

Abstract
The renal collecting duct (CD) contains two major cell types, intercalated (ICs) and principal cells (PCs). A previous report showed that deletion of β1-integrin in the entire renal CD causes defective CD morphogenesis resulting in kidney dysfunction. However, subsequent deletion of β1-integrin specifically in ICs and PCs, respectively, did not cause any morphological defects in the CDs. The discrepancy between these studies prompts us to reinvestigate the role of β1-integrin in CD cells, specifically in the PCs. We conditionally deleted β1-integrin in mouse CD PCs using a specific aquaporin-2 (AQP2) promoter Cre-LoxP system. The resulting mutant mice, β-1f/fAQP2-Cre+, had lower body weight, failed to thrive, and died around 8–12 wk. Their CD tubules were dilated, and some of them contained cellular debris. Increased apoptosis and proliferation of PCs were observed in the dilated CDs. Trichrome staining and electron microscopy revealed the presence of peritubular and interstitial fibrosis that is associated with increased production of extracellular matrix proteins including collagen type IV and fibronectin, as detected by immunoblotting. Further analysis revealed a significantly increased expression of transforming growth factor-β (TGF-β)-induced protein, fibronectin, and TGF-β receptor-1 mRNAs and concomitantly increased phosphorylation of SMAD-2 that indicates the activation of the TGF-β signaling pathway. Therefore, our data reveal that normal expression of β1-integrin in PCs is a critical determinant of CD structural and functional integrity and further support the previously reported critical role of β1-integrin in the development and/or maintenance of the CD structure and function.
Keywords: aquaporin-2, β1-integrins, collecting ducts, renal fibrosis, TGF-β
integrins are transmembrane receptors known for their role in sensing and monitoring extracellular matrix (ECM) components to sustain ECM remodeling and homeostasis (21, 33). Failure in managing ECM remodeling is considered to be a major contributor to fibrotic-associated kidney disorders, and integrins have been proposed to be major players in renal fibrosis (54). β1-Integrin is the most abundantly expressed β-integrin subunit in the kidney, and previous studies have shown that β1-integrin is required for collecting duct (CD) development and function (47, 82). β1-Integrin can convey ECM signals into intracellular signaling pathways influencing a wide variety of vital cell behaviors including cell migration, proliferation, differentiation, adhesion, and apoptosis (33). β1-Integrin can also initiate intracellular signaling pathways from several growth factors, in particular, transforming growth factor-β (TGF-β) (3, 4, 7, 19, 58). The roles of integrins in TGF-β-associated fibrosis have been well established in various organs, including the kidney, and both in vitro and in vivo renal studies have clearly shown the effects of TGF-β in renal fibrosis (12, 29, 31, 35, 44, 45, 56, 77). Moreover, the loss of β1-integrin in a human kidney tubular cell line enhances TGF-β-induced collagen expression, leading to renal cell fibrogenesis (30).
The CDs of the kidney connect the distal part of the nephron to the ureter. They are comprised of two cell types, the intercalated cells (ICs), which are involved primarily in urine acidification and regulation of acid/base balance, and the principal cells (PCs), which express aquaporin water channels (AQP2, AQP3, and AQP4) and the epithelial sodium channel, regulating water and sodium transport in the CDs (8–10, 15, 23, 32, 61). AQP2 is a classic water channel that acts as the central player for vasopressin (VP)-regulated water homeostasis in mammals. It is well understood that proper AQP2 trafficking is critical for water transport in the CDs (8, 49, 50). Our group and others have made significant efforts to understand the cellular and molecular basis of AQP2 trafficking and function. Recently, we and others have shown that AQP2 interacts with β1-integrin through an Arg-Gly-Asp (RGD) integrin-binding motif, and modulates integrin trafficking and turnover at the focal adhesions, therefore promoting cell migration (13, 14, 69). Consequently, we wanted to further investigate the functional interaction between AQP2 and integrin signaling in kidney tubular function.
Recent studies conditionally deleted β1-integrin from the entire CD system using a HoxB7 promoter Cre-LoxP system (80, 84). This caused retardation of kidney growth, CD degeneration, and significant renal dysfunction leading to premature death. In contrast to this dramatic phenotype, the loss of β1-integrin in ICs alone did not cause any tubular damage but only distal renal tubular acidosis (24). Surprisingly, two previous attempts to delete β1-integrin specifically in PCs did not reveal any kidney phenotype either, unless a kidney injury was introduced (39, 84). Although the AQP2 promoter is routinely used to drive the expression of the Cre recombinase in PCs, the patterns of its transgene activity critically depend on the AQP2 promoter fragment used (25, 48, 86). Using a different AQP2 Cre recombinase system with stronger expression in CD but decreasing levels along the connecting tubule (CNT) toward the early CNT (60), we conditionally deleted β1-integrin specifically in PCs. Under this particular AQP2 Cre-Lox system, mice lacking β1-integrin in PCs developed significant CD tubular damage and kidney fibrosis, which led to premature death shortly after 2 mo of age, probably due to kidney failure. These findings indicate that the timing and level of β1-integrin expression in the PCs of the CD system are important in maintaining normal kidney structure and renal function after birth.
MATERIALS AND METHODS
Animals.
All animal experiments were conducted according to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care. B6;129-Itgb1tm1Efu/J mice possessing a LoxP site on either side of exon 3 in the β1-integrin gene (55), donated by Dr. Elaine Fuchs from The Rockefeller University, were obtained from The Jackson Laboratory (Bar Harbor, ME). To generate mice lacking β1-integrin in PCs, homozygous β1-integrin-floxed (β-1f/f) male mice were mated with female mice carrying AQP2 Cre recombinase (AQP2-Cre+) obtained from Dr. Wenzheng Zhang of Albany Medical Center (Albany, NY), previously described by Ronzaud et al. (60) and Wu et al. (79) and generated by the group of Dr. Günther Schütz at the German Cancer Research Center (Heidelberg, Germany). This deletion generated homozygous knockouts (β-1f/fAQP2-Cre+), heterozygous knockouts (β-1f/+AQP2-Cre+), and control littermates/wild-type (β-1f/fAQP2-Cre−). For genotyping, DNA was isolated from tail biopsies using the Mouse Direct PCR Kit (Cat. No. B40015; Biotool.com, Houston, TX). Mice were genotyped for the presence of the floxed β1-integrin allele by using the primers 5′-CGG CTC AAA GCA GAG TGT CAG TC-3′ and 5′-CCA CAA CTT TCC CAG TTA GCT CTC-3′ and for the presence of AQP2 Cre recombinase by using the primers 5′-AAG TGC CCA CAG TCT AGC CTC T-3′ and 5′-CCT GTT CAG CTT GCA CCA G-3′.
Metabolic cage studies.
Five-week-old wild-type (WT) and β-1f/+AQP2-Cre+ mice were subjected to 8-h metabolic cage monitoring. Spot urine samples were collected, and water and food intake were recorded. After 2 h of monitoring, the mice received intraperitoneal injection of either, vehicle alone (saline), or desmopressin (ddAVP; 1 μg/kg; Sigma-Aldrich). Urine osmolality was analyzed with a Vapor Pressure Osmometer 5520 (Wescor, Logan, UT).
Serum creatinine measurements and assessment of kidney function.
Blood was collected immediately after mice were euthanized and serum was separated using a BD Microtainer Ref. No. 365956 (Becton-Dickinson, Franklin Lakes, NJ) and immediately stored at −80°C. Serum creatinine levels were measured with a QuantiChrom Creatinine Assay Kit Cat. No. DICT-500 (BioAssay Systems, Hayward, CA), following the manufacturer’s protocol.
Glomerular filtration rate can be calculated as creatinine clearance using the Cockcroft-Gault equation: creatinine clearance = sex × (140 − age) × body weight/(72 × serum creatinine) (16). This formula was determined for humans and is not tailored for use in small rodents. However, regardless of the values of the constant terms, creatinine clearance will be directly proportional to the body weight-to-serum creatinine ratio. To compare kidney function in WT and β-1f/+AQP2-Cre+ mice of same sex and similar age, one would have to compare the ratio of body weight to serum creatinine. We thus calculated this ratio in our mice as an assessment of kidney function.
Kidney tissue collection.
