

Abstract
Objective
Bruton’s Tyrosine Kinase (BTK) is a B cell signaling protein that also contributes to innate immunity. BTK-inhibitors prevent autoimmune arthritis, but have off-target effects, and the mechanisms of protection remain unknown. These studies used genetic deletion to investigate the role of BTK in adaptive and innate immune responses that drive inflammatory arthritis.
Methods
Btk-deficient K/BxN mice were generated to study the role of BTK in a spontaneous model that requires both adaptive and innate immunity. The K/BxN serum transfer model was used to bypass the adaptive system and elucidate the role of BTK in innate immune contributions to arthritis.
Results
Btk-deficiency conferred disease protection to K/BxN mice, confirming BTK-inhibitor outcomes. B lymphocytes were profoundly reduced, more than in other Btk-deficient models. Subset analysis revealed loss at all developmental stages. Germinal center B cells were also decreased, with downstream effects on T follicular helper numbers, and greatly reduced autoantibodies. In contrast, total IgG was only mildly decreased. Strikingly, and in contrast to small molecule inhibitors, Btk-deficiency had no effect on the serum transfer model of arthritis.
Conclusions
BTK contributes to autoimmune arthritis primarily via its role in B cell signaling, not innate immune components.
Rheumatoid arthritis (RA) is an autoantibody-mediated inflammatory disease characterized by synovial inflammation and destruction of cartilage and bone. Both innate and adaptive immunity play crucial roles (1). Though initial treatment for RA remains non-biologic disease-modifying anti-rheumatic drugs (DMARDs) (2), targeted biologic agents are used with increasing frequency. Targets include cytokines and cytokine receptors, signaling kinases, co-stimulation of T-lymphocytes, and B-lymphocytes (3). One possible target for small molecule inhibitors is Bruton’s tyrosine kinase (BTK), a protein that propagates both antigen-specific and TLR-related signaling in B lymphocytes, macrophages, dendritic cells, mast cells and neutrophils. BTK is critical to cellular development and activation via the B cell receptor (BCR), toll-like receptor 4 (TLR4) and CD40 (4–9). BTK also functions in myeloid cells, where it propagates signaling by certain TLRs, FcεR, and FcγRs (10–12). BTK likely plays an important role in regulating autoimmunity in B cells. Increased BTK expression increases the propensity for autoreactivity, while decreased levels improve tolerance in autoimmune models (13, 14). Autoreactive-prone B cell subsets, such as B1a B cells, rely more on BTK than normal follicular B cells (7), and we recently reported that anergic autoreactive cells require BTK for development (15). A role for BTK in RA is hypothesized based on studies demonstrating that BTK inhibitors are effective in arthritis models (12, 16–20), but these drugs can have off-target effects, including T cell alterations secondary to interleukin-2-inducible T cell kinase (ITK) inhibition (21). Genetic deletion of BTK can provide insight into its role in arthritis and enhance drug development by exploring the mechanism of protection mediated specifically by loss of BTK. One early study using a small number of xid mice showed protection against collagen-induced arthritis (CIA), but no further cellular, histologic, or molecular mechanistic studies have been performed using genetic models (22).
The K/BxN model of arthritis provides tools to test contributions from both innate and adaptive immunity. K/BxN mice are generated by crossing KRN mice, which express a transgenic TCR, with mice expressing MHC class II IAg7 characteristic of non-obese diabetic (NOD) mice. In the context of the IAg7 the KRN TCR recognizes a peptide from glucose-6-phosphate isomerase (GPI) (23, 24). This interaction causes robust, spontaneous arthritis, more severe in males, that requires both lymphoid and myeloid immune cells (25–29). In addition, serum transfer from K/BxN mice induces immune complex-mediated arthritis in recipients, relying on innate immunity and bypassing the adaptive immune response (25, 30). We generated Btk-deficient K/BxN males and assessed the progression of arthritis compared to Btk-sufficient littermate controls. Our studies were intended to determine the effect of BTK deficiency on spontaneous and serum transfer arthritis, and to discover the relative roles of B lymphocytes and innate immune mechanisms in arthritis development.
