

This research is the first to demonstrate enhanced nitric oxide-dependent vasodilation that limits increased vasoconstrictor reactivity in neonatal pulmonary hypertension. These results suggest that augmented vasoconstriction in this setting reflects changes in smooth muscle reactivity rather than a reduction in nitric oxide-dependent pulmonary vasodilation.
Keywords: neonatal pulmonary hypertension, in situ perfused lung, nitric oxide, endothelial nitric oxide synthase phosphorylation
Abstract
Augmented vasoconstrictor reactivity is thought to play an important role in the development of chronic hypoxia (CH)-induced neonatal pulmonary hypertension. However, whether this response to CH results from pulmonary endothelial dysfunction and reduced nitric oxide (NO)-mediated vasodilation is not well understood. We hypothesized that neonatal CH enhances basal tone and pulmonary vasoconstrictor sensitivity by limiting NO-dependent pulmonary vasodilation. To test this hypothesis, we assessed the effects of the NO synthase (NOS) inhibitor Nω-nitro-l-arginine (l-NNA) on baseline pulmonary vascular resistance (PVR) and vasoconstrictor sensitivity to the thromboxane mimetic U-46619 in saline-perfused lungs (in situ) from 2-wk-old control and CH (12-day exposure, 0.5 atm) Sprague-Dawley rats. Basal tone was defined as that reversed by exogenous NO (spermine NONOate). CH neonates displayed elevated right ventricular systolic pressure (in vivo) and right ventricular hypertrophy, indicative of pulmonary hypertension. Perfused lungs from CH rats demonstrated greater baseline PVR, basal tone, and U-46619-mediated vasoconstriction compared with control rats in the absence of l-NNA. l-NNA markedly increased baseline PVR and reactivity to U-46619 in lungs from CH neonates, further augmenting vasoconstrictor sensitivity compared with control lungs. Exposure to CH also enhanced NO-dependent vasodilation to arginine vasopressin, pulmonary expression of NOS III [endothelial NOS (eNOS)], and eNOS phosphorylation at activation residue Ser1177. However, CH did not alter lung nitrotyrosine levels, a posttranslational modification reflecting scavenging of NO. We conclude that, in contrast to our hypothesis, enhanced basal tone and agonist-induced vasoconstriction after neonatal CH is limited by increased NO-dependent pulmonary vasodilation resulting from greater eNOS expression and phosphorylation at activation residue Ser1177.
NEW & NOTEWORTHY This research is the first to demonstrate enhanced nitric oxide-dependent vasodilation that limits increased vasoconstrictor reactivity in neonatal pulmonary hypertension. These results suggest that augmented vasoconstriction in this setting reflects changes in smooth muscle reactivity rather than a reduction in nitric oxide-dependent pulmonary vasodilation.
neonatal pulmonary hypertension (pHTN) affects 1–2 of every 1,000 live births in the United States with 20% of cases being fatal. Surviving patients face significant morbidity associated with detriments to neural development and chronic lung disease (2, 60). Chronic hypoxia (CH) is implicated in the pathogenesis of pHTN in infants with chronic cardiorespiratory conditions (4, 30). CH is thought to elevate postnatal pulmonary vascular resistance (PVR) by increasing basal arterial tone and by augmenting reactivity to endogenous vasoconstrictor agonists (5, 11, 56). Enhanced pulmonary vasoconstriction after neonatal CH may result from an imbalance in the production of endothelium-derived contractile and relaxing factors or changes in smooth muscle responsiveness to these factors (6, 25, 27, 31).
CH may contribute to pHTN by limiting the normal postnatal increases in O2 tension that stimulate pulmonary vascular endothelial nitric oxide (NO) synthase (eNOS)-dependent NO production and relief of fetal pulmonary vascular tone (1, 29). Phenotypic changes in endothelial function have additionally been implicated in the development of neonatal pHTN. Such changes include increased production of endothelium-derived vasoconstrictor compounds (3, 28, 37) and disruption of NO signaling through reactive oxygen species (ROS)-dependent scavenging of NO (19, 24, 33, 39), eNOS uncoupling (38), decreased eNOS expression (14, 27), and reduced activity due to altered phosphorylation (38). However, the contribution of endothelial dysfunction to the development of basal arterial tone and enhanced receptor-mediated vasoconstriction in neonatal pHTN resulting from CH is not well understood. We hypothesized that neonatal CH increases basal tone and pulmonary vasoconstrictor sensitivity by attenuating NO-dependent pulmonary vasodilation.
To test our hypothesis, we used an isolated (in situ) perfused lung preparation to evaluate the contribution of endogenous NO to basal and agonist-induced pulmonary vasoconstriction in normoxic and CH neonatal (2-wk-old) rats. Exposure of neonatal rats to CH is an established approach to model neonatal pHTN that occurs in human infants (33, 69). Additional experiments examined vasodilatory responsiveness to the receptor-mediated, NO-dependent pulmonary vasodilator arginine vasopressin (AVP) and assessed pulmonary eNOS expression and phosphorylation status by Western blot analysis. Our findings demonstrate the novel effects of neonatal CH augmenting NO-dependent vasodilation and increasing pulmonary eNOS expression and phosphorylation at activation residue Ser1177, responses that lessen enhanced basal tone and vasoconstrictor sensitivity to the thromboxane analog U-46619.
METHODS
Animals and CH Exposure Protocol
Timed-pregnant Sprague-Dawley (Harlan Industries) rats were allowed to acclimate for 1 wk before giving birth at ambient normobaric pressure (~630 mmHg in Albuquerque, NM). Litters selected for exposure to CH were housed in a hypobaric hypoxic chamber with barometric pressure maintained at 380 ± 5 mmHg for 12 days [beginning on postnatal day 2 of life]. Age-matched normoxic control rats were housed in similar sham conditions at ambient barometric pressure. Neonates were housed with their birthing dam. The chamber was opened 2 times/wk to provide animals with food, water, and clean bedding. Animals were housed on a 12:12-h light-dark cycle. Animal protocols used for this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico Health Sciences Center.
Assessment of Right Ventricular Systolic Pressure, Right Ventricular Weight, and Hematocrit
Right ventricular (RV) systolic pressure (RVSP) and RV hypertrophy were evaluated as indexes of pHTN (8, 61). Rats from each group were anesthetized with ketamine (40 mg/kg) and xylazine (6 mg/kg) (41). An upper transverse laparotomy was performed, and RVSP was assessed using a closed-chest transdiaphragmatic approach with a 25-gauge needle attached to a pressure transducer (Hugo Sachs Electronik-Harvard Apparatus). Entry into the RV was confirmed by monitoring the pressure waveform. RVSP and heart rate were obtained using AT-CODAS data-acquisition software (Dataq Instruments). Fulton’s index, expressed as the percent ratio of RV to left ventricular (LV) and interventricular septum (LV + S) weight, was used to assess the degree of CH-induced RV hypertrophy. The polycythemic response to CH was determined by measuring hematocrit from blood samples taken by direct cardiac puncture at the time of lung isolation.
In Situ Perfused Rat Lung Preparation
To evaluate the effect of neonatal CH on baseline as well as agonist-dependent changes in PVR, neonatal rats were anesthetized with pentobarbital sodium (200 mg/kg ip). The animal was placed on a heating block, the trachea was cannulated, and the lungs were ventilated with a positive-pressure mouse ventilator (Hugo Sachs Electronik-Harvard Apparatus) at a tidal volume of 5 ml/kg body wt and a rate of 100 breaths/min with a warmed and humidified gas mixture (21% O2-6% CO2-balance N2). End-expiratory pressure was maintained at 3 cmH2O. After removal of the frontal ribcage, heparin (5 units, 50 μl) was injected into the RV, and the pulmonary artery was cannulated with custom-fitted PE-90 tubing (Becton Dickinson). The preparation was immediately perfused with a peristaltic pump (Ismatec) at a rate of 0.1 ml/min with physiological saline solution (PSS) containing the following (in mM): 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 19 NaHCO3, 1.8 CaCl2, and 5.5 glucose with 4% (wt/vol) BSA (all from Sigma). After cannulation of the pulmonary artery, the LV was cannulated with custom-fitted PE-160 tubing. The perfusion rate was gradually increased to a maximum of 15 ml·min−1·kg body wt−1 (15). Perfusion was nonrecirculating until the perfusate exiting the lung was nearly free of blood. Recirculation was then initiated and maintained for the duration of the experiment. Total recirculating volume was ~3.5 ml. After a 20-min equilibration period, an effluent sample of PSS was collected for the determination of pH, Pco2, and Po2 using a handheld iSTAT 1 blood gas analyzer (Abbott) and iSTAT test cartridges (G3+, Abbott). Zone 3 conditions were achieved by maintaining venous pressure at 3 mmHg and airway pressure at 1 mmHg (15). Pulmonary arterial pressure (Pa) and venous pressure (Pv) were recorded continuously. Pressures were measured using a P-75 pressure transducer (Hugo Sachs Electronik-Harvard Apparatus) connected to the pulmonary arterial and venous lines. Data were recorded and processed using data-acquisition software and hardware (AT-CODAS, Dataq Instruments). Total PVR was calculated as (Pa − Pv)/Q, where Q is the perfusion rate.
Assessment of Segmental Vascular Resistances
The effects of neonatal CH on basal and agonist-induced changes in arterial and venous resistance were determined using a double occlusion method as previously reported (21, 52). Briefly, the lungs were isolated as described above, and inflow and outflow lines were simultaneously occluded, allowing Pa and Pv to rapidly equilibrate to an approximation of microvascular capillary pressure (Pc). This double occlusion method provides estimates of Pc that closely agree with values of Pc assessed by isogravimetric methods (18, 53, 66). Pulmonary arterial resistance was calculated as (Pa − Pc)/Q, and pulmonary venous resistance was calculated as (Pc − Pv)/Q.
