

Abstract

The efficient integration of photoactive and catalytic materials is key to promoting photoelectrochemical water splitting as a sustainable energy technology built on solar power. Here, we report highly stable water splitting photoanodes from BiVO4 photoactive cores decorated with CoFe Prussian blue-type electrocatalysts (CoFe-PB). This combination decreases the onset potential of BiVO4 by ∼0.8 V (down to 0.3 V vs reversible hydrogen electrode (RHE)) and increases the photovoltage by 0.45 V. The presence of the catalyst also leads to a remarkable 6-fold enhancement of the photocurrent at 1.23 V versus RHE, while keeping the light-harvesting ability of BiVO4. Structural and mechanistic studies indicate that CoFe-PB effectively acts as a true catalyst on BiVO4. This mechanism, stemming from the adequate alignment of the energy levels, as showed by density functional theory calculations, allows CoFe-PB to outperform all previous catalyst/BiVO4 junctions and, in addition, leads to noteworthy long-term stability. A bare 10–15% decrease in photocurrent was observed after more than 50 h of operation under light irradiation.
Keywords: photoelectrocatalysis, water splitting, oxygen evolution, electrochemistry, computational chemistry, Prussian blue
Introduction
The production of solar fuels (by converting the energy of solar photons into chemical bonds) stands as a promising technology to power the planet with sunlight. This approach elegantly overcomes the problems related to the intrinsically intermittent solar flux, by transforming the solar energy into compounds, which can be stored, transported, and used upon demand.1 In this context, photoelectrochemical (PEC) water splitting offers a reliable strategy to generate solar H2, which can directly power fuel cells. Alternatively, H2 can be combined with CO2 for the synthesis of complex hydrocarbons, leading to C-neutral energy schemes.2 From a technoeconomical perspective, the key performance indicators, to consider the technology competitive, can be summarized as follows: (i) solar-to-hydrogen (STH) efficiency of 10%, (ii) durability of 10 years, and (iii) cost of $2–4/kg of dispensed hydrogen.3
These stringent requirements impose the use of Earth-abundant materials and low-cost synthetic procedures for the fabrication of photoelectrochemical (PEC) devices. In this context, n-type metal oxide semiconductor materials (TiO2,4,5 Fe2O3,6−9 WO3,10−63 BiVO4,13−19 etc.) have been extensively studied as promising candidates for the development of the technology due to their relatively good stability under operation in harsh environments. From this family, BiVO4 holds the record of performance with 8.1% solar-to-hydrogen (STH) efficiency, when combined with a double-junction GaAs/InGaAsP photovoltaic device.21 In tandem with a single perovskite solar cell, unassisted water splitting with a solar-to-hydrogen conversion efficiency of up to 6.2% for more than 10 h has been recently demonstrated.22 Because there are some concerns on the relative abundance of bismuth in the Earth crust,23 different compositional modifications have been explored to achieve a competitive metal vanadate based on this system.24−26 In all of these arrangements, a water oxidation catalyst (WOC) is deposited on top of the photoactive semiconductor material to overcome the thermodynamic and kinetic barriers of the sluggish water oxidation reaction at the BiVO4 surface, boosting the performance of the photoanode. Consequently, the deposition of an efficient, stable, and cost-effective WOC on the photoactive semiconductor material is key to achieving the targeted technoeconomical requirements.
Moreover, the state of the art in heterogeneous catalysis for oxygen evolution is dominated by noble metals (Ir or Ru), but their high price and scarcity preclude large technological impact. Abundant and inexpensive oxides of first-row transition metals are also competent WOCs, although exclusively at a very high pH or with the help of ancillary electrolytes (i.e., phosphates).27,28 Alternatively, Prussian blue (PB)-type materials have emerged as promising catalysts for water oxidation catalysis.29−31 These coordination polymers are (i) easy to obtain and to process by soft chemistry methods; (ii) available as nanoparticles or thin films; (iii) and stable and active in a very large pH range, from neutral down to extremely acidic conditions;32 (iv) while being non-toxic. The unique performance of these catalysts is based on their structural and electronic features. They are built from hexacyanometallate anionic complexes and a metal dication, in stoichiometric excess. These coordination networks are very robust due to the strength of cyanide bridging, with a rather high covalent character. Their structure is built following an ideal face-centered cubic (FCC) network (Scheme 1), but their nonstoichiometric nature creates multiple voids for solvent (water) and countercations (if needed). Typically, the dication is in excess and coordinatively unsaturated due to missing [M(CN)6]n− moieties. This opens two to three accessible coordination sites for solvent molecules, where water oxidation catalysis is supposedly taking place. At the same time, these semiconductor materials possess versatile redox properties, allowing the metals to be in multiple oxidation states.33 Their low absorption in the UV–vis region also makes them very good candidates to be combined with chromophores for a photocatalytic process. Indeed, light-driven water oxidation was already proven in suspension of a Co-containing PB material, using a Ru chromophore and a sacrificial electron acceptor.34
Scheme 1. Representation of the Ideal Face-Centered Cubic Structure of the CoFe-PB Prussian Blue-Type Catalyst.

In the present study, we have been able to successfully combine the unique properties of these two materials, producing a BiVO4 photoanode coupled to a Prussian blue-type WOC (CoFe-PB/BiVO4). This heterostructure appears to be superior to previous examples, with a significant and robust photoelectrocatalytic performance, arising from the optimum matching of the photoactive BiVO4 core and the PB system, which does not alter the capacitive properties of BiVO4. This is distinct from other metal oxide WOCs on semiconductor surfaces, which appear to work mainly as charge accumulators.35,36 Furthermore, our results are supported with state-of-the-art theoretical density functional theory (DFT) calculations that demonstrate the correct alignment of the electronic levels between the different photocatalytic units. Besides the remarkable increase of performance, the CoFe-PB/BiVO4 system exhibits outstanding stability thanks to the intimate interfacial connectivity achieved via a soft wet processing method.
Experimental Section
Materials
Chemicals bismuth(III) nitrate (Bi(NO3)3·5H2O ≥ 98.0%), vanadyl acetylacetonate (VO(acac)2 ≥ 97.0%), potassium ferri(III)cyanide (K3[Fe(CN)6] ≥ 99.0%), and potassium hydroxide (KOH flakes ≥ 90%) were purchased from Sigma-Aldrich and cobalt chloride hexahydrate (CoCl2·6H2O ≥ 98.0%) from Fluka Analytical. Solvents dimethylsulfoxide (DMSO ≥ 99.9%) and ethylene glycol (ethane-1,2-diol ≥ 99%) were obtained from Sigma-Aldrich. The buffer solution was prepared from potassium phosphate monobasic and dibasic (KH2PO4 ≥ 99.0% and K2HPO4 ≥ 98.0%, Sigma-Aldrich). As a hole scavenger, Na2SO3 from J.T. Baker was used. High-purity (milliQ) water was obtained with a millipore purification system (Synergy) and used for all solutions. Fluorine-doped tin oxide (FTO)-coated glass slides were purchased from Hartford glass (15 Ω/cm2).
Synthesis of BiVO4 Electrodes
Thin-film BiVO4 electrodes were prepared following a simple and cost-efficient electrodeposition method, described by Kang et al.64 Prior to deposition, fluorine-doped tin oxide (FTO) electrodes were ultrasonicated and then thoroughly cleaned with water and ethanol (isopropanol). Metallic Bi was deposited from a solution of 20 mM Bi(NO3)3·5H2O in ethylene glycol by applying a repetitive sequence of passing 0.04 C/cm2 at a potential of −1.8 V versus Ag/AgCl (3 M KCl), followed by a resting time of 2 s eight times. Thus, a total charge of 0.32 C/cm2 was passed. After carefully rinsing the films with milliQ water, 60 μL/cm2 of 0.15 M VO(acac)2 in DMSO was dropped on the Bi surface and heated up to about 80 °C on a hot plate to evaporate the DMSO. The electrodes were calcined in air by heating them with a rate of 2 °C/min up to 500 °C and then at 500 °C for 2 h. During heating, metallic Bi and VO2+ oxidize and form BiVO4. Excess V2O5, which is formed during the calcination process, was removed by leaching the electrodes in 1 M KOH for 20–30 min under vigorous stirring.