Materials and Methods
Mice and Disease Studies
KRN mice were a generous gift from Christophe Benoist and Diane Mathis. Btk-deficient NOD mice were derived as previously described (4). Mice are bred and maintained under specific pathogen-free conditions. KRN males were bred to Btk+/− NOD females, producing Btk-sufficient and Btk-deficient K/BxN males for spontaneous arthritis studies, assessed weekly for 5 weeks post weaning. Serum from 8–9 week old arthritic Btk-sufficient K/BxN males was pooled and injected (200μL, intraperitoneal injection) to produce arthritis in male Btk-sufficient and Btk-deficient NOD mice. Mice were injected on days 0 and 2 and assessed for arthritis for two weeks. All studies are approved by the Vanderbilt University Animal Care and Use Committee.
Arthritis Scoring
The Chondrex mouse arthritis scoring system was used to assess arthritis progression in the K/BxN and serum-transfer models (https://www.chondrex.com/documents/Mouse%20CIA.pdf). Hind foot pad paw thickness was measured by a Swiss Precision Instrument (SPI) dial gauge (13-159-9).
Flow Cytometry and Antibodies
Single cell suspensions from spleens, popliteal lymph nodes (LN), and peritoneal cavity were obtained as previously described (4) and stained using fluorochrome or biotin-conjugated antibodies against B220 (RA3-6B2), IgM (μ-chain specific, Life Technologies), IgD (11-26c.2a), CD21 (76G), CD23 (B3B4), CD4 (RM4-5), CD8a (53-6.7), CD11b (M1/70), CD11c (HL3, BD Biosciences or N418, eBioscience), CD5 (53-7.3), CD19 (ID3), Fas (Jo2), BCL6 (K11291), GL7, CD44 (IM7, eBioscience), CXCR5 (2G8), PD1 (J43), ICOS (C398.4A, eBioscience), F4/80 (BM8, eBioscience), Ly6G (IA8), and/or CCR7 (4B12). Unless otherwise stated, antibodies are from BD Biosciences. Biotin-conjugated antibodies were secondarily stained with fluorochrome-conjugated streptavidin (BD Bioscience). Dead cells were excluded using 7 Aminoactinomycin D (BD Biosciences), fixable viability dye eFluor® 450 (eBioscience) or Alexa Fluor® 700 Succinimidyl Ester (Life Technologies). Samples were read on a LSRII flow cytometer (BD Biosciences) and data analyzed using FlowJo (Tree Star) software.
Bone Marrow-derived Macrophages
Murine bone marrow cells harvested from femurs were differentiated in RPMI 1640 media (Corning) with 10% FCS (Gibco), 1% antibiotic-antimycotic (Gibco), and 10ng/mL macrophage colony stimulating factor (R&D) for 7 days in non-TC treated polystyrene plates (Fisher). On day seven, attached macrophages were harvested, transferred to 96-well flat-bottom NUNC plates, allowed to adhere, then incubated without stimulus, with 1/20 K/BxN serum, or with 100ng/mL LPS (DIFCO Laboratories) overnight at 37° C. Supernatants were frozen for analysis.
ELISA
Serum IgG (total) and anti-GPI IgG from 8–9 week old Btk-sufficient and Btk-deficient K/BxN were measured. 96-well flat-bottom NUNC plates were coated with 1μg/mL recombinant mouse GPI (Cloud-Clone Corp) or 2μg/mL goat anti-mouse Ig (Southern Biotech) in PBS overnight at 4°C. Plates were blocked with 1% BSA in PBS or 10% non-fat dry milk in PBS+0.5% Tween-20 (PBST). Diluted sera (1:3000 or 1:5000) were added to plates. IgG antibodies were detected using goat anti-mouse IgG-alkaline phosphatase (AP) (Southern Biotech). p-Nitrophenyl Phosphate (PNPP) was added to the plate and O.D. read on a Microplate Autoreader (Bio-Tek Instruments) at 405nm. Mouse TNFα Ready-Set-Go!® ELISAs (eBioscience) were performed on BMDM supernatants according to manufacturer’s protocol and O.D. read at 450nm.
Histology
Hind paws collected from 8–9 week old Btk-sufficient and Btk-deficient K/BxN mice were processed as previously described (31). Four blinded observers scored histological samples for inflammation (0, normal; 1, minimal; 2, mild; 3, moderate; 4, severe), cartilage destruction and bone erosion (0, no destruction/erosion; 1, moderate 2, severe).