Regulation of Basal Tone and Agonist-Induced Vasoconstriction by Endogenous NO
To assess the role of endogenous NO to baseline PVR, lungs from each group were perfused with PSS containing either saline vehicle or the NOS inhibitor Nω-nitro-l-arginine (l-NNA; 300 μM). We have previously demonstrated that this dose of l-NNA effectively inhibits NO-dependent pulmonary vasodilation in isolated, perfused lungs from adult rats (54). After a 20-min equilibration period, Pc was assessed by the double occlusion technique. The contribution of active tone to baseline PVR was assessed by administration of the NO donor 1,3-propanediamine,N-{4-[1-(3-aminopropyl)-2-hydroxy-2-nitro-sohydrazino]butyl} (spermine NONOate; 100 μM) to maximally dilate the pulmonary vasculature. A second double occlusion was performed after stabilization of the response to spermine NONOate. Basal tone is expressed as the change in resistance to spermine NONOate.
The effect of endogenous NO limiting vasoconstrictor reactivity was determined by performing cumulative concentration-response curves to the thromboxane analog 9,11-dideoxy-9α,11α-methanoepoxy PGF2α (U-46619) in the presence and absence of l-NNA. Double occlusions were performed to assess Pc under baseline conditions and after the development of a stable pressor response to each concentration of U-46619.
Effect of CH on NO-Dependent Pulmonary Vasodilation
Endothelium-dependent vasodilatory responses.
Receptor-mediated vasodilation to the endothelium-derived NO (EDNO)-dependent vasodilator AVP (52) was measured in the lungs from control and CH neonatal rats. After a 20-min equilibration period, lungs were preconstricted with the thromboxane analog U-46619 to achieve a stable arterial pressor response of ∼10 mmHg. AVP (25 nM) was then added to the perfusate to induce vasodilation. Parallel protocols were performed in the presence of l-NNA to verify the contribution of endogenous NO to this response. Vasodilatory responses are expressed as the percent reversal of U-46619-mediated constriction.
Responses to exogenous NO.
Considering that EDNO-dependent responses after CH may be influenced by altered NO bioavailability or vascular smooth muscle (VSM) sensitivity to NO, we assessed concentration-response curves to the NO donor spermine NONOate in lungs from each group of rats. After the equilibration period, lungs were preconstricted with U-46619 to ~10 mmHg above baseline Pa. Once the constriction stabilized, a cumulative concentration-response curve to spermine NONOate was performed. Considering that lungs from neonates exposed to CH display greater basal pulmonary arterial constriction than control lungs, we repeated these experiments in lungs preconstricted with U-46619 to achieve similar overall Pa between groups. All experiments were conducted in the presence of l-NNA to eliminate the contribution of endogenous NO to these responses. Segmental vascular resistances were evaluated by the double occlusion technique at baseline, after stabilization of the U-46619 constriction, and after stabilization of the response to each concentration of NONOate.
Western Blot Analysis
NOS isoforms.
The effects of CH on the expression of the NOS isoforms NOS I [neuronal NOS (nNOS)], NOS II [inducible NOS (iNOS)], and NOS III (eNOS) as well as the posttranslational phosphorylation of eNOS at Ser1177, a modification associated with increased eNOS-dependent NO production (20), were also investigated. Western blots were performed using whole lung homogenates from both control and CH neonates as previously described (52). Lungs were snap frozen in liquid nitrogen and homogenized in ice-cold homogenization buffer {10 mM Tris·HCl (pH 7.4), 255 mM sucrose, 2 mM EDTA, 12 μM leupeptin, 4 μM pepstatin A, 1 μM aprotinin, and 1% (vol/vol) phosphatase inhibitor cocktail 3 [cantharidin, (−)-p-bromolevamisole oxalate, and calyculin A] from Sigma}. Tissue homogenates were then sonicated on ice with an ultrasonic homogenizer (Cole Parmer). Homogenates were spun at 1,500 g for 10 min in a desktop centrifuge (Beckman) at 4°C. Sample protein concentrations were assessed using a NanoDrop (Thermo Fisher Scientific). Samples (45 μg protein/lane) were resolved by SDS-PAGE with 7.5% acrylamide along with molecular weight standards (Bio-Rad). The separated proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad) and blocked for 1 h at room temperature with 5% nonfat milk (Carnation) and 0.05% Tween 20 (Bio-Rad) in Tris-buffered saline (TBS) containing 10 mM Tris·HCl and 50 mM NaCl (pH 7.5). Blots were then incubated overnight at 4°C with slight agitation in primary antibody. Antibodies used to probe for NOS isoforms included mouse monoclonal antibodies against eNOS (1:1,000, BD Biosciences 610296, lot 4077705), iNOS (1:1,000, BD Biosciences 610328, lot 5051954), nNOS (1:1,000, BD Biosciences 611852, lot 3224661), and phospho(Ser1177)-eNOS (1:1,000, BD Biosciencess 612392, lot 4164690) in TBS with 0.05% Tween 20 (TTBS) and 5% nonfat milk. Blots were then probed with a horseradish peroxidase-conjugated goat anti-mouse secondary antibody (1:3,000, Bio-Rad 1721011, lot 350002594) in TTBS plus 5% nonfat milk to achieve immunochemical labeling. Protein loading was determined by reprobing blots with β-actin primary antibody (1:14,000, Abcam ab8227, lot GR186254-1) for 1 h at room temperature followed by probing with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:3,000, Bio-Rad 1721019, lot 64026773). Chemiluminescence labeling was performed according to the manufacturer's instructions (ECL substrate, Thermo Scientific). Protein bands were detected using chemiluminescent-sensitive film, and band density was quantified by densitometric analysis using ImageJ (National Institutes of Health). Band density was compared after normalization to each of two protein loading controls: 1) β-actin bands on the same blot and 2) total protein content in the membrane as assessed by Coomassie brilliant blue (Bio-Rad) staining (47, 59).
Nitrotyrosine residues.
Nitrotyrosine levels were additionally detected by Western blot analysis as a measure of ONOO− formation in lungs from both CH and control neonates similar to that previously described (45). Samples (10 μg protein/lane) were prepared with Laemmli sample buffer in the absence of β-mercaptoethanol and resolved by SDS-PAGE with 4–15% gradient acrylamide gels (Bio-Rad). Blots were incubated overnight at 4°C and rocked in primary antibody against nitrotyrosine (1:1,000, Abcam ab7048, lot GR273163-4) in TTBS plus 5% nonfat milk and then probed the following day with horseradish peroxidase-conjugated goat anti-mouse secondary antibody (1:3,000, Bio-Rad 1721011, lot 350002594). Specific nitrotyrosine bands at ~30–35 kDa were identified as those that were undetected in the presence of β-mercaptoethanol as well as a secondary-alone negative control. Protein loading was determined by stripping and reprobing this blot with β-actin primary antibody (1:14,000, Abcam ab8227, lot GR186254-1) for 1 h at room temperature followed by probing with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:3,000, Bio-Rad 1721019, lot 64026773). For a nitrotyrosine positive control, whole lung homogenates were treated for 1 h on ice (covered with aluminum foil) with 1 mM peroxynitrite (Millipore) before Western blot analysis. A standard dilution of protein concentrations revealed detectable nitrotyrosine bands in the 30- to 35-kDa region of interest (not shown).
Calculations and Statistics
Data are expressed as means ± SE; n values of numbers of animals per experimental group. A t-test, one-way ANOVA, or two-way ANOVA was used to make comparisons between groups when appropriate. If differences were detected using ANOVA, individual groups were further evaluated using a Student-Newman-Keuls post hoc comparison. Percent data were arcsine transformed before statistical comparison. P < 0.05 was considered significant.
RESULTS
Evidence for pHTN and RV Hypertrophy in Neonatal CH Rats
CH neonates developed higher RVSP compared with normoxic control rats (Fig. 1, A and B). Heart rate was not significantly different between groups (Table 1). Consistent with elevated RVSP, the ratio of RV to LV + S weight was increased by CH exposure, demonstrating RV hypertrophy indicative of pHTN (Fig. 1C). Similar results were obtained for RV weight normalized to either body weight or total heart weight (Table 1). CH also increased LV + S weight normalized to body mass, which may reflect the lower body weight of CH neonates compared with age-matched controls (Table 1). Hematocrit was greater in rats exposed to CH compared with control rats, reflecting the polycythemic response to CH (Table 1).
Fig. 1.

Chronic hypoxia (CH) increases peak right ventricular (RV) systolic pressure (RVSP) and induces RV hypertrophy in neonatal rats. A and B: sample pressure traces (A) and summary data (B) for peak RVSP in control and CH neonates. C: ratio of RV mass to left ventricular (LV) plus septal (LV + S) mass for each group. Values are means ± SE; n = 4–11/group (indicated in bars). *P < 0.05 vs. control, analyzed by unpaired t-test.
Table 1.
Body weight, heart weight, hematocrit, and heart rate from control and CH neonatal rats
| Control | CH | |
|---|---|---|
| Final body weight, g | 26.2 ± 0.5 (17) | 20.0 ± 0.8* (9) |
| Right ventricle/body weight, mg/g | 1.12 ± 0.05 (11) | 4.40 ± 0.39* (9) |
| Left ventricle + septum/body weight, mg/g | 3.15 ± 0.07 (11) | 5.85 ± 0.25* (9) |
| Right ventricle/total heart weight, mg/mg | 0.26 ± 0.01 (11) | 0.43 ± 0.02* (9) |
| Hematocrit, % | 31 ± 1 (11) | 37 ± 1* (9) |
| Heart rate, beats/min | 255 ± 11 (6) | 301 ± 26 (4) |
Values are means ± SE; n = 4−11 animals/group (shown in parentheses). CH, chronic hypoxia.
