

Abstract
Both zinc (Zn2+) and reactive oxygen species (ROS) have been shown to accumulate during hypoxic-ischemic stress and play important roles in pathological processes. To understand the cross talk between the two of them, here we studied Zn2+ and ROS accumulation by employing fluorescent probes in HeLa cells to further the understanding of the cause and effect relationship of these two important cellular signaling systems during chemical-ischemia, stimulated by oxygen and glucose deprivation (OGD). We observed two Zn2+ rises that were divided into four phases in the course of 30 min of OGD. The first Zn2+ rise was a transient, which was followed by a latent phase during which Zn2+ levels recovered; however, levels remained above a basal level in most cells. The final phase was the second Zn2+ rise, which reached a sustained plateau called Zn2+ overload. Zn2+ rises were not observed when Zn2+ was removed by TPEN (a Zn2+ chelator) or thapsigargin (depleting Zn2+ from intracellular stores) treatment, indicating that Zn2+ was from intracellular storage. Damaging mitochondria with FCCP significantly reduced the second Zn2+ rise, indicating that the mitochondrial Zn2+ accumulation contributes to Zn2+ overload. We also detected two OGD-induced ROS rises. Two Zn2+ rises preceded two ROS rises. Removal of Zn2+ reduced or delayed OGD- and FCCP-induced ROS generation, indicating that Zn2+ contributes to mitochondrial ROS generation. There was a Zn2+-induced increase in the functional component of NADPH oxidase, p47phox, thus suggesting that NADPH oxidase may mediate Zn2+-induced ROS accumulation. We suggest a new mechanism of cross talk between Zn2+ and mitochondrial ROS through positive feedback processes that eventually causes excessive free Zn2+ and ROS accumulations during the course of ischemic stress.
Keywords: ischemia, free zinc, reactive oxygen species, hypoxia, mitochondria, NADPH oxidase
zinc (zn2+) is an important element in physiology and is recognized to play a role in hypoxia/ischemia-induced cytotoxicity as Zn2+ accumulation precedes cell death (39, 50, 69). The removal of Zn2+ with a chelator has been shown to reduce cytotoxicity (26, 40, 62). Reactive oxygen species (ROS) produced by mitochondria have been of interest for many years because of their involvement in cellular signaling at moderate levels and in pathological mechanisms of cell death at high levels (17, 54, 68, 75). Many studies on hypoxia/ischemia-induced cytotoxicity have focused on ROS because hypoxic exposure induces oxidative stress and excessive ROS accumulation, which subsequently damages cells. Given its importance in cell stress signaling, the sources, mechanisms, and time course of ROS generation during ischemia and reoxygenation continue to receive intensive investigation. Intracellular Zn2+ accumulation has been associated with cytotoxicity and has been observed shortly after the onset of ischemia. We recently showed that hypoxia exposure induced a rapid Zn2+ transient (Zn2+ wave) that preceded mitochondrial ROS induction and accumulation (66). We are thus compelled to further study possible cross talk between these two prominent signaling systems during ischemia-like conditions of oxygen and glucose deprivation (OGD), which may lead to a better understanding of the regulation mechanism between the two important events.
Total Zn2+ concentration in human cells is in the range of several hundred micromolar (µM), with most Zn2+ being bound to proteins or sequestered into intracellular organelles (44). Zn2+ is tightly regulated in a healthy cell where labile or free Zn2+ is maintained in picomolar (pM) range, because free Zn2+ is toxic to the cells (26, 40, 62, 65). Intracellular compartments and Zn2+ binding proteins are involved in keeping Zn2+ homeostasis. Among these, mitochondria are the key intracellular organelles for buffering Zn2+ levels in neurons (62). Studies demonstrated that Zn2+ overload in mitochondria would induce multiple mitochondrial injuries (28, 50, 63) and activate mitochondrial-mediated proapoptotic factors (25, 35, 61). Zn2+ is also reported to activate mitochondrial outer membrane channels (6, 31) and cytochrome c discharge from mitochondria (12, 24). Studies also show that other organelle stores such as endoplasmic reticulum (ER) (42, 70) contain free and mobile Zn2+ pools. Together with ubiquitous metallothioneins (MT), they modulate Zn2+ homeostasis by serving as either sources or sinks of Zn2+.
The understanding of contributing factors in ROS production has been evolving in recent years, with mitochondria, xanthine oxidase, and NADPH oxidase playing an important role in ROS generation (5, 78). For example, the activity of NADPH oxidase and ROS production can be seen increasing with the duration of hypoxia (10) and mitochondria are in communication with NADPH oxidase-produced ROS (2, 18). Recent studies have highlighted the notion that Zn2+ and ROS signaling systems are intimately integrated such that Zn2+-dependent regulation of components of ROS homeostasis might influence intracellular redox balance, and vice versa. Zn2+-induced cell death is accompanied by increased levels of ROS and is attenuated by various antioxidative measures. On one hand, a number of ROS-generating and antioxidant systems of living cells have been shown to be Zn2+ dependent (62, 65). If both play key roles in regulating cellular stress responses, than the question is, what is the interaction or cross talk between Zn2+ and ROS? The objective of the present study was to identify the fundamental processes that determine Zn2+ and ROS accumulation and to explore the cause and effect relationship between Zn2+ rises and mitochondrial ROS production in cells during OGD. On the basis of the data presented here, we propose a novel positive feedback mechanism of cross talk between these two intracellular signals.
EXPERIMENTAL PROCEDURES
Materials and reagents.
Most chemicals were purchased from Sigma-Aldrich (St. Louis, MO), with exception of apocynin, which was purchased from Santa Cruz Biotechnology (Dallas, TX). Fluorescent dyes were purchased from MitoSOX Red dye (Molecular Probes, Eugene, OR) and FluoZin-3, AM (Life Technologies, Grand Island, NY). HeLa cells were purchased from ATCC (Manassas, VA). Western blot materials and reagents were purchased from Bio-Rad (Des Plaines, IL) and Invitrogen (Carlsbad, CA). Antibodies were from Cell Signaling (Danvers, MA).
Cell culture.
HeLa cells were used between passages 4 and 14. They were split every other day using the standard trypsinization method and maintained in EMEM medium supplemented with 5% fetal bovine serum (ATCC) in 5% CO2-95% humid air at 37°C (as suggested by ATCC).
Fluorescent experiments.
HeLa cells were trypsinized and seeded at medium density onto glass-bottom petri dishes (P35G-4.5-14-C; MatTek, Ashland, MA). Cells were incubated in 5% CO2-95% humid air at 37°C for at least 24 h before experimentation. On the day of the experiment, the cells were washed three times with 1 ml of physiological buffer (in mM): 25 HEPES, 125 NaCl, 3 KCl, 1.28 CaCl2, 1.1 MgCl2, 5 glucose, pH 7.4. For mitochondrial superoxide detection, MitoSOX Red dye was used at final concentration of 5 µM and cells were loaded for 10 min at 37°C. For Zn2+ detection, FluoZin-3, AM was used at final concentration of 1 µM and loaded onto cells for 60 min at room temperature. After incubation with respective dyes for each treatment, cells were washed three times with physiological buffer and left to “rest” at room temperature for 10 min before experimentation. Images were collected with a Motic AE31 microscope using Olympus U Plan FL 40X, 075 NA, with QImaging Retiga 1300i camera. Image-Pro Plus 6.2 (Media Cybernetics, Rockville, MD) was used to collect and analyze the data.
Chemical ischemia and OGD.
Chemical ischemia was induced with hypoxic buffer by using 4 mM final concentration of sodium dithionite in oxygen and glucose-deprived (OGD) physiological buffer (66). To achieve OGD, nitrogen gas was bubbled through the physiological buffer for at least 10 min before the experiment. This OGD and 4 mM sodium dithionite buffer was added by pipetting it as a 2× concentrated solution into the petri dish holding the cells, to induce rapid and reliable hypoxia-like condition (23, 43, 77). After at least 1 min of the baseline fluorescence was observed. The recordings lasted for 30 min.
Colocalization experiments.
HeLa cells were simultaneously preloaded with mitochondrial ROS indicator MitoSOX Red (5 μM) and free Zn2+ indicator Zinpyr-1 (5 μM) at 37°C for 10 min; the same protocol was then followed as described above. Confocal system Nikon A1R was used with Nikon Eclipse Ti microscope, and Nikon NIS- Elements, version 4 software. Zinpyr-1 (11, 74) (KD = 0.7 ± 0.1 nM) was chosen for this experiment because this sensor is lipophilic and easily penetrates plasma and mitochondrial membranes, which yielded a relatively strong signal of Zn2+ fluorescence. The colocalization was also analyzed by using region of interest (ROI) and NIS-element (Nikon) colocalization software. We carefully outlined multiple randomly selected mitochondria and calculated Pearson’s correlation coefficient and Mander’s overlap coefficient of the same mitochondria, using the software. We used Dunn’s suggestions in interpreting the colocalization data (19).
NADPH oxidase and xanthine oxidase inhibition.
HeLa cells were prepared using the same protocols as above (fluorescent experiments). Cells were loaded with MitoSOX Red (5 µM). The NADPH oxidase inhibitor apocynin was dissolved in DMSO and used at 60 µM final concentration with exogenous Zn2+ (50 µM) and sodium pyrithione (10 µM). Apocynin was added at the same time as the other solutions. All the solutions had 0.1% final concentration of DMSO, as a control. Xanthine oxidase was inhibited by 60 μM oxypurinol, and experiments were performed as for NADPH oxidase inhibition. Fluorescent intensity was measured before the addition of treatment solutions and was measured again after 30 min of treatment.
Immunoblotting (Western blot analysis).