Whole-body fluorescence imaging
Fluorescence imaging was performed as previously described (32). Cy5-PEG-folate (Nanocs Inc., NY; excitation wavelength - 650 nm, emission - 670 nm) was injected intravenously (500nmol/kg). Fluorescent imaging was performed after 4 hours by a Pearl Impulse system (LI-COR, Lincoln, NE). Data were collected and analyzed using Pearl Impulse software (LI-COR).
Statistics
Statistics were performed using GraphPad Prism version 6.00 for Windows, (GraphPad Software, La Jolla California USA). P-values for disease curves and FolRβ imaging were calculated using a two-way ANOVA. All other p-values were calculated by unpaired T tests with Welch’s correction or the Holm-Sidak method of multiple T tests, as appropriate.
Results
Loss of BTK protects against development of arthritis in K/BxN mice
To determine BTK contributions to arthritis development, Btk-deficiency was introduced to the K/BxN mouse model. Btk loss significantly protected against arthritis, as assessed by clinical score and paw thickness (Fig 1a,b). By 5 weeks post-weaning, Btk-sufficient K/BxN had mean clinical scores of 15.7 (±0.816), compared to Btk-deficient K/BxN scores of 9.75 (±2.49), (p<0.0001). Paw thickness averaged 3.03mm (±0.156) in Btk-sufficient K/BxNs and 2.33mm (±0.271) in Btk-deficient K/BxN (p<0.0001). H&E stained histologic sections from right hind paws were assessed for inflammation, cartilage destruction, and bone erosion. Fig 1c shows a representative, non-arthritic NOD control (left), Btk-sufficient K/BxN (middle), and Btk-deficient K/BxN (right). Fig 1d shows pooled scores of Btk-sufficient and –deficient K/BxNs. All three measures show significant differences between genotypes, with Btk-deficient K/BxNs exhibiting lower inflammation (Btk-sufficient 3.5±0.548, Btk-deficient 1.6±0.548, p=0.0003), no cartilage destruction (Btk-sufficient 1.5±0.548, Btk-deficient 0±0, p=0.0011), and little bone erosion (Btk-sufficient 1.5±0.548, Btk-deficient 0.2±0.447 p=0.0019). These data demonstrate that lymphocytic infiltration and arthritic damage, and thus arthritis progression, is significantly reduced by BTK loss.
Figure 1. Btk deficiency is protective against the development of autoimmune arthritis in K/BxN mice.

A) and B) Btk-sufficient (circles, n=6) and Btk-deficient (squares, n=12) K/BxN mice were scored for arthritis for 5 weeks post weaning. Clinical scores (A) were assigned on a scale of 0–4 for each limb and pooled for a total possible score of 16. In addition, paw thickness (mm) was measured by caliper (B). Mean values are shown ± standard deviation. C) Representative H&E staining from right hind paws of WT NOD (left), Btk-sufficient K/BxN (middle), and Btk-deficient K/BxN (right). 2X magnification (top), 10X magnification (bottom). Arrows indicate areas of bone erosion, triangles indicate cartilage loss. D) Scoring of Btk-sufficient (circles, n=6) or –deficient (squares, n=5) for inflammation (top), cartilage loss (middle), and bone erosion (bottom). Inflammation was scored on a scale of 0 to 4, while cartilage loss and bone erosion were scored as 0 to 2. P values were calculated by a 2-way AVOVA with Sidak correction (A, B) **p≤0.01, ***p≤0.001, by unpaired T test with Welch’s correction (D).
Innate and adaptive immune cell populations are decreased in Btk-deficient K/BxNs
To determine the effect of Btk-deficiency on immune cell development and survival in K/BxN mice, we used flow cytometry to enumerate T and B lymphocytes, macrophages, neutrophils, and dendritic cells. Fig 2a shows representative flow plots of live-gated splenocytes from Btk-sufficient (left) and Btk-deficient (right) K/BxN, gated to CD4 and CD8 T cells (top) and IgM versus B220 B cells (bottom). In Fig 2b, the cell populations are quantified as percent of live lymphocytes (top) or total number of cells (bottom). The percentages of B cells were significantly decreased in Btk-deficient K/BxNs (37.8±4.44) compared to Btk-sufficient (47.9±6.32) (p=0.0098). Percentages of both CD4+ and CD8+ T cells were significantly increased in Btk-deficient K/BxNs (8.46±1.74, 4.65±1.03) compared to Btk-sufficient (6.12±1.08, 3.15±0.971) (p=0.0206, p=0.0271), reciprocal to the loss of large numbers of B cells (Btk-sufficient=7.18e6±2.37e6, Btk-deficient=1.86e6±8.50e5, p=0.0004). However, though T cells do not express BTK, T cells numbers were significantly reduced, (CD4+ Btk-sufficient=1.26e6±4.44e5, Btk-deficient=5.39e5±2.34e5, p=0.0056; CD8+ Btk-sufficient=6.19e5±1.75e5, Btk-deficient=2.94e5±1.23e5, p=0.0040), suggesting T cell expansion during arthritic progression in Btk-sufficient K/BxNs.