Tissue sections were prepared as previously described (51). Briefly, 2-mo-old WT and mutant mice were anesthetized using isofluorane. Kidneys were harvested, and 5-mm slices were carefully cut and immersed in 4% paraformaldehyde-lysine-periodate (PLP) fixative for an overnight fixation at 4°C. PLP-fixed kidney slices were washed three times for a total of 3 h on a rocker with PBS containing 0.02% sodium azide. Slices were incubated in PBS with 30% sucrose overnight at 4°C and embedded in Tissue-Tek OCT compound 4583 (Sakura Finetek, Torrance, CA). Five-micrometer sections were cut on a CM3050S cryostat (Leica Microsystems, Bannockburn, IL), collected onto Superfrost Plus microscope slides (Fisher Scientific, Pittsburgh, PA), and stored at −20°C until use.
Antibodies, immunofluorescence staining, and immunoblotting.
For immunofluorescence, AQP2 was detected using a goat polyclonal antibody, sc-9882 (1:500 dilution) from Santa Cruz Biotechnology (Dallas, TX). AQP4 was detected using a rabbit polyclonal antibody from EMD Millipore (Darmstadt, Germany), AB3594 (1:400 dilution). β1-Integrin was detected using a hamster monoclonal antibody CBL1348, clone HM β1.1 (1:400 dilution), and caspase-3 with a rabbit polyclonal antibody against active/cleaved caspase-3, AB3623 (1:300 dilution), both from EMD Millipore. Immunoblotting was performed as previously reported (42), using the following antibodies: AQP2 goat polyclonal antibody, sc-9882 (1:5,000 dilution), fibronectin sc-9068 (1:1,000 dilution), and Smad2/3 sc-8332 (1:200 dilution) rabbit polyclonal antibodies from Santa Cruz Biotechnology; and mouse anti-GAPDH monoclonal antibody AM4300 (at 1:20,000) from Ambion/Thermo Fisher Scientific. Rabbit phospho-Smad2 (Ser465/467) No. 3101 (1:1,000 dilution), phospho-Smad3 (Ser423/425) No. 9520 (1:1,000 dilution), and β-actin (13E5) rabbit No. 4970 (1:10,000) antibodies were from Cell Signaling Technology (Danvers, MA).
Kidney slides stored at −20°C were rehydrated with PBS and treated with 1% SDS for 4 min for antigen retrieval (11). Immunofluorescence staining was then performed as previously reported (42). The evaluation was done with a Nikon 80i fluorescence microscope or a Nikon A1 confocal microscope (Nikon Instruments, Melville, NY). ImageJ (NIH; https://imagej.nih.gov/) and Volocity 5.0 (Perkin Elmer, Waltham, MA) software were used for image analysis.
Quantitative RT-PCR.
RNA was extracted from 5-wk-old and 2-mo-old mouse renal medulla using QIAshredder and RNeasy purification kits (Qiagen, Valencia, CA). cDNA synthesis was carried out using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative real-time PCR was performed by QuantStudio 3 Real-Time PCR system (Thermo Fisher Scientific), using Power SYBR Green PCR Master Mix (Life Technology, Austin, TX). mRNA levels for each gene were normalized to the expression levels of the housekeeping gene β-actin, and the relative mRNA expression was determined using the ΔΔCt method. For the list of primer sequences, see Table 1.
Table 1.
List of primers used for quantitative real-time PCR
| Gene | Forward | Reverse |
|---|---|---|
| Fibronectin | 5′-CTG GAG TCA AGC CAG ACA CA-3′ | 5′-CGA GGT GAC AGA GAC CAC AA-3′ |
| TGF-β R1 | 5′-CAG CTC CTC ATC GTG TTG GTG-3′ | 5′-GCA CAT ACA AAT GGC CTG TCT C-3′ |
| TGF-βi | 5′-CCT CAC CTC CAT GTA CCA GAA-3′ | 5′-TGG AAA TGA CCTTGT CAATGA GAG-3′ |
Transmission electron microscopy.
Transmission electron microscopy was carried out as previously described (52, 62). Briefly, kidney slices fixed as described above were rinsed in PBS and postfixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer for a minimum of 24 h at 4°C. The slices were rinsed several times in 0.1 M sodium cacodylate buffer and infiltrated with 1% osmium tetroxide in cacodylate buffer for 1 h at room temperature. Tissue blocks were rinsed again in cacodylate buffer, dehydrated through a graded series of ethanols to 100%, dehydrated briefly in 100% propylene oxide, and preinfiltrated in a 1:1 mix of Eponate resin (Ted Pella, Redding, CA) and propylene oxide overnight on a gentle rotator. The following day, specimens were infiltrated with fresh Eponate resin for several hours, embedded in flat molds with fresh Eponate and allowed to polymerize for 24–48 h at 60°C. Thin sections were cut using an EM UC7 ultramicrotome (Leica Microsystems), collected onto formvar-coated grids, stained with uranyl acetate and lead citrate, and examined in a JEM 1011 transmission electron microscope (JEOL, Tokyo, Japan) at 80 kV. Images were collected using an AMT digital imaging system (Advanced Microscopy Techniques, Danvers, MA).
H&E and Masson’s trichrome staining.
Hematoxylin and eosin staining was carried out as previously described (71). For trichrome staining, 5 μm PLP-fixed, paraffin-embedded kidney sections were stained with Trichrome Stain (Masson) Kit No. HT15–1KT (Sigma-Aldrich, St. Louis, MO), which stains collagenous material in blue, cytoplasm and muscle fibers in red, and nuclei in black, following the manufacturer’s protocol. Quantification of collagen density on Masson staining was performed with ImageJ software (NIH; https://imagej.nih.gov/).
Statistics.
Renal injury scores were analyzed by the Mann-Whitney nonparametric test. Data are shown as means ± SE, of a variable number of experiments, detailed in the figure legends. Data were analyzed with an unpaired design (Student’s t-test for two groups or one-way ANOVA for more) using GraphPad Prism version 6.00 (GraphPad Software, San Diego, CA). P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001).
RESULTS
Deletion of β1-integrin in mouse CD PCs led to premature death.
We generated mice lacking β1-integrin specifically in CD PCs by mating mice harboring LoxP sites around exon 3 of β1-integrin with mice carrying AQP2 promoter Cre-LoxP system previously reported by Ronzaud et al. (60). Upon birth, no obvious differences were detected between the newly generated β1-integrin homozygous knockout mice (β-1f/fAQP2-Cre+) (KO) and their WT control littermates (β-1f/fAQP2-Cre−). The KO animals had normal-sized bodies, and their kidneys had no gross morphological abnormality when compared with their control littermates. Starting around 5–6 wk of age, we noticed a rapid and progressive decline of the KO mice, characterized by hunched posture and ruffled fur. The KO mice were less active, with minimal movements around the cage, thus accessing less food and water than their WT littermates. Their growth appeared significantly impaired toward 2 mo of age when they weighed about one-half of the weight of their control littermates. KO animals died between 8 and 12 wk of age (Fig. 1A). In comparison, the heterozygous animals (β-1f/+AQP2-Cre+ mice) appeared healthy over 6 mo of observation. After tissue dissection, the kidneys obtained from the KO mice were smaller, pale, and had a rough, granular surface compared with the kidneys from their WT littermates (Fig. 1B).
Fig. 1.

A: the exterior appearance of 2-mo-old wild-type (WT) and β-1f/fAQP2-Cre+ mice [knockout (KO)]. B: image showing a normal WT kidney and kidneys from 2 different KO mice, showing differences in phenotype severity between mice of the same age (the middle kidney is from the KO mouse shown in Fig. 1A). C: PCR analysis of tail DNA showing genotyping results of the floxed band at 280 bp and the WT band at 160 bp. Both WT and KO mice express aquaporin-2 (AQP2) Cre (310-bp band).
Further examination of the KO mice revealed that the onset of this clinical decline differed from mouse to mouse. Around 10% of the KO mice appeared very sick and had to be killed as early as 4 wk postnatal, while around 30% of the KO mice delayed their phenotype onset up to 2 mo of age. They rapidly lost weight and appeared dehydrated. Most KO animals developed a gross phenotype between 5 and 7 wk after birth (Fig. 2, A and F).
Fig. 2.