HeLa cells were treated and plated as described in fluorescent experiments. For these experiments, 35-mm sterile culture dishes were used instead of glass-bottom dishes. After 24 h of culture, the HeLa cells were washed three times with physiological buffer and solutions with appropriate treatments were added to the cells. Cells were incubated for 30 min in a 37°C incubator. After the incubation, the experimental solutions were washed out once with physiological buffer and removed from the samples. Laemmli sample buffer (1.5×; Bio-Rad) was added at 100 μl per each 35-mm culture dish. Cells were scraped off and homogenized in the sample buffer. Samples were boiled for 5 min. Proteins were separated immediately by SDS-PAGE electrophoresis on 10% acrylamide/bis gel with XCell II module (Bio-Rad). Polyvinylidene difluoride (PVDF) 0.45 μm membrane (Invitrogen) was used to transfer the proteins using XCell II module at 300 mA constant voltage for 2 h at room temperature. The membrane was blocked with 5% bovine serum albumin (BSA) in tris(hydroxymethyl)aminomethane buffered saline (TBS) with 5% Triton X-100 (TBST) for 2 h at room temperature. Rabbit monoclonal antibody against p47phox was diluted 1:1,000 in 2% BSA-TBST, and the membrane was incubated with primary antibody solution overnight at 4°C. The p47phox antibody was purchased from Cell Signaling (no. 4301, lot 1) and is specific to total exogenous p47phox (NCF1). Horseradish peroxidase (HRP)-labeled secondary antibody anti- rabbit was used (no. 7074, Cell Signaling). ChemiDoc system (Bio-Rad) was used to visualize the signal with Clarity Western ECL Substrate (Bio-Rad); this reagent was used as specified by the manufacturer. Total protein staining on the same blot was used as a loading control. To stain total proteins on the blot, Coomassie Brilliant Blue R-250 (Bio-Rad) was used. The membrane was incubated for 1 min and the stain was then washed off three times, as previously described (76). The advantage of using total protein over the traditional housekeeping proteins has been previously described (76). The washed membrane was dried completely and imaged with ChemiDoc imaging system with colorimetric blot setting.
Brain slices.
Brain slices were prepared from male Sprague-Dawley rats (4-6 mo old), raised in normal laboratory conditions with standard rat food. Animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC). The hippocampus was dissected out of the brain and sliced to 200-µm thickness and incubated immediately with artificial cerebral spinal fluid (ACSF, in mM: 121 NaCl; 1.75 KCl; 1.3 MgCl2; 2.5 CaCl2; 1.25 KH2PO4; 26 NaHCO3 10 glucose) and gassed with 95%-O2-5% CO2, pH 7.2–7.3, for 1 h to recover from slicing in interface chamber. After recovery, slices were transferred to a dye incubation chamber, containing 2 ml of ACSF, and bubbled with 95%-O2-5% CO2 and loaded with 20 µM HEt dye for 30 min. After loading, the dye was removed and slices were washed three times with ACSF before the experiments. For oxygen-glucose deprivation (OGD), all slice manipulations were performed in 35-mm glass-bottomed dishes, where slices were fully submerged in circulated oxygen-glucose-deprived ACSF solution, which was prebubbled with 95% N2-5% CO2 gas for 30 min before slice immersion, to ensure oxygen removal. Baseline fluorescence was measured for 5–10 min before exposure to OGD. OGD exposure was implemented for 30 min, where slices were perfused continually. This treatment was followed by subsequent reperfusion with normal ACSF for 40 min (reoxygenation or reperfusion). The fluorescence was recorded before reoxygenation and 30 min after reoxygenation. The regions of interest (ROIs) in the CA1 region of the hippocampus were highlighted and fluorescence was quantified using Image Pro software, as in above described fluorescent experiments. Apocynin, an NADPH oxidase inhibitor (180 µM), or N,N,N′,N′-tetrakis-(2-pyridylmethyl)ethylenediamine (TPEN), a Zn2+-specific chelator (35 µM), was added to the perfusion during reoxygenation only and was not present during hypoxia. The fluorescent measurements were compared with the brain slices without any treatment during hypoxia and reoxygenation.
Data analyses.
For Zn2+ and mitochondrial ROS transients, the cytosol excluding the nucleus was analyzed for each cell. The changes in florescence (∆ F) in the cytosol were background corrected by subtracting fluorescence from area without cells. Each relative fluorescence trace was normalized to baseline by the following formula ∆ F = (F measure − F0)/F0, where F measure is a recorded data point and F0 is relative fluorescence at baseline. The averages of the traces were plotted on the graph with standard deviation bars or a representative trace was selected and plotted. For Zn2+-induced ROS experiments and enzyme inhibitors, the percent change in fluorescence was calculated using the following formula: [(F − F0)/F0] × 100, where F0 is averaged baseline fluorescence intensity before addition of experimental solutions and F is fluorescence intensity value after 30 min of incubation with the experimental solutions. The significance was measured by simple two-group comparison and analyzed by Student’s paired t- test (or single-factor ANOVA), with P < 0.05 considered significant.
RESULTS
OGD-induced intracellular Zn2+ concentration rises and their relationships with mitochondrial ROS accumulation production.
To induce a hypoxia-like response in live cells, an oxygen scavenger and reducing agent, sodium dithionite, was used. This method of “chemical” hypoxia has been widely used to rapidly induce a low-oxygen environment with reliability (23, 29, 56, 60, 77). Others used this method of hypoxia induction paralleled with oxygen deprivation (like we did in brain slice experiments, discussed further) (41, 43). To simulate an ischemia-like condition, in addition to chemical hypoxia, we removed glucose from the solutions as well, inducing a state of oxygen and glucose deprivation (OGD). To measure the changes in intracellular Zn2+ concentration ([Zn2+]i) during the course of OGD, cells were labeled with the intracellular fluorescent free Zn2+ indicator FluoZin-3, AM (1 µM) with KD ~15 nM. Basal cytosolic Zn2+ distribution with FluoZin-3, AM appeared generally very faint although in some cells a brighter Zn2+ fluorescence was observed around the nucleus. The nucleus had no detectible free Zn2+ and appeared as a dark oval structure in the middle of the cell. Cells in the OGD treatment were continuously bathed in OGD buffer. Changes in fluorescence intensity indicated changes in intracellular free Zn2+ concentration. Note that the extracellular solutions during OGD experiments did not contain detectible amounts of free Zn2+. We observed two Zn2+ elevations in the course of 30 min of hypoxic treatment (Fig. 1, A and B). On the other hand, cells in control tests were continuously bathed in physiological buffer and yielded no significant change in the fluorescence. The time course of intracellular Zn2+ elevations can be divided into four phases (Fig. 1, A–C). Phase I is the ascending part of the first Zn2+ rise (Zn2+ transient) observed and is defined by a sharp increase in [Zn2+]i, beginning as early as 60 s after onset of OGD treatment, and continued to ascend with a sharp slope for 90 s. Phase I was quickly followed by Phase II, which is the descending part of cytosolic Zn2+ transient and characterized by Zn2+ returning toward basal level (Fig. 1A). Because cells were continuously bathed in OGD buffer, the majority of examined cells (20/33, 61%) had rather a short phase II and Zn2+ did not return to basal level but maintained a sustained increase in [Zn2+]i (Fig. 1A-1). There were ~40% cells in which Zn2+ transient returned close to the baseline and the phase II lasted for ~2 min (Fig. 1A-2). The third phase (III) is a latent phase, during which Zn2+ transient gradually elevated above basal levels (Fig. 1A). The duration of the latent phase was variable but generally ranging around 10 min. The latent phase was followed by phase IV, which was the second sharp Zn2+ elevation characterized by a substantial increase in [Zn2+]i to a sustained plateau, which we have also termed Zn2+ overload. Phase IV or Zn2+ overload was observed in all examined cells (Fig. 1A), and onset of the overload started consistently around 19–22 min of OGD exposure. Overall, its amplitude was about twice as large as the amplitude of phase I. It did not exhibit a wavelike transient and only had an ascending component that continued for ~4 min before reaching a plateau, where there was continued increase in [Zn2+]i throughout the observation time.
Fig. 1.