Figure 2. Immune cell numbers are decreased in the spleen of Btk-deficient K/BxNs compared to Btk-sufficient controls.

A) Representative flow plots for Btk-sufficient (left) and Btk-deficient (right) K/BxN, showing total T cells (top) and B cells (bottom). Cells are pre-gated on single live lymphocytes. T cells are gated as CD4+ and CD8+, while B cells are designated as B220+/IgM+. B) Quantification of T and B cell percentages of lymphocytes (top) and total cell number (bottom) in Btk-sufficient (circles, n=6) or –deficient (squares, n=6). C) Innate immune cells are shown by flow cytometry for a representative Btk-sufficient (top) or –deficient (bottom) K/BxN. Cells were gated as single live mononuclear cells. CD11b+CD11c− cells were gated as F4/80+Ly6G− for macrophages or F4/80−Ly6G+ for neutrophils. CD11b+CD11c+ cells were designated myeloid dendritic cells (DCs) and plasmacytoid dendritic cells (pDCs) were designated as CD11b−CD11c+B220+. D) Quantification of percentages (top) or total numbers (bottom) of innate immune cells in Btk-sufficient (n=3–7) or Btk-deficient (n=6–8) K/BxN. *p≤0.05, **p≤0.01, ***p≤0.001, as calculated by the Holm-Sidak method of multiple T tests.
Innate cells were also quantified using CD11b, CD11c, F4/80, Ly6G, and B220 to identify macrophages, neutrophils, myeloid dendritic cells (DCs), and plasmacytoid dendritic cells (pDCs) (Fig 2c,d). Btk-deficient K/BxN had significantly increased percentages of neutrophils (Btk-sufficient=3.26±0.843, Btk-deficient=10.3±3.57) and macrophages (Btk-sufficient=2.15±0.491, Btk-deficient=4.00±1.05) (p=0.0140, p=0.0258); however, absolute numbers were not significantly altered. Thus, the higher percentage again reflects the substantial loss in numbers of B cells. Myeloid dendritic cell numbers in the spleen were significantly decreased in Btk-deficient K/BxN, (Btk-sufficient=3.41e5±6.09e4, Btk-deficient=1.97e5±4.98e4 p=0.0064), as were plasmacytoid dendritic cells (Btk-sufficient=8.62e4±2.16e4, Btk-deficient=3.01e4±4.43e3 p<0.0001).
Btk-deficiency reduces mature B cell subsets in K/BxN mice
Btk-deficiency results in a block in the late transitional (T2) stage of B cell development in NOD and C57Bl/6 mice (4, 33). To determine the developmental stage at which B cells are reduced in Btk-deficient K/BxN mice, we evaluated B cell subsets by expression of IgM, IgD, CD21, and CD23. Fig 3a shows representative samples of Btk-sufficient (left) and Btk-deficient (right) K/BxN B cells designated as early transitional (T1), late transitional (T2), follicular (FO,), pre-marginal zone (PMZ), and marginal zone (MZ). Fig 3b shows quantification of B cell subsets by percentages (left) and total cell numbers (right). The percentages of T1 and T2 B cells are significantly higher in Btk-deficient K/BxNs (37.1±6.30, 27.6±0.571) compared to Btk-sufficient K/BxN (10.7±3.75, 11.5±1.205) (p<0.0001, p<0.0001). A corresponding significant decrease in the percentage of FO cells (Btk-sufficient=37.3±6.65, Btk-deficient=9.03±3.57, p<0.0001) points to a developmental block at T1 and T2 B cell stages. However, there is no increase in total T1 or T2 B cell numbers as seen in other Btk-deficient models, suggesting that B cells are lost at early and late transitional stages as well. Furthermore, the decrease in the number of FO (Btk-sufficient=2.71e6±1.27e6, Btk-deficient=1.85e5±1.69e5, p=0.0007), pre-MZ (Btk-sufficient=4.86e5±2.83e5, Btk-deficient=8.99e4±4.22e4, p=.0069) and MZ B cells (Btk-sufficient=6.47e5±1.50e5, Btk-deficient=4.67e4±1.73e4, p<0.0001) in Btk-deficient K/BxNs is severe, with substantially greater cell loss than is seen in Btk-deficient NOD or C57BL/6 mice, more typical of when BTK is removed from autoreactive B cells, as we have recently reported (15). Thus, B cells in K/BxN mice greatly rely on BTK-mediated signaling at all developmental stages. B1a B cells were also severely depleted, typical of BTK-deficient models (data not shown)(4, 7).