Bar graphs showing results from metabolic cage studies of C57BL/6 wild-type (WT, Control) and β-1f/fAQP2-Cre+ (KO) mice. A: a Kaplan-Meier curve showing the survival pattern of the KO mice compared with their WT littermates (n = 22). B: bar graph showing serum creatinine levels in KO mice as compared with control between 5 and 10 wk of age (n = 10). C: comparison of kidney function in WT and KO mice (n = 5) using the ratio of body weight (BW) to serum creatinine (Scr). D: urine osmolality in the KO mice was significantly lower compared with WT (n ≥ 10). E: there were no significant differences in water intake between KO mice and their control littermates (n ≥ 10). F: the average body weight of KO mice was roughly one-half of that of their control littermates between 5 and 10 wk of age (n ≥ 10). Shown are combined mean values ± SE (*P < 0.05 and ****P < 0.0001).
Mice lacking β1-integrin in CD PCs developed polyuria and kidney failure.
Despite the KO mice being quite sick clinically with kidney atrophy, their baseline serum creatinine measured by a commercial ELISA assay kit was not significantly different from that of WT animals. However, we did observe a trend of elevation of serum creatinine in the KO animals compared with the WT controls (Fig. 2B). Similarly, there was also no significant elevation of serum creatinine in the HoxB7 Cre-driven β1-integrin KO animals despite the presence of dramatic kidney pathology (80). This is not surprising since a previous report showed that the level of serum creatinine has a poor association with kidney pathology and does not correlate with the degree of fibrosis in patient biopsies (36). In addition, serum creatinine can be affected by age, dietary protein intake, and lean mass, especially in patients with low muscle mass (20, 46). This could be particularly true in the β1-integrin KO animals given their significantly reduced body weight. Therefore, the baseline “normal creatinine” in the β1-integrin KO mice compared with their WT littermates in fact suggests an abnormal kidney. Consistent with the Cockcroft-Gault equation for glomerular filtration rate calculation (see materials and methods), we calculated the ratio of body weight-to-serum creatinine to assess kidney function. Accordingly, kidney function was significantly, nearly 50% reduced in β1-integrin KO mice (Fig. 2C).
In addition, we observed a significantly reduced urine osmolality in KO animals averaging ~1,000 mosmol/kgH2O at 5 wk of age to ~400 mosmol/kgH2O at 8 wk of age, compared with 2,000 mosmol/kgH2O in their WT littermates (Fig. 2D). However, total urine volume of KO animals could not be measured by metabolic cage experiments due to their fragility, especially in the late stage of their disease. To safely assess their urine output, we monitored 5-wk-old WT and KO mice for 8 h in metabolic cages. Eight hours of monitoring confirmed the presence of polyuria in 5-wk-old KO animals. There was no significant difference in water intake between the KO and WT animals (Fig. 2E). We also performed ddAVP treatment in an attempt to reverse the polyuria in 5-wk-old KO animals. ddAVP treatment caused a significant urinary concentration in WT mice from the average baseline urine osmolality of 1,964 to 5,380 mosmol/kgH2O. In contrast, ddAVP treatment failed to increase the urine osmolality in KO animals. The average baseline urine osmolality in the KO mice was as low at 1,102 mosmol/kgH2O, and it remained low at an average of 1,138 mosmol/kgH2O post-ddAVP administration. Therefore, the polyuria seen in KO mice at 5 wk could not be reversed by ddAVP treatment.
It was difficult to confirm and quantify the deletion of β1-integrin in the kidney CDs of KO mice by either Western blotting or quantitative RT-PCR. This is not surprising, since β1-integrin is abundantly expressed in almost every cell type and the PC-specific β1-integrin knockdown effect would be masked by β1-integrin expression in the nearby tissues and cells. It is also possible that PC-specific β1-integrin deletion may cause compensatory upregulation of β1-integrin expression in nearby cells and tissues. Therefore, we performed careful immunostaining of the CDs and revealed an ~75% reduction of β1-integrin in PCs of the CDs, thus confirming a successful PC-specific β1-integrin deletion (Fig. 3).
Fig. 3.
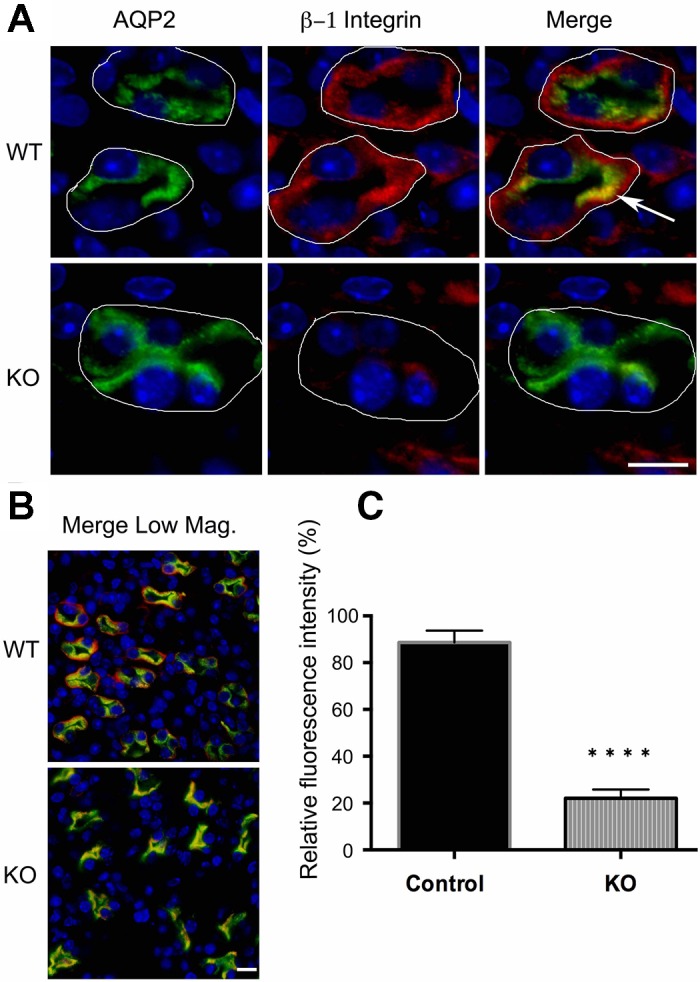
A: immunostaining of 3-wk-old WT and β-1f/fAQP2-Cre+ mice showing β1-integrin deletion in renal medullary collecting ducts. WT mice show colocalization (arrow) of AQP2 (green) and β1-integrin (red) and continuous staining of β1-integrin around the basement membrane of all collecting duct cells, while β-1f/fAQP2-Cre+ (KO) medullary collecting ducts show only patches of β1-integrin staining indicating that β1-integrin is only expressed in non-AQP2-expressing cells (i.e., in intercalated cells). White lines outline the basement membrane of the collecting duct cells. Scale bar = 20 μm. B: a lower magnification image (Merge Low Mag.) taken from the tip of the papilla shows an almost complete loss of β1-integrin in the collecting duct principal cell basement membrane. Scale bar = 20 μm. C: quantification of β1-integrin expression. Bar values represent means ± SE. Statistical analysis was performed with Student’s t-test. ****P < 0.0001; n ≥ 3.
CD PCs lacking β1-integrin had reduced AQP2 expression.
Around the time when KO mice developed a detectable phenotype (5–6 wk of age), we also started noticing a decrease in AQP2 expression in the CDs. This reduction in AQP2 expression continued and AQP2 almost disappeared in some CDs at advanced stages (~2 mo of age). Immunofluorescence staining for AQP4 showed that some KO CDs still express APQ4 but without AQP2, indicating a specific loss of AQP2 expression in PCs during the advanced stage of KO mouse phenotype. The reduction of AQP2 expression in KO mouse medulla was also confirmed by Western blotting (Fig. 4E). Reduced AQP2 expression at this late stage could account for the polyuria seen in the KO mice.
Fig. 4.