P < 0.05 vs. the control group (analyzed by unpaired t-test).
Endogenous NO Limits CH-Dependent Increases in Basal PVR
In situ lungs from neonates exposed to CH displayed elevated total and arterial baseline vascular resistance compared with normoxic controls (Fig. 2, A and B). In contrast, venous resistance was unaltered by CH exposure (Fig. 2C). l-NNA increased total, arterial, and venous baseline PVR in lungs from CH neonates, while having no effect in control pups (Fig. 2).
Fig. 2.
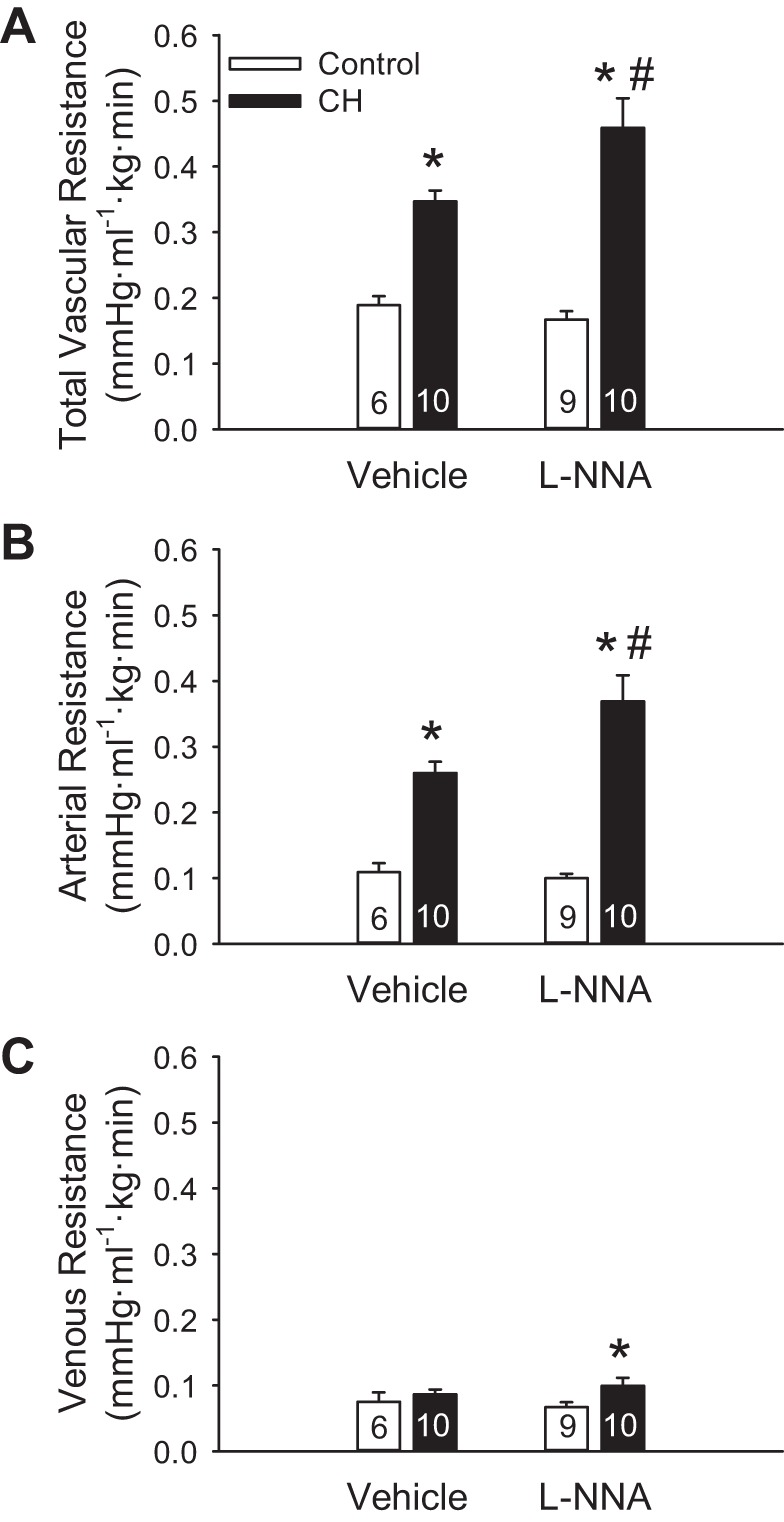
Endogenous nitric oxide (NO) limits chronic hypoxia (CH)-dependent increases in baseline pulmonary vascular resistance. A−C: total (A), arterial (B), and venous (C) baseline vascular resistances in lungs (in situ) from control and CH neonatal rats in the presence or absence of the NO synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (l-NNA; 300 μM). Resistances are in mmHg·ml−1.kg·min. Values are means ± SE; n = 6–10/group (indicated in bars). *P < 0.05 vs. control; #P < 0.05 vs. vehicle, analyzed by two-way ANOVA followed by Student-Newman-Keuls post hoc comparison.
The contribution of basal tone to CH-dependent increases in PVR was assessed by evaluating changes in total and segmental resistances to the NO donor spermine NONOate in the lungs from each group. CH increased basal tone (Fig. 3A) in the absence of NOS inhibition. This effect of CH appeared to result from arterial constriction since changes in arterial resistance to spermine NONOate tended to be greater in lungs from CH rats compared with control rats (Fig. 3B), although this response did not reach statistical significance. NOS inhibition increased basal pulmonary vascular tone in CH rats (Fig. 3A) by further augmenting arterial constriction (Fig. 3B) while having no significant effect in control lungs. In contrast, venous tone was not apparent in lungs from either group of rats whether in the presence or absence of l-NNA (Fig. 3C).
Fig. 3.

Endogenous nitric oxide (NO) limits chronic hypoxia (CH)-dependent increases in basal pulmonary arterial tone. Contributions of basal tone to total (A), arterial (B), and venous (C) resistance are expressed as changes in resistance to spermine NONOate (100 μM) in lungs (in situ) from control and CH neonates. Experiments were conducted in the presence or absence of Nω-nitro-l-arginine (l-NNA; 300 μM). Values are means ± SE; n = 6–10/group (indicated in or above bars). *P < 0.05 vs. control; #P < 0.05 vs. vehicle, analyzed by two-way ANOVA followed by Student-Newman-Keuls post hoc comparison.
Endogenous NO Attenuates U-46619-Induced Pulmonary Vasoconstriction
In addition to the effect of CH increasing basal tone (Fig. 3), total, arterial, and venous constrictor responses to U-46619 were greater in lungs from CH compared with normoxic neonates in the absence of NOS inhibition (Fig. 4). l-NNA markedly augmented vasoconstrictor reactivity to U-46619 in the lungs from CH rats while having a more modest effect in control lungs. Enhanced vasoconstrictor sensitivity to U-46619 after CH was primarily a function of greater arterial constriction (Fig. 4B), as venous responses to U-46619 were minimal (Fig. 4C).
Fig. 4.
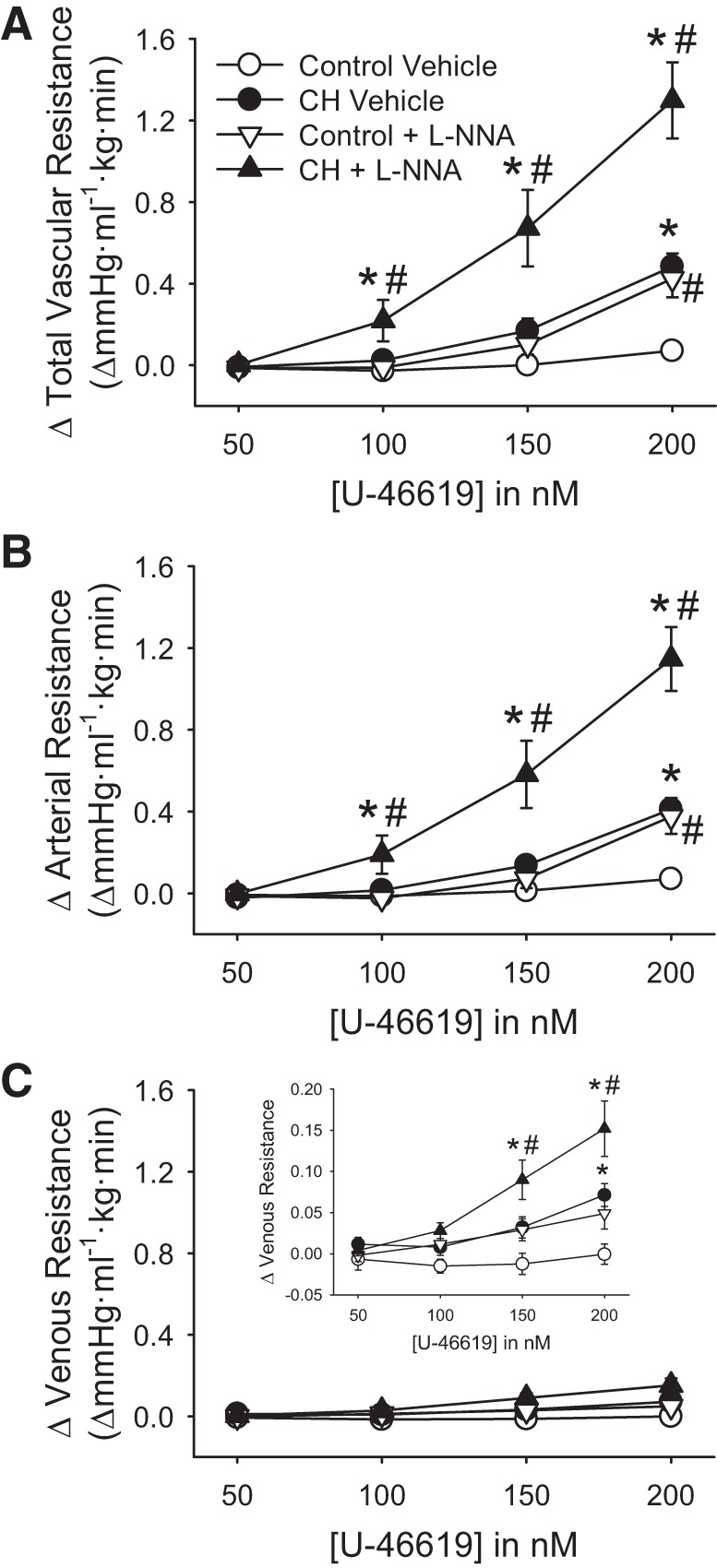
Endogenous nitric oxide (NO) limits chronic hypoxia (CH)-dependent increases in vasoconstrictor sensitivity to U-46619. A−C: changes in total (A), arterial (B), and venous (C) resistance to U-46619 in lungs (in situ) from control and CH neonates. Experiments were conducted in the presence or absence of Nω-nitro-l-arginine (l-NNA; 300 μM). Values are means ± SE; n = 8 for control vehicle, n = 10 for CH vehicle, n = 9 for control l-NNA, and n = 9 for CH l-NNA. *P < 0.05 vs. control; #P < 0.05 vs. vehicle, analyzed by two-way ANOVA at each concentration of U-46619 followed by Student-Newman-Keuls post hoc comparison.