Figure 3. Btk-deficiency reduces mature B cell subsets in K/BxNs.
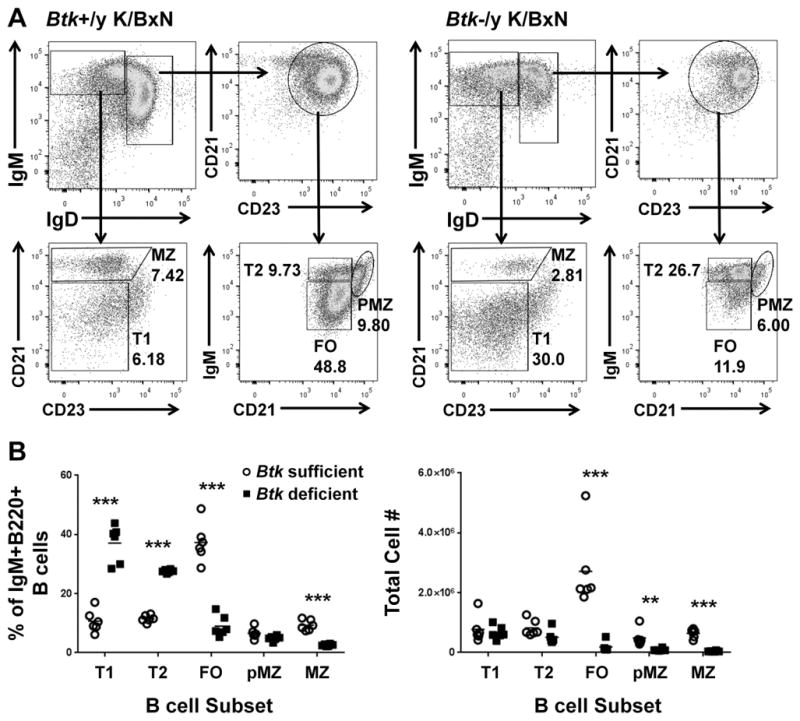
A) Representative flow plots for Btk-sufficient (left) and Btk-deficient (right) K/BxN, showing gating scheme for B cell subsets. Cells are gated as single, live, B220+ lymphocytes. B) Quantification of B cell subsets in Btk-sufficient (circles, n=6) or –deficient (squares, n=6) K/BxN by percentage of total IgM+IgD+ B cells (left) and by total cell number (right). **p≤0.01, ***p≤0.001, as calculated by the Holm-Sidak method of multiple T tests.
Germinal center B cells are decreased in spleens and popliteal lymph nodes of Btk-deficient K/BxNs
Formation of germinal centers (GCs) in spleen and draining lymph nodes is central to development of high-affinity anti-GPI IgG autoantibodies. GC formation was therefore assessed. CD19+/B220+/IgM+/IgDlo live lymphocytes were defined as GC B cells using GL7, FAS, and BCL6 (Fig 4). Percentages of GC B cells in the spleens were significantly reduced in Btk-deficient K/BxN (0.1086±0.043) versus Btk-sufficient K/BxN (1.18±0.5524) (p=0.0051). This decreased percentage corresponded to dramatic reduction in cell number (Btk-sufficient=1.69e5±1.46e5, Btk-deficient=3.63e3±1.68e3, p=0.0393). GC B cells were similarly reduced in draining popliteal LNs of Btk-deficient K/BxN (1.14±0.544, 4.73e3±5.44e3) compared to Btk-sufficient counterparts (1.83±0.273, 7.08e4±3.80e4) (p=0.026, p=0.0076). Thus, BTK contributions to autoimmune arthritis include development or expansion of GC B cells.