Deletion of β1-integrin in principal cells causes a significant decrease in AQP2 expression in certain tubules. AQP2 (green) shows normal expression and distribution in the inner (A) and outer medulla (C) of WT mice. AQP2 immunostaining is significantly reduced in the β-1f/fAQP2-Cre+ (KO) medulla (green in B and D), while AQP4 immunostaining is preserved in most KO collecting ducts (red in D). Scale bar = 100 μm for A and B and 40 μm for C and D. E: the loss of AQP2 expression in KO mouse collecting ducts was confirmed by Western blot analysis. F: a bar graph representing the total AQP2 protein expression levels from Western blotting results. There was a significant decrease in AQP2 protein in 5 wk old KO animals, which continues to decrease significantly to almost total loss by ~2 mo of age. Bar values represent means ± SE. Statistical analysis was performed with one-way ANOVA. **P < 0.01 and ****P < 0.0001; n ≥ 3.
The loss of β1-integrin in CD PCs led to PC apoptosis.
Along with the decrease in AQP2 protein expression, activated/cleaved caspase-3 immunostaining showed that some of the PCs lacking β1-integrin underwent apoptosis. This activation of caspase-3 and thus apoptosis was observed after the onset of clinical decline (around 5–6 wk of age) and continued to the advanced stage of the KO mouse phenotype (Fig. 5). Most apoptotic CD cells during the disease onset coexpressed both caspase-3 and AQP2 antibodies, suggesting that the dying cells in the CD are PCs. At the age of 5 wk, we observed apoptosis more in the CDs in the inner medulla than in the cortex and outer medulla. The percentage of cells stained with cleaved caspase-3 in the inner medulla was ~6% of total CD cells in the KO mice. There was no apoptosis seen in the WT CDs at this age (Fig. 5C).
Fig. 5.

Confocal microscopy images of 5-wk-old β-1f/fAQP2-Cre+ (KO) and wild type (WT) mouse renal medulla. A: there is no active/cleaved caspase-3 expression (red) in WT medulla. B: active/cleaved caspase was detected in KO collecting duct (CD) principal cells (PCs). C: bar graphs showing the percentage of active/cleaved caspase-3 positive PCs per CD. Scale bar = 20 μm. Bar values represent means ± SE. Statistical analysis was performed with Student’s t-test. ***P < 0.001; n ≥ 3.
Tubular injury, peritubular ECM deposition, and interstitial fibrosis in β-1f/fAQP2-Cre+ mouse kidneys.
Histological analysis of kidneys from 2-mo-old KO mice revealed the presence of dilated CDs and disrupted overall architecture and organization of interstitium in both inner and outer medulla, as revealed by hematoxylin and eosin staining (Fig. 6). Despite the significant damage of tubules and interstitium in the cortical region in the KO mice, the majority of glomeruli appeared morphologically intact. Masson’s trichrome staining showed the presence of excessive collagen deposition in the interstitium in the KO mouse kidneys (Fig. 7).
Fig. 6.

Histology of perfusion-fixed mouse kidneys showing significantly disrupted organization in the cortex and outer medulla and severely altered interstitial and tubular architecture in the inner medulla of 2-mo-old β-1f/fAQP2-Cre+ (KO) mice as compared with wild type (WT) littermates. Despite the advanced damage in the cortical region, the morphology of the glomeruli appeared intact in KO mice. Scale bar = 50 μm for cortex and 100 μm for inner and outer medulla.
Fig. 7.

Diffuse interstitial fibrosis in β-1f/fAQP2-Cre+ (KO) kidneys by trichrome stain (Masson). A: representative photomicrograph of a Masson-stained kidney section of a 2-mo-old WT mouse showing no collagen deposition in the outer medulla. B: the degree of collagenous material in blue, cytoplasm and muscle fibers in red and nuclei in black. Collagen deposition was significantly higher in KO mouse outer medulla than in WT littermates. C: histological injury score for medulla samples stained with Hematoxylin and eosin. Injury scale: 1.0 - 3.0, with 1.0 being normal. D: quantification of collagen deposition in Masson-stained kidney section. Bar values represent means ± SE. Statistical analysis was performed with Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; n ≥ 3.
Further examination by electron microscopy clearly showed CD tubular injury in the KO kidney. During the onset of KO phenotype, some ICs and PCs appeared to be morphologically intact. The cilia of PCs and microvilli of the ICs appeared intact (data not shown). However, as the disease advanced, PCs and subsequently ICs underwent significant morphological changes, characterized by the presence of disrupted cell-to-cell junctions, detachment of tubule cells from the basement membrane, and cell fragmentation. In addition to this dramatic tubular injury we observed significant deposition of ECM around the CD tubules in the cortical region as well as in the medullar interstitium of the KO mouse kidneys (Fig. 8). This peritubular and interstitial ECM accumulation was clearly observed in both 5-wk-old and 2-mo-old KO kidneys.
Fig. 8.

Collecting duct tubular injury and peritubular fibrosis as revealed by transmission electron microscopy. Compared with the 2-mo-old wild-type animals (A and D), a disruption of intercellular junctions was detected in 5 wk old and more obviously 2 mo-old β-1f/fAQP2-Cre+ (KO) animals (arrows in B and C). Collecting duct cell debris was clearly seen in the lumen of 2-mo-old KO mouse collecting ducts (green arrow in C). Significant peritubular extracellular matrix deposition and interstitial fibrosis were visible in 5-wk-old (B and E) and more evident in 2-mo-old KO mice (C and F). Scale bar = 4 μm (A–C) and 10 μm (D–F). Blue arrows indicate basolateral extracellular matrix space. Red arrows indicate collecting duct cell detachment from neighboring cells. The green arrow indicates cell debris in the collecting duct lumen.
β-1f/fAQP2-Cre+ mouse CDs express markers of TGF-β-induced renal fibrosis.
PC-specific β1-integrin KO mouse kidneys showed tubular injury and peritubular and interstitial fibrosis. The loss of β1-integrin has been shown to induce the activation of TGF-β signaling pathways in renal cells (30). Nonetheless, renal fibrosis results from excessive accumulation of ECM induced by TGF-β signaling (22). To investigate the involvement of TGF-β signaling in KO mouse disease onset, we examined three known markers of active TGF-β signaling in renal fibrosis: 1) transforming growth factor-β-induced protein (TGF-βi or βigH3), a protein secreted into the ECM upon induction of TGF-β signaling (27); 2) fibronectin, a protein known to be synthesized along with collagen I during TGF-β-induced renal fibrosis (6, 83); and 3) TGF-β receptor-1, known to be upregulated during TGF-β-induced renal fibrosis (76). We found that all three, TGF-βi, fibronectin, and TGF-βi receptor-1 mRNAs, are significantly overexpressed in KO renal medulla compared with WT medulla. TGF-βi and TGF-β receptor-1 mRNAs were significantly overexpressed at the onset of the KO disease (5 wk of age) and remained overexpressed throughout disease progression to the advanced stage. In conjunction, both mRNA and protein levels of fibronectin, which is known to be overexpressed in the ECM upon TGF-β-associated fibrosis (26, 64, 70, 75), were significantly overexpressed during the disease onset and remained elevated in the KO medulla until the advanced stage of the disease (Fig. 9). We next performed immunoblotting analysis of phospho-SMAD-2 and phospho-SMAD-3, which are downstream key players of the canonical TGF-β signaling pathway (37). We found a significant increase in phospho-SMAD-2 in the KO medulla (Fig. 10). Although SMAD3 phosphorylation was not significantly upregulated, there was also a trend of SMAD3 phosphorylation increase in the KO medulla. These findings suggest that the observed peritubular matrix deposition and interstitial fibrosis observed in KO medulla are associated with the upregulation of TGF-β signaling pathway.
Fig. 9.

Results from quantitative RT-PCR showing fibronectin, TGF-βi (βigH3) and TGF-β receptor-1 mRNA expression in WT and β-1f/fAQP2-Cre+ mice (KO) renal medulla. A: fibronectin transcript was elevated in the 5-wk-old and more significantly in the 2-mo-old KO animals (**P < 0.01 and ***P < 0.001). B: TGF-βi mRNA expression was also significantly increased in KO mice (**P < 0.01 and ****P < 0.0001). C; similar to TGF-βi, TGF-β receptor-1 mRNA was also increased significantly in both 5 wk- and 2 mo-old KO animals (*P < 0.05 and **P < 0.01). For each point shown, data are normalized to the expression level in 5-wk-old WT mice. #P < 0.05 and ##P < 0.01 compared with 5-wk-old mice.
Fig. 10.