CH Augments EDNO-Dependent Pulmonary Vasodilation
Lungs from control and CH neonates were preconstricted with a concentration of U-46619 sufficient to achieve similar pressor responses between groups (Table 2) and then treated with AVP to dilate the pulmonary circulation. Vasodilatory responses to AVP were greatly augmented after exposure to CH (Fig. 5A). l-NNA nearly abolished AVP-induced pulmonary vasodilation in lungs from each group, demonstrating that this response depends on NOS-derived NO.
Table 2.
Pulmonary vascular resistances in lungs from control and CH neonates treated with AVP in the presence or absence of l-NNA
| Control |
CH |
|||
|---|---|---|---|---|
| Vehicle | l-NNA | Vehicle | l-NNA | |
| Baseline R | 0.18 ± 0.01 (14) | 0.22 ± 0.02 (6) | 0.50 ± 0.03* (6) | 0.70 ± 0.04*† (5) |
| Constricted R | 0.75 ± 0.06 (14) | 0.98 ± 0.13 (6) | 1.35 ± 0.1* (6) | 1.53 ± 0.16* (5) |
| ΔR to U-46619 | 0.57 ± 0.05 (14) | 0.76 ± 0.13 (6) | 0.84 ± 0.1* (6) | 0.83 ± 0.16 (5) |
| ΔR to AVP | −0.17 ± 0.03 (14) | −0.05 ± 0.02† (6) | −0.76 ± 0.08* (6) | −0.08 ± 0.01*† (5) |
Values are means ± SE; n = 5−14 animals/group (shown in parentheses). CH, chronic hypoxia; AVP, arginine vasopressin; l-NNA, Nω-nitro-l-arginine; R, resistance (in mmHg·ml−1·kg·min).
P < 0.05 vs. control;
P < 0.05 vs. vehicle (analyzed by two-way ANOVA; individual groups were further evaluated using a Student-Newman-Keuls post hoc comparison).
Fig. 5.
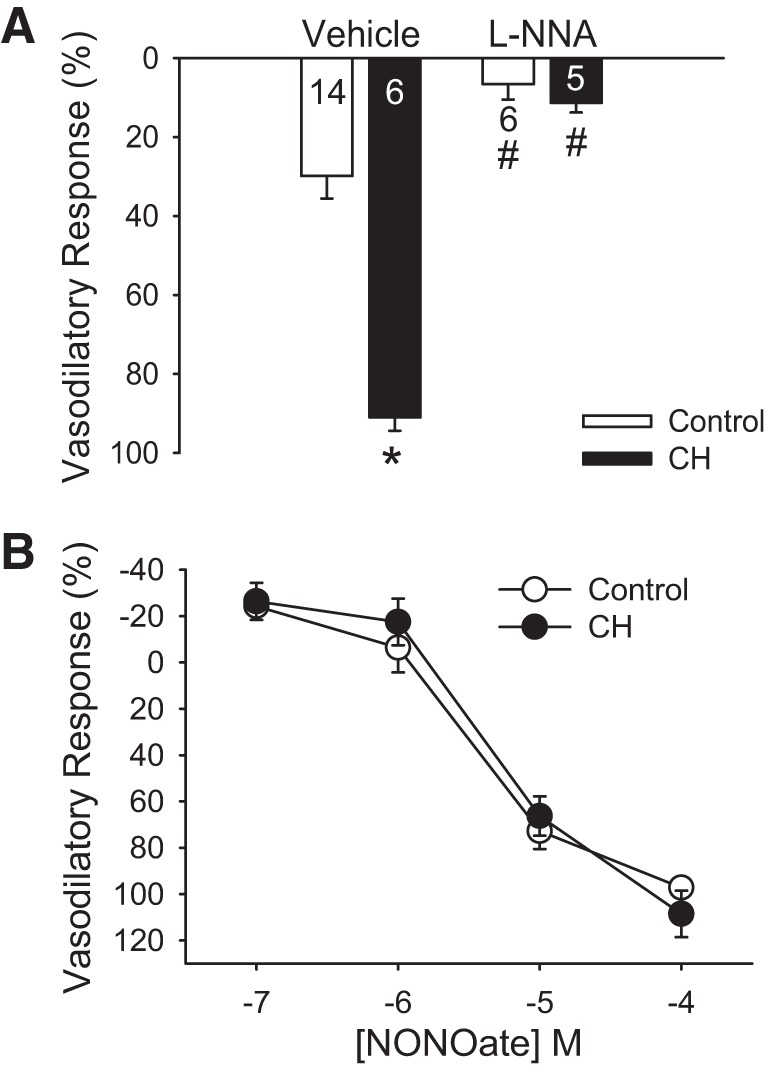
Chronic hypoxia (CH) augments endothelium-derived nitric oxide (EDNO)-dependent vasodilation to arginine vasopressin (AVP) without altering smooth muscle sensitivity to NO. A: total vasodilatory responses (percent reversal of U-46619-induced constriction) to AVP (25 nM) in lungs (in situ) from control and CH neonates. Experiments were conducted in the presence or absence of Nω-nitro-l-arginine (l-NNA; 300 μM). Values are means ± SE; n = 5–14/group (indicated in or below bars). *P < 0.05 vs. control; #P < 0.05 vs. vehicle, analyzed by two-way ANOVA followed by Student-Newman-Keuls post hoc comparison. B: total vasodilatory responses (percent reversal of U-46619-induced constriction) to spermine NONOate. Experiments were conducted in the presence of l-NNA to limit the contribution of endogenous NO production. Values are means ± SE; n = 5 for control and n = 5 for CH. Results were analyzed by two-way ANOVA.
CH Does Not Alter Vasodilatory Responsiveness to Exogenous NO
In contrast to the effect of CH augmenting EDNO-dependent vasoreactivity (Fig. 5A), vasodilatory responses to exogenous NO (spermine NONOate) in l-NNA-pretreated lungs were not different between lungs from control and CH neonatal rats (Fig. 5B). Similar responses to spermine NONOate were observed whether U-46619 was added to achieve similar changes in PVR or total PVR between groups (Table 3).
Table 3.
Pulmonary vascular resistances in lungs from control and CH neonates treated with spermine NONOate
| U-46619 Added to Achieve a Similar Change in Pulmonary Vascular Resistance Between Groups |
U-46619 Added to Achieve a Similar Total Pulmonary Vascular Resistance Between Groups |
|||
|---|---|---|---|---|
| Control | CH | Control | CH | |
| Baseline R | 0.20 ± 0.03 (5) | 0.49 ± 0.04* (5) | 0.2 ± 0.01 (4) | 0.49 ± 0.05* (4) |
| Constricted R | 0.87 ± 0.07 (5) | 1.26 ± 0.09* (5) | 1.13 ± 0.93 (4) | 1.33 ± 0.08 (4) |
| ΔR to U-46619 | 0.67 ± 0.06 (5) | 0.77 ± 0.10 (5) | 0.93 ± 0.08 (4) | 0.84 ± 0.09 (4) |
| ΔR to 10−7 M NONOate | 0.16 ± 0.04 (5) | 0.22 ± 0.09 (5) | 0.25 ± 0.03 (4) | 0.26 ± 0.01 (4) |
| ΔR to 10−6 M NONOate | 0.04 ± 0.11 (5) | 0.16 ± 0.09 (5) | 0.22 ± 0.07 (4) | 0.21 ± 0.10 (4) |
| ΔR to 10−5 M NONOate | −0.53 ± 0.03 (5) | −0.53 ± 0.11 (5) | −0.55 ± 0.06 (4) | −0.63 ± 0.06 (4) |
| ΔR to 10−4 M NONOate | −0.65 ± 0.05 (5) | −0.86 ± 0.14 (5) | −0.88 ± 0.07 (4) | −0.98 ± 0.08 (4) |
Values are means ± SE; n = 4−5 animals/group (shown in parentheses). All experiments were performed in the presence of l-NNA (300 μM). CH, chronic hypoxia; R, resistance (in mmHg·ml−1·kg·min).
P < 0.05 vs. control (analyzed by unpaired t-test).