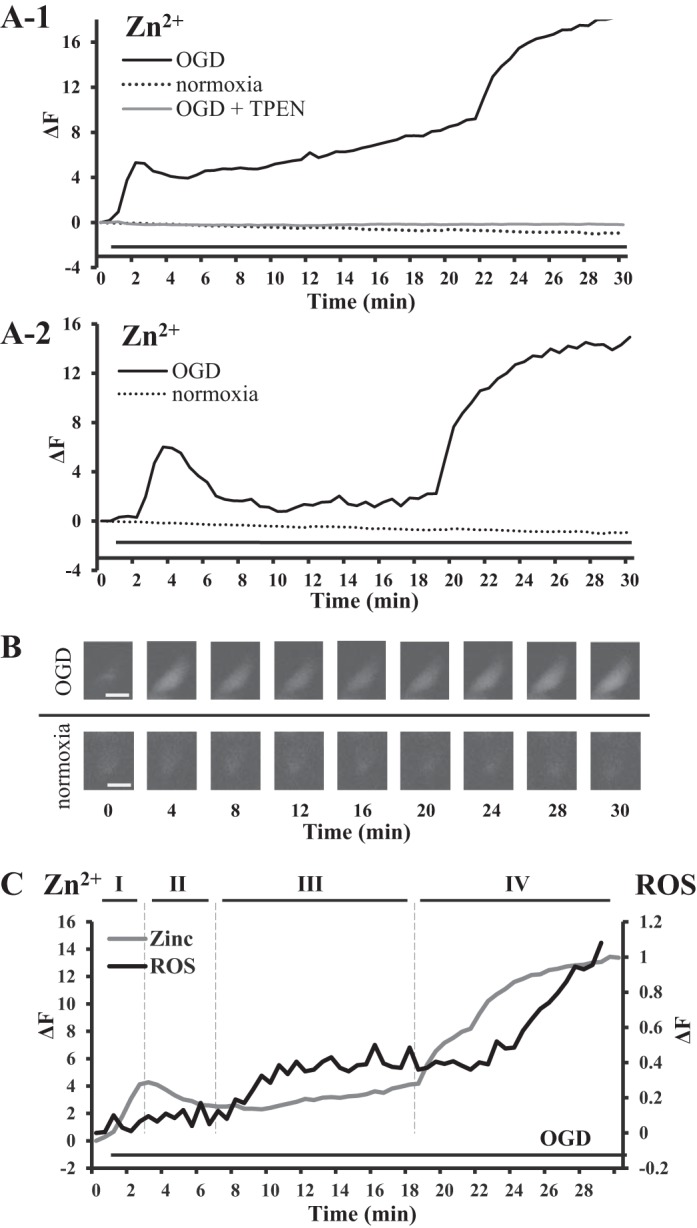
Zinc rises during oxygen glucose deprivation (OGD) and the temporal relationship with mitochondrial reactive oxygen species (ROS) accumulation. A: representative traces of Zn2+ fluorescence (FluoZin-3, AM) in HeLa cells, showing increases in [Zn2+]i during hypoxic treatment. Cells were continuously bathed in hypoxic buffer throughout fluorescence measurement as indicated by the solid horizontal lines underneath all of the traces. There are two distinct Zn2+ rises (Zn2+ transient and Zn2+ overload) and a latent phase between two elevations, which can be divided into four phases (as depicted in C). A-1: representative trace of OGD-induced Zn2+ rises observed in 61% (n = 33 in 6 separate experiments) of examined cells, where the first Zn2+ increase or Zn2+ transient does not return to the basal level with a short phase II and elevated phase III, which was caused by sustained increases in [Zn2+]i due to continuous OGD. Gray line trace represents cells treated with OGD buffer in the presence of the Zn2+ chelator TPEN. Dotted line traces (A-1 and A-2) represent control cells treated with physiological buffer, under normoxic conditions. A-2: representative of OGD-induced Zn2+ increases observed in 39% of examined cells, where Zn2+ transient returns to the basal level. B: representative Zn2+ fluorescent images of HeLa cells during OGD (FluoZin-3, AM); control is HeLa cells exposed to physiological buffer. Minute 0 is the baseline fluorescence before exposure to OGD, where the Zn2+ fluorescence is very faint. At 4 min after OGD, there is a significant increase in fluorescence. At minute 8, the fluorescence is lower and remains low at minute 12, 16. The Zn2+ fluorescence starts to rises at minute 20. Scale bar, 10 µm. C: representative traces of Zn2+ (FluoZin-3, AM) and mitochondrial ROS (MitoSox Red) during OGD, plotted on the same graph to show temporal relationship of the two important phenomena. The lines above representative traces and the roman numerals indicate the phases of OGD-induced Zn2+ rises.
Fig. 2.

Thapsigargin (TG) and FCCP-sensitive intracellular sources of OGD-induced increases in [Zn2+]i. A: average traces of Zn2+ fluorescence during OGD treatment in HeLa cells with or without TG pretreatment (2 µM). Black trace represents cells pretreated with TG for 20 min before the induction with OGD (TG + OGD), where the baseline is normalized to the elevated Zn2+ before OGD exposure (n = 12 in two separate experiments). Gray trace represents control (DMSO + OGD) cells pretreated with 0.1% DMSO before induction of OGD (TG was dissolved in DMSO; n = 15 cells in two separate experiments). The black line under the traces represents the duration of OGD treatment. Error bars are standard deviation. B: average trace of Zn2+ fluorescence (FluoZin-3, AM), showing FCCP-induced [Zn2+]i rises during normoxic conditions (1 µM FCCP; n = 12 cells from two separate experiments); baseline is normalized to starting elevated Zn2+ level. The black line under the traces represents FCCP treatment. Error bars are standard deviation. C: average traces of Zn2+ fluorescence in HeLa cells that were pretreated with or without FCCP before OGD treatment. Black trace represents cells pretreated with FCCP for 10 min and after washout of FCCP before OGD treatment (n = 20 from three separate experiments). Gray trace represents control cells pretreated with 0.1% DMSO before exposure to OGD (control, n = 10 cells from two separate experiments). D: bar graph showing ratio of peak fluorescence intensity of the second OGD-induced Zn2+ rise divided by peak fluorescence intensity of the first Zn2+ rise. Error bars represent standard deviation (*P = 0.0017; FCCP, n = 20; control, n = 10).
The OGD-induced increases in [Zn2+]i was compared with mitochondrial ROS accumulation ([ROS]mito) to explore the possible temporal relationship of these two important phenomena during ischemia-like conditions of OGD. The mitochondrial superoxide indicator MitoSOX Red (5 µM) was used to study ROS accumulation in mitochondria. The probe’s distribution resembled mitochondrial distribution, described previously, where mitochondria was labeled with the mitochondria-specific fluorescent dye MitoFluor Red (42). When cells were exposed to OGD under the same condition as in Zn2+ fluorescence-labeled cells, [ROS]mito started to rapidly rise after 5 min of exposure and continued to rise for 4 min, after which the ROS levels plateaued and remained elevated until 22–23 min of exposure (Fig. 1C). The plateau resembled the latent phase seen in OGD-induced Zn2+ elevation and was followed by the second ROS increase. During the period of delayed Zn2+ overload, the mitochondrial ROS sharply increased and continued to increase during the remaining observation. To show the temporal relationship of these two signals, Zn2+ fluorescence and mitochondrial ROS fluorescence were plotted together on one plot (Fig. 1C). The first Zn2+ elevation (Zn2+ transient) precedes the beginning of [ROS]mito increase that corresponds with Zn2+ transient on the late phase II. While steady and sustained ROS production is parallel to the latent phase and is before the second sharp Zn2+ elevation, the latter precedes the second sharp ROS increase.
Intracellular sources of OGD-induced [Zn2+]i rises depend on TPEN, thapsigargin, and FCCP-sensitive stores.
To explore the possible contributions of intracellular Zn2+ sources to OGD-induced Zn2+ rises, we treated cells with TPEN, a membrane-permeable Zn2+ chelator. TPEN (35 µM) was applied 5 min before the application of OGD buffer and was also included in OGD buffer throughout the hypoxic treatment. As shown in Fig. 1A-1, TPEN completely removed the OGD-induced Zn2+ increases, including the first and second rises. This result not only supported that the OGD-induced Zn2+ rise detected with the fluorescent Zn2+ indicator was a Zn2+-dependent phenomenon, but also indicated that they originated from intracellular Zn2+ storage. Zn2+ is found in cellular organelles such as endoplasmic reticulum (ER) (14, 42, 59, 70, 72), with ER requirements of Zn2+ for its normal function (21, 22). Thapsigargin (TG) is an inhibitor of the sarco/ER Ca2+-ATPase (SERCA) and is widely used for Ca2+ store depletion by preincubation with low concentrations of TG. We showed previously that TG also increased [Zn2+]i by releasing Zn2+ from TG-sensitive and inositol (1,4,5)-trisphosphate receptor-mediated stores of the ER (42, 70). In this study, we incubated cells with TG (2 µM) for 20 min before inducting OGD conditions. Since TG was dissolved in DMSO, cells exposed to DMSO (0.1%) were used as a control for this set of experiments. Control cells responded to OGD treatment with smaller Zn2+ rises (Fig. 2A); however, we still observed two Zn2+ rises with overall response resembling OGD-induced Zn2+ changes seen in Fig. 1. In comparison, the cells that were treated with TG showed little Zn2+ increases in response to hypoxia treatment (Fig. 2A). Baseline of thapsigargin-treated cells was normalized to elevated Zn2+ levels before the induction of OGD. These results show that TG had a significant influence on the OGD-induced Zn2+ response in HeLa cells, which suggests that ER is a potential major source of free Zn2+ and that ER can contribute to Zn2+ dyshomeostasis. Thus, the ability of both TPEN and TG to limit OGD-induced Zn2+ rises supports that the observed Zn2+ during chemical ischemia is of an intracellular origin and is derived from intracellular stores.
To determine whether mitochondrial Zn2+ pool was contributing to OGD-induced Zn2+ increases, we mobilized mitochondrial Zn2+ by the application of FCCP, a mitochondrial uncoupler that dissipates the proton gradient across the inner mitochondrial membrane. Prior studies have found that FCCP releases Zn2+ from mitochondria and also disrupts the uptake of Zn2+ into the mitochondria (50, 62). In this study we observed a significant FCCP-induced [Zn2+]i rise (measured with FluoZin-3, AM) when cells were bathed in a physiological buffer with FCCP (1 µM) (Fig. 2B). These results support that Zn2+ can be released into the cytosol by FCCP. Next, to learn whether the release of Zn2+ from mitochondria by impairing mitochondrial function was responsible for OGD-induced Zn2+ rises, we preincubated cells with FCCP (before OGD treatment). These experiments were independent from the above described experiments and were done separately. Cells were pretreated with FCCP (1 µM) for 10 min; FCCP was then washed out before the start of OGD treatment. The data from FCCP-pretreated cells were then compared with the data from cells without FCCP pretreatment, where the FCCP-treated cells were normalized to increased Zn2+ during baseline. Under this condition, we still observed the increases in [Zn2+]i with two sharp rises. The first Zn2+ transient was not affected by FCCP pretreatment and remained generally unchanged in onset, duration, and amplitude. The second OGD-induced Zn2+ rise, however, was significantly reduced compared with the cells without FCCP pretreatment (Fig. 2C), indicating that the release of mitochondrial Zn2+ may be a major contributor to the second rise of [Zn2+]i. To quantify the change in the second Zn2+ rise, the fluorescent intensity of the second rise was divided by the intensity of the first rise of [Zn2+]i in the same cell and the data are presented as a ratio (Fig. 2D). Overall, the second OGD-induced [Zn2+]i rise is greater than the first OGD-induced rise with ratio of 2nd to 1st > 1. In the cells pretreated with FCCP the second Zn2+ rise was less than half the amplitude of the first Zn2+ rise with ratio 2nd to 1st < 0.5.