Western blot results showing the following: A: significant increase in pSMAD2 but not pSMAD3 in 5-wk-old KO mouse medulla. Total SMAD2/3 was used as control; B: significant increase in fibronectin deposition in both 5-wk-old and 2 mo-old KO mouse CDs. β-Actin was used as fibronectin protein loading control. Quantification of Western blot results for phosphorylated SMAD2 and 3 (C) and fibronectin (D). Bar values represent means ± SE. Statistical analysis was performed by one-way ANOVA. ***P < 0.001 and ****P < 0.0001; n ≥ 3.
DISCUSSION
The complete removal of β1-integrin in the renal CDs causes defective CD morphogenesis resulting in severe kidney dysfunction (80). Since the renal CD consists of two major cell types, ICs and PCs, this implies an important role of β1-integrin in at least one of these two cell types. Surprisingly, deletion of β1-integrin specifically from ICs (24) and PCs (39, 86), respectively, did not cause any morphological defects of the CD. These results are inconsistent with the previously reported critical role of β1-integrin in the CD structure and function. However, it is known that patterns of AQP2 Cre recombinase activity depend on the location and size of the AQP2 promoter fragment used in designing the Cre-Lox system (25, 48, 86). To address this discrepancy, we reexamined the role of β1-integrin in the CD using a different AQP2-Cre promoter system. This AQP2 promoter Cre-Lox system was previously reported by Ronzaud et al. (60) and harbors a 156-kb genomic fragment containing the regulatory elements of the mouse AQP2 gene, with an ~125-kb 5′-upstream region. Unlike the previous attempts, deletion of β1-integrin using our AQP2 promoter Cre-Lox system caused morphological and functional defects of the CDs that resulted in broad interstitial fibrosis and kidney dysfunction. We found that under our AQP2 promoter Cre-Lox system, β1-integrin was almost completely deleted in the CD PCs as early as 21 days postnatal (Fig. 3), while in the previous attempts, β1-integrin was still present in PCs at that time point. We believe that the timing and extent of β1-integrin excision are critical in the development of this phenotype.
When under the control of the ureteric bud-specific HoxB7 promoter Cre-Lox system (HoxB7-Cre), β1-integrin is deleted from the entire CD system as early as embryonic day 9.5 (E9.5) and the KO animals present with significant CD defects and early mortality (80). However, when under the control of the PC-specific AQP2 promoter, whose expression begins around E18.5 (2), the deletion of β1-integrin occurs late during kidney development. Particularly, the AQP2 mRNA expression starts to increase significantly around day 3 of postnatal life; thus, unlike the HoxB7 Cre mutant, in our β-1f/fAQP2-Cre mice β1-integrin is deleted mostly after birth or at least as late as E18.5 (2). Therefore, developmental defects may not occur, or not significantly, in the β-1f/fAQP2-Cre mice. However, our study shows that β1-integrin deletion at this early postnatal stage is still detrimental and supports the critical function of β1-integrin in maintaining the structure and function of the CD.
Inhibition of β1-integrin in ICs did not cause any of the phenotypes seen in either our β-1f/fAQP2-Cre+ mutant or the HoxB7 Cre mutants. Inhibition of β1-integrin in ICs only induced distal renal tubular acidosis (24). This suggests an important role for PCs in maintaining the structural integrity of the CD. The PC-to-IC ratio varies along the CD (18, 28, 43, 74). PCs comprise >80% of total cells in the inner medullary CD (28). Thus there is a possibility that the severe phenotype we observe in the β-1f/fAQP2-Cre+ mutant inner medulla is due to the higher PC-to-IC ratio. However, we still noticed severe damage in cortical and outer medullary CDs, where the PC-to-IC ratio is markedly lower (28, 74), suggesting that the ratio between PCs and ICs may not be the reason for the observed phenotype. On the other hand, β1-integrin is known to interact with AQP2, and this interaction is believed to play a role in modulating integrin trafficking and turnover at the focal adhesions thus promoting cell migration (13, 14, 69). Moreover, AQP2 knockout mice develop severe kidney morphological and functional abnormalities that lead to early mortality (59, 81). It is reasonable to hypothesize that complex interplay between β1-integrin and AQP2 in the PCs may be important in maintaining the CD integrity and function.
Cells are well known to communicate with ECM proteins such as fibronectin through integrin receptors. β1-Integrin is one of the major fibronectin receptors; thus, integrin–fibronectin interactions are important in regulating cell behavior including cell adhesion and wound healing (78, 85). It has been reported that the loss of β1-integrin leads to loss of cell-to-cell and cell-to-ECM contact and cell death (65, 67, 68). We have detected apoptosis of AQP2-expressing PCs in 5-wk-old and 2-mo-old KO mice. Apoptosis is critical in kidney organogenesis and cell death in the nondifferentiating mesenchyme during kidney development; however, very little apoptosis is seen in a normal adult renal medulla (1). Apoptosis occurs under various pathological conditions and is believed to contribute in the progression of tubular atrophy and to play an important role in the pathogenesis of tubulointerstitial scarring (72). In our KO animals, we did observe an increase in PC apoptosis; even though most of the detectable apoptotic cells were located in the papillary area, it was significant enough to trigger a profound fibrotic signaling and lead to interstitial fibrosis. This progressed further, and eventually, collagen and fibronectin deposition in the ECM and the interstitial compartment became excessive, indicating a severe renal medullary fibrosis (5, 41, 53).
To investigate whether TGF-β was involved in this KO mouse renal medullary fibrosis, we examined the mRNA expression of three reported markers of active TGF-β signaling in renal fibrosis: TGF-β receptor-1, TGF-βi, and fibronectin. TGF-βi is a newly described gene whose mRNA expression and protein secretion into the ECM are induced by active TGF-β signaling (27, 38, 66). Moreover, TGF-βi expression is almost entirely blocked by anti-TGF-β antibody in the renal proximal tubule (40). Since its expression is very moderate in the kidney, it is a useful index of TGF-β bioactivity in the renal system (27, 34). Alternatively, fibronectin is a protein known to be synthesized and secreted in the ECM along with collagen during TGF-β-induced renal fibrosis (6, 83). Our results show that mRNAs for all three markers (TGF-β receptor-1, TGF-βi, and fibronectin) were significantly overexpressed at 5 wk and 2 mo of age in β-1f/fAQP2-Cre+ renal medulla compared with controls, which showed moderate expression of all three genes. In conjunction, we found that phosphorylation of SMAD2, a critical downstream mediator of the canonical TGF-β signaling, was also significantly upregulated in our KO model, suggesting an upregulation of the TGF-β signaling pathway subsequent to the loss of β1-integrin in CD PCs.
The role of integrins in modulating fibrosis has been well established in the kidney and other organs. There is convincing evidence of TGF-β signaling involvement in the pathogenesis of renal dysfunction (63) and that β1-integrin signaling has been associated with TGF-β activation in different organs (3). Recent studies also suggest that the loss of β1-integrin can induce the activation of profibrotic TGF-β signaling pathways in the renal system (30). In addition, more recent reports reveal that β1-integrin ablation can increase the expression of other β-integrins that are critical in activating the TGF-β signaling pathway (17, 57, 73). To determine whether this is the case in our PC specific β1-integrin KO animals will require further investigations.
In summary, our data demonstrate that the loss of β1-integrin specifically in kidney CD PCs causes significant interstitial fibrosis and CD injury, probably through upregulation of the TGF-β-associated signaling pathway. Our study highlights the importance of β1-integrin specifically in PCs, and the importance of PCs in maintaining the structural and functional integrity of the entire CD system after birth.
GRANTS
F. A. Mamuya is supported by National Cancer Institute T32 Training Grant 5T32CA079443-15. This work was supported by the NephCure Foundation, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant DK-096015, a Gottschalk Research grant, Massachusetts General Hospital ECOR support, and the S&R Foundation Ryuji Ueno Award (to H. A. Lu). The Microscopy Core facility of the Program in Membrane Biology receives additional support from the Boston Area Diabetes and the Endocrinology Research Center (NIDDK Grant DK-57521) and the Center for the Study of Inflammatory Bowel Disease (NICCK Grant DK-43351).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.A.M., T.G.P. and H.A.J.L. conceived and designed research; F.A.M., D.X., L.L., M.H., K.T., and D.E.C. performed the experiments; F.A.M., D.X., L.L., M.H., D.E.C., T.G.P. and H.A.J.L. analyzed data; F.A.M., D.X., L.L., M.H., K.T., D.E.C., T.G.P., and H.A.J.L. interpreted results of experiments; F.A.M., L.L., M.H., and D.E.C. prepared figures; F.A.M., T.G.P., and H.A.J.L. drafted manuscript; F.A.M., T.G.P., and H.A.J.L. edited and revised manuscript; F.A.M., B.Y., R.W., T.G.P., and H.A.J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. W. Zhang from the University of Texas Health Science Center at Houston for the AQP2 Cre mice and Dr. Dennis Brown from Massachusetts General Hospital and Harvard Medical School for valuable scientific insight.