CH Increases Levels of Pulmonary eNOS as well as Ser1177-Phosphorylated eNOS
Both total eNOS and the ratio of Ser1177-phosphorylated eNOS to total eNOS were greater in lung samples from CH compared with control neonates (Fig. 6) as determined by Western blot analysis. In contrast, CH was without effect on pulmonary nNOS or iNOS levels (nNOS/β-actin: control 1.3 ± 0.4 vs. CH 0.6 ± 0.2, n = 6/group, P = 0.10; and iNOS/β-actin: control 1.6 ± 0.3 vs. CH 1.2 ± 0.4, n = 6/group, P = 0.36). Results were similar whether NOS band density was normalized to β-actin (Fig. 6) or total protein as determined by Coomassie staining (not shown).
Fig. 6.
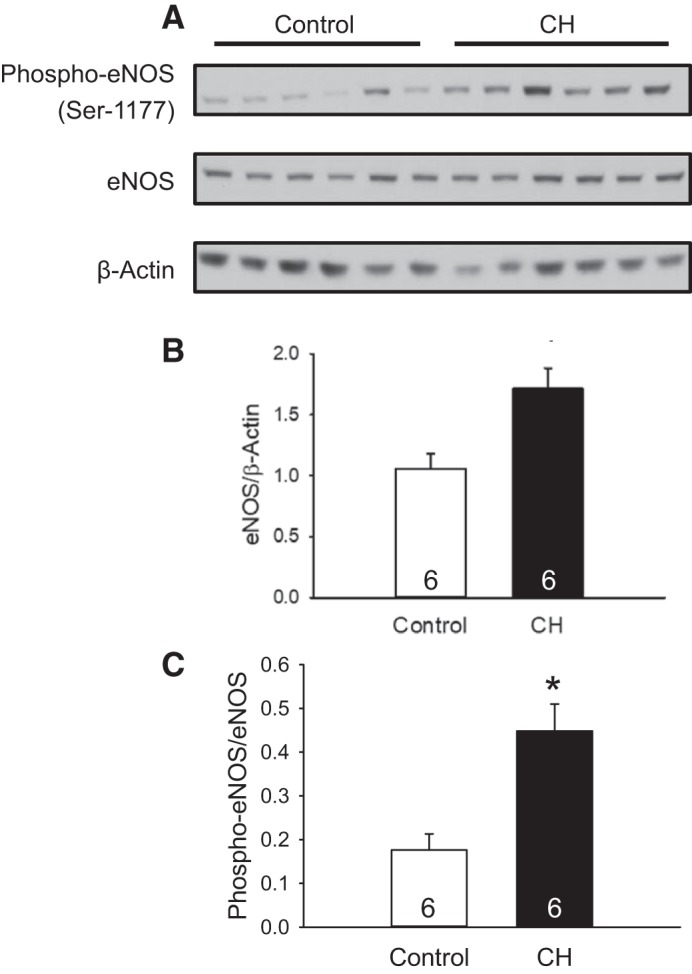
Chronic hypoxia (CH) increases levels of endothelial nitric oxide synthase (eNOS) and phosphorylated eNOS (Ser1177) in neonatal rat lungs. A−C: Western blots for eNOS and phospho-eNOS (A) and summary data for total eNOS (B and C) and the ratio of phosphorylated eNOS to total eNOS in lungs from control and CH neonatal rats. Values are means ± SE; n = 6/group. *P < 0.05 vs. control, analyzed by unpaired t-test.
CH Does Not Alter Lung Nitrotyrosine Levels
Despite the effect of CH augmenting EDNO-dependent pulmonary vasodilation, this response may be limited by the reaction of with NO to form ONOO−. We therefore measured levels of nitrotyrosine residues as an index of ONOO− in whole lung homogenates from control and CH neonates (45). Specific nitrotyrosine bands were identified as a doublet at ~30–35 kDa. No significant differences were detected between groups (Fig. 7).
Fig. 7.
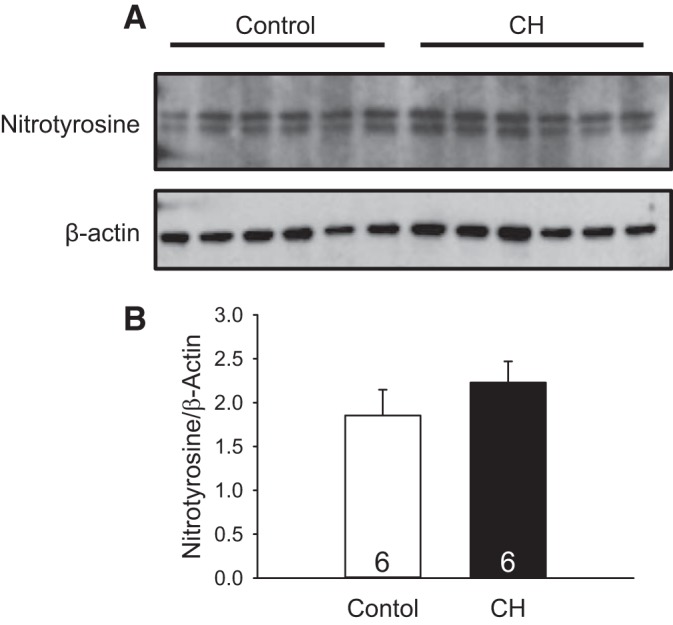
Chronic hypoxia (CH) does not alter levels of nitrotyrosine residues in lungs from neonatal rats. A: nitrotyrosine and β-actin bands in whole lung homogenates from normoxic and CH neonatal rats. B: relative nitrotyrosine protein levels quantified by ImageJ, normalized to β-actin. Values are means ± SE; n = 6/group. Results were analyzed by unpaired t-test.
DISCUSSION
Vasoconstrictor mechanisms play an important role in the development of CH-induced neonatal pHTN (5, 11, 28, 31). However, whether vasoconstriction in this setting is a consequence of diminished endothelium-dependent vasodilatory capacity, enhanced production of a contractile factor, or an inherent increase in VSM contractility is poorly understood. Considering that endothelial dysfunction is implicated in the development of neonatal pHTN (27, 31), the present study examined the hypothesis that neonatal CH enhances basal tone and pulmonary vasoconstrictor sensitivity by limiting NO-dependent pulmonary vasodilation. The major findings from this study are that 1) neonatal CH enhances both basal pulmonary arterial tone and agonist-induced vasoconstrictor sensitivity; 2) endogenous NO limits these responses to CH while having little to no effect in lungs from normoxic control animals; 3) CH augments receptor-mediated, EDNO-dependent pulmonary arterial dilation but is without effect on vasoreactivity to exogenous NO; 4) CH increases pulmonary expression of eNOS as well as eNOS phosphorylation at Ser1177, a posttranslational modification associated with enhanced NO generation (20); and 5) CH was without effect on pulmonary protein tyrosine nitration, a posttranslational modification reflective of NO scavenging by . Collectively, these data demonstrate an effect of CH enhancing both basal pulmonary arterial tone and receptor-mediated vasoconstrictor sensitivity despite a compensatory increase in eNOS expression and activity.
Exposure to CH leads to neonatal pHTN in a variety of animals models (11, 25, 36) by elevating PVR through increased basal arterial tone and augmented reactivity to endogenous vasoconstrictor agonists (5, 11, 56). Direct evidence supporting an important contribution of vasoconstriction to this response includes findings that Rho kinase (ROK) inhibition (41) or inhaled NO (22) acutely reverse PVR in neonatal CH rats. Furthermore, pulmonary vasoconstrictor sensitivity to serotonin is enhanced in newborn lambs housed at high altitude (3,801 m) relative to low-altitude (335 m) lambs in a manner sensitive to ROK inhibition (11). After exposure to chronic normobaric hypoxia in neonatal piglets, endothelium-derived thromboxane products lead to pulmonary arterial vasoconstriction that is absent in normoxic controls (28). In a neonatal lamb model of pHTN that involves banding of the ductus arteriosus, acute increases in pulmonary blood flow lead to vasoconstriction in a manner consistent with a myogenic response (62). Although enhanced pulmonary vasoconstriction in the setting of neonatal pHTN may be due to increased production of and sensitivity to endogenous vasoconstrictor agonists, there is also evidence for diminished production of endothelium-derived relaxing factors (25, 58). However, the relative contributions of diminished endothelium-dependent vasodilation and increased VSM contractile responsiveness to the vasoconstrictor component of neonatal pHTN are not well defined.
One potential mechanism by which neonatal pHTN may occur is through limitation of the normal NO-dependent fall in PVR. During gestation, the fetal pulmonary circulation is marked by high resistance to pulmonary blood flow that is maintained by hypoxic pulmonary vasoconstriction (60). At birth, the increase in O2 tension stimulates pulmonary arterial eNOS-dependent vasodilation that relieves fetal pulmonary arterial tone (1, 29). However, even after delivery, the pulmonary circulation retains residual arterial tone that can be uncovered by eNOS inhibition. For example, in a study (15) using isolated, perfused lungs from neonatal rats, acute administration of l-NNA increased baseline PVR by threefold, with the majority of the response in the pulmonary arterial circulation. Furthermore, occlusion of the ductus arteriosus in neonatal lambs after NOS inhibition leads to an increase in PVR in a manner consistent with a myogenic response (63). Considering the importance of NO-dependent pulmonary vasodilation in mediating the transition from high-resistance fetal to low-resistance antenatal pulmonary circulation, one mechanism by which enhanced vasoconstriction may occur in neonatal pHTN is via reduction in NO-dependent vasodilatory capacity.