Zn2+ mediates mitochondrial ROS accumulation.
We previously showed that Zn2+ fluorescence was colocalized in mitochondria when it was labeled with the mitochondrial membrane indicator MitoFluor Red (42) and MitoTracker Red (52). It is consistent with the notion that mitochondria are Zn2+ storing organelles containing free Zn2+ (62, 65). Figure 3A shows the colocalization of Zn2+ and mitochondrial ROS fluorescence. For this set of experiments, to show the mitochondrial origin of ROS production, mitochondria were labeled with MitoSOX Red (5 μM), a ROS indicator sensitive to mitochondrial superoxide production, where oxidation of MitoSOX Red by mitochondrial superoxide produces red fluorescence. Mitochondrial Zn2+ fluorescence was detected with Zinpyr-1, which is lipophilic and easily penetrates plasma and mitochondrial membranes, which yielded a relatively strong signal of Zn2+ fluorescence. We observed a strong colocalization of two fluorescent signals in merged images (Fig. 3A), which resembled previous colocalization study of Zn2+ and mitochondria (42). In addition, the colocalization was also analyzed using NIS-element (Nikon) colocalization software, where we carefully outlined multiple randomly selected mitochondria and calculated colocalization with Mander’s overlap coefficient of 0.95 ± 0.02 and Pearson’s correlation coefficient of 0.85 ± 0.05. Taking into account the visual inspection of merged images (Fig. 3A) and coefficients of colocalization (both Pearson’s and Mander’s), we concluded that mitochondria labeled with Zn2+ fluorescence were also undergoing mitochondrial ROS production.
Fig. 3.
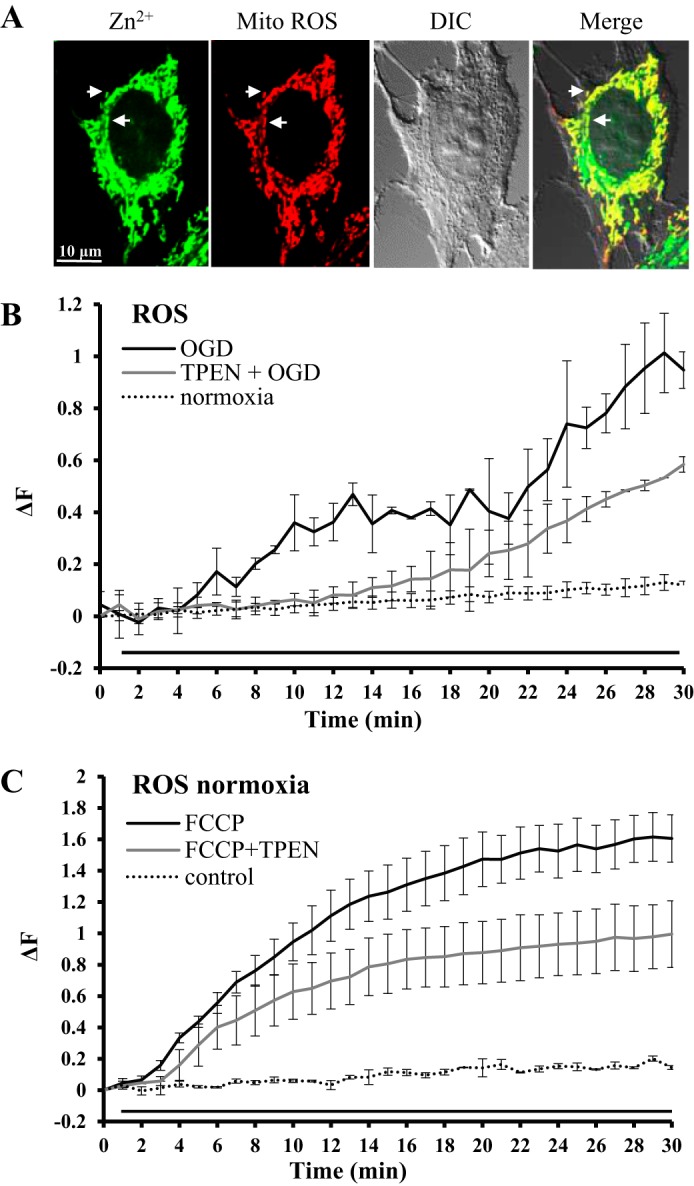
Mitochondrial ROS production reduced by Zn2+ chelation. A: confocal images of a HeLa cell double-labeled with Zn2+ and mitochondrial ROS fluorescent indicators. Zinc: Zinpyr-1 (1 µM) green fluorescence. Mito ROS: MitoSOX Red (5 µM) red fluorescence. DIC: differential interference contrast image of the same cell by transmitted light to show overall structure of the cell. Merge: composite overlay fluorescence image. The yellow structures of the merged image show colocalization of Zn2+ and mitochondrial ROS fluorescence. All images were captured at ×100. Scale bar, 10 µm. Arrows highlight a couple of mitochondria with colocalized signal. B: average traces show the effect of TPEN, a Zn2+-specific chelator, on OGD-induced ROS production in cells loaded with MitoSOX Red (5 µM). Black line trace represents OGD-induced ROS accumulation (n = 15 from five separate experiments). Gray line trace represents OGD-induced ROS in the presence of TPEN (35 µM; n = 6 from two separate experiments). TPEN reduced OGD-induced ROS generation. The black line under the traces represents hypoxic treatment. Dotted line trace represents control cells loaded with MitoSOX Red but without hypoxic treatment (n = 10 from five separate experiments). C: average traces show the effect of TPEN, a Zn2+-specific chelator, on FCCP-induced ROS productions in cells loaded with MitoSOX Red (5 µM) under normoxic conditions. Black line trace represents FCCP (1 µM)-induced ROS accumulation (n = 15 from two separate experiments). Gray line trace represents FCCP-induced ROS in the presence of TPEN (35 µM; n = 10 from two separate experiments). TPEN reduced FCCP-induced ROS generation. The black line under the traces represents the duration of FCCP treatment. Dotted line trace represents control cells loaded with MitoSOX Red but without FCCP treatment (n = 10 from five separate experiments).
Next, we wanted to determine whether mitochondrial ROS production was sensitive to Zn2+ accumulation. We used MitoSOX Red as a mitochondrial ROS fluorescent probe. The control cells were exposed to physiological buffer under normoxic conditions. When cells were treated with OGD buffer, the MitoSOX Red fluorescence increased significantly within 5 min after the start of OGD, indicating the rising of [ROS]mito production. There were generally two [ROS]mito rises during 30 min of OGD treatment (Fig. 3B; also see Fig. 1C). When we removed intracellular Zn2+ with TPEN during OGD, we observed only one mitochondrial ROS rise. Overall, in the presence of TPEN, OGD-induced mitochondrial ROS rise was delayed almost 10 min and was significantly smaller in amplitude. These data suggest that [ROS]mito production under OGD conditions is sensitive to Zn2+ accumulation. In another set of experiments, FCCP application (1 µM) induced a steady increase of ROS in cells labeled with MitoSOX Red (Fig. 3C). In the presence of TPEN, FCCP induced a significantly smaller ROS accumulation, supporting further that Zn2+ was an important factor in sustained mitochondrial ROS production.
NADPH oxidase contributes to [ROS]mito accumulation induced by Zn2+.
In following study, we examined the generation of ROS in response to the application of exogenous Zn2+ with the purpose of identifying the possible mechanism in which accumulation of [Zn2+]i leads to ROS generation. HeLa cells were labeled with MitoSOX Red and exposed to exogenous Zn2+ (50 µM) along with the Zn2+ ionophore sodium pyrithione, which transports Zn2+ across a cell membrane and increases intracellular Zn2+ concentration. In reality, by adding the zinc/ionophore combination, we are studying a rapid intracellular Zn2+ increase during normoxia, or in other words, Zn2+-induced changes. As a contrast, the control cells were bathed in physiological buffer. All of these experiments were done under normoxic conditions. Zn2+ addition caused significant increases in [ROS]mito production, which started 10–15 min after addition of Zn2+. Further in the paper we will be calling this Zn2+-induced [ROS]mito, to differentiate these data from OGD-induced ROS. Next, we examined Zn2+-induced ROS generation in the presence of oxipyrinol, a xanthine oxidase inhibitor, or apocynin, a NADPH oxidase inhibitor (56a). Both enzymes have been shown to be significant contributors to ROS generation. As show in Fig. 4A, the inhibition of xanthine oxidase by oxipyrinol (60 µM) did not affect Zn2+-induced ROS accumulation. On the other hand, Zn2+-induced ROS production was significantly reduced when NADPH oxidase was inhibited by apocynin (60 µM) (Fig. 4A), suggesting that Zn2+-induced ROS accumulation involves the activation of NADPH oxidase.
Fig. 4.
