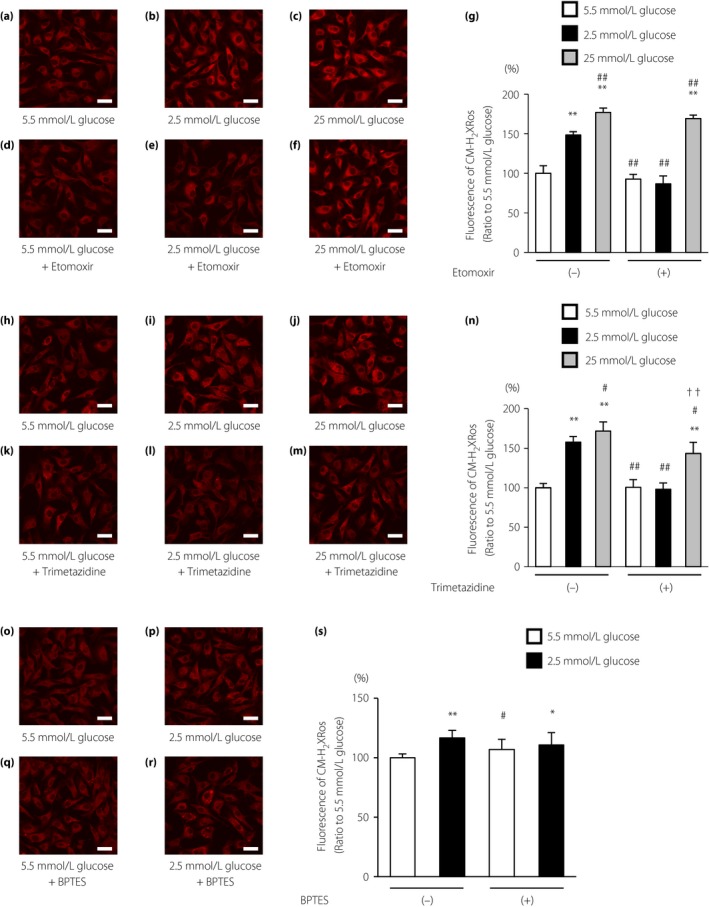
Low glucose increased mitochondrial reactive oxygen species (mtROS) generation using fatty acid oxidation. (a–f) Effects of etomoxir (100 μmol/L) on mtROS generation under low glucose conditions. Cells were incubated under the indicated conditions. Scale bars, 30 μm. (g) The relative fluorescence intensity of the MitoTracker Red CM‐H2XRos probe was measured. (h–m) Effects of trimetazidine (50 μmol/L) on mtROS generation under low glucose conditions. Cells were incubated under the indicated conditions. Scale bars, 30 μm. (n) The relative fluorescence intensity of the MitoTracker Red CM‐H2XRos probe was measured. (o–r) Effects of bis‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl) ethyl sulfide (BPTES; 10 μmol/L), a selective inhibitor of glutaminase, on mtROS generation under low glucose conditions. Cells were incubated under the indicated conditions. Scale bars, 30 μm. (s) The relative fluorescence intensity of the MitoTracker Red CM‐H2XRos probe was measured. White bars, 5.5 mmol/L glucose; black bars, 2.5 mmol/L glucose; gray bars, 25 mmol/L glucose. Data are four independent experiments carried out in duplicate (mean ± SD). *P < 0.05, **P < 0.01 compared with 5.5 mmol/L glucose; # P < 0.05, ## P < 0.01 compared with 2.5 mmol/L glucose; †† P < 0.01 compared with 25 mmol/L glucose.
We also evaluated the effects of bis‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl) ethyl sulfide, a selective inhibitor of glutaminase. Glutaminase converts glutamine to glutamate, and can be further oxidized to α‐ketoglutarate to feed the tricarboxylic acid cycle. Bis‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl) ethyl sulfide had no effect on mtROS production under LG conditions (Figure 3o–s).
LG inhibited eNOS activation and increased the expression of vascular adhesion molecules
To determine whether LG stimulation affected endothelial function, we examined eNOS activation by measuring its phosphorylation at Ser1177. As shown in Figure 4a, LG stimulation inhibited the phosphorylation of eNOS, and this effect was suppressed by MnSOD overexpression or etomoxir treatment. We next examined the expression of the vascular adhesion molecules, VCAM‐1 and ICAM‐1, in ECs. VCAM‐1 and ICAM‐1 messenger RNA expression levels were significantly increased by incubation with LG compared with those in cells cultured under normal glucose conditions. In addition, LG‐induced messenger RNA induction was suppressed by MnSOD overexpression or incubation with etomoxir (Figure 4b,c). These results suggested that activated FAO induced by LG stimulation caused endothelial dysfunction.