REFERENCES
- 1.Araki T, Saruta T, Okano H, Miura M. Caspase activity is required for nephrogenesis in the developing mouse metanephros. Exp Cell Res 248: 423–429, 1999. doi: 10.1006/excr.1999.4424. [DOI] [PubMed] [Google Scholar]
- 2.Baum MA, Ruddy MK, Hosselet CA, Harris HW. The perinatal expression of aquaporin-2 and aquaporin-3 in developing kidney. Pediatr Res 43: 783–790, 1998. doi: 10.1203/00006450-199806000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem 276: 46707–46713, 2001. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 4.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med 342: 1350–1358, 2000. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 5.Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol 6: 643–656, 2010. doi: 10.1038/nrneph.2010.120. [DOI] [PubMed] [Google Scholar]
- 6.Border WA, Noble NA. TGF-beta in kidney fibrosis: a target for gene therapy. Kidney Int 51: 1388–1396, 1997. doi: 10.1038/ki.1997.190. [DOI] [PubMed] [Google Scholar]
- 7.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med 331: 1286–1292, 1994. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 8.Brown D. The ins and outs of aquaporin-2 trafficking. Am J Physiol Renal Physiol 284: F893–F901, 2003. doi: 10.1152/ajprenal.00387.2002. [DOI] [PubMed] [Google Scholar]
- 9.Brown D, Bouley R, Păunescu TG, Breton S, Lu HA. New insights into the dynamic regulation of water and acid-base balance by renal epithelial cells. Am J Physiol Cell Physiol 302: C1421–C1433, 2012. doi: 10.1152/ajpcell.00085.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown D, Fenton R. The cell biology of vasopressin action. In: Brenner and Rector’s The Kidney, edited by Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu AS, Brenner BM. Philadelphia, PA: Elsevier, 2015, p. 302. [Google Scholar]
- 11.Brown D, Lydon J, McLaughlin M, Stuart-Tilley A, Tyszkowski R, Alper S. Antigen retrieval in cryostat tissue sections and cultured cells by treatment with sodium dodecyl sulfate (SDS). Histochem Cell Biol 105: 261–267, 1996. doi: 10.1007/BF01463929. [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Li R, Ross RS, Manso AM. Integrins and integrin-related proteins in cardiac fibrosis. J Mol Cell Cardiol 93: 162–174, 2016. doi: 10.1016/j.yjmcc.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Rice W, Li W, Kong Y, Fenton R, Li J, Hsu V, Brown D, Lu HJ. AQP2 affects renal epithelial cell adhesion, migration and tubule formation by interacting with integrin β1 via an external RGD motif (Abstract). J Am Soc Nephrol 21: SA-PO2112, 2010. [Google Scholar]
- 14.Chen Y, Rice W, Gu Z, Li J, Huang J, Brenner MB, Van Hoek A, Xiong J, Gundersen GG, Norman JC, Hsu VW, Fenton RA, Brown D, Lu HA. Aquaporin 2 promotes cell migration and epithelial morphogenesis. J Am Soc Nephrol 23: 1506–1517, 2012. doi: 10.1681/ASN.2012010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clapp WL, Madsen KM, Verlander JW, Tisher CC. Morphologic heterogeneity along the rat inner medullary collecting duct. Lab Invest 60: 219–230, 1989. [PubMed] [Google Scholar]
- 16.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 16: 31–41, 1976. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 17.Danen EH, Sonneveld P, Brakebusch C, Fassler R, Sonnenberg A. The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J Cell Biol 159: 1071–1086, 2002. doi: 10.1083/jcb.200205014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Groot T, Alsady M, Jaklofsky M, Otte-Höller I, Baumgarten R, Giles RH, Deen PM. Lithium causes G2 arrest of renal principal cells. J Am Soc Nephrol 25: 501–510, 2014. doi: 10.1681/ASN.2013090988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Palo S, Ferrari G, Castoldi R, Fiacco E, Cristallo M, Staudacher C, Chiesa R, Di Carlo V. Surgical septic complications in diabetic patients. Acta Diabetol Lat 25: 49–54, 1988. doi: 10.1007/BF02581245. [DOI] [PubMed] [Google Scholar]
- 20.Doi K, Negishi K, Ishizu T, Katagiri D, Fujita T, Matsubara T, Yahagi N, Sugaya T, Noiri E. Evaluation of new acute kidney injury biomarkers in a mixed intensive care unit. Crit Care Med 39: 2464–2469, 2011. doi: 10.1097/CCM.0b013e318225761a. [DOI] [PubMed] [Google Scholar]
- 21.Espersen GT, Ernst E, Vestergaard M, Pedersen JO, Grunnet N. Changes in PMN leukocyte migration activity and complement C3d levels in RA patients with high disease activity during steroid treatment. Scand J Rheumatol 18: 51–56, 1989. doi: 10.3109/03009748909095403. [DOI] [PubMed] [Google Scholar]
- 22.Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int 56: 1455–1467, 1999. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- 23.Fenton RA, Praetorius J. Molecular physiology of the medullary collecting duct. Compr Physiol 1: 1031–1056, 2011. doi: 10.1002/cphy.c100064. [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Eladari D, Leviel F, Tew BY, Miró-Julià C, Cheema FH, Miller L, Nelson R, Păunescu TG, McKee M, Brown D, Al-Awqati Q. Deletion of hensin/DMBT1 blocks conversion of beta- to alpha-intercalated cells and induces distal renal tubular acidosis. Proc Natl Acad Sci USA 107: 21872–21877, 2010. [Erratum. Proc Natl Acad Sci USA 109: 18625, 2012]. doi: 10.1073/pnas.1010364107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge Y, Ahn D, Stricklett PK, Hughes AK, Yanagisawa M, Verbalis JG, Kohan DE. Collecting duct-specific knockout of endothelin-1 alters vasopressin regulation of urine osmolality. Am J Physiol Renal Physiol 288: F912–F920, 2005. doi: 10.1152/ajprenal.00432.2004. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh Choudhury G, Abboud HE. Tyrosine phosphorylation-dependent PI 3 kinase/Akt signal transduction regulates TGFbeta-induced fibronectin expression in mesangial cells. Cell Signal 16: 31–41, 2004. doi: 10.1016/S0898-6568(03)00094-9. [DOI] [PubMed] [Google Scholar]
- 27.Gilbert RE, Wilkinson-Berka JL, Johnson DW, Cox A, Soulis T, Wu LL, Kelly DJ, Jerums G, Pollock CA, Cooper ME. Renal expression of transforming growth factor-beta inducible gene-h3 (beta ig-h3) in normal and diabetic rats. Kidney Int 54: 1052–1062, 1998. doi: 10.1046/j.1523-1755.1998.00081.x. [DOI] [PubMed] [Google Scholar]
- 28.Guo Q, Wang Y, Tripathi P, Manda KR, Mukherjee M, Chaklader M, Austin PF, Surendran K, Chen F. Adam10 mediates the choice between principal cells and intercalated cells in the kidney. J Am Soc Nephrol 26: 149–159, 2015. doi: 10.1681/ASN.2013070764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayashida T. Integrins modulate cellular fibrogenesis at multiple levels; Regulation of TGF-β signaling. Endocr Metab Immune Disord Drug Targets 10: 302–319, 2010. doi: 10.2174/1871530311006040302. [DOI] [PubMed] [Google Scholar]
- 30.Hayashida T, Jones JC, Lee CK, Schnaper HW. Loss of beta1-integrin enhances TGF-beta1-induced collagen expression in epithelial cells via increased alphavbeta3-integrin and Rac1 activity. J Biol Chem 285: 30741–30751, 2010. doi: 10.1074/jbc.M110.105700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henderson NC, Sheppard D. Integrin-mediated regulation of TGFβ in fibrosis. Biochim Biophys Acta 1832: 891–896, 2013. doi: 10.1016/j.bbadis.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huber SM, Braun GS, Horster MF. Expression of the epithelial sodium channel (ENaC) during ontogenic differentiation of the renal cortical collecting duct epithelium. Pflugers Arch 437: 491–497, 1999. doi: 10.1007/s004240050806. [DOI] [PubMed] [Google Scholar]