Sequential CoFe-PB Coating
The catalyst was deposited by sequentially dipping the BiVO4 electrodes in reactant solutions of 0.02 M K3[Fe(CN)6] in H2O and 0.04 M CoCl2 in H2O. First, the electrodes were dipped in a [Fe(CN)6]3– solution for 10–15 min under slow stirring, so that the negatively charged iron cyanide complexes can bind to the BiVO4 surface. Afterwards, the electrodes were thoroughly rinsed with milliQ water and then dipped in the Co2+ solution, again for 10–15 min under stirring, to form CoFe-PB complex structures. The sequence was repeated at least four times (four to eight times) to reach optimum photoelectrocatalytic enhancement.
Photoelectrochemical (PEC) Measurements
PEC experiments were performed with an Eco Chemie Autolab potentiostat coupled with the NOVA electrochemical software. A typical three-electrode cell consisted of the BiVO4 photoanode as the working electrode, a Pt-wire or mesh as the counter electrode, and a Ag/AgCl (3 M KCl) reference electrode. All potentials were converted to the pH-independent reversible hydrogen electrode (RHE) by using the Nernst equation (eq 1)
| 1 |
To normalize the measured current (in ampere, A) to current density j (in mA/cm2), the electrode geometrical areas were determined by the graphical software ImageJ 1.50i. If not stated otherwise, the experiments were performed in a 0.1 M solution of potassium phosphate (KH2PO4) buffer at pH = 7(±0.1). The pH was determined with a CRISON Basic 2° pH meter. Some experiments were carried out in a 1 M Na2SO3 solution in buffer (pH = 8), which served as a hole scavenger. A 450 W Xe arc lamp with an AM 1.5 solar filter (Sciencetech Inc.) was used to simulate sunlight of 100 mW/cm2 (1 sun). Cyclic voltammetry (CV) was performed at a scan rate of 50 mV/s, and for chopped light experiments, the shutter frequency was 1 Hz. Because it takes typically 2–4 CVs to reach a stable signal, all experiments were generally repeated five times and the fifth scan is presented here. All BiVO4 electrodes were illuminated through the FTO substrate and not, as it is usual for hematite, from the electrolyte. This ensures a small mean free path for excited electrons because conduction in BiVO4 is well known to be limited by its small electron diffusion length (around 70–100 nm)65,66 in contrast to that of hematite, where it is limited by its small hole diffusion length of 2–4 nm.67−69 It is to be noted that the shown CVs were systematically taken after stabilization of the system. Impedance data were collected between 10–2 and 106 Hz using a 20 mV amplitude voltage perturbation and analyzed with ZView software (Scribner associates). Incident photon-to-current conversion efficiencies (IPCEs) were obtained by collecting the photocurrent recorded under monochromatic light irradiation (Newport spectrometer) using the same three-electrode setup as described above. IPCE, as a function of wavelength, is given by the ratio of the measured photocurrent, jphoto, to the incident monochromatic light intensity, Pmono
| 2 |
with 1239.87/λ as the wavelength-to-electronvolt conversion factor.
Structural and Optical Characterization
Morphologies, particle sizes, and chemical compositions were determined by scanning electron microscopy (SEM) with a JSM-7000F JEOL FEG-SEM system (Tokyo, Japan) equipped with an INCA 400 Oxford EDS analyzer (Oxford, U.K.) operating at 15 kV and a JEM-2100 JEOL transmission electron microscope (TEM) operating at 200 kV. Prior to the SEM experiment, the samples were sputtered with a 2 nm thick layer of Pt. To evaluate the effect of aging on the microstructural features of the samples, they were irradiated at 100 mW/cm2 at 1.23 V versus RHE for 1 h. X-ray photoelectron spectroscopy (XPS) was used to analyze the surface and possible changes of electronic coordination before and after the electrochemical treatment. The analyses were performed with a Specs SAGE 150 instrument using nonmonochrome Al Kα irradiation (1486.6 eV) at 20 mA and 13 kV, a constant energy pass of 75 eV for overall analysis and 30 eV for analysis in the specific binding energy ranges of each element, and a measurement area of 1 × 1 mm2. The pressure in the analysis chamber was 8 × 10–9 hPa. The data were evaluated using Casa XPS software. The energy corrections of the spectra were performed considering a reference value of C 1s from the organic matter at 284.8 eV. UV–vis spectra of the electrodes were recorded with a Cary 300 Bio spectrometer (UV0911 M213). Infrared absorption spectroscopy was performed with a Thermo-Scientific NICOLET iS50 Fourier transform infrared (FT-IR) spectrometer. For the measurement, the substrate (<1 mg) was scratched from several electrodes, finely ground with large excess of KBr, and pressed to obtain a thin transparent disk. Electrolyte solutions were analyzed for trace metals using inductively coupled plasma optical emission spectrometry (ICP-OES).
Oxygen Detection
The faradaic efficiency of the photoanodes was calculated during a chronoamperometric measurement at constant potential (1.23 V vs RHE) and under illumination (100 mW/cm2), where total oxygen evolution was determined by gas chromatography (GC). An Agilent Technologies 490 Micro GC device was connected to the electrochemical cell, which was constantly purged with Ar. After a first blank measurement (without applying any voltage and in the dark), periodic measurements of the O2 content were repeated every 5 min before and after switching on the light. The detected O2 amount is correlated to the measured photocurrent by its faradaic efficiency (FE) (more details in the Supporting Information)
| 3 |
Computational Details
Density functional theory (DFT) calculations were carried out using the Vienna ab initio simulation package.70,71 Pure density functional theory is insufficient in correctly describing the complex electronic structure of Prussian blue-type materials. In contrast, hybrid functionals, which include 20–25% of exact exchange from Hartree–Fock (HF) theory, often overcorrect the DFT-inherent self-interaction error and yield exceedingly large band splittings.72 Therefore, the required amount of exact exchange was optimized to be 13% for the hybrid functional HSE03, by fitting the optical band gap, and will be referred to as HSE03-13 in the following text. HSE03-13 gives a good description of the electronic structure for both CoFe-PB and BiVO4 (see Computational Modeling in Supporting Information). Projector augmented waves (PAWs) with small cores, expanding valence-subshell s- and p-electrons, ensure accuracy and were used for all metal atoms in the lattice.73 The valence electrons were expanded in plane waves with kinetic energies up to 500 eV. The Brillouin zone was sampled using a Monkhorst–Pack k-point mesh with 3 × 3 × 3 k-points for CoFe-PB and 6 × 3 × 4 k-points for BiVO4, ensuring similar k-point samplings in both compounds. Water was calculated with the same scheme, and the water solvent was represented through the MGCM method (see the Supporting Information for more details). ). All structures and calculations have been uploaded to the ioChem-BD database (http://www.iochem-bd.org), where they are openly accessible.
Results and Discussion
The nanostructured BiVO4 films were modified with a cobalt hexacyanoferrate (CoFe-PB) catalyst by a sequential coating method, which has shown to be advantageous over other deposition methods (see, Supporting Information, Figure S1, for details). Mild deposition conditions were employed (aqueous solution at pH 7, room temperature, and no applied potential) to ensure that the underlying photoanode is not damaged. The optimum photoelectrochemical performance was obtained after four dipping cycles (Figure S2). Higher catalyst loading did not further improve photocurrent. This ensures a very thin deposited layer, favoring fast charge and mass transport through the catalyst.
Figure 1a,b shows zenithal SEM images of the nanostructured BiVO4 film with and without the CoFe-PB catalyst, respectively. Figure 1c shows the cross section of the modified BiVO4 photoanode, with a thickness of about 200–250 nm. The electrochemical treatment (100 mW/cm2 irradiation at an applied bias of 1.23 V vs RHE for 1 h), which was done to detect the effects of aging, did not alter the morphology significantly (Figure S3).