Figure 4.
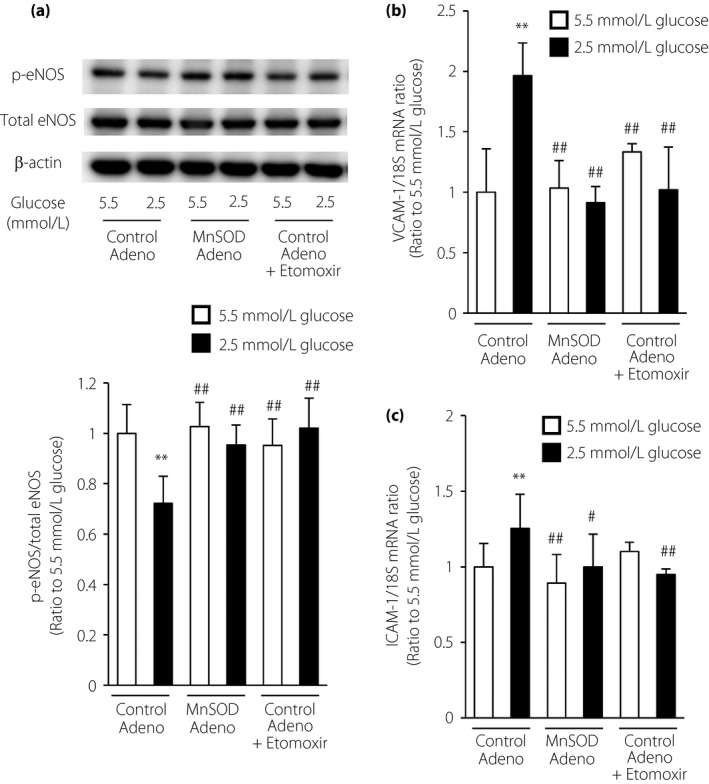
Etomoxir and manganese superoxide dismutase (MnSOD) overexpression ameliorated the endothelial dysfunction induced by low glucose. (a) Representative images of protein expression determined by western blotting in bovine aortic endothelial cells (BAECs) under the indicated conditions for 1 h. eNOS, endothelial nitric oxide synthase; p‐eNOS, phosphorylated eNOS (Ser1177). (b,c) Effects of etomoxir and MnSOD overexpression on (b) vascular cell adhesion molecule‐1 (VCAM‐1) and (c) intercellular adhesion molecule‐1 (ICAM‐1) messenger ribonucleic acid (mRNA) expression. Cells were incubated under the indicated conditions for 1 h. The expression levels of VCAM‐1 and ICAM‐1 mRNA were measured by quantitative reverse transcription polymerase chain reaction analysis. Control Adeno, control adenovirus; MnSOD Adeno, MnSOD adenovirus. White bars, 5.5 mmol/L glucose; black bars, 2.5 mmol/L glucose. Data are six independent experiments carried out in duplicate (mean ± SD). **P < 0.01 compared with 5.5 mmol/L glucose and control adenovirus; # P < 0.05, ## P < 0.01 compared with 2.5 mmol/L glucose and control adenovirus.
Discussion
In the present study, we found that LG conditions increased the production of mtROS in ECs. Although downregulation of intermediate metabolites in glycolysis was observed in ECs under LG conditions by metabolome analysis, there were no changes on intracellular acetyl‐CoA, adenosine triphosphate or tricarboxylic acid cycle metabolites, suggesting that other pathways without glycolysis were involved in maintaining the tricarboxylic acid cycle. Additionally, we found that blockade of FAO prevented the increased oxygen consumption rate with palmitic acid stimulation and decreased LG‐mediated mtROS production. In general, FAO generates just 5% of the total amount of adenosine triphosphate of ECs under normal glucose conditions17, and many cancer cells increase glucose and glutamine uptake to fuel the tricarboxylic acid cycle18, 19. However, our novel findings suggested that the tricarboxylic acid cycle was maintained by increasing FAO, thereby increasing mtROS production in ECs under LG conditions. Accelerated beta‐oxidation of palmitate caused excess electron flux in the mitochondrial respiratory chain, resulting in increased ROS generation in H4IIEC3 hepatocytes20. Taken together, these findings suggested that excess fatty acid oxidation could, at least in part, induce ROS generation in ECs. In contrast, Nakamura et al.20 reported that palmitate might induce the CPT1 gene expression, and LG conditions had no significant effect on CPT1 expression in the present study. Therefore, the mechanism for increasing FAO under LG conditions remains unclear, and further studies are necessary to determine this mechanism.