- 33.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687, 2002. doi: 10.1016/S0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 34.Ivanov SV, Ivanova AV, Salnikow K, Timofeeva O, Subramaniam M, Lerman MI. Two novel VHL targets, TGFBI (BIGH3) and its transactivator KLF10, are up-regulated in renal clear cell carcinoma and other tumors. Biochem Biophys Res Commun 370: 536–540, 2008. doi: 10.1016/j.bbrc.2008.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanwar YS. TGF-β and renal fibrosis: a Pandora’s box of surprises. Am J Pathol 181: 1147–1150, 2012. doi: 10.1016/j.ajpath.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labban B, Arora N, Restaino S, Markowitz G, Valeri A, Radhakrishnan J. The role of kidney biopsy in heart transplant candidates with kidney disease. Transplantation 89: 887–893, 2010. doi: 10.1097/TP.0b013e3181cd4abb. [DOI] [PubMed] [Google Scholar]
- 37.Lan HY. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int J Biol Sci 7: 1056–1067, 2011. doi: 10.7150/ijbs.7.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LeBaron RG, Bezverkov KI, Zimber MP, Pavelec R, Skonier J, Purchio AF. Beta IG-H3, a novel secretory protein inducible by transforming growth factor-beta, is present in normal skin and promotes the adhesion and spreading of dermal fibroblasts in vitro. J Invest Dermatol 104: 844–849, 1995. doi: 10.1111/1523-1747.ep12607024. [DOI] [PubMed] [Google Scholar]
- 39.Lee K, Boctor S, Barisoni LM, Gusella GL. Inactivation of integrin-β1 prevents the development of polycystic kidney disease after the loss of polycystin-1. J Am Soc Nephrol 26: 888–895, 2015. doi: 10.1681/ASN.2013111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SH, Bae JS, Park SH, Lee BH, Park RW, Choi JY, Park JY, Ha SW, Kim YL, Kwon TH, Kim IS. Expression of TGF-beta-induced matrix protein betaig-h3 is up-regulated in the diabetic rat kidney and human proximal tubular epithelial cells treated with high glucose. Kidney Int 64: 1012–1021, 2003. doi: 10.1046/j.1523-1755.2003.00158.x. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7: 684–696, 2011. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu H, Sun TX, Bouley R, Blackburn K, McLaughlin M, Brown D. Inhibition of endocytosis causes phosphorylation (S256)-independent plasma membrane accumulation of AQP2. Am J Physiol Renal Physiol 286: F233–F243, 2004. doi: 10.1152/ajprenal.00179.2003. [DOI] [PubMed] [Google Scholar]
- 43.Madsen KM, Verlander JW, Tisher CC. Relationship between structure and function in distal tubule and collecting duct. J Electron Microsc Tech 9: 187–208, 1988. doi: 10.1002/jemt.1060090206. [DOI] [PubMed] [Google Scholar]
- 44.Mamuya FA, Duncan MK. aV integrins and TGF-β-induced EMT: a circle of regulation. J Cell Mol Med 16: 445–455, 2012. doi: 10.1111/j.1582-4934.2011.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep 11: 97–105, 2010. doi: 10.1038/embor.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mårtensson J, Martling CR, Bell M. Novel biomarkers of acute kidney injury and failure: clinical applicability. Br J Anaesth 109: 843–850, 2012. doi: 10.1093/bja/aes357. [DOI] [PubMed] [Google Scholar]
- 47.Mathew S, Lu Z, Palamuttam RJ, Mernaugh G, Hadziselimovic A, Chen J, Bulus N, Gewin LS, Voehler M, Meves A, Ballestrem C, Fässler R, Pozzi A, Sanders CR, Zent R. β1 integrin NPXY motifs regulate kidney collecting-duct development and maintenance by induced-fit interactions with cytosolic proteins. Mol Cell Biol 32: 4080–4091, 2012. doi: 10.1128/MCB.00568-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson RD, Stricklett P, Gustafson C, Stevens A, Ausiello D, Brown D, Kohan DE. Expression of an AQP2 Cre recombinase transgene in kidney and male reproductive system of transgenic mice. Am J Physiol Cell Physiol 275: C216–C226, 1998. [DOI] [PubMed] [Google Scholar]
- 49.Nielsen S, Frør J, Knepper MA. Renal aquaporins: key roles in water balance and water balance disorders. Curr Opin Nephrol Hypertens 7: 509–516, 1998. doi: 10.1097/00041552-199809000-00005. [DOI] [PubMed] [Google Scholar]
- 50.Nielsen S, Kwon TH, Frøkiaer J, Knepper MA. Key roles of renal aquaporins in water balance and water-balance disorders. News Physiol Sci 15: 136–143, 2000. [DOI] [PubMed] [Google Scholar]
- 51.Păunescu TG, Da Silva N, Marshansky V, McKee M, Breton S, Brown D. Expression of the 56-kDa B2-subunit isoform of the vacuolar H+-ATPase in proton-secreting cells of the kidney and epididymis. Am J Physiol Cell Physiol 287: C149–C162, 2004. doi: 10.1152/ajpcell.00464.2003. [DOI] [PubMed] [Google Scholar]
- 52.Păunescu TG, Lu HA, Russo LM, Pastor-Soler NM, McKee M, McLaughlin MM, Bartlett BE, Breton S, Brown D. Vasopressin induces apical expression of caveolin in rat kidney collecting duct principal cells. Am J Physiol Renal Physiol 305: F1783–F1795, 2013. doi: 10.1152/ajprenal.00622.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peleg I, Greenfeld Z, Cooperman H, Shoshan S. Type I and type III collagen mRNA levels in kidney regions of old and young rats. Matrix 13: 281–287, 1993. doi: 10.1016/S0934-8832(11)80023-5. [DOI] [PubMed] [Google Scholar]
- 54.Pozzi A, Zent R. Integrins in kidney disease. J Am Soc Nephrol 24: 1034–1039, 2013. doi: 10.1681/ASN.2013010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E. Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol 150: 1149–1160, 2000. doi: 10.1083/jcb.150.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reed NI, Jo H, Chen C, Tsujino K, Arnold TD, DeGrado WF, Sheppard D. The αvβ1 integrin plays a critical in vivo role in tissue fibrosis. Sci Transl Med 7: 288ra79, 2015. doi: 10.1126/scitranslmed.aaa5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Retta SF, Cassarà G, D’Amato M, Alessandro R, Pellegrino M, Degani S, De Leo G, Silengo L, Tarone G. Cross talk between beta(1) and alpha(V) integrins: beta(1) affects beta(3) mRNA stability. Mol Biol Cell 12: 3126–3138, 2001. doi: 10.1091/mbc.12.10.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roberts CJ, Birkenmeier TM, McQuillan JJ, Akiyama SK, Yamada SS, Chen WT, Yamada KM, McDonald JA. Transforming growth factor beta stimulates the expression of fibronectin and of both subunits of the human fibronectin receptor by cultured human lung fibroblasts. J Biol Chem 263: 4586–4592, 1988. [PubMed] [Google Scholar]
- 59.Rojek A, Füchtbauer EM, Kwon TH, Frøkiaer J, Nielsen S. Severe urinary concentrating defect in renal collecting duct-selective AQP2 conditional-knockout mice. Proc Natl Acad Sci USA 103: 6037–6042, 2006. doi: 10.1073/pnas.0511324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ronzaud C, Loffing J, Bleich M, Gretz N, Gröne HJ, Schütz G, Berger S. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 18: 1679–1687, 2007. doi: 10.1681/ASN.2006090975. [DOI] [PubMed] [Google Scholar]