Figure 1.

Electron micrographs obtained by SEM (2a–d) and TEM (2e–i): SEM images (25000× magnified) of (a) bare and (b) CoFe-PB-modified BiVO4 surfaces and (c) cross section of CoFe-PB/BiVO4 with (d) large (250 000×) magnification of one surface particle. High-resolution (HR)-TEM of scratched CoFe-PB/BiVO4 particles before (e, f, h) and after (g, i) photoelectrochemical treatment (100 mW/cm2 for 1 h at 1.23 V vs RHE). Amorphous CoFe-PB particles on the BiVO4 surfaces before (f) and after (i) photoelectrochemical treatment (2 h) showing the nanocrystalline BiVO4 structure at the surface.
Energy-dispersive spectrometry (EDS) microanalysis confirms the presence of the CoFe-PB catalyst showing about 1–2% of Fe and Co on the electrode surface. (Figures S3 and S4). High resolution transmission electron microscopy (HR-TEM) shows the presence of <50 nm nanoparticles on the surface (Figure 1e–i). The Co/Fe ratio is not homogeneous, varying between 1:1 and 3:2 (Figure S5), corresponding to the two limiting compositions KCo[Fe(CN)6] and Co3[Fe(CN)6]2.37 The measured lattice spacings 2.8 and 3.1 Å of BiVO4 are consistent with the (1̅21) and (040) planes of the monoclinic scheelite structure, respectively (Figure S6).38 No lattice fringes from the CoFe-PB layer were detected, probably due to a lack of long-range order, along with its very small size (Figure 1e–i). More detailed surface characterization was carried out by X-ray photoelectron spectroscopy (XPS) on different BiVO4 electrodes (bare and CoFe-PB-modified; see Table S1 and Figures S7–S11). Quantitative analysis confirms the presence of Co and Fe on the surface of the electrodes with a higher amount of Co. Moreover, an excess of Bi with an average Bi/V ratio of 1.6 was found, in agreement with EDS/TEM data and recent mechanistic studies, which detected structural destabilization and chemical attack via vanadium loss into solution upon hole accumulation at the BiVO4 surface.39 XPS of CoFe-PB/BiVO4 reveals a predominant Fe 2p peak at a binding energy of 708.3–708.4 eV, which can be attributed to Fe(II) as in [Fe(CN)6]4–.40 Co binding energies cannot be unambiguously deconvoluted. However, an obvious peak maximum at around 780–781 eV is attributed to Co(III) in the structure. This means that upon CoFe-PB formation, starting reagents [Fe(CN)6]3– and Co2+(aq) undergo (partial) electron transfer to yield a majority of Fe2+ and Co3+ centers in the fresh electrodes (Table S1 and Figures S7–S11). XPS analysis was also carried out after photoelectrochemical treatment (1 h at 1.23 V vs RHE under 1 sun irradiation). No significant changes were detected for the Fe and Co centers, confirming the redox stability of the CoFe-PB material under water oxidation conditions.
Additional evidence on the formation of a PB solid structure on the surface of the BiVO4 electrodes comes from infrared spectroscopy (FT-IR). The spectra (Figure S12) clearly show the characteristic and unique C–N stretching mode at frequencies in the 2070–2150 cm–1 range. The multiple bands also confirm the presence of both metals in multiple oxidation states, typical of these nonstoichiometric solids.
Cyclic voltammetry (CV) using CoFe-PB/BiVO4 photoanodes was carried out under chopped (Figure 2a) and constant (Figure 2b) illumination (100 mW/cm2) in a neutral (pH 7) KPi buffer (0.1 M) solution. The photocurrent density is significantly improved when CoFe-PB is present, particularly at the low-voltage region, accompanied by a large cathodic shift of the onset potential. The transient cathodic dark current observed in the CoFe-PB/BiVO4 sample in Figure 2a originates from back-reduction of oxidized Co centers by electrons from the BiVO4 conduction band, after turning off the light. A similar phenomenon was previously reported for CoPi-modified hematite photoanodes.36 The onset potentials were determined from quasi steady-state j–V curves obtained at 1 mV/s (Figure 2b). The voltage needed to attain 0.1 mA/cm2 photocurrent was taken as the onset potential (Von).41 The obtained values were 0.3 V versus RHE for CoFe-PB/BiVO4 and 1.1 V versus RHE for bare BiVO4, reflecting a 0.8 V gain. This is directly connected to the estimated photovoltage from open circuit measurements in the dark and under illumination. The photovoltage increases from 0.15 V for bare BiVO4 to 0.60 V upon deposition of CoFe-PB (Figure S13). This behavior has been previously ascribed to passivation of surface states or release of Fermi-level pinning at the semiconductor–liquid junction.6,42
Figure 2.

Anodic CV scans of CoFe-PB/BiVO4 (red) compared to those of bare BiVO4 (black) under (a) chopped (νshutter = 1 Hz) and (b) constant light irradiation (100 mW/cm2) at scan rates of 50 mV/s (solid lines) and 1 mV/s (dashed lines) in 0.1 M KPi buffer (pH 7). Incident photon-to-current conversion efficiency (IPCE) obtained in the (c) buffer (pH 7) and (d) hole scavenger (pH 7.9) at 1.23 V vs RHE for bare BiVO4 (black circles) and CoFe-PB-modified BiVO4 (red triangles). Chronoamperometric (CA) measurements of a CoFe-PB-coated BiVO4 photoanode at 1.23 V vs RHE under (e) constant and (f) chopped (νshutter = 0.03 Hz) irradiation (100 mW/cm2) in 0.1 M KPi buffer (pH 7). Different colors indicate different measurements of the same CoFe-PB electrode at different days.
The spectral signature of the photocurrent, characterized by the incident photon-to-current conversion efficiency (IPCE), was obtained for both pristine and CoFe-PB-decorated BiVO4 photoelectrodes (Figure 2c). The 6-fold enhancement of the photocurrent observed between 350 and 450 nm is fully consistent with the results obtained by cyclic voltammetry. Indeed, the integrated photocurrents (0.38 mA/cm2 for BiVO4 and 0.92 mA/cm2 for CoFe-PB/BiVO4) perfectly match those obtained at 1.23 V versus RHE by cyclic voltammetry (0.40 and 0.95 mA/cm2, respectively). Moreover, the full spectral absorption range of BiVO4 is not affected by the presence of the CoFe-PB catalyst. This should be related to the very low catalyst coverage (1−2%). Thus, the CoFe-PB absorption band at 520–550 nm is not fully developed and does not interfere. Indeed, it is not even detected (Figure S14). The calculated BiVO4 band gap, between 2.40 and 2.45 eV, is not affected by the catalyst either, and it is in good agreement with other reports.43 On the other hand, IPCE measurements of CoFe-PB/BiVO4 in the presence of a hole scavenger (1 M Na2SO3) (Figure 2d) show almost identical values compared to those for water oxidation (in buffer), suggesting a 100% faradaic efficiency for the CoFe-PB catalyst.
The long-term stability of the CoFe-PB/BiVO4 photoanodes was assessed by chronoamperometric measurements at 1.23 V versus RHE under constant (Figure 2e) and chopped (Figure 2f) illumination (100 mW/cm2). After an initial transient decrease of the photocurrent during 1 h, the photoanodes show a remarkable stability, with a mere decrease of about 10% current density over a time scale of more than 50 h. Remarkably, an analogous decrease is observed for bare BiVO4 photoanodes. Thus, we can assign this decay to BiVO4 deactivation39 and not to catalytic fatigue. Additionally, no catalyst leaching was detected by ion plasma chromatography (IPC) analysis of the supernatant buffer solutions after water splitting, suggesting excellent catalytic stability for this system (Tables S2 and S3), in agreement with previous studies.29,32
The high ex situ stability of this catalyst is worth mentioning. The photoelectrodes can be stored and dried in air, for several days, while retaining their photocatalytic activity, as confirmed by subsequent measurements (Figure 2f). This outstanding stability in air constitutes a clear advantage over the widely known cobalt oxide (CoPi or CoOx) systems, which easily crack upon drying, leading to fatal catalytic loss (Figure S15).36,44
To benchmark the performance of the CoFe-PB catalyst under the same experimental conditions, CoOx and FeOOH (as related WOCs containing Fe or Co) were deposited on top of our nanostructured BiVO4 films (see Supporting Information for details). The photoelectrochemical behavior of CoFe-PB clearly outperforms both catalysts (Figure 3a). Because deposition conditions were not specifically optimized, we cannot quantitatively use these data to sustain the overall superior catalytic activity. However, it is clear that CoFe-PB is at least photoelectrocatalytically competitive, with the additional advantages described above.