Reduced NO-dependent vasodilatory responsiveness in neonatal pHTN has been attributed to both endothelial dysfunction and impaired VSM sensitivity to NO. In neonatal lambs with pHTN induced by banding of the ductus arteriosus, pulmonary eNOS levels are reduced (58). Additionally, in this lamb model of neonatal pHTN, protein expression of phosphodiesterase type 5 (PDE5) in the pulmonary circulation is elevated (10). PDE5 scavenges cGMP, a key second messenger that promotes NO-induced vasodilation (55). Increased protein expression of PDE5 may therefore limit the vasodilatory effect of NO in the setting of neonatal pHTN due to disruption of the NO signaling cascade (10). Furthermore, in a study (49) using chronic catheterization of neonatal lambs with pHTN to measure PVR in vivo, there is diminished endothelium-dependent pulmonary vasodilation to infusions of acetylcholine and ATP. In animal models of CH-induced neonatal pHTN, NO and eNOS levels are also diminished (13, 25, 31). Given evidence for endothelial dysfunction in the setting of neonatal pHTN, the purpose of the present study was to address whether neonatal CH elevates PVR through a similar mechanism related to diminished NO-dependent pulmonary vasodilation.
Our present findings demonstrate that, in contrast to the evidence in the literature of diminished NO-dependent pulmonary vasodilation, CH exposure enhanced EDNO-dependent vasodilation in the pulmonary circulation of newborn rats. In this study, we observed increased basal tone and vasoconstrictor sensitivity to U-46610 in lungs from neonatal CH rats that were greatly enhanced after inhibition of NOS, supporting an effect of endogenous NO limiting CH-dependent vasoconstriction. In addition, the majority of the CH-dependent increases in PVR resulted from arterial constriction as determined by the double occlusion technique. These results suggest that neonatal CH preferentially impacts arterial function, perhaps by increasing arterial smooth muscle contractility due to enhanced Ca2+-dependent mechanisms (62) and/or by enhancing Ca2+ sensitivity via ROK-dependent signaling (41, 65).
Consistent with the effects of NOS inhibition to markedly augment basal tone and agonist-induced arterial constriction after neonatal CH, we found that CH also augments endogenous NO-dependent pulmonary vasodilation to AVP while having no effect on reactivity to the NO donor spermine NONOate. These findings suggest that neonatal CH augments EDNO-dependent pulmonary vasodilation without altering responsiveness to exogenous NO and are consistent with previous observations from our group and others demonstrating that CH selectively increases pulmonary arterial eNOS expression and enhances NO-dependent pulmonary vasodilation in adult rats (39a, 50, 52, 57). Our present results also support studies examining NO-related pathways in human resident multigenerational and first-generation high-altitude populations. For example, NO was found to be elevated in lungs of Tibetan and Andean highlanders compared with a low-altitude reference population (7). Moreover, among Tibetans, higher exhaled NO levels are correlated with increased pulmonary blood flow (32). This finding suggests that pulmonary blood flow is elevated in high-altitude adapted Tibetan populations, a response potentially promoted by NO. Human newborns demonstrate a similar response to high-altitude exposure, as exhaled NO levels are remarkably higher in infants born at altitude Lhasa (3,658-m elevation) compared with those from lower elevations (68). Interestingly, exhaled NO levels in high-altitude-born infants were similar between multigenerational and first-generation high-altitude groups (68), suggesting that greater NO production at altitude represents an acute physiological adaptation. Taken together, these findings are consistent with an effect of CH facilitating NO signaling (52), a response that may improve hypoxic tolerance and limit the development of CH-induced pHTN.
Our study further suggests a potential mechanism by which CH elevates endogenous NO production through CH-dependent increases in protein expression of eNOS as well as levels of phosphorylated eNOS at Ser1177 while having no detectable effect on levels of nNOS or iNOS. The lack of CH-dependent changes in iNOS protein expression is interesting considering that iNOS is a hypoxia-inducible gene (46) that is increased in lungs from adult rats exposed to CH (39b, 51). However, in isolated, pressurized pulmonary arteries from neonatal piglets exposed to either CH or normoxia, selective iNOS inhibition with aminoguanidine had no effect on baseline arterial diameter in arteries from either group, suggesting that iNOS does not play a role in the regulation of baseline tone in the neonatal pulmonary circulation (25).
The mechanism by which CH increases pulmonary eNOS expression and Ser1177 phosphorylation is not clear but may result from increased arterial shear stress associated with CH exposure (64). Ser1177 phosphorylation of eNOS occurs mainly due to activity of the serine/threonine kinase Akt (20). Akt-dependent Ser1177 phosphorylation of eNOS is increased by shear stress on endothelial cells in vitro (48), and shear stress increases eNOS mRNA and protein expression in fetal lambs (9).
Our findings of enhanced endothelium-dependent pulmonary vasodilation and associated increases in eNOS protein expression/Ser1177 phosphorylation differ from previous reports of diminished NO-dependent pulmonary vasodilation in neonatal pHTN (10, 25). Some of this difference could be attributable to study design as well as species difference. Whereas the present study used a CH model of pHTN involving chronic exposure to hypobaric hypoxia, others have studied the effects of chronic exposure to normobaric hypoxia (6, 36) or occlusion of the ductus arteriosus (3, 42). Other reports have described pulmonary endothelial dysfunction in response to chronic hypobaric hypoxia in alternate species, including piglets (25) or lambs (11), suggesting a possible species difference in the neonatal response to hypobaric CH.
The effect of neonatal CH augmenting basal and agonist-induced vasoconstriction after NOS blockade may be explained by an increase in intrinsic VSM contractility or possibly greater sensitivity to endogenous contractile factors. In neonatal piglets exposed to CH, endothelium-derived thromboxane products contribute to pulmonary vasoconstriction (28). Additionally, in neonatal lambs with pHTN, Ca2+ influx through voltage-gated Ca2+ channels facilitates a myogenic response (62). Furthermore, enhanced Ca2+ entry via a store-operated mechanism contributes to vasoconstriction in adult mice with CH-induced pHTN (40, 44, 67). In addition to Ca2+-dependent mechanisms of enhancing pulmonary VSM contractility, ROK-induced myofilament Ca2+ sensitization has been implicated both in the fetal pulmonary myogenic response (65) and in mediating enhanced vasoconstrictor reactivity in neonatal pHTN (11, 41, 69). Similar effects of ROK-dependent Ca2+ sensitization contribute to enhanced VSM contractility and pressure-dependent tone in lungs from adult CH rats (12, 35).
Whereas our present findings indicate that CH augments EDNO-dependent pulmonary vasodilation, this effect of CH may be suppressed by the reaction of with NO to produce ONOO−, thereby limiting NO bioavailability. However, we observed no significant effect of CH to alter lung protein tyrosine nitration indicative of ONOO− production. The lack of effect of CH altering pulmonary vasodilatory responsiveness to exogenous NO is consistent with this observation, as greater ROS-mediated scavenging of NO in this setting would be expected to attenuate reactivity to exogenous NO. Although these findings argue against an effect of CH increasing pulmonary ONOO− levels, they do not preclude the potential involvement of ROS in enhanced vasoconstrictor reactivity after CH through direct contractile effects on VSM.
The levels of ROS resulting from NADPH oxidase (19), xanthine oxidase (33), uncoupled eNOS (38), and mitochondria (3) are elevated in models of neonatal pHTN. ROS signaling has been linked to increases in ROK-dependent Ca2+ sensitization and subsequent potentiation of myofilament contractility (35). Although not directly tested, ROS-induced ROK-dependent Ca2+ sensitization may play a role in mediating enhanced VSM contractility and subsequent augmentation of pulmonary arterial vasoconstrictor sensitivity in the setting of neonatal pHTN.
It is notable that CH caused growth restriction in the present study as evidenced by lower body weight in CH neonates compared with control pups. The effect of CH limiting growth is consistent with previous reports in animal models of CH-induced neonatal pHTN (8, 23, 26, 70) and in humans where newborns delivered at altitude weigh less at birth than those born at sea level (43). This response to CH may result from a direct effect of hypoxia limiting fetal growth (13, 23, 34) or disrupting maternal nursing behavior (13). However, the long-term impacts of CH-induced growth restriction on health outcomes is poorly characterized.
In conclusion, this study demonstrated a previously undescribed effect of neonatal CH augmenting both basal pulmonary arterial tone and receptor-mediated vasoconstrictor sensitivity despite a compensatory increase in NO-mediated pulmonary vasodilation and eNOS expression and activity. These results advance our understanding of protective mechanisms that may limit the severity of neonatal pHTN and are consistent with an effect of CH increasing basal VSM tone and contractile responsiveness rather than to impair endothelium-dependent vasodilation. Future investigation into mechanisms of enhanced vasoconstriction may yield potential avenues for therapeutic management of infants with neonatal pHTN.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants K12-GM-088021 (to A. Wandinger-Ness), F31-HL-131334 (to J. R. Sheak), T32-HL-007736 (to T. C. Resta), R01-HL-132883 (to T. C. Resta), and R01-HL-088192 (to T. C. Resta) and by American Heart Association Grant 16GRNT27700010 (to T. C. Resta).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.R.S., B.R.W., N.L.J., and T.C.R. conceived and designed research; J.R.S., L.W.-C., and R.J.D. performed experiments; J.R.S. and L.W.-C. analyzed data; J.R.S., L.W.-C., B.R.W., N.L.J., and T.C.R. interpreted results of experiments; J.R.S. and L.W.-C. prepared figures; J.R.S. drafted manuscript; J.R.S., L.W.-C., R.J.D., B.R.W., N.L.J., and T.C.R. edited and revised manuscript; J.R.S., L.W.-C., R.J.D., B.R.W., N.L.J., and T.C.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Minerva Murphy (University of New Mexico) for technical support, Dr. Charles Norton (University of Missouri) for assistance in developing the in situ lung preparation, and both Dr. Matthew Campen (University of New Mexico) and Dr. Kevin O’Hair (University of New Mexico) for assistance with the anesthesia protocol in neonatal rats.