Acute infusion of free fatty acids (FFAs) induces oxidative stress and reduces endothelium‐dependent vasodilation in humans in vivo 21. Furthermore, treatment with FFAs impairs endothelial function by several mechanisms, including increased production of ROS, stimulation of pro‐inflammatory signaling, and reduction of eNOS activity and nitric oxide (NO) production22, 23, 24. Furthermore, overexpression of uncoupling protein‐2, an important regulator of intracellular ROS production, improves the endothelial dysfunction induced by ROS‐mediated FFA toxicity25. Therefore, accumulation of FFAs might mediate endothelial dysfunction under hypoglycemic condition.
Hypoglycemia, mediated by intensive glucose‐lowering interventions, is a common side‐effect in patients with diabetes, and might increase the risk of poor outcomes26, 27. Additionally, ECs play important roles not only as vessel barriers, but also in the regulation of vascular tone, coagulation, leukocyte adhesion and vascular permeability. Vascular tone is modulated by various factors, such as NO. The earliest and most important factor mediating endothelial dysfunction is a reduction in NO bioactivity, which is mediated by ROS. Therefore, ROS generation in vascular cells plays a key role in endothelial dysfunction and subsequent atherosclerotic lesion formation23. Furthermore, hypoglycemia induces endothelial dysfunction through the production of several inflammatory factors, including VCAM‐1, ICAM‐1, vascular endothelial growth factor, interleukin‐8, interleukin‐6, endothelin‐1 and tumor necrosis factor‐alpha, resulting in increased risk of diabetic macro‐angiopathy11, 28, 29, 30. In particular, Jin et al.31 reported that hypoglycemia increases serum adrenaline and levels of adhesion molecules, such as VCAM‐1 and ICAM‐1, in the endothelium of the rat aorta. In addition, increased ROS also activates nuclear factor‐κB, which mediates the overexpression of adhesion molecules, such as VCAM‐1, ICAM‐1 and E‐selectin32. In the present study, we observed that LG stimulation decreased the bioavailability of NO, and increased the expression of VCAM‐1 and ICAM‐1 in ECs. Furthermore, blockade of FAO or suppression of mtROS ameliorated the LG‐induced bioavailability of NO, and expression of VCAM‐1 and ICAM‐1. These results suggested that hypoglycemia‐induced reduction of NO bioactivity, and overproduction of VCAM‐1 and ICAM‐1 messenger RNA were mediated by the overproduction of mtROS and activation of FAO in ECs.
The present findings might have implications in both diabetic macro‐angiopathy and diabetic micro‐angiopathy. In fact, our preliminary data showed that etomoxir decreased ROS generation and prevented vascular permeability caused by recurrent hypoglycemia in the retinas of diabetic mice (unpubl. data). Interestingly, Schoors et al.33 reported that etomoxir also reduces pathological angiogenesis in a model of oxygen‐induced retinopathy. We therefore speculate that mtROS and FAO might be involved in the progression of microvascular complications caused by recurrent hypoglycemia.
The present analysis was limited by the fact that we did not examine whether LG induced ROS generation, and whether ROS‐mediated endothelial dysfunction was inhibited by blockade of FAO in animal models. Therefore, further studies are required to evaluate the association of hypoglycemia and diabetic complications in vivo. Furthermore, ketone bodies play an important role as alternative energy sources to neurons during hypoglycemia34. Rains et al.35 reported that hyperketonemia increases monocyte adhesion to ECs. Based on these previous reports, additional studies are also required to investigate the effects of ketone bodies on ECs during hypoglycemia in vivo. Such studies are necessary to determine whether arteriosclerosis progression as a result of monocyte adhesion or macrophage infiltration is mediated by enhancement of fatty acid metabolism in the context of recurrent hypoglycemia in diabetic mice in vivo.
In conclusion, we showed that LG conditions exacerbated mtROS production through activation of FAO in ECs (Figure 5). In addition, we revealed that blockade of FAO suppressed LG‐mediated mtROS production and ameliorated LG‐induced endothelial dysfunction. Collectively, the present findings suggested that inhibition of FAO and subsequent suppression of mtROS generation could be novel therapeutic approaches for the prevention of endothelial dysfunction and diabetic macrovascular complications under hypoglycemic conditions.
Figure 5.