Figure 3.

(a) Cyclic voltammetry curves under illumination (100 mW/cm2) for bare BiVO4 (black) and coated with different state-of-the-art catalysts: CoFe-PB (red), CoOx prepared by dip-coating (green) and electrodeposition (yellow),45 and FeOOH (blue).46 (b) Theoretical (black line) and measured (red triangles) O2 evolution, as well as faradaic efficiency (black circles), of the CoFe-PB/BiVO4 photoanode during chronoamperometry (CA) at 1.23 V vs RHE under 100 mW/cm2 irradiation in 0.1 M KPi buffer (pH 7).
The CoFe-PB/BiVO4 photoelectrochemical behavior was also characterized in the presence of an efficient hole scavenger. Under these conditions, it is assumed that no electron–hole recombination takes place at the semiconductor–liquid interface.45,47,48 The experiments were carried out in a 1 M sodium sulfite (Na2SO3) solution (pH = 7.9), identified as the optimum concentration of the hole scavenger in solution (see Supporting Information, Figure S16, for details). Figure 4a shows the photocurrent densities obtained in both, buffer solution and 1 M Na2SO3 for bare and Co-Fe-PB/BiVO4 anodes. In good agreement with previous studies, a large difference between the obtained photocurrents with and without the hole scavenger is observed for pristine BiVO4, highlighting its poor water oxidation kinetics.44 Indeed, BiVO4 yields only <15% charge transfer efficiency even at high anodic potentials (>1.4 V vs RHE), where the large electric field precludes surface recombination (Figure 4b). Upon deposition of the CoFe-PB catalyst, the difference between the obtained photocurrents with and without the hole scavenger is significantly narrowed, indicating enhanced charge transfer kinetics. Nonetheless, both curves do not overlap, suggesting that further optimization is still possible (some guidelines can be extracted from DFT calculations, as shown below). The charge transfer efficiency for both photoanodes together with a comparison to that of a CoOx catalyst is shown in Figure 4b. In agreement with CV data (Figure 3a), CoOx exhibits lower charge transfer/catalytic efficiency for water oxidation up to 1.4 V versus RHE when compared to that of the CoFe-PB catalyst. The maximum charge transfer efficiency for the CoFe-PB/BiVO4 system (≈80%) is obtained in the 1.1–1.2 V versus RHE range (Table S4). The charge separation efficiency was also evaluated, and a 5–10% enhancement is obtained after CoFe-PB deposition (see the Supporting Information, Figure S17).
Figure 4.

(a) Anodic scans (50 mV/s) of CoFe-PB-modified (red) and bare BiVO4 (black) photoelectrodes under 1 sun irradiation in 0.1 M KPi buffer (solid lines) and after addition of hole scavenger Na2SO3 (dashed lines). (b) Calculated charge transfer efficiencies for both photoelectrodes as compared to those of CoOx/BiVO4 (dashed blue).
To confirm that measured photocurrents result from oxygen production at the photoanode and to exclude any contribution from side reactions, gas chromatography measurements under an inert argon atmosphere were carried out. Figure 3b shows the amount of detected oxygen (red triangles) at 1.23 V versus RHE after switching on the light source and compares it to the theoretical oxygen evolution (black line), which can be determined by the total amount of charge passed through the cell. The measured O2 evolution coincides well with that theoretically estimated from the measured photocurrent by Faraday’s law, maintaining >95% faradaic efficiency.
The enhanced photoelectrocatalytic behavior obtained upon deposition of the CoFe-PB layer can be due to several factors, such as (i) a stronger electric field at the interface leading to more favorable recombination kinetics;49 (ii) the development of a capacitive layer,50 which can act as a hole reservoir; (iii) the passivation of surface states;51 (iv) a cathodic shift of the semiconductor bands due to a surface dipole;52 and/or (v) suppression of surface recombination.49,53 To gain insight into this issue, Electrochemical impedance spectroscopy (EIS) measurements were carried out on BiVO4 and CoFe-PB/BiVO4 electrodes at 0.1–1.2 V versus RHE under 100 mW/cm2 illumination. Different CoFe-PB deposition cycles (×6, ×8, and ×10 cycles) were carried out to identify the effect of the catalyst loading on the measured response. The obtained Nyquist plots systematically showed a single arc (Figure S18) and, consequently, the data were fitted to a simple Randles circuit.54 Independent of the CoFe-PB layer thickness, the capacitance of all electrodes is identical, within experimental error (Figure 5a), ruling out any significant participation of a capacitive mechanism. This is in contrast with the conclusions previously claimed for IrOx and CoPi when deposited on top of α-Fe2O3.35,36,56 On BiVO4, Durrant et al. concluded that CoPi does not contribute significantly to the overall water oxidation current (<5%) but was essentially just retarding the electron-hole recombination.49 In the same line, a recent study by Van de Krol et al. also claims that the photocurrent of BiVO4 is limited by surface recombination rather than by surface catalysis.53
Figure 5.
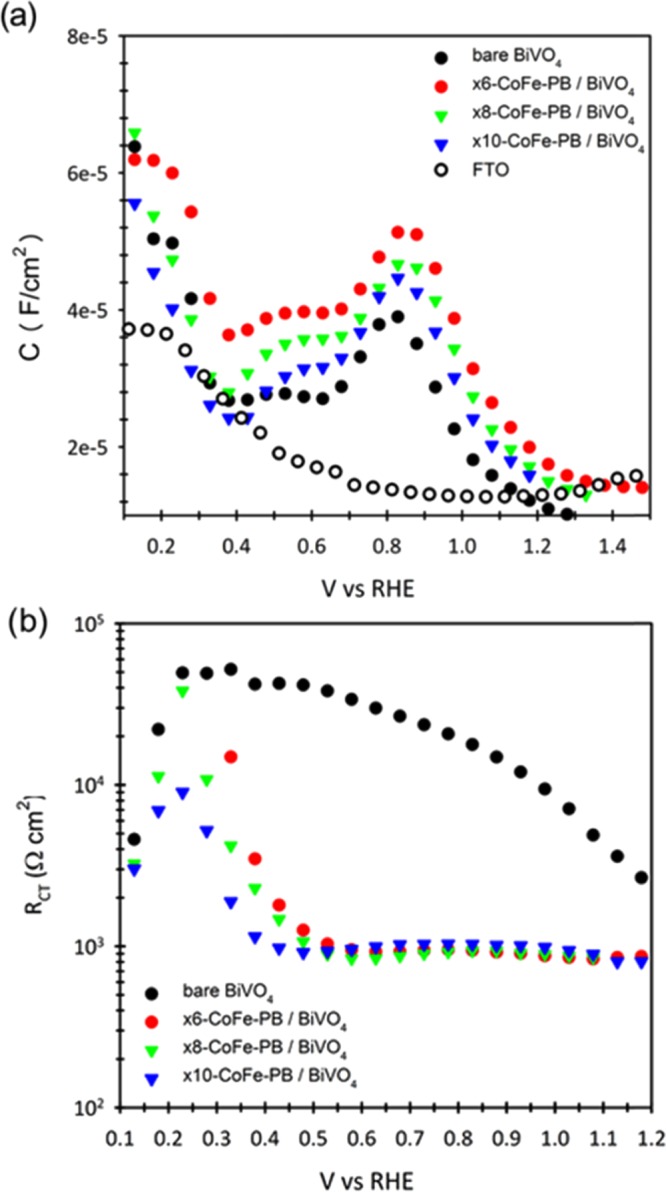
(a) Capacitance and (b) charge transfer resistance for bare (black solid circles) and different CoFe-PB-modified BiVO4 electrodes, obtained by modeling the electrochemical system to a simple Randles circuit. The capacitance of the FTO substrate is also included in (a).