REFERENCES
- 1.Abman SH, Chatfield BA, Hall SL, McMurtry IF. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol Heart Circ Physiol 259: H1921–H1927, 1990. [DOI] [PubMed] [Google Scholar]
- 2.Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, Hanna BD, Rosenzweig EB, Raj JU, Cornfield D, Stenmark KR, Steinhorn R, Thébaud B, Fineman JR, Kuehne T, Feinstein JA, Friedberg MK, Earing M, Barst RJ, Keller RL, Kinsella JP, Mullen M, Deterding R, Kulik T, Mallory G, Humpl T, Wessel DL; American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Surgery and Anesthesia; American Thoracic Society . Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation 132: 2037–2099, 2015. doi: 10.1161/CIR.0000000000000329. [DOI] [PubMed] [Google Scholar]
- 3.Afolayan AJ, Eis A, Teng R-J, Bakhutashvili I, Kaul S, Davis JM, Konduri GG. Decreases in manganese superoxide dismutase expression and activity contribute to oxidative stress in persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 303: L870–L879, 2012. doi: 10.1152/ajplung.00098.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen J, Zwerdling R, Ehrenkranz R, Gaultier C, Geggel R, Greenough A, Kleinman R, Klijanowicz A, Martinez F, Ozdemir A, Panitch HB, Nickerson B, Stein MT, Tomezsko J, Van Der Anker J, Van den Anker J; American Thoracic Society . Statement on the care of the child with chronic lung disease of infancy and childhood. Am J Respir Crit Care Med 168: 356–396, 2003. doi: 10.1164/rccm.168.3.356. [DOI] [PubMed] [Google Scholar]
- 5.Allen SW, Chatfield BA, Koppenhafer SA, Schaffer MS, Wolfe RR, Abman SH. Circulating immunoreactive endothelin-1 in children with pulmonary hypertension. Association with acute hypoxic pulmonary vasoreactivity. Am Rev Respir Dis 148: 519–522, 1993. doi: 10.1164/ajrccm/148.2.519. [DOI] [PubMed] [Google Scholar]
- 6.Ambalavanan N, Bulger A, Murphy-Ullrich J, Oparil S, Chen YF. Endothelin-A receptor blockade prevents and partially reverses neonatal hypoxic pulmonary vascular remodeling. Pediatr Res 57: 631–636, 2005. doi: 10.1203/01.PDR.0000159512.55862.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beall CM, Laskowski D, Strohl KP, Soria R, Villena M, Vargas E, Alarcon AM, Gonzales C, Erzurum SC. Pulmonary nitric oxide in mountain dwellers. Nature 414: 411–412, 2001. doi: 10.1038/35106641. [DOI] [PubMed] [Google Scholar]
- 8.Bierer R, Nitta CH, Friedman J, Codianni S, de Frutos S, Dominguez-Bautista JA, Howard TA, Resta TC, Bosc LV. NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. Am J Physiol Lung Cell Mol Physiol 301: L872–L880, 2011. doi: 10.1152/ajplung.00405.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black SM, Johengen MJ, Ma ZD, Bristow J, Soifer SJ. Ventilation and oxygenation induce endothelial nitric oxide synthase gene expression in the lungs of fetal lambs. J Clin Invest 100: 1448–1458, 1997. doi: 10.1172/JCI119665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Black SM, Sanchez LS, Mata-Greenwood E, Bekker JM, Steinhorn RH, Fineman JR. sGC and PDE5 are elevated in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 281: L1051–L1057, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Blood AB, Terry MH, Merritt TA, Papamatheakis DG, Blood Q, Ross JM, Power GG, Longo LD, Wilson SM. Effect of chronic perinatal hypoxia on the role of Rho-kinase in pulmonary artery contraction in newborn lambs. Am J Physiol Regul Integr Comp Physiol 304: R136–R146, 2013. doi: 10.1152/ajpregu.00126.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Broughton BRS, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 294: L797–L806, 2008. doi: 10.1152/ajplung.00253.2007. [DOI] [PubMed] [Google Scholar]
- 13.Chen F, Du S, Bian J, You ZB, Wu Y. Chronic hypoxia exposure during pregnancy is associated with a decreased active nursing activity in mother and an abnormal birth weight and postnatal growth in offspring of rats. Horm Behav 61: 504–511, 2012. doi: 10.1016/j.yhbeh.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Chicoine LG, Avitia JW, Deen C, Nelin LD, Earley S, Walker BR. Developmental differences in pulmonary eNOS expression in response to chronic hypoxia in the rat. J Appl Physiol (1985) 93: 311–318, 2002. doi: 10.1152/japplphysiol.01083.2001. [DOI] [PubMed] [Google Scholar]
- 15.Chicoine LG, Paffett ML, Girton MR, Metropoulus MJ, Joshi MS, Bauer JA, Nelin LD, Resta TC, Walker BR. Maturational changes in the regulation of pulmonary vascular tone by nitric oxide in neonatal rats. Am J Physiol Lung Cell Mol Physiol 293: L1261–L1270, 2007. doi: 10.1152/ajplung.00235.2006. [DOI] [PubMed] [Google Scholar]
- 18.Dawson CA, Linehan JH, Rickaby DA. Pulmonary microcirculatory hemodynamics. Ann N Y Acad Sci 384: 90–106, 1982. doi: 10.1111/j.1749-6632.1982.tb21365.x. [DOI] [PubMed] [Google Scholar]
- 19.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L596–L607, 2009. doi: 10.1152/ajplung.90568.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399: 601–605, 1999. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 21.Eichinger MR, Walker BR. Enhanced pulmonary arterial dilation to arginine vasopressin in chronically hypoxic rats. Am J Physiol Heart Circ Physiol 267: H2413–H2419, 1994. . [DOI] [PubMed] [Google Scholar]
- 22.Enomoto M, Jain A, Pan J, Shifrin Y, Van Vliet T, McNamara PJ, Jankov RP, Belik J. Newborn rat response to single vs. combined cGMP-dependent pulmonary vasodilators. Am J Physiol Lung Cell Mol Physiol 306: L207–L215, 2014. doi: 10.1152/ajplung.00164.2013. [DOI] [PubMed] [Google Scholar]
- 23.Farahani R, Kanaan A, Gavrialov O, Brunnert S, Douglas RM, Morcillo P, Haddad GG. Differential effects of chronic intermittent and chronic constant hypoxia on postnatal growth and development. Pediatr Pulmonol 43: 20–28, 2008. doi: 10.1002/ppul.20729. [DOI] [PubMed] [Google Scholar]
- 24.Farrow KN, Lakshminrusimha S, Reda WJ, Wedgwood S, Czech L, Gugino SF, Davis JM, Russell JA, Steinhorn RH. Superoxide dismutase restores eNOS expression and function in resistance pulmonary arteries from neonatal lambs with persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L979–L987, 2008. doi: 10.1152/ajplung.90238.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fike CD, Aschner JL, Zhang Y, Kaplowitz MR. Impaired NO signaling in small pulmonary arteries of chronically hypoxic newborn piglets. Am J Physiol Lung Cell Mol Physiol 286: L1244–L1254, 2004. doi: 10.1152/ajplung.00345.2003. [DOI] [PubMed] [Google Scholar]
- 26.Fike CD, Kaplowitz MR. Effect of chronic hypoxia on pulmonary vascular pressures in isolated lungs of newborn pigs. J Appl Physiol 77: 2853–2862, 1994. [DOI] [PubMed] [Google Scholar]
- 27.Fike CD, Kaplowitz MR, Thomas CJ, Nelin LD. Chronic hypoxia decreases nitric oxide production and endothelial nitric oxide synthase in newborn pig lungs. Am J Physiol Lung Cell Mol Physiol 274: L517–L526, 1998. [DOI] [PubMed] [Google Scholar]
- 28.Fike CD, Pfister SL, Kaplowitz MR, Madden JA. Cyclooxygenase contracting factors and altered pulmonary vascular responses in chronically hypoxic newborn pigs. J Appl Physiol 92: 67–74, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Gao Y, Raj JU. Regulation of the pulmonary circulation in the fetus and newborn. Physiol Rev 90: 1291–1335, 2010. doi: 10.1152/physrev.00032.2009. [DOI] [PubMed] [Google Scholar]
- 30.Haworth SG, Hislop AA. Lung development-the effects of chronic hypoxia. Semin Neonatol 8: 1–8, 2003. doi: 10.1016/S1084-2756(02)00195-1. [DOI] [PubMed] [Google Scholar]
- 31.Hislop AA, Springall DR, Oliveira H, Pollock JS, Polak JM, Haworth SG. Endothelial nitric oxide synthase in hypoxic newborn porcine pulmonary vessels. Arch Dis Child Fetal Neonatal Ed 77: F16–F22, 1997. doi: 10.1136/fn.77.1.F16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoit BD, Dalton ND, Erzurum SC, Laskowski D, Strohl KP, Beall CM. Nitric oxide and cardiopulmonary hemodynamics in Tibetan highlanders. J Appl Physiol (1985) 99: 1796–1801, 2005. doi: 10.1152/japplphysiol.00205.2005. [DOI] [PubMed] [Google Scholar]
- 33.Jankov RP, Kantores C, Pan J, Belik J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol 294: L233–L245, 2008. doi: 10.1152/ajplung.00166.2007. [DOI] [PubMed] [Google Scholar]
- 34.Jensen GM, Moore LG. The effect of high altitude and other risk factors on birthweight: independent or interactive effects? Am J Public Health 87: 1003–1007, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kantores C, McNamara PJ, Teixeira L, Engelberts D, Murthy P, Kavanagh BP, Jankov RP. Therapeutic hypercapnia prevents chronic hypoxia-induced pulmonary hypertension in the newborn rat. Am J Physiol Lung Cell Mol Physiol 291: L912–L922, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Konduri GG, Bakhutashvili I, Eis A, Afolayan A. Antenatal betamethasone improves postnatal transition in late preterm lambs with persistent pulmonary hypertension of the newborn. Pediatr Res 73: 621–629, 2013. doi: 10.1038/pr.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konduri GG, Ou J, Shi Y, Pritchard KA Jr. Decreased association of HSP90 impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 285: H204–H211, 2003. doi: 10.1152/ajpheart.00837.2002. [DOI] [PubMed] [Google Scholar]