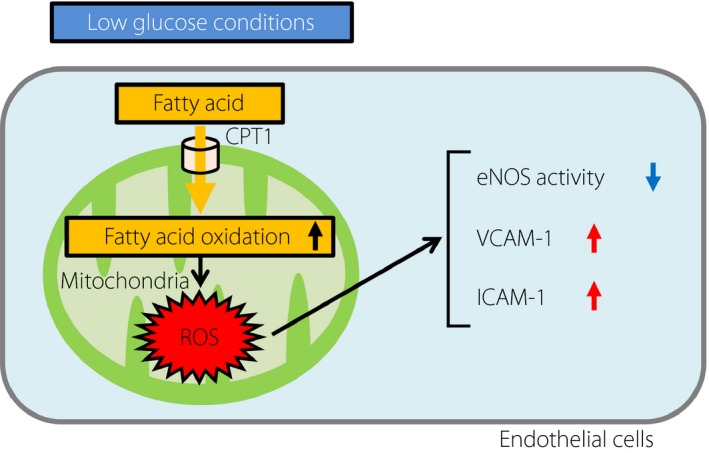
Schematic model of the proposed mechanisms of mtROS induction by low glucose conditions. Low glucose conditions increased mtROS generation through fatty acid oxidation in endothelial cells (ECs). Low glucose‐induced mtROS inhibited the phosphorylation of endothelial nitric oxide synthase (eNOS) and increased the expression of vascular cell adhesion molecule‐1 (VCAM‐1) or intercellular adhesion molecule‐1 (ICAM‐1). CTP1, carnitine palmitoyltransferase I; ROS, reactive oxygen species.
Disclosures
The authors declare no conflict of interest.
Supporting information
Figure S1 ¦ Carnitine palmitoyltransferase I expression and the mitophagy status of bovine aortic endothelial cells under low glucose conditions.
Acknowledgments
This work was supported by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science, Japan (grant nos. 15K09393 to DK and 26461340 to TN).We thank Professor Shokei Kim‐Mitsuyama (Department of Pharmacology and Molecular Therapeutics, Graduate School of Medical Sciences, Faculty of Life Sciences, Kumamoto University) and Professor Motohiro Takeya (Department of Cell Pathology, Graduate School of Medical Sciences, Faculty of Life Sciences, Kumamoto University) for helpful discussions.
J Diabetes Investig 2017; 8: 750–761
References
- 1. Panth N, Paudel KR, Parajuli K. Reactive oxygen species: a key hallmark of cardiovascular disease. Adv Med 2016; 2016: 9152732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ceriello A, Testa R, Genovese S. Clinical implications of oxidative stress and potential role of natural antioxidants in diabetic vascular complications. Nutr Metab Cardiovasc Dis 2016; 26: 285–292. [DOI] [PubMed] [Google Scholar]
- 3. Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci (Lond) 2005; 109: 143–159. [DOI] [PubMed] [Google Scholar]
- 4. Sheetz MJ, King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA 2002; 288: 2579–2588. [DOI] [PubMed] [Google Scholar]
- 5. The Diabetes Control and Complications Trial Research Group . The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes 1995; 44: 968–983. [PubMed] [Google Scholar]
- 6. UK Prospective Diabetes Study (UKPDS) Group . Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33. Lancet 1998; 352: 837–853. [PubMed] [Google Scholar]
- 7. Ohkubo Y, Kishikawa H, Araki E, et al Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non‐insulin‐dependent diabetes mellitus: a randomized prospective 6‐year study. Diabetes Res Clin Pract 1995; 28: 103–117. [DOI] [PubMed] [Google Scholar]
- 8. Nishikawa T, Edelstein D, Du XL, et al Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787–790. [DOI] [PubMed] [Google Scholar]
- 9. Skyler JS, Bergenstal R, Bonow RO, et al Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA Diabetes Trials: a position statement of the American Diabetes Association and a Scientific Statement of the American College of Cardiology Foundation and the American Heart Association. J Am Coll Cardiol 2009; 53: 298–304. [DOI] [PubMed] [Google Scholar]
- 10. Zoungas S, Patel A, Chalmers J, et al Severe hypoglycemia and risks of vascular events and death. N Engl J Med 2010; 363: 1410–1418. [DOI] [PubMed] [Google Scholar]
- 11. Desouza CV, Bolli GB, Fonseca V. Hypoglycemia, diabetes, and cardiovascular events. Diabetes Care 2010; 33: 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ceriello A, Novials A, Ortega E, et al Evidence that hyperglycemia after recovery from hypoglycemia worsens endothelial function and increases oxidative stress and inflammation in healthy control subjects and subjects with type 1 diabetes. Diabetes 2012; 61: 2993–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cardoso S, Santos RX, Correia SC, et al Insulin‐induced recurrent hypoglycemia exacerbates diabetic brain mitochondrial dysfunction and oxidative imbalance. Neurobiol Dis 2013; 49: 1–12. [DOI] [PubMed] [Google Scholar]
- 14. Kukidome D, Nishikawa T, Sonoda K, et al Activation of AMP‐activated protein kinase reduces hyperglycemia‐induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006; 55: 120–127. [PubMed] [Google Scholar]