In the present study, the capacitance values obtained for all samples lie in the 10–5–10–4 F/cm2 region, suggesting an important contribution of the double layer capacitance of the electrode. For this reason, the capacitance of the bare FTO substrate is also included in Figure 5a. At applied voltages below 0.4 V and above 1.2 V versus RHE, the capacitance of the photoelectrodes is dominated by FTO. At intermediate voltages, the capacitance is dominated by BiVO4. Additionally, in agreement with previous studies, a capacitive peak at 0.8 V versus RHE is observed, which has been attributed to the V4+/V5+ redox couple.50 On the other hand, the charge transfer resistance drops to a constant value of ∼1 kΩ at 0.5 V versus RHE for the samples coated with the CoFe-PB catalyst, whereas significantly higher voltages (>0.8 V) are needed for pristine BiVO4 photoelectrodes (Figure 5b). Both the constant capacitance and the charge-transfer resistance drop at lower applied potentials, strongly suggests that CoFe-PB is acting as a true catalyst, enhancing charge transfer kinetics to the solution, although suppression of surface recombination cannot be ruled out at this stage.
The synergistic interaction between both BiVO4 and CoFe-PB was assessed by hybrid density functional theory (DFT) calculations. Both the bulk monoclinic scheelite bismuth vanadate (ms-BiVO4) and the CoFe-PB catalyst (topologically described as the ideal FCC structure-type KCoFe[(CN)6], Scheme 1) were modeled as well as a solvated single water molecule,57 using a modified HSE03-13 functional (see the Supporting Information for a methodological discussion). Figure 6 shows the aligned densities of states (DOS) of the photoanode, the catalyst, and water. The BiVO4 valence band (VB) edge consists of mainly O 2p and, to a smaller extent, Bi 6s contributions. This s–p hybridization is crucial as it enables ms-BiVO4 to be a good hole conductor.58−61CoFe-PB, on the right-hand side, has filled valence band levels above the BiVO4 VB edge, namely, the nonbonding t2g states of Co (purple) and Fe (blue). The HOMO (1b1) level of water, which lies about 0.34 eV above the Co t2g band, is also presented in Figure 6.
Figure 6.

Densities of states of BiVO4 (left), KCoFe[(CN)6] (middle), and solvated H2O molecule54 (right) aligned by their O 2s bands. The CoFe-PB valence band edge is set as zero energy level, and filled electronic states are represented by filled areas. A simplified representation is given as the inset (top right). Above band gap light excitation creates a hole (h+) in the BiVO4 VB, which is filled by electron transfer from the CoFe-PB catalyst, which subsequently oxidizes water.
The photoelectrochemical experiments can be understood as illustrated in the simplified energy diagram in Figure 6: Upon supra band gap illumination from the FTO substrate (2.55 eV from HSE03-13 calculation, in good agreement with the 2.40–2.45 eV experimental value, Figure S14) from the FTO substrate, an electron−hole pair is created at BiVO4 close to the BiVO4/FTO interface. Because of the applied positive bias, the electron moves to the FTO and enters the external circuit, whereas the photogenerated hole moves towards the CoFe-PB/BiVO4 interface, where it is transferred to the CoFe-PB catalyst, being available for water oxidation. The [Fe(CN)6] units are coordinatively saturated and robust, as CN is a strong ligand when bonded from the C-end. Therefore, the interface between BiVO4 and CoFe-PB is likely to be formed by the interaction of O atoms from the oxide and surface Co centers from CoFe-PB. The filled Co t2g lies about 0.9 eV higher in energy compared with the VB of BiVO4, which energetically favors the electron transfer from Co to the BiVO4 VB, which is mainly formed by the O 2p band. The difference between the VB of the BiVO4 substrate and the Co levels is in good agreement with the observed cathodic shift of the onset potential of 0.8 V. The subsequently created hole in the Co t2g set is now available for water oxidation.
The cathodic shift of the onset potential, together with the increase of photovoltage due to the CoFe-PB layer, can then be interpreted as follows: Considering BiVO4, the distance between the Bi 6s states and the HOMO position of water is about 1.3 eV, causing a relatively high potential needed to initiate water oxidation (1.1 V vs RHE). Thus, the BiVO4 states do not overlap with the HOMO and and hence charge transfer between those states is kinetically hampered, which implies that more external energy is needed in order to overcome this barrier. In the presence of CoFe-PB, the situation changes and the photogenerated hole at the BiVO4 surface is filled by electrons from the catalyst. In CoFe-PB, the energy difference to the water HOMO is reduced to 0.3–0.4 eV, facilitating water oxidation at a lower onset (0.3 V vs RHE). This smaller energy difference (as well as symmetry considerations) allows good overlap of Co t2g and the water HOMO (1b1). Nevertheless, there is still an energy mismatch of about 0.3 eV to be overcome, and this situation can be related to a <100% charge transfer efficiency of CoFe-PB/BiVO4 (See Figure 4b). A more efficient charge-transfer catalyst would require catalytically active states to lie as close as possible to the HOMO level of water, while possessing an uncoordinated site with matching symmetry and further keeping the good electronic overlap with the light absorber.
Conclusions
In summary, we have demonstrated that integrated photoelectrocatalytic systems with earth-abundant materials and low-cost synthetic procedures based on the CoFe-PB/BiVO4 system exhibit excellent performance with (i) remarkable increase of photocurrent, (ii) low onset potentials, and (iii) excellent stability. Impedance spectroscopy analysis suggests that CoFe-PB is acting as a true catalyst, enhancing charge transfer kinetics to the solution, as inferred by the constant capacitance and decreased charge transfer resistance observed. CoFe-PB appears to be the main catalyst in these CoFe-PB/BiVO4 photoelectrodes, resulting in faster oxygen evolution and significantly lower onset potentials when compared to those of bare electrodes.
Our results also highlight the importance of the interface between the catalyst and photoanode. Under general conditions, CoFe-PB has not been systematically faster than CoPi or CoOx.28,31 However, it is intrinsically better matching the activity of the BiVO4 semiconductor. Indeed, the performance of our photoanodes also exceeds those results obtained with any other decorated BiVO4 photoelectrode, as discussed through the text.
Furthermore, the outstanding performance of this system can be understood on the basis of the adequate alignment of the valence levels of the BiVO4 light-harvesting material and the CoFe-PB catalyst together with the matching symmetry and small energy difference between the Co levels in the catalyst and the HOMO of water. DFT also provides a useful guideline for further catalyst optimization to achieve 100% charge transfer efficiency for water oxidation, by achieving a closer proximity of the Co states and the HOMO level of water, while still maintaining an uncoordinated site with matching symmetry and good electronic overlap with the light absorber. Finally, the outcome of this study can be extrapolated to other metal oxides, providing that a good adsorption of CoFe-PB on the metal oxide surface, which leads to high mechanic stability and efficient interfacial charge transfer, is favored by the energy level alignment and orbital symmetry considerations.
Acknowledgments
We would like to acknowledge financial support from the University Jaume I through the P11B2014-51 project, and from the Generalitat Valenciana through the Santiago Grisolia Program, grant 2015-031. Serveis Centrals at UJI (SCIC) are also acknowledged. This work was also supported by the European Union (project ERC StG grant CHEMCOMP no. 279313); the Spanish Ministerio de Economía y Competitividad (MINECO) through projects CTQ2015-71287-R, CTQ2015-68770-R, and the Severo Ochoa Excellence Accreditation 2014-2018 SEV-2013-0319; the Generalitat de Catalunya (2014SGR-797 and 2014SGR-199), and the CERCA Programme/Generalitat de Catalunya. Additionally, the “LaCaixa”-Severo Ochoa International Programme of Ph.D. Scholarships (Programa internacional de Becas “LaCaixa”-Severo Ochoa) is acknowledged for F.S. Hegner’s predoctoral grant. We thank BSC-RES for generous computational resources.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.7b09449.