- 39.Lakshminrusimha S, Wiseman D, Black SM, Russell JA, Gugino SF, Oishi P, Steinhorn RH, Fineman JR. The role of nitric oxide synthase-derived reactive oxygen species in the altered relaxation of pulmonary arteries from lambs with increased pulmonary blood flow. Am J Physiol Heart Circ Physiol 293: H1491–H1497, 2007. doi: 10.1152/ajpheart.00185.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39a.Le Cras TD, Tyler RC, Horan MP, Morris KG, Tuder RM, McMurtry IF, Johns RA, Abman SH. Effects of chronic hypoxia and altered hemodynamics on endothelial nitric oxide synthase expression in the adult rat lung. J Clin Invest 101: 795–801, 1998. doi: 10.1172/JCI786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39b.Le Cras TD, Xue C, Rengasamy A, Johns RA. Chronic hypoxia upregulates endothelial and inducible NO synthase gene and protein expression in rat lung. Am J Physiol Lung Cell Mol Physiol 270: L164–L170, 1996. [DOI] [PubMed] [Google Scholar]
- 40.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 41.McNamara PJ, Murthy P, Kantores C, Teixeira L, Engelberts D, van Vliet T, Kavanagh BP, Jankov RP. Acute vasodilator effects of Rho-kinase inhibitors in neonatal rats with pulmonary hypertension unresponsive to nitric oxide. Am J Physiol Lung Cell Mol Physiol 294: L205–L213, 2008. doi: 10.1152/ajplung.00234.2007. [DOI] [PubMed] [Google Scholar]
- 42.McQueston JA, Kinsella JP, Ivy DD, McMurtry IF, Abman SH. Chronic pulmonary hypertension in utero impairs endothelium-dependent vasodilation. Am J Physiol Heart Circ Physiol 268: H288–H294, 1995. [DOI] [PubMed] [Google Scholar]
- 43.Niermeyer S, Andrade-M MP, Vargas E, Moore LG. Neonatal oxygenation, pulmonary hypertension, and evolutionary adaptation to high altitude (2013 Grover Conference series). Pulm Circ 5: 48–62, 2015. doi: 10.1086/679719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR, Jernigan NL. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 306: H41–H52, 2014. doi: 10.1152/ajpheart.00269.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norton CE, Jernigan NL, Kanagy NL, Walker BR, Resta TC. Intermittent hypoxia augments pulmonary vascular smooth muscle reactivity to NO: regulation by reactive oxygen species. J Appl Physiol 111: 980–988, 2011. doi: 10.1152/japplphysiol.01286.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmer LA, Semenza GL, Stoler MH, Johns RA. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am J Physiol Lung Cell Mol Physiol 274: L212–L219, 1998. [DOI] [PubMed] [Google Scholar]
- 47.Plomaritas DR, Herbert LM, Yellowhair TR, Resta TC, Gonzalez Bosc LV, Walker BR, Jernigan NL. Chronic hypoxia limits H2O2-induced inhibition of ASIC1-dependent store-operated calcium entry in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 307: L419–L430, 2014. doi: 10.1152/ajplung.00095.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ranjan V, Xiao Z, Diamond SL. Constitutive NOS expression in cultured endothelial cells is elevated by fluid shear stress. Am J Physiol Heart Circ Physiol 269: H550–H555, 1995. [DOI] [PubMed] [Google Scholar]
- 49.Reddy VM, Wong J, Liddicoat JR, Johengen M, Chang R, Fineman JR. Altered endothelium-dependent responses in lambs with pulmonary hypertension and increased pulmonary blood flow. Am J Physiol Heart Circ Physiol 271: H562–H570, 1996. [DOI] [PubMed] [Google Scholar]
- 50.Resta TC, Gonzales RJ, Dail WG, Sanders TC, Walker BR. Selective upregulation of arterial endothelial nitric oxide synthase in pulmonary hypertension. Am J Physiol Heart Circ Physiol 272: H806–H813, 1997. [DOI] [PubMed] [Google Scholar]
- 51.Resta TC, O’Donaughy TL, Earley S, Chicoine LG, Walker BR. Unaltered vasoconstrictor responsiveness after iNOS inhibition in lungs from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 276: L122–L130, 1999. [DOI] [PubMed] [Google Scholar]
- 52.Resta TC, Walker BR. Chronic hypoxia selectively augments pulmonary arterial vasodilation. Am J Physiol Heart Circ Physiol 270: H888–H896, 1996. [DOI] [PubMed] [Google Scholar]
- 53.Rippe B, Parker JC, Townsley MI, Mortillaro NA, Taylor AE. Segmental vascular resistances and compliances in dog lung. J Appl Physiol 62: 1206–1215, 1987. [DOI] [PubMed] [Google Scholar]
- 54.Russ RD, Walker BR. Maintained endothelium-dependent pulmonary vasodilation following chronic hypoxia in the rat. J Appl Physiol 74: 339–344, 1993. [DOI] [PubMed] [Google Scholar]
- 55.Sanchez LS, de la Monte SM, Filippov G, Jones RC, Zapol WM, Bloch KD. Cyclic-GMP-binding, cyclic-GMP-specific phosphodiesterase (PDE5) gene expression is regulated during rat pulmonary development. Pediatr Res 43: 163–168, 1998. doi: 10.1203/00006450-199802000-00002. [DOI] [PubMed] [Google Scholar]
- 56.Santhosh KT, Sikarwar AS, Hinton M, Chelikani P, Dakshinamurti S. Thromboxane receptor hyper-responsiveness in hypoxic pulmonary hypertension requires serine 324. Br J Pharmacol 171: 676–687, 2014. doi: 10.1111/bph.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaul PW, North AJ, Brannon TS, Ujiie K, Wells LB, Nisen PA, Lowenstein CJ, Snyder SH, Star RA. Prolonged in vivo hypoxia enhances nitric oxide synthase type I and type III gene expression in adult rat lung. Am J Respir Cell Mol Biol 13: 167–174, 1995. doi: 10.1165/ajrcmb.13.2.7542896. [DOI] [PubMed] [Google Scholar]
- 58.Shaul PW, Yuhanna IS, German Z, Chen Z, Steinhorn RH, Morin FC 3rd. Pulmonary endothelial NO synthase gene expression is decreased in fetal lambs with pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 16: L1005–L1012, 1997. [DOI] [PubMed] [Google Scholar]
- 59.Snow JB, Gonzalez Bosc LV, Kanagy NL, Walker BR, Resta TC. Role for PKCβ in enhanced endothelin-1-induced pulmonary vasoconstrictor reactivity following intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol 301: L745–L754, 2011. doi: 10.1152/ajplung.00020.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinhorn RH. Pharmacotherapy for pulmonary hypertension. Pediatr Clin North Am 59: 1129–1146, 2012. doi: 10.1016/j.pcl.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steudel W, Scherrer-Crosbie M, Bloch KD, Weimann J, Huang PL, Jones RC, Picard MH, Zapol WM. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest 101: 2468–2477, 1998. doi: 10.1172/JCI2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Storme L, Parker TA, Kinsella JP, Rairigh RL, Abman SH. Chronic hypertension impairs flow-induced vasodilation and augments the myogenic response in fetal lung. Am J Physiol Lung Cell Mol Physiol 282: L56–L66, 2002. [DOI] [PubMed] [Google Scholar]
- 63.Storme L, Rairigh RL, Parker TA, Kinsella JP, Abman SH. In vivo evidence for a myogenic response in the fetal pulmonary circulation. Pediatr Res 45: 425–431, 1999. doi: 10.1203/00006450-199903000-00022. [DOI] [PubMed] [Google Scholar]
- 64.Su Z, Tan W, Shandas R, Hunter KS. Influence of distal resistance and proximal stiffness on hemodynamics and RV afterload in progression and treatments of pulmonary hypertension: A computational study with validation using animal models. Comput Math Methods Med 2013: 618326, 2013. doi: 10.1155/2013/618326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tourneux P, Chester M, Grover T, Abman SH. Fasudil inhibits the myogenic response in the fetal pulmonary circulation. Am J Physiol Heart Circ Physiol 295: H1505–H1513, 2008. doi: 10.1152/ajpheart.00490.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Townsley MI, Korthuis RJ, Rippe B, Parker JC, Taylor AE. Validation of double vascular occlusion method for Pc,i in lung and skeletal muscle. J Appl Physiol 61: 127–132, 1986. [DOI] [PubMed] [Google Scholar]
- 67.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 68.Wu P, Shanminna, Liang K, Yue H, Qian L, Sun B. Exhaled nitric oxide is associated with postnatal adaptation to hypoxia in Tibetan and non-Tibetan newborn infants. Acta Paediatr 105: 475–482, 2016. doi: 10.1111/apa.13331. [DOI] [PubMed] [Google Scholar]
- 69.Xu EZ, Kantores C, Ivanovska J, Engelberts D, Kavanagh BP, McNamara PJ, Jankov RP. Rescue treatment with a Rho-kinase inhibitor normalizes right ventricular function and reverses remodeling in juvenile rats with chronic pulmonary hypertension. Am J Physiol Heart Circ Physiol 299: H1854–H1864, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ziino AJ, Ivanovska J, Belcastro R, Kantores C, Xu EZ, Lau M, McNamara PJ, Tanswell AK, Jankov RP. Effects of rho-kinase inhibition on pulmonary hypertension, lung growth, and structure in neonatal rats chronically exposed to hypoxia. Pediatr Res 67: 177–182, 2017. [DOI] [PubMed] [Google Scholar]