- 15. Esposti MD, Hatzinisiriou I, McLennan H, et al Bcl‐2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species‐sensitive probes. J Biol Chem 1999; 274: 29831–29837. [DOI] [PubMed] [Google Scholar]
- 16. Saha AK, Ruderman NB. Malonyl‐CoA and AMP‐activated protein kinase: an expanding partnership. Mol Cell Biochem 2003; 253: 65–70. [DOI] [PubMed] [Google Scholar]
- 17. De Bock K, Georgiadou M, Schoors S, et al Role of PFKFB3‐driven glycolysis in vessel sprouting. Cell 2013; 154: 651–663. [DOI] [PubMed] [Google Scholar]
- 18. Vander Heiden MG. Exploiting tumor metabolism: challenges for clinical translation. J Clin Invest 2013; 123: 3648–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DeBerardinis RJ, Mancuso A, Daikhin E, et al Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 2007; 104: 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakamura S, Takamura T, Matsuzawa‐Nagata N, et al Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem 2009; 284: 14809–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tripathy D, Mohanty P, Dhindsa S, et al Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 2003; 52: 2882–2887. [DOI] [PubMed] [Google Scholar]
- 22. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005; 54: 1615–1625. [DOI] [PubMed] [Google Scholar]
- 23. Potenza MA, Gagliardi S, Nacci C, et al Endothelial dysfunction in diabetes: from mechanisms to therapeutic targets. Curr Med Chem 2009; 16: 94–112. [DOI] [PubMed] [Google Scholar]
- 24. Wang XL, Zhang L, Youker K, et al Free fatty acids inhibit insulin signaling‐stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 2006; 55: 2301–2310. [DOI] [PubMed] [Google Scholar]
- 25. Lee KU, Lee IK, Han J, et al Effects of recombinant adenovirus‐mediated uncoupling protein 2 overexpression on endothelial function and apoptosis. Circ Res 2005; 96: 1200–1207. [DOI] [PubMed] [Google Scholar]
- 26. Action to Control Cardiovascular Risk in Diabetes Study Group ; Gerstein HC, Miller ME, Byington RP, et al Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358: 2545–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duckworth W, Abraira C, Moritz T, et al VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360: 129–139. [DOI] [PubMed] [Google Scholar]
- 28. Sommerfield AJ, Wilkinson IB, Webb DJ, et al Vessel wall stiffness in type 1 diabetes and the central hemodynamic effects of acute hypoglycemia. Am J Physiol Endocrinol Metab 2007; 293: E1274–E1279. [DOI] [PubMed] [Google Scholar]
- 29. Fisman EZ, Motro M, Tenenbaum A, et al Is hypoglycaemia a marker for increased long‐term mortality risk in patients with coronary artery disease? An 8‐year follow‐up. Eur J Cardiovasc Prev Rehabil 2004; 11: 135–143. [DOI] [PubMed] [Google Scholar]
- 30. Joy NG, Tate DB, Younk LM, et al Effects of acute and antecedent hypoglycemia on endothelial function and markers of atherothrombotic balance in healthy humans. Diabetes 2015; 64: 2571–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jin WL, Azuma K, Mita T, et al Repetitive hypoglycaemia increases serum adrenaline and induces monocyte adhesion to the endothelium in rat thoracic aorta. Diabetologia 2011; 54: 1921–1929. [DOI] [PubMed] [Google Scholar]
- 32. Kim SR, Bae YH, Bae SK, et al Visfatin enhances ICAM‐1 and VCAM‐1 expression through ROS‐dependent NF‐kappaB activation in endothelial cells. Biochim Biophys Acta 2008; 1783: 886–895. [DOI] [PubMed] [Google Scholar]
- 33. Schoors S, Bruning U, Missiaen R, et al Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015; 520: 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amaral AI. Effects of hypoglycaemia on neuronal metabolism in the adult brain: role of alternative substrates to glucose. J Inherit Metab Dis 2013; 36: 621–634. [DOI] [PubMed] [Google Scholar]
- 35. Rains JL, Jain SK. Hyperketonemia increases monocyte adhesion to endothelial cells and is mediated by LFA‐1 expression in monocytes and ICAM‐1 expression in endothelial cells. Am J Physiol Endocrinol Metab 2011; 301: E298–E306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 ¦ Carnitine palmitoyltransferase I expression and the mitophagy status of bovine aortic endothelial cells under low glucose conditions.