Additional computational details, electrochemical data, and extensive information on the structural and photoelectrochemical characterization of the employed materials (SEM, TEM, EDS, XPS, IR, IPC, electrochemical measurements, comparison to other catalysts, EIS, and computational methods); present DFT calculations have been uploaded to the ioChem-BD database62,100 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gimenez S.; Bisquert J.. Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices; Springer International Publishing: Cham, 2016. [Google Scholar]
- Walter M. G.; Warren E. L.; McKone J. R.; Boettcher S. W.; Mi Q.; Santori E. A.; Lewis N. S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- Pinaud B. A.; Benck J. D.; Seitz L. C.; Forman A. J.; Chen Z.; Deutsch T. G.; James B. D.; Baum K. N.; Baum G. N.; Ardo S.; Wang H.; Miller E.; Jaramillo T. F. Technical and Economic Feasibility of Centralized Facilities for Solar Hydrogen Production via Photocatalysis and Photoelectrochemistry. Energy Environ. Sci. 2013, 6, 1983–2002. 10.1039/c3ee40831k. [DOI] [Google Scholar]
- Fujishima A.; Honda K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- Yang C.; Wang Z.; Lin T.; Yin H.; Lü X.; Wan D.; Xu T.; Zheng C.; Lin J.; Huang F.; Xie X.; Jiang M. Core-Shell Nanostructured “Black” Rutile Titania as Excellent Catalyst for Hydrogen Production Enhanced by Sulfur Doping. J. Am. Chem. Soc. 2013, 135, 17831–17838. 10.1021/ja4076748. [DOI] [PubMed] [Google Scholar]
- Du C.; Yang X.; Mayer M. T.; Hoyt H.; Xie J.; McMahon G.; Bischoping G.; Wang D. Hematite-Based Water Splitting with Low Turn-On Voltages. Angew. Chem., Int. Ed. 2013, 52, 12692–12695. 10.1002/anie.201306263. [DOI] [PubMed] [Google Scholar]
- Jang J.-W.; Du C.; Ye Y.; Lin Y.; Yao X.; Thorne J.; Liu E.; McMahon G.; Zhu J.; Javey A.; Guo J.; Wang D. Enabling Unassisted Solar Water Splitting by Iron Oxide and Silicon. Nat. Commun. 2015, 6, 7447 10.1038/ncomms8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay A.; Cesar I.; Graetzel M. New Benchmark for Water Photooxidation by Nanostructured α-Fe2O3 Films. J. Am. Chem. Soc. 2006, 128, 15714–15721. 10.1021/ja064380l. [DOI] [PubMed] [Google Scholar]
- Kim J. Y.; Magesh G.; Youn D. H.; Jang J.-W.; Kubota J.; Domen K.; Lee J. S. Single-Crystalline, Wormlike Hematite Photoanodes for Efficient Solar Water Splitting. Sci. Rep. 2013, 3, 2681 10.1038/srep02681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillet J.; Yum J.-H.; Cornuz M.; Hisatomi T.; Solarska R.; Augustynski J.; Graetzel M.; Sivula K. Highly Efficient Water Splitting by a Dual-Absorber Tandem Cell. Nat. Photonics 2012, 6, 824–828. 10.1038/nphoton.2012.265. [DOI] [Google Scholar]
- Alexander B. D.; Kulesza P. J.; Rutkowska I.; Solarska R.; Augustynski J. Metal Oxide Photoanodes for Solar Hydrogen Production. J. Mater. Chem. 2008, 18, 2298–2303. 10.1039/b718644d. [DOI] [Google Scholar]
- Deb S. K. Opportunities and Challenges in Science and Technology of WO3 for Electrochromic and Related Applications. Sol. Energy Mater. Sol. Cells 2008, 92, 245–258. 10.1016/j.solmat.2007.01.026. [DOI] [Google Scholar]
- Ping Y.; Goddard W. A. III; Galli G. A. Energetics and Solvation Effects at the Photoanode/Catalyst Interface: Ohmic Contact versus Schottky Barrier. J. Am. Chem. Soc. 2015, 137, 5264–5267. 10.1021/jacs.5b00798. [DOI] [PubMed] [Google Scholar]
- Abdi F. F.; Han L.; Smets A. H. M.; Zeman M.; Dam B.; van de Krol R. Efficient Solar Water splitting by Enhanced Charge Separation in a Bismuth Vanadate-Silicon Tandem Photoelectrode. Nat. Commun. 2013, 4, 2195 10.1038/ncomms3195. [DOI] [PubMed] [Google Scholar]
- Chatchai P.; Murakami Y.; Kishioka S.-Y.; Nosaka A. Y.; Nosaka Y. FTO/SnO2/BiVO4 composite Photoelectrode for Water Oxidation under Visible Light Irradiation. Electrochem. Solid-State Lett. 2008, 11, H160–H163. 10.1149/1.2906109. [DOI] [Google Scholar]
- Kim T. W.; Choi K.-S. Nanoporous BiVO4 Photoanodes with Dual-Layer Oxygen Evolution Catalysts for Solar Water Splitting. Science 2014, 343, 990–994. 10.1126/science.1246913. [DOI] [PubMed] [Google Scholar]
- Park Y.; McDonald K. J.; Choi K.-S. Progress in Bismuth Vanadate Photoanodes for Use in Solar Water Oxidation. Chem. Soc. Rev. 2013, 42, 2321–2337. 10.1039/C2CS35260E. [DOI] [PubMed] [Google Scholar]
- Shi X.; Zhang K.; Shin K.; Ma M.; Kwon J.; Choi I. T.; Kim J. K.; Kim H. K.; Wang D. H.; Park J. H. Unassisted PhotoelectroChemical Water Splitting beyond 5.7% Solar-to-Hydrogen Conversion Efficiency by a Wireless Monolithic Photoanode/Dye-Sensitised Solar Cell Tandem Device. Nano Energy 2015, 13, 182–191. 10.1016/j.nanoen.2015.02.018. [DOI] [Google Scholar]
- Shi X.; Choi Y.; Zhang K.; Kwon J.; Kim D. Y.; Lee J. K.; Oh S. H.; Kim J. K.; Park J. H. Efficient Photoelectrochemical Hydrogen Production from Bismuth Vanadate-Decorated Tungsten Trioxide Helix Nanostructures. Nat. Commun. 2014, 5, 4775 10.1038/ncomms5775. [DOI] [PubMed] [Google Scholar]
- Chhetri M.; Dey S.; Rao C. N. R. Photoelectrochemical Oxygen Evolution Reaction Activity of Amorphous Co–La Double Hydroxide-BiVO4 Fabricated by Pulse Plating Electrodeposition. ACS Energy Lett. 2017, 2, 1062–1069. 10.1021/acsenergylett.7b00247. [DOI] [Google Scholar]
- Pihosh Y.; Turkevych I.; Mawatari K.; Uemura J.; Kazoe Y.; Kosar S.; Makita K.; Sugaya T.; Matsui T.; Fujita D.; Tosa M.; Kondo M.; Kitamori T. Photocatalytic Generation of Hydrogen by Core-Shell WO3/BiVO4 Nanorods with Ultimate Water Splitting Efficiency. Sci. Rep. 2015, 5, 11141 10.1038/srep11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y.; Liu W.; Chen W.; Zhou G.; Hsu P.-C.; Zhang R.; Liang Z.; Fan S.; Zhang Y.; Cui Y.; et al. Efficient Solar-Driven Water Splitting by Nanocone BiVO4-perovskite Tandem Cells. Sci. Adv. 2016, 2, e1501764 10.1126/sciadv.1501764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesborg P. C. K.; Jaramillo T. F. Addressing the Terawatt Challenge: Scalability in the Supply of Chemical Elements for Renewable Energy. RSC Adv. 2012, 2, 7933–7947. 10.1039/c2ra20839c. [DOI] [Google Scholar]