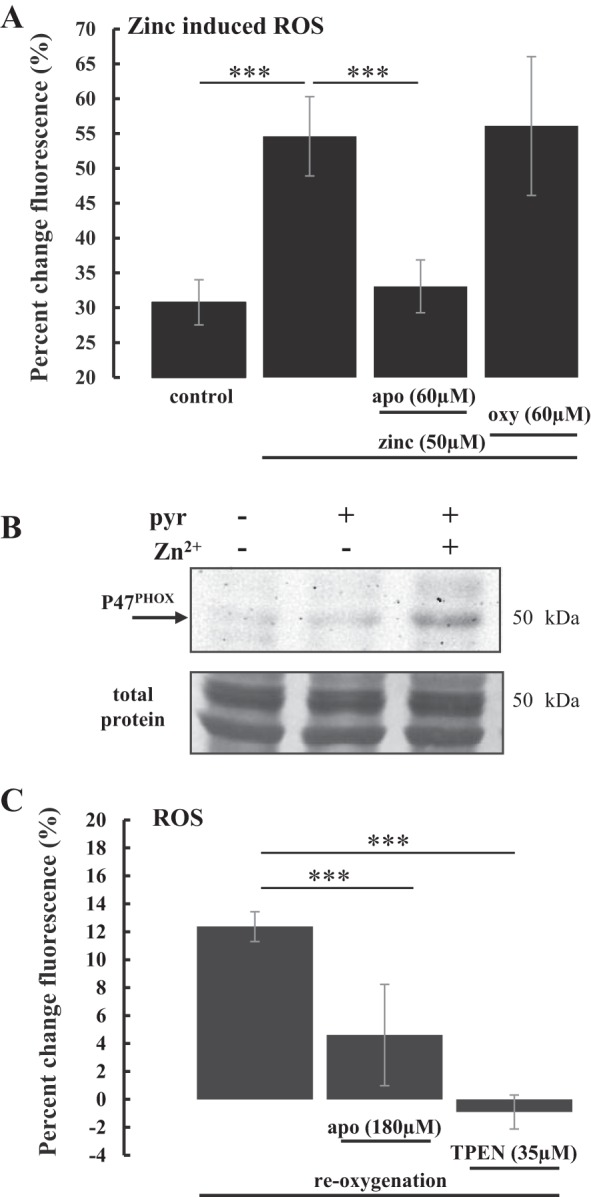
Interplay between NADPH oxidase and Zn2+ in ROS accumulation during induced intracellular Zn2+ increase. A: bar graph of Zn2+-induced mitochondrial ROS in HeLa cells loaded with ROS indicator MitoSOX Red. Fluorescence was measured at the end of 30 min of treatments with exogenous Zn2+ (n = 12 from two separate experiments), Zn2+ with apocynin, a NADPH oxidase inhibitor (apo, n = 12 from two separate experiments), or Zn2+ with oxypurinol, a xanthine oxidase inhibitor (oxy, n = 12 from two separate experiments). Zn2+ was coapplied with the Zn2+ ionophore pyrithione (10 μM), which induces rapid intracellular Zn2+ increase. ROS levels (means ± SD) are expressed as percent change in mean fluorescence intensity from baseline before treatment. Control is cells exposed to physiological buffer (n = 9 from three separate experiments) under normoxic conditions. B: representative Western blot of HeLa cell total cell homogenate, probed with anti-p47phox antibody, which detects endogenous levels of p47phox, a functional subunit of NADPH oxidase. The first lane of the blot is control cells exposed to physiological buffer. The second lane is cells treated with sodium pyrithione (pyr, 10 μM) alone. The third lane is cells treated with combination of Zn2+ (50 μM) and pyrithione (10 μM), showing that intracellular Zn2+ increase leads to higher level of p47phox. Bottom panel is the loading control, where the blot was stained with Coomassie Blue R-250 to show total protein on the blot after the probing with antibodies (n = 2). C: bar graph showing changes in baseline ROS fluorescence (HEt) in rat hippocampal slices after OGD and reoxygenation. Fluorescence was measured at the end of 30 min of OGD and the end of 30 min of reoxygenation. ROS levels (means ± SD) are expressed as percent change of fluorescence intensity before reoxygenation and 30 min after reoxygenation. Reoxygenation caused a significant increase in HEt fluorescence (12.4% ± 1.1%, n = 10 from two separate experiments). NADPH oxidase inhibitor apocynin (apo) reduced ROS accumulation to 4.6% ± 3.6%. TPEN, a Zn2+ chelator, caused greater reduction in ROS accumulation, resulting in fluorescence −1.0% ± 1.2%. ***P < 0.001.
To further study if NADPH oxidase mediated Zn2+-induced ROS accumulation, we examined whether the application of Zn2+ will alter the amount of the functional component of NADPH oxidase, p47phox (NCF1), in HeLa cells. NADPH oxidase subunit p47phox has been used as a proxy of NADPH oxidase amount (8, 37). In this experiment, HeLa cells were exposed to following treatments: physiological buffer, pyrithione alone, and pyrithione plus Zn2+. A representative Western blot is shown in Fig. 4B. The antibody used detects endogenous total p47phox and as expected, we did not detect a lot of p47phox in physiological or pyrithione alone conditions; however, there were significantly increased levels of the NADPH oxidase subunit p47phox in Zn2+-treated cells. Interestingly, these detectable changes were observed within 30 min of treatment, which is a very short time for protein induction, suggesting that Zn2+ action on NADPH oxidase is very fast.
Both Zn2+ and NADPH oxidase contribute to ROS accumulation during re-oxygenation.
The accumulation of ROS during reoxygenation has been well described as a possible contributor to reperfusion or reoxygenation injury (4). There is evidence that NADPH oxidase contributes significantly to ROS generation following reoxygenation or reperfusion (1, 9, 30). Here, we examined the effect of apocynin, an NADPH oxidase inhibitor, on ROS generation during reoxygenation, which was compared with the effect of Zn2+ removal by TPEN on the ROS production. The experiments were carried out in freshly prepared rat hippocampal brain slices. The advantage of the hippocampal brain slice is that it is an established in situ model for neural function and injury and the hippocampal tissue is particularly sensitive to ischemia (OGD) and reoxygenation. Ischemia-like conditions were achieved by perfusing brain slices with oxygen and glucose deprived (OGD) physiological buffer (artificial cerebral spinal fluid, ACSF); subsequently, slices were reperfused with normal ACSF to achieve re-oxygenation. The slices were loaded with the ROS indicator dihydroethidium (HEt) (see details in experimental procedures). Apocynin (180 µM) or TPEN (35 µM) was administrated in the perfusate during reoxygenation. As summarized in Fig. 4C, reoxygenation caused a significant increase in HEt fluorescence compared with HEt fluorescence with OGD. Both treatments with apocynin and TPEN significantly reduced the generation of cellular ROS during reoxygenation. The inhibition of ROS production by TPEN was significantly greater than that by apocynin (Fig. 4C). Taken together, we have demonstrated that NADPH oxidase mediates Zn2+-induced ROS accumulation (Fig. 4A) and that Zn2+ enhances the amount of functional subunit p47phox of NADPH oxidase (Fig. 4B). The results indicate that both Zn2+ and NADPH oxidase are contributing to ROS accumulation during reoxygenation with Zn2+ accumulation possibly precipitating ROS accumulation by activating NADPH oxidase.
DISCUSSION
Two distinct Zn2+ rises in multiphase responses during OGD.
In the present study, we applied sodium dithionite to live cells, which rapidly binds oxygen in solution, to induce a consistent hypoxic-like state in cells, as many other groups have done (23, 29, 41, 43, 56, 60, 67, 77). We observed changes in cytosolic free Zn2+ that followed a consistent pattern that could be divided into four phases of two distinct Zn2+ rises (Fig. 1). The first phase (I) is a rapid rising of Zn2+ transient elicited soon after induction of OGD condition. Zn2+ then recovered toward basal levels during the second phase (II) as a descending phase of Zn2+ transient, indicating that increased Zn2+ was not necessary to sustain homeostasis changes if OGD was brief. Therefore, the first Zn2+ rise is a transient increase of cytosolic free Zn2+. It was observed in all recorded cells, suggesting a uniform response to OGD, and the response was consistent in the onset of phase I. However, there was a difference in phase II observed among cells, where the descending part of the Zn2+ transient came back toward the baseline. In most cells the Zn2+ level reduced but stayed elevated. Since cells were continuously under the OGD in this study, the Zn2+ rise did not completely return to basal levels in the most cells. There was a latent phase (III) between first and second Zn2+ rises, during which intracellular free Zn2+ level maintained at above the basal level. The forth phase (IV) was marked by a quick increase in the intracellular free Zn2+ to a sustained plateau in the significantly higher amplitude than the first phase from which Zn2+ increases did not recover. We have termed this delayed Zn2+ elevation as Zn2+ overload.
The principal finding of this study is the occurrence of early phases (phases I and II) and a latent phase (phase III) of Zn2+ response following OGD. The early Zn2+ transient increase is an adaptive and protective response to hypoxic treatment. Studies suggest that the increased Zn2+ concentration had a proantioxidant effect by Zn2+ binding to thiols and preventing their oxidation, and/or by activating antioxidant response elements (46, 57). Many radical scavengers such as Cu-Zn-SOD are Zn2+-containing proteins; therefore the moderate increase in cytosolic Zn2+ may be protective to cells. The key feature of phase II is that the Zn2+ transient appears to descend toward basal levels in all cells, which indicates that a short hypoxic treatment does not irreversibly change Zn2+ homeostasis and suggests that Zn2+ buffering and homeostatic mechanisms were intact. Since maintaining basal Zn2+ requires the concerted efforts of a number of regulatory and metabolic processes, the finding that this system is functional following OGD suggests that the OGD-induced cellular process may be reversible during a short exposure to OGD. As OGD continues, we observe a latent phase (III), during which Zn2+ remains elevated (Fig. 1). At this point the Zn2+ buffering systems may become overwhelmed, and in addition, the increased Zn2+ may trigger other adverse cellular responses, such as ROS production (40, 44), which in turn may further disturb the buffering systems, causing total loss of Zn2+ homeostasis, which is marked by Zn2+ overload (phase IV). This second significant zinc increase was almost double the amplitude of the first increase and rapidly reached a plateau, suggesting irreversible dysfunction of Zn2+ homeostasis (3, 13).
Sources of intracellular Zn2+ contributing to Zn2+ transient.
The presence of such a consistent and large Zn2+ response to OGD was in itself a fascinating observation; however, we wanted to explain a source for these two Zn2+ responses (Figs. 1 and 2). When TPEN was applied, before cells were exposed to OGD, both the first and second rise were abolished (Fig. 1A-1). During the hypoxic experiments the extracellular solutions did not have a detectible amount of Zn2+, suggesting that both rises we observed during OGD without TPEN were of intracellular origin, which is not surprising due to mounting evidence that Zn2+ is bound to proteins and stored in cellular organelles (44).