- 61.Roy A, Al-bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10: 305–324, 2015. doi: 10.2215/CJN.08880914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Russo LM, McKee M, Brown D. Methyl-beta-cyclodextrin induces vasopressin-independent apical accumulation of aquaporin-2 in the isolated, perfused rat kidney. Am J Physiol Renal Physiol 291: F246–F253, 2006. doi: 10.1152/ajprenal.00437.2005. [DOI] [PubMed] [Google Scholar]
- 63.Schnaper HW, Jandeska S, Runyan CE, Hubchak SC, Basu RK, Curley JF, Smith RD, Hayashida T. TGF-beta signal transduction in chronic kidney disease. Front Biosci (Landmark Ed) 14: 2448–2465, 2009. doi: 10.2741/3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma K, Ziyadeh FN. The emerging role of transforming growth factor-beta in kidney diseases. Am J Physiol Renal Physiol 266: F829–F842, 1994. [DOI] [PubMed] [Google Scholar]
- 65.Simirskii VN, Wang Y, Duncan MK. Conditional deletion of beta1-integrin from the developing lens leads to loss of the lens epithelial phenotype. Dev Biol 306: 658–668, 2007. doi: 10.1016/j.ydbio.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD, Purchio AF. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol 11: 511–522, 1992. doi: 10.1089/dna.1992.11.511. [DOI] [PubMed] [Google Scholar]
- 67.Sträter J, Wedding U, Barth TF, Koretz K, Elsing C, Möller P. Rapid onset of apoptosis in vitro follows disruption of beta 1-integrin/matrix interactions in human colonic crypt cells. Gastroenterology 110: 1776–1784, 1996. doi: 10.1053/gast.1996.v110.pm8964403. [DOI] [PubMed] [Google Scholar]
- 68.Sugiyama H, Kashihara N, Maeshima Y, Okamoto K, Kanao K, Sekikawa T, Makino H. Regulation of survival and death of mesangial cells by extracellular matrix. Kidney Int 54: 1188–1196, 1998. doi: 10.1046/j.1523-1755.1998.00116.x. [DOI] [PubMed] [Google Scholar]
- 69.Tamma G, Lasorsa D, Ranieri M, Mastrofrancesco L, Valenti G, Svelto M. Integrin signaling modulates AQP2 trafficking via Arg-Gly-Asp (RGD) motif. Cell Physiol Biochem 27: 739–748, 2011. doi: 10.1159/000330082. [DOI] [PubMed] [Google Scholar]
- 70.Tang O, Chen XM, Shen S, Hahn M, Pollock CA. MiRNA-200b represses transforming growth factor-β1-induced EMT and fibronectin expression in kidney proximal tubular cells. Am J Physiol Renal Physiol 304: F1266–F1273, 2013. doi: 10.1152/ajprenal.00302.2012. [DOI] [PubMed] [Google Scholar]
- 71.Tanwar PS, Lee HJ, Zhang L, Zukerberg LR, Taketo MM, Rueda BR, Teixeira JM. Constitutive activation of Beta-catenin in uterine stroma and smooth muscle leads to the development of mesenchymal tumors in mice. Biol Reprod 81: 545–552, 2009. doi: 10.1095/biolreprod.108.075648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thomas GL, Yang B, Wagner BE, Savill J, El Nahas AM. Cellular apoptosis and proliferation in experimental renal fibrosis. Nephrol Dial Transplant 13: 2216–2226, 1998. doi: 10.1093/ndt/13.9.2216. [DOI] [PubMed] [Google Scholar]
- 73.Truong HH, Xiong J, Ghotra VP, Nirmala E, Haazen L, Le Dévédec SE, Balcioğlu HE, He S, Snaar-Jagalska BE, Vreugdenhil E, Meerman JH, van de Water B, Danen EH. β1 integrin inhibition elicits a prometastatic switch through the TGFβ-miR-200-ZEB network in E-cadherin-positive triple-negative breast cancer. Sci Signal 7: ra15, 2014. doi: 10.1126/scisignal.2004751. [DOI] [PubMed] [Google Scholar]
- 74.Vedovelli L, Rothermel JT, Finberg KE, Wagner CA, Azroyan A, Hill E, Breton S, Brown D, Păunescu TG. Altered V-ATPase expression in renal intercalated cells isolated from B1 subunit-deficient mice by fluorescence-activated cell sorting. Am J Physiol Renal Physiol 304: F522–F532, 2013. doi: 10.1152/ajprenal.00394.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Viedt C, Bürger A, Hänsch GM. Fibronectin synthesis in tubular epithelial cells: up-regulation of the EDA splice variant by transforming growth factor beta. Kidney Int 48: 1810–1817, 1995. doi: 10.1038/ki.1995.479. [DOI] [PubMed] [Google Scholar]
- 76.Wang B, Jha JC, Hagiwara S, McClelland AD, Jandeleit-Dahm K, Thomas MC, Cooper ME, Kantharidis P. Transforming growth factor-β1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int 85: 352–361, 2014. doi: 10.1038/ki.2013.372. [DOI] [PubMed] [Google Scholar]
- 77.Worthington JJ, Klementowicz JE, Travis MA. TGFβ: a sleeping giant awoken by integrins. Trends Biochem Sci 36: 47–54, 2011. doi: 10.1016/j.tibs.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 78.Wu C, Bauer JS, Juliano RL, McDonald JA. The alpha 5 beta 1 integrin fibronectin receptor, but not the alpha 5 cytoplasmic domain, functions in an early and essential step in fibronectin matrix assembly. J Biol Chem 268: 21883–21888, 1993. [PubMed] [Google Scholar]
- 79.Wu H, Chen L, Zhou Q, Zhang X, Berger S, Bi J, Lewis DE, Xia Y, Zhang W. Aqp2-expressing cells give rise to renal intercalated cells. J Am Soc Nephrol 24: 243–252, 2013. doi: 10.1681/ASN.2012080866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu W, Kitamura S, Truong DM, Rieg T, Vallon V, Sakurai H, Bush KT, Vera DR, Ross RS, Nigam SK. Beta1-integrin is required for kidney collecting duct morphogenesis and maintenance of renal function. Am J Physiol Renal Physiol 297: F210–F217, 2009. doi: 10.1152/ajprenal.90260.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang B, Zhao D, Qian L, Verkman AS. Mouse model of inducible nephrogenic diabetes insipidus produced by floxed aquaporin-2 gene deletion. Am J Physiol Renal Physiol 291: F465–F472, 2006. doi: 10.1152/ajprenal.00494.2005. [DOI] [PubMed] [Google Scholar]
- 82.Yazlovitskaya EM, Tseng HY, Viquez O, Tu T, Mernaugh G, McKee KK, Riggins K, Quaranta V, Pathak A, Carter BD, Yurchenco P, Sonnenberg A, Böttcher RT, Pozzi A, Zent R. Integrin α3β1 regulates kidney collecting duct development via TRAF6-dependent K63-linked polyubiquitination of Akt. Mol Biol Cell 26: 1857–1874, 2015. doi: 10.1091/mbc.E14-07-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yokoi H, Mukoyama M, Sugawara A, Mori K, Nagae T, Makino H, Suganami T, Yahata K, Fujinaga Y, Tanaka I, Nakao K. Role of connective tissue growth factor in fibronectin expression and tubulointerstitial fibrosis. Am J Physiol Renal Physiol 282: F933–F942, 2002. doi: 10.1152/ajprenal.00122.2001. [DOI] [PubMed] [Google Scholar]
- 84.Zhang X, Mernaugh G, Yang DH, Gewin L, Srichai MB, Harris RC, Iturregui JM, Nelson RD, Kohan DE, Abrahamson D, Fässler R, Yurchenco P, Pozzi A, Zent R. beta1 integrin is necessary for ureteric bud branching morphogenesis and maintenance of collecting duct structural integrity. Development 136: 3357–3366, 2009. doi: 10.1242/dev.036269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Z, Vuori K, Reed JC, Ruoslahti E. The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc Natl Acad Sci USA 92: 6161–6165, 1995. doi: 10.1073/pnas.92.13.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zharkikh L, Zhu X, Stricklett PK, Kohan DE, Chipman G, Breton S, Brown D, Nelson RD. Renal principal cell-specific expression of green fluorescent protein in transgenic mice. Am J Physiol Renal Physiol 283: F1351–F1364, 2002. doi: 10.1152/ajprenal.0224.2001. [DOI] [PubMed] [Google Scholar]