- Guo W.; Chemelewski W. D.; Mabayoje O.; Xiao P.; Zhang Y.; Mullins C. B. Synthesis and Characterization of CuV2O6 and Cu2V2O7: Two Photoanode Candidates for Photoelectrochemical Water Oxidation. J. Phys. Chem. C 2015, 119, 27220–27227. 10.1021/acs.jpcc.5b07219. [DOI] [Google Scholar]
- Seabold J. A.; Neale N. R. All First Row Transition Metal Oxide Photoanode for Water Splitting Based on Cu3V2O8. Chem. Mater. 2015, 27, 1005–1013. 10.1021/cm504327f. [DOI] [Google Scholar]
- Cardenas-Morcoso D.; Peiro-Franch A.; Herraiz-Cardona I.; Gimenez S. Chromium Doped Copper Vanadate Photoanodes for Water Splitting. Catal. Today 2017, 290, 65–72. 10.1016/j.cattod.2016.11.002. [DOI] [Google Scholar]
- Galán-Mascarós J. R. Water Oxidation at Electrodes Modified with Earth-Abundant Transition-Metal Catalysts. ChemElectroChem 2015, 2, 37–50. 10.1002/celc.201402268. [DOI] [Google Scholar]
- Gerken J. B.; McAlpin J. G.; Chen J. Y. C.; Rigsby M. L.; Casey W. H.; Britt R. D.; Stahl S. S. Electrochemical Water Oxidation with Cobalt-Based Electrocatalysts from pH 0–14: The Thermodynamic Basis for Catalyst Structure, Stability, and Activity. J. Am. Chem. Soc. 2011, 133, 14431–14442. 10.1021/ja205647m. [DOI] [PubMed] [Google Scholar]
- Pintado S.; Goberna-Ferrón S.; Escudero-Adán E. C.; Galan-Mascaros J. R. Fast and Persistent Electrocatalytic Water Oxidation by Co–Fe Prussian Blue Coordination Polymers. J. Am. Chem. Soc. 2013, 135, 13270–13273. 10.1021/ja406242y. [DOI] [PubMed] [Google Scholar]
- Yamada Y.; Oyama K.; Gates R.; Fukuzumi S. High Catalytic Activity of Heteropolynuclear Cyanide Complexes Containing Cobalt and Platinum Ions: Visible-Light Driven Water Oxidation. Angew. Chem., Int. Ed. 2015, 54, 5613–5617. 10.1002/anie.201501116. [DOI] [PubMed] [Google Scholar]
- Aksoy M.; Nune S. V. K.; Karadas F. A Novel Synthetic Route for the Preparation of an Amorphous Co/Fe Prussian Blue Coordination Compound with High Electrocatalytic Water Oxidation Activity. Inorg. Chem. 2016, 55, 4301–4307. 10.1021/acs.inorgchem.6b00032. [DOI] [PubMed] [Google Scholar]
- Han L.; Tang P. Y.; Reyes-Carmona A.; Rodriguez-Garcia B.; Torrens M.; Morante J. R.; Arbiol J.; Galan-Mascaros J. R. Enhanced Activity and Acid pH Stability of Prussian Blue-type Oxygen Evolution Electrocatalysts Processed by Chemical Etching. J. Am. Chem. Soc. 2016, 138, 16037–16045. 10.1021/jacs.6b09778. [DOI] [PubMed] [Google Scholar]
- de Tacconi N. R.; Rajeshwar K.; Lezna R. O. Metal Hexacyanoferrates: Electrosynthesis, in Situ Characterization, and Applications. Chem. Mater. 2003, 15, 3046–3062. 10.1021/cm0341540. [DOI] [Google Scholar]
- Goberna-Ferrón S.; Hernández W. Y.; Rodríguez-García B.; Galan-Mascaros J. R. Light-Driven Water Oxidation with Metal Hexacyanometallate Heterogeneous Catalysts. ACS Catal. 2014, 4, 1637–1641. 10.1021/cs500298e. [DOI] [Google Scholar]
- Badia-Bou L.; Mas-Marza E.; Rodenas P.; Barea E. M.; Fabregat-Santiago F.; Gimenez S.; Peris E.; Bisquert J. Water Oxidation at Hematite Photoelectrodes with an Iridium-Based Catalyst. J. Phys. Chem. C 2013, 117, 3826–3833. 10.1021/jp311983n. [DOI] [Google Scholar]
- Klahr B.; Gimenez S.; Fabregat-Santiago F.; Bisquert J.; Hamann T. W. Photoelectrochemical and Impedance Spectroscopic Investigation of Water Oxidation with “Co–Pi”-Coated Hematite Electrodes. J. Am. Chem. Soc. 2012, 134, 16693–16700. 10.1021/ja306427f. [DOI] [PubMed] [Google Scholar]
- Kang D.; Park Y.; Hill J. C.; Choi K.-S. Preparation of Bi-Based Ternary Oside Photoanodes BiVO4, Bi2WO6, Bi2Mo3O12 Using Dendritic Bi Metal Electrodes. J. Phys. Chem. Lett. 2014, 5, 2994–2999. 10.1021/jz501544k. [DOI] [PubMed] [Google Scholar]
- Abdi F. F.; Savenije T. J.; May M. M.; Dam B.; van de Krol R. The Origin of Slow Carrier Transport in BiVO4 Thin Film Photoanodes: A Time-Resolved Microwave Conductivity Study. J. Phys. Chem. Lett. 2013, 4, 2752–2757. 10.1021/jz4013257. [DOI] [Google Scholar]
- Rettie A. J. E.; Lee H. C.; Marshall L. G.; Lin J.-F.; Capan C.; Lindemuth J.; McCloy J. S.; Zhou J.; Bard A. J.; Mullins C. B. Combines Charge Carrier Transport and Photoelectrochemical Characterization of BiVO4 Single Crystals: Intrinsic Behavior of a Complex Metal Oxide. J. Am. Chem. Soc. 2013, 135, 11389–11396. 10.1021/ja405550k. [DOI] [PubMed] [Google Scholar]
- Ahn H.-J.; Kwak M.-J.; Lee J.-S.; Yoon K.-Y.; Jang J.-H. Nanoporous Hematite Structures to Overcome Short Diffusion Lengths in Water Splitting. J. Mater. Chem. A 2014, 2, 19999–20003. 10.1039/C4TA04890C. [DOI] [Google Scholar]
- Dare-Edwards M.-P.; Goodenough J. B.; Hamnett A.; Trevellick P. R. Electrochemistry and Photoelectrochemistry of Iron(III) Oxide. J. Chem. Soc., Faraday Trans. 1 1983, 79, 2027–2041. 10.1039/f19837902027. [DOI] [Google Scholar]
- Kennedy J.-H.; Frese K. W. Photooxidation of Water at alpha-Fe2O3 Electrodes. Chem. Informationsdienst 1977, 124, C130–C130. 10.1002/chin.197834031. [DOI] [Google Scholar]
- Hafner J.; Kresse G.. The Vienna Ab-Initio Simulation Program VASP: An Efficient and Versatile Tool for Studying the Structural, Dynamic and Electronics Properties of Materials. Plenum Press Div Plenum Publishing Corp.: New York, 1997. [Google Scholar]
- Kresse G.; Furthmuller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Hegner F. S.; Galan-Mascaros J. R.; Lopez N. A. Database of the Structural and Electronic Properties of Prussian Blue, Prussian White, and Berlin Green Compounds through Density Functional Theory. Inorg. Chem. 2016, 55, 12851–12862. 10.1021/acs.inorgchem.6b02200. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Lezna R. O.; Romagnoli R.; de Tacconi N. R.; Rajeshwar K. Cobalt Hexacyanoferrate: Compound Stoichiometry, Infrared Spectroelectrochemistry, and Photoinduced Electron Transfer. J. Phys. Chem. B 2002, 106, 3612–3621. 10.1021/jp013991r. [DOI] [Google Scholar]