Emerging evidence has shown that Zn2+ transporters are found on the plasma membrane of cellular organelles, providing molecular evidence that Zn2+ is transported among discrete subcellular compartments (34, 40). Zn2+ can accumulate in the mitochondrial matrix (see below). ER has been emerging as a site of Zn2+ storage (14, 42, 59, 70, 72), where Zn2+ is required for normal ER function (21, 22). Worthy of notice is that ER and mitochondria are key organelles involved in the storage and release of intracellular Ca2+ (but also see refs. 47, 69). In the present study, the timescale of Zn2+ transient observed soon after starting hypoxic treatment is similar to Ca2+ waves, which is attributed to the release and uptake of Ca2+ by the ER. We previously showed that this ER Zn2+ pool can be readily mobilized when cells are treated with TG (70). Consistent with previous studies, the present study found that TG pretreatment abolishes Zn2+ rise in cells undergoing OGD (Fig. 2A). Thus, the ability of both TPEN and TG to limit OGD-induced Zn2+ elevation supports the hypothesis that the observed Zn2+ increases are of intracellular origin and are derived from intracellular stores.
An interesting question is how the elevated Zn2+ is so quickly buffered as seen in phase II or the descending phase of the first Zn2+ transient. The considerably quick response observed in hypoxic treatment also suggests a fast kinetics of Zn2+ mobilization and removal. We suggest that intracellular Zn2+ storage, such as MTs, ER, and mitochondria, can play dual roles as both a Zn2+ source and a Zn2+ sink. For example, mammalian Zn2+ transporter ZIP7 proteins are present in the ER, where they contribute to the release of free Zn2+ into the cytosol from ER stores (32, 72). On the other hand, Zn2+ transporters ZnT5/ZnT6/ZnT7 are involved in ER homeostasis by transporting Zn2+ into the lumen (33, 71) and may also function as bidirectional transporters (20, 21, 58). Furthermore, elevated levels of Zn2+ cause the biological system to store the extra Zn2+ ions in the mitochondrial matrix, another crucial player in Zn2+ clearance (see below). The prolonged OGD exposure causes the continuous release of Zn2+ from the above discussed stores, which may contribute to Zn2+ and ROS accumulation seen in the latent phase (phase III).
Mitochondrial dyshomeostasis and MTs are a major sources of the second Zn2+ increase.
There has been considerable focus on mitochondria taking up cytosolic Zn2+ to maintain intracellular Zn2+ concentration (62). We previously reported that both Zn2+ and mitochondria colocalize in healthy live cells (42). Other groups showed that application of FCCP mobilized the mitochondrial Zn2+ pool (50, 63). In the present study we observed a substantial increase in cytosolic Zn2+ after 2 min of treatment with FCCP (Fig. 2B), which supports that mitochondria may serve as a source of free Zn2+. Further tests indicate that mitochondrial dyshomeostasis acts as a source of second Zn2+ increase or Zn2+ overload (Fig. 2, C and D). Taking into account the visual inspection of merged images of cells double-stained with Zn2+ and mitochondrial ROS dyes, our study supports the notion that mitochondria are Zn2+-storing organelles and are also undergoing mitochondrial ROS production during stress under OGD (62, 65). Studies have shown that Zn2+ transporters, or the mitochondrial calcium uniporter, are involved in Zn2+ uptake by mitochondria (20, 51). The mitochondrial Zn2+ uptake and membrane depolarization is also associated with Zn2+ accumulation following OGD (65) (see Fig. 5C). Thus, Zn2+ uptake by mitochondria may be responsible for phase II or the descending phase of early Zn2+ transient (Figs. 1 and 5). While the Zn2+ uptake may provide clearance of cytosolic Zn2+ in cells under OGD, continuous and consistent Zn2+ accumulation in mitochondrial lumen alters or, consequently, impairs mitochondrial function, leading to the opening of the mitochondrial permeability transition pore (mPTP) (7, 50). A consequence of mPTP opening is the efflux of Zn2+ from mitochondria (50), which contributes to Zn2+ elevation in the phase III and is a causal factor of massive Zn2+ overload seen in the phase IV. Hence, there is a biphasic control of cytosolic Zn2+ by mitochondria in response to the rising Zn2+: early uptake, to remove cytosolic Zn2+ and late release, due to mitochondrial dysfunction, which causes Zn2+ overload. This provides a novel basis for complex pathological patterns of intracellular Zn2+ signaling. This study shows that TPEN application delayed the OGD-induced mitochondrial ROS accumulation as well as reduced FCCP-induced mitochondrial ROS production (Fig. 3, B and C), supporting the notion that the mitochondrial Zn2+ uptake and accumulation may lead to a loss of mitochondrial membrane potential and a subsequent increase in ROS production (50, 51).
Fig. 5.

Summary of cross talk between OGD-induced Zn2+ and ROS increases that involve intracellular Zn2+ storage, mitochondria, and NADPH oxidase. A: schematic drawing of intracellular Zn2+ storage including metallothioneins (MT), organelle apparatus such as endoplasmic reticulum, and mitochondria, showing the release and uptake of Zn2+ by them in four described phases of Zn2+ rises during OGD. B: diagram of temporal relationship of OGD-induced Zn2+ and ROS accumulation during the course of prolonged hypoxic exposure (30 min in the present study). Green area represents Zn2+ increases; red line shows ROS accumulation. Roman numerals and vertical dashed lines show four phases of Zn2+ transient during OGD. C: schematic drawing showing mitochondrial Zn2+ storage and release during OGD in relationship with mitochondrial ROS production and NADPH oxidase-mediated ROS production. C,a: before OGD. C,b: mitochondria uptake and store Zn2+ during OGD. Drawing shows the positive feedback between mitochondrial Zn2+ stress facilitating ROS production, which further releases Zn2+. C,c: the large release of Zn2+ and ROS from mitochondria. Zn2+ may activate NADPH oxidase and consequently further increase ROS accumulation.
We hypothesize that another critical source of this Zn2+ may be the abundant metallothioneins (MTs) that bind as much as 5–10% of all cellular Zn2+, from which Zn2+ could be released rapidly by OGD-induced ROS (45). The sequestration and storage of Zn2+ in MTs have been extensively investigated. MT have been shown to be the sink as well as a source of Zn2+, and they help to maintain Zn2+ homeostasis (15). In OGD, there are abnormal cellular conditions such as acidosis, nitrosylation, lipid peroxidation products, and glutathione disulfide, all of which favor Zn2+ dissociation from MTs, resulting in increases in intracellular free Zn2+ (26, 40, 49). For example, prior studies have found that Zn2+ binding to MTs is decreased at acidic pH (36). Zn2+ dissociation from MTs may contribute to the Zn2+ overload (phase IV) of Zn2+ transient seen in prolonged OGD. These data will need to be confirmed with inhibitors specific to MTs.
Mechanisms of Zn2+-induced ROS accumulation.
OGD-induced Zn2+ rise was not the only focus of this study. Our results indicate that Zn2+ is a causal factor of OGD-induced ROS accumulation. We examine the temporal relationship between the two important phenomena, where early Zn2+ transient (phases I and II) preceded the early ROS accumulation. Interestingly, the second Zn2+ rise (phase IV) appears also to precede the second ROS accumulation. As discussed above, mitochondria can shape and maintain Zn2+ homeostasis by actively taking up Zn2+ into the mitochondria matrix, which is a protective mechanism to curtail Zn2+ imbalance caused by a short OGD. However, an excess of Zn2+ in the mitochondrial matrix may disrupt most of the enzymes of the oxidative phosphorylation and leads to oxidative stress and membrane impairment. A consequence of excessive Zn2+ accumulation is the increased generation of ROS (7, 50). We found that Zn2+ facilitates ROS production based on multiple lines of evidence. The application of Zn2+ induced steady ROS production (Fig. 4A). TPEN significantly delayed OGD-induced and FCCP-induced ROS increases (Fig. 3, B and C). It is important to note that TPEN did not completely remove ROS increases due to possibly Zn2+ independent ROS production within mitochondria. Taking all the above data into account, we suggest that Zn2+ is required for the induction of mitochondrial ROS accumulation and precedes its onset. To our knowledge we are the first to report this kind of temporal cross talk between Zn2+ and mitochondrial ROS.
OGD-induced ROS production is a very complicated cellular response with the mechanism and key factors that are still unclear. By applying exogenous Zn2+ to cells under normoxic condition and observing the mitochondrial ROS, we collected valuable data because we were only changing Zn2+ concentration and these data are more straightforward to interpret. The application of Zn2+ induced a large mitochondrial ROS increase (Fig. 4A), which was consistent with results that Zn2+ removal by TPEN delayed or reduced mitochondrial ROS production induced by OGD or FCCP (Fig. 3, B and C), as discussed above. Besides mitochondria, Zn2+ also appears to interact with NADPH oxidase, because Zn2+-induced ROS accumulation was significantly reduced with inhibition of NADPH oxidase (Fig. 4A). To further solidify these findings, we used Western blot technique to quantify the amount of p47phox, which is one of the functionally important subunits of NADPH oxidase in HeLa cells that were exposed to exogenous Zn2+ treatment with addition of Zn2+ ionophore, which when combined, translates to intracellular Zn2+ increase. Intracellular Zn2+ increase induced higher levels of p47phox, which creates the possibility for an increase in NADPH oxidase (Nox) in the cells. The increase happened in very short period of time (30 min), while control cells had very low expression in both physiological control as well as ionophore alone control (Fig. 4B). p47phox has been shown to be an important component of Nox1 (3, 73), Nox2 (5), and Nox3 (73); however, a recent comprehensive review suggests that Nox2 is clearly associated with p47phox (55). In light of previous reports of zinc and NADPH oxidase interaction (38, 48, 53) and taking into account data reported above, we suggest that NADPH oxidase is a major contributor to Zn2+-induced ROS in HeLa cells and that the Zn2+ induces ROS through the upregulation of p47phox, a functional subunit of NADPH oxidase (Nox2). Recently, there is emerging evidence that NADPH oxidase contributes significantly to ROS generation following reperfusion in neurons (1, 9, 30). However, the molecular mechanism for so-called “burst” of ROS generation remains largely uncertain. In this study we report that both NADPH oxidase inhibitor and Zn2+ removal by TPEN significantly inhibited reoxygenation-induced ROS increases (Fig. 4C), which further solidifies the importance of Zn2+ and NADPH oxidase interaction.