- Sleight A. W.; Chen H. Y.; Ferretti A.; Cox D. E. Crystal Growth and Structure of BiVO4. Mater. Res. Bull. 1979, 14, 1571–1581. 10.1016/0025-5408(72)90227-9. [DOI] [Google Scholar]
- Toma F. M.; Cooper J. K.; Kunzelmann V.; McDowell M. T.; Yu J.; Larson D. M.; Borys N. J.; Abelyan C.; Beeman J. W.; Yu K. M.; Yang J. H.; Chen L.; Shaner M. R.; Spurgeon J.; Houle F. A.; Persson K. A.; Sharp I. D. Mechanistic Insights into Chemical and Photochemical Transformations of Bismuth Vanadate Photoanodes. Nat. Commun. 2016, 7, 12012 10.1038/ncomms12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cris B. V.Handbook of Monochromatic XPS Spectra; Wiley: Weinheim, 2000. [Google Scholar]
- Bourgeteau T.; Tondelier D.; Geffroy B.; Brisse R.; Cornut R.; Artero V.; Jousselme B. Enhancing the Performances of P3HT:PCBM-MoS3-Based H2-Evolving Photocathodes with Interfacial Layers. ACS Appl. Mater. Interfaces 2015, 7, 16395–16403. 10.1021/acsami.5b03532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzesniewski B. J.; Smith W. A. Photocharged BiVO4 Photoanodes for Improved Solar Water Splitting. J. Mater. Chem. A 2016, 4, 2919–2926. 10.1039/C5TA04716A. [DOI] [Google Scholar]
- Cooper J. K.; Gul S.; Toma F. M.; Chen L.; Liu Y.-S.; Guo J.; Ager J. W.; Yano J.; Sharp I. D. Indirect Bandgap and Optical Properties of Monoclinic Bismuth Vanadate. J. Phys. Chem. C 2015, 119, 2969–2974. 10.1021/jp512169w. [DOI] [Google Scholar]
- Jeon T. H.; Choi W.; Park H. Cobalt-Phosphate Complexes Catalyze the Photoelectrochemical Water Oxidation of BiVO4 electrodes. Phys. Chem. Chem. Phys. 2011, 13, 21392–21401. 10.1039/c1cp23135a. [DOI] [PubMed] [Google Scholar]
- Zhong D. K.; Choi S.; Gamelin D. R. Near-Complete Suppression of Surface Recombination in Solar Photoelectrolysis by “Co-Pi” Catalyst-Modified W:BiVO4. J. Am. Chem. Soc. 2011, 133, 18370–18377. 10.1021/ja207348x. [DOI] [PubMed] [Google Scholar]
- Seabold J. A.; Choi K.-S. Efficient and stable Photo-Oxidation of Water by a Bismuth Vanadate Photoanode Coupled with an Iron Oxyhydroxide Oxygen Evolution Catalyst. J. Am. Chem. Soc. 2012, 134, 2186–2192. 10.1021/ja209001d. [DOI] [PubMed] [Google Scholar]
- Dotan H.; Sivula K.; Graetzel M.; Rothschild A.; Warren S. C. Probing the Photoelectrochemical Properties of Hematite (α-Fe2O3) Electrodes Using Hydrogen Peroxide as a Hole Scavenger. Energy Environ. Sci. 2011, 4, 958–964. 10.1039/C0EE00570C. [DOI] [Google Scholar]
- Shaddad M. N.; Ghanem M. A.; Al-Mayouf A. M.; Gimenez S.; Bisquert J.; Herraiz-Cardona I. Cooperative Catalytic Effect of ZrO2 and α-Fe2O3 Nanoparticles on BiVO4 Photoanodes for Enhanced Photoelectrochemical Water Splitting. ChemSusChem 2016, 9, 2779–2783. 10.1002/cssc.201600890. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Kafizas A.; Pendlebury S. R.; Le Formal F.; Durrant J. R. Photoinduced Absorption Spectroscopy of CoPi on BiVO4: The Function of CoPi during Water Oxidation. Adv. Funct. Mater. 2016, 26, 4951–4960. 10.1002/adfm.201600711. [DOI] [Google Scholar]
- Trześniewski B. J.; Digdaya I. A.; Nagaki T.; Ravishankar S.; Herraiz-Cardona I.; Vermaas D. A.; Longo A.; Gimenez S.; Smith W. A. Near-Complete Suppression of Surface Losses and Total Internal Quantum Efficiency in BiVO4 Photoanodes. Energy Environ. Sci. 2017, 10, 1517–1529. 10.1039/C6EE03677E. [DOI] [Google Scholar]
- Steier L.; Herraiz-Cardona I.; Gimenez S.; Fabregat-Santiago F.; Bisquert J.; Tilley S. D.; Graetzel M. Understanding the Role of Underlayers and Overlayers in Thin Film Hematite Photoanodes. Adv. Funct. Mater. 2014, 24, 7681–7688. 10.1002/adfm.201402742. [DOI] [Google Scholar]
- Xu J.; Herraiz-Cardona I.; Yang X.; Gimenez S.; Antonietti M.; Shalom M. The Complex Role of Carbon Nitride as a Sensitizer in Photoelectrochemical Cells. Adv. Opt. Mater. 2015, 3, 1052–1058. 10.1002/adom.201500010. [DOI] [Google Scholar]
- Zachäus C.; Abdi F. F.; Peter L. M.; van de Krol R. Photocurrent of BiVO4 Is Limited by Surface Recombination, not Surface Catalysis. Chem. Sci. 2017, 8, 3712–3719. 10.1039/C7SC00363C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randles J. E. B. Kinetics of Rapid Electrode Reactions. Discuss. Faraday Soc. 1947, 1, 11–19. 10.1039/df9470100011. [DOI] [Google Scholar]
- Barroso M.; Cowan A. J.; Pendlebury S. R.; Grätzel M.; Klug D. R.; Durrant J. R. The Role of Cobalt Phosphate in Enhancing the Photocatalytic Activity of α-Fe2O3 toward Water Oxidation. J. Am. Chem. Soc. 2011, 133, 14868–14871. 10.1021/ja205325v. [DOI] [PubMed] [Google Scholar]
- Garcia-Ratés M.; López N. Multigrid-Based Methodology for Implicit Solvation Models in Periodic DFT. J. Chem. Theory Comput. 2016, 12, 1331–1341. 10.1021/acs.jctc.5b00949. [DOI] [PubMed] [Google Scholar]
- Cooper J. K.; Gul S.; Toma F. M.; Chen L.; Glans P.-A.; Guo J.; Ager J. W.; Yano J.; Sharp I. D. Electronic Structure of Monoclinic BiVO4. Chem. Mater. 2014, 26, 5365–5373. 10.1021/cm5025074. [DOI] [Google Scholar]
- Zhao Z.; Li Z.; Zou Z. Electronic Structure and Optical Properties of Monoclinic Clinobisvanite BiVO4. Phys. Chem. Chem. Phys. 2011, 13, 4746–4753. 10.1039/c0cp01871f. [DOI] [PubMed] [Google Scholar]
- Walsh A.; Yan Y.; Huda M. N.; Al-Jassim M. M.; Wei S.-H. Band Edge Electronic Structure of BiVO4: Elucidating the Role of the Bi and V d Orbitals. Chem. Mater. 2009, 21, 547–551. 10.1021/cm802894z. [DOI] [Google Scholar]
- Kweon K. E.; Hwang G. S. Structural Phase-Dependent Hole Localization and Transport in Bismuth Vanadate. Phys. Rev. B 2013, 87, 205202 10.1103/PhysRevB.87.205202. [DOI] [Google Scholar]
- Álvarez-Moreno M.; de Graaf C.; López N.; Maseras F.; Poblet J. M.; Bo C. Managing the Computational Chemistry Big Data Problem: The ioChem BD Platform. J. Chem. Inf. Model. 2015, 55, 95–103. 10.1021/ci500593j. [DOI] [PubMed] [Google Scholar]
- DOI: 10.19061/iochem-bd-1-50. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.