In summary, we present here for the first time that there are multiple intracellular Zn2+ rises during hypoxic exposure, revealing a rather interesting and complicated cross talk between ROS and Zn2+ that involves multiple factors. As depicted in Fig. 5, OGD triggers Zn2+ release from the ER stores. It is possible that the early Zn2+ transient may be protective in response to short OGD, but, at the same time, it makes cells vulnerable if OGD continues. Zn2+ surge was quickly buffered by normal cellular sequestration processes, and Zn2+ may return to basal level if OGD ceases. Specifically, Zn2+ is sequestered in mitochondria through the activation of a cation-permeable channel or other unidentified independent pathway (16, 27, 28, 35, 50). Excessive and prolonged intramitochondrial Zn2+ overload inhibits the activity of complex III of the electron transport chain, or by interfering with complex I and α-ketoglutarate dehydrogenase (62), and the activation of the mitochondrial permeability transition pore (mPTP) (6, 7, 13, 28, 64). Therefore, mitochondria may become the first victim of prolonged Zn2+ increase. In return, injured mitochondria release ROS and Zn2+, which worsen dyshomeostasis and trigger the proapoptotic signaling cascade pathways. Extensive increase in [Zn2+]i may be a causal factor of burst ROS generation seen in oxidation as Zn2+ interacts with NADPH oxidase and causes further ROS increase and Zn2+ overload.
GRANTS
This research was supported in part by NIH National Institute of Neurological Disorders and Stroke Grant NS-081629 (to Y. V. Li).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.G.S. and Q.L. performed experiments; K.G.S. and Q.L. analyzed data; K.G.S., Q.L., and Y.V.L. interpreted results of experiments; K.G.S., Q.L., and Y.V.L. prepared figures; K.G.S. drafted manuscript; K.G.S., Q.L., and Y.V.L. edited and revised manuscript; K.G.S., Q.L., and Y.V.L. approved final version of manuscript; Y.V.L. conceived and designed research.
ACKNOWLEDGMENTS
We acknowledge the use of the confocal microscope facility at Heritage College of Osteopathic Medicine, Ohio University.
REFERENCES
- 1.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 27: 1129–1138, 2007. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babu D, Leclercq G, Goossens V, Vanden Berghe T, Van Hamme E, Vandenabeele P, Lefebvre RA. Mitochondria and NADPH oxidases are the major sources of TNF-α/cycloheximide-induced oxidative stress in murine intestinal epithelial MODE-K cells. Cell Signal 27: 1141–1158, 2015. doi: 10.1016/j.cellsig.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 3.Bánfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem 278: 3510–3513, 2003. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 4.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res 61: 461–470, 2004. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 5.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 6.Bonanni L, Chachar M, Jover-Mengual T, Li H, Jones A, Yokota H, Ofengeim D, Flannery RJ, Miyawaki T, Cho CH, Polster BM, Pypaert M, Hardwick JM, Sensi SL, Zukin RS, Jonas EA. Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J Neurosci 26: 6851–6862, 2006. doi: 10.1523/JNEUROSCI.5444-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bossy-Wetzel E, Talantova MV, Lee WD, Schölzke MN, Harrop A, Mathews E, Götz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 41: 351–365, 2004. doi: 10.1016/S0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- 8.Brandes RP, Miller FJ, Beer S, Haendeler J, Hoffmann J, Ha T, Holland SM, Görlach A, Busse R. The vascular NADPH oxidase subunit p47phox is involved in redox-mediated gene expression. Free Radic Biol Med 32: 1116–1122, 2002. doi: 10.1016/S0891-5849(02)00789-X. [DOI] [PubMed] [Google Scholar]
- 9.Braunersreuther V, Montecucco F, Ashri M, Pelli G, Galan K, Frias M, Burger F, Quinderé AL, Montessuit C, Krause KH, Mach F, Jaquet V. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol 64: 99–107, 2013. doi: 10.1016/j.yjmcc.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci 12: 857–863, 2009. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burdette SC, Walkup GK, Spingler B, Tsien RY, Lippard SJ. Fluorescent sensors for Zn2+ based on a fluorescein platform: synthesis, properties and intracellular distribution. J Am Chem Soc 123: 7831–7841, 2001. doi: 10.1021/ja010059l. [DOI] [PubMed] [Google Scholar]
- 12.Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci 24: 9903–9913, 2004. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capasso M, Jeng JM, Malavolta M, Mocchegiani E, Sensi SL. Zinc dyshomeostasis: a key modulator of neuronal injury. J Alzheimers Dis 8: 93–108, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Chabosseau P, Tuncay E, Meur G, Bellomo EA, Hessels A, Hughes S, Johnson PR, Bugliani M, Marchetti P, Turan B, Lyon AR, Merkx M, Rutter GA. Mitochondrial and ER-targeted eCALWY probes reveal high levels of free Zn2+. ACS Chem Biol 9: 2111–2120, 2014. doi: 10.1021/cb5004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colvin RA, Holmes WR, Fontaine CP, Maret W. Cytosolic zinc buffering and muffling: their role in intracellular zinc homeostasis. Metallomics 2: 306–317, 2010. doi: 10.1039/b926662c. [DOI] [PubMed] [Google Scholar]
- 16.Csordás G, Hajnóczky G. Plasticity of mitochondrial calcium signaling. J Biol Chem 278: 42273–42282, 2003. doi: 10.1074/jbc.M305248200. [DOI] [PubMed] [Google Scholar]
- 17.Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol 6: 472–485, 2015. doi: 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51: 1289–1301, 2011. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300: C723–C742, 2011. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eide DJ. Zinc transporters and the cellular trafficking of zinc. Biochim Biophys Acta 1763: 711–722, 2006. doi: 10.1016/j.bbamcr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Ellis CD, Macdiarmid CW, Eide DJ. Heteromeric protein complexes mediate zinc transport into the secretory pathway of eukaryotic cells. J Biol Chem 280: 28811–28818, 2005. doi: 10.1074/jbc.M505500200. [DOI] [PubMed] [Google Scholar]
- 22.Ellis CD, Wang F, MacDiarmid CW, Clark S, Lyons T, Eide DJ. Zinc and the Msc2 zinc transporter protein are required for endoplasmic reticulum function. J Cell Biol 166: 325–335, 2004. doi: 10.1083/jcb.200401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Englander SW, Calhoun DB, Englander JJ. Biochemistry without oxygen. Anal Biochem 161: 300–306, 1987. doi: 10.1016/0003-2697(87)90454-4. [DOI] [PubMed] [Google Scholar]
- 24.Feng P, Li TL, Guan ZX, Franklin RB, Costello LC. Direct effect of zinc on mitochondrial apoptogenesis in prostate cells. Prostate 52: 311–318, 2002. doi: 10.1002/pros.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng P, Liang JY, Li TL, Guan ZX, Zou J, Franklin R, Costello LC. Zinc induces mitochondria apoptogenesis in prostate cells. Mol Urol 4: 31–36, 2000. [PubMed] [Google Scholar]
- 26.Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci 6: 449–462, 2005. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 27.Frederickson CJ, Maret W, Cuajungco MP. Zinc and excitotoxic brain injury: a new model. Neuroscientist 10: 18–25, 2004. doi: 10.1177/1073858403255840. [DOI] [PubMed] [Google Scholar]
- 28.Gazaryan IG, Krasinskaya IP, Kristal BS, Brown AM. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition. J Biol Chem 282: 24373–24380, 2007. doi: 10.1074/jbc.M611376200. [DOI] [PubMed] [Google Scholar]
- 29.Gebhardt C, Heinemann U. Anoxic decrease in potassium outward currents of hippocampal cultured neurons in absence and presence of dithionite. Brain Res 837: 270–276, 1999. doi: 10.1016/S0006-8993(99)01616-9. [DOI] [PubMed] [Google Scholar]
- 30.Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol 6: 524–551, 2015. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guan Z, Kukoyi B, Feng P, Kennedy MC, Franklin RB, Costello LC. Kinetic identification of a mitochondrial zinc uptake transport process in prostate cells. J Inorg Biochem 97: 199–206, 2003. doi: 10.1016/S0162-0134(03)00291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang L, Kirschke CP, Zhang Y, Yu YY. The ZIP7 gene (Slc39a7) encodes a zinc transporter involved in zinc homeostasis of the Golgi apparatus. J Biol Chem 280: 15456–15463, 2005. doi: 10.1074/jbc.M412188200. [DOI] [PubMed] [Google Scholar]
- 33.Ishihara K, Yamazaki T, Ishida Y, Suzuki T, Oda K, Nagao M, Yamaguchi-Iwai Y, Kambe T. Zinc transport complexes contribute to the homeostatic maintenance of secretory pathway function in vertebrate cells. J Biol Chem 281: 17743–17750, 2006. doi: 10.1074/jbc.M602470200. [DOI] [PubMed] [Google Scholar]
- 34.Jeong J, Eide DJ. The SLC39 family of zinc transporters. Mol Aspects Med 34: 612–619, 2013. doi: 10.1016/j.mam.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang D, Sullivan PG, Sensi SL, Steward O, Weiss JH. Zn2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem 276: 47524–47529, 2001. doi: 10.1074/jbc.M108834200. [DOI] [PubMed] [Google Scholar]
- 36.Jiang LJ, Vasák M, Vallee BL, Maret W. Zinc transfer potentials of the alpha - and beta-clusters of metallothionein are affected by domain interactions in the whole molecule. Proc Natl Acad Sci USA 97: 2503–2508, 2000. doi: 10.1073/pnas.97.6.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson JL, Park JW, Benna JE, Faust LP, Inanami O, Babior BM. Activation of P47PHOX, a cytosolic subunit of the leukocyte NADPH oxidase. Phosphorylation of Ser-359 or Ser-370 precedes phosphorylation at other sites and is required for activity. J Biol Chem 273: 35147–35152, 1998. doi: 10.1074/jbc.273.52.35147. [DOI] [PubMed] [Google Scholar]
- 38.Kim YH, Koh JY. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol 177: 407–418, 2002. doi: 10.1006/exnr.2002.7990. [DOI] [PubMed] [Google Scholar]
- 39.Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272: 1013–1016, 1996. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 40.Li YV. Zinc overload in stroke. In: Metal Ion in Stroke, edited by Li YV, Zhang JH. New York: Springer Science+Business Media, 2012, p. 167–189. [Google Scholar]
- 41.López-Hernández B, Ceña V, Posadas I. The endoplasmic reticulum stress and the HIF-1 signalling pathways are involved in the neuronal damage caused by chemical hypoxia. Br J Pharmacol 172: 2838–2851, 2015. doi: 10.1111/bph.13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu Q, Haragopal H, Slepchenko KG, Stork C, Li YV. Intracellular zinc distribution in mitochondria, ER and the Golgi apparatus. Int J Physiol Pathophysiol Pharmacol 8: 35–43, 2016. [PMC free article] [PubMed] [Google Scholar]
- 43.Mancarella S, Wang Y, Deng X, Landesberg G, Scalia R, Panettieri RA, Mallilankaraman K, Tang XD, Madesh M, Gill DL. Hypoxia-induced acidosis uncouples the STIM-Orai calcium signaling complex. J Biol Chem 286: 44788–44798, 2011. doi: 10.1074/jbc.M111.303081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maret W. Analyzing free zinc(II) ion concentrations in cell biology with fluorescent chelating molecules. Metallomics 7: 202–211, 2015. doi: 10.1039/C4MT00230J. [DOI] [PubMed] [Google Scholar]
- 45.Maret W. Metallothionein redox biology in the cytoprotective and cytotoxic functions of zinc. Exp Gerontol 43: 363–369, 2008. doi: 10.1016/j.exger.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 46.Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal 8: 1419–1441, 2006. doi: 10.1089/ars.2006.8.1419. [DOI] [PubMed] [Google Scholar]
- 47.Martin JL, Stork CJ, Li YV. Determining zinc with commonly used calcium and zinc fluorescent indicators, a question on calcium signals. Cell Calcium 40: 393–402, 2006. doi: 10.1016/j.ceca.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 48.Matsunaga Y, Kawai Y, Kohda Y, Gemba M. Involvement of activation of NADPH oxidase and extracellular signal-regulated kinase (ERK) in renal cell injury induced by zinc. J Toxicol Sci 30: 135–144, 2005. doi: 10.2131/jts.30.135. [DOI] [PubMed] [Google Scholar]
- 49.McCord MC, Aizenman E. The role of intracellular zinc release in aging, oxidative stress, and Alzheimer’s disease. Front Aging Neurosci 6: 77, 2014. doi: 10.3389/fnagi.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Medvedeva YV, Lin B, Shuttleworth CW, Weiss JH. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J Neurosci 29: 1105–1114, 2009. doi: 10.1523/JNEUROSCI.4604-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medvedeva YV, Weiss JH. Intramitochondrial Zn2+ accumulation via the Ca2+ uniporter contributes to acute ischemic neurodegeneration. Neurobiol Dis 68: 137–144, 2014. doi: 10.1016/j.nbd.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagaraj R, Gururaja-Rao S, Jones KT, Slattery M, Negre N, Braas D, Christofk H, White KP, Mann R, Banerjee U. Control of mitochondrial structure and function by the Yorkie/YAP oncogenic pathway. Genes Dev 26: 2027–2037, 2012. doi: 10.1101/gad.183061.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noh KM, Koh JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci 20: RC111, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev 39: 443–455, 2007. doi: 10.1080/03602530701468516. [DOI] [PubMed] [Google Scholar]
- 55.Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol 12: 5–23, 2015. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paquet-Durand F, Bicker G. Hypoxic/ischaemic cell damage in cultured human NT-2 neurons. Brain Res 1011: 33–47, 2004. doi: 10.1016/j.brainres.2004.02.060. [DOI] [PubMed] [Google Scholar]
- 56a.Petrônio MS, Zeraik ML, Fonseca LM, Ximenes VF. Apocynin: chemical and biophysical properties of a NADPH oxidase inhibitor. Molecules 18: 2821–2839, 2013. doi: 10.3390/molecules18032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Powell SR. The antioxidant properties of zinc. J Nutr 130, Suppl: 1447S–1454S, 2000. [DOI] [PubMed] [Google Scholar]
- 58.Qiao W, Ellis C, Steffen J, Wu CY, Eide DJ. Zinc status and vacuolar zinc transporters control alkaline phosphatase accumulation and activity in Saccharomyces cerevisiae. Mol Microbiol 72: 320–334, 2009. doi: 10.1111/j.1365-2958.2009.06644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin Y, Dittmer PJ, Park JG, Jansen KB, Palmer AE. Measuring steady-state and dynamic endoplasmic reticulum and Golgi Zn2+ with genetically encoded sensors. Proc Natl Acad Sci USA 108: 7351–7356, 2011. doi: 10.1073/pnas.1015686108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rootwelt T, Dunn M, Yudkoff M, Itoh T, Almaas R, Pleasure D. Hypoxic cell death in human NT2-N neurons: involvement of NMDA and non-NMDA glutamate receptors. J Neurochem 71: 1544–1553, 1998. doi: 10.1046/j.1471-4159.1998.71041544.x. [DOI] [PubMed] [Google Scholar]
- 61.Rudolf E, Rudolf K, Cervinka M. Zinc induced apoptosis in HEP-2 cancer cells: the role of oxidative stress and mitochondria. Biofactors 23: 107–120, 2005. doi: 10.1002/biof.5520230206. [DOI] [PubMed] [Google Scholar]
- 62.Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci 10: 780–791, 2009. doi: 10.1038/nrn2734. [DOI] [PubMed] [Google Scholar]
- 63.Sensi SL, Ton-That D, Weiss JH, Rothe A, Gee KR. A new mitochondrial fluorescent zinc sensor. Cell Calcium 34: 281–284, 2003. doi: 10.1016/S0143-4160(03)00122-2. [DOI] [PubMed] [Google Scholar]
- 64.Shahbaz AU, Zhao T, Zhao W, Johnson PL, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT. Calcium and zinc dyshomeostasis during isoproterenol-induced acute stressor state. Am J Physiol Heart Circ Physiol 300: H636–H644, 2011. doi: 10.1152/ajpheart.00900.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shuttleworth CW, Weiss JH. Zinc: new clues to diverse roles in brain ischemia. Trends Pharmacol Sci 32: 480–486, 2011. doi: 10.1016/j.tips.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Slepchenko KG, Lu Q, Li YV. Zinc wave during the treatment of hypoxia is required for initial reactive oxygen species activation in mitochondria. Int J Physiol Pathophysiol Pharmacol 8: 44–51, 2016. [PMC free article] [PubMed] [Google Scholar]
- 67.Solaini G, Baracca A, Lenaz G, Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta 1797: 1171–1177, 2010. doi: 10.1016/j.bbabio.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 68.Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann NY Acad Sci 1147: 37–52, 2008. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stork CJ, Li YV. Intracellular zinc elevation measured with a “calcium-specific” indicator during ischemia and reperfusion in rat hippocampus: a question on calcium overload. J Neurosci 26: 10430–10437, 2006. doi: 10.1523/JNEUROSCI.1588-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stork CJ, Li YV. Zinc release from thapsigargin/IP3-sensitive stores in cultured cortical neurons. J Mol Signal 5: 5, 2010. doi: 10.1186/1750-2187-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suzuki T, Ishihara K, Migaki H, Matsuura W, Kohda A, Okumura K, Nagao M, Yamaguchi-Iwai Y, Kambe T. Zinc transporters, ZnT5 and ZnT7, are required for the activation of alkaline phosphatases, zinc-requiring enzymes that are glycosylphosphatidylinositol-anchored to the cytoplasmic membrane. J Biol Chem 280: 637–643, 2005. doi: 10.1074/jbc.M411247200. [DOI] [PubMed] [Google Scholar]
- 72.Taylor KM, Hiscox S, Nicholson RI, Hogstrand C, Kille P. Protein kinase CK2 triggers cytosolic zinc signaling pathways by phosphorylation of zinc channel ZIP7. Sci Signal 5: ra11, 2012. doi: 10.1126/scisignal.2002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol 26: 2160–2174, 2006. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Walkup GK, Burdette SC, Lippard SJ, Tsien RY. A new cell-permeable fluorescent probe for Zn2+. J Am Chem Soc 122: 5644–5645, 2000. doi: 10.1021/ja000868p. [DOI] [Google Scholar]
- 75.Wang CC, Fang KM, Yang CS, Tzeng SF. Reactive oxygen species-induced cell death of rat primary astrocytes through mitochondria-mediated mechanism. J Cell Biochem 107: 933–943, 2009. doi: 10.1002/jcb.22196. [DOI] [PubMed] [Google Scholar]
- 76.Welinder C, Ekblad L. Coomassie staining as loading control in Western blot analysis. J Proteome Res 10: 1416–1419, 2011. doi: 10.1021/pr1011476. [DOI] [PubMed] [Google Scholar]
- 77.Yuan XJ, Goldman WF, Tod ML, Rubin LJ, Blaustein MP. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol 264: L116–L123, 1993. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Y, Tocchetti CG, Krieg T, Moens AL. Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radic Biol Med 53: 1531–1540, 2012. doi: 10.1016/j.freeradbiomed.2012.07.010. [DOI] [PubMed] [Google Scholar]