Figure 4. GC B cells and T follicular helper cells are decreased in Btk-deficient K/BxNs.

A) Representative flow plots of Btk-sufficient and –deficient K/BxN B220+CD19+ live splenocytes. IgMhiIgDlo gating was applied (not shown) and GL7+Fas+ cells (white) or GL7−Fas− controls (gray) evaluated for GC marker BCL6. GC B cells (GL7+Fas+IgMhiIgDloBCL6+) were quantified as percent of B cells, and total number in spleen (A, right panel) and popliteal LNs (B). C) Representative plots of splenic CD4+ live lymphocytes stained for Tfh markers PD-1 and CXCR5. Expression levels of BCL6, CD44 and ICOS in GC Tfh (PD1hiCXCR5hi) and Tfh (PD1+CXCR5+), shown as fold change over Btk-sufficient K/BxN non-Tfh cells (middle panel). GC Tfh, Tfh and non-Tfh were quantified as percentage of CD4+ cells (top) or total cell number (bottom) for spleen (C, right) and popliteal LNs (D). E) Serum anti-GPI IgG (top) and total IgG (bottom) were quantified by ELISA. Btk-sufficient, circles, n=6 for A–D; n=7–8 for E; Btk-deficient, squares, n=6 for A–D, n=7–9 for E. *p≤0.05, **p≤0.01, ***p≤0.001, calculated by unpaired T test with Welch’s correction (A, B, E), the Holm-Sidak method of multiple T tests (C right, D) or by a multi-way AVOVA with Sidak correction (C middle).
Germinal center T follicular helper cells are decreased in spleens and popliteal lymph nodes of Btk-deficient K/BxN mice, while non-Tfh T cells are unchanged
Though T cells do not rely on BTK for cell signaling, we found that their numbers were decreased in Btk-deficient K/BxN mice (Fig 2). B cell interactions with T cells drive T follicular helper (Tfh) cell formation and maintenance at several checkpoints, both at the T-B zone and within GCs (34). We therefore assessed Tfh cells in this model. Fig 4c (left) shows representative flow plots of Btk-sufficient K/BxN (top) and Btk-deficient K/BxN (bottom) splenocytes, gated on live, CD4+ lymphocytes. The Tfh markers PD-1 and CXCR5 were used for initial analysis. The double negative population is defined as non-Tfh, PD-1midCXCR5+ as Tfh, and PD-1hiCXCR5+ cells as GC Tfh (35). These cell subsets were additionally characterized by their expression of Tfh markers BCL6, CD44, and ICOS. Expression levels were quantified by flow cytometry and shown in Fig 4c (middle) as fold change compared to the Btk-sufficient K/BxN non-Tfh. BCL6 (top), the transcription factor that is associated with GC B and Tfh cells, was significantly increased in the GC Tfh compartment compared to non-Tfh cells. CD44 and ICOS were also significantly increased in both the Tfh and GC Tfh compartments, further confirming the cells’ classification. Fig 4c (right) shows each subset as a percentage of total CD4+ (top) or as number of total cells (bottom). Btk-deficient K/BxNs had increased percentage of non-Tfh cells (45.5±3.76) over Btk-sufficient K/BxN (32.8±8.76) (p=0.0087); however, the total number of non-Tfh cells was not significantly different. Therefore, the loss of Btk does not impact non-Tfh cells. The most dramatic phenotype was the GC Tfhs, decreased in both percentage and number in Btk-deficient K/BxNs (10.5±7.04, 9.91e4±6.41e4) compared to Btk-sufficient controls (27.7±11.3, 4.98e5±1.82e5) (p=0.0103, p=0.0005). Popliteal LN Tfh were determined identically to splenocytes (Fig 4d), and also showed reduced GC Tfh cell numbers in Btk-deficient (1.22e4±5.22e3) versus Btk-sufficient K/BxNs (6.15e4±4.14e4) (p=0.0159). These data demonstrate that a defect in B cells, the loss of BTK, affects germinal center T cells in K/BxN arthritis.
BTK-deficiency reduces anti-GPI antibodies
GCs are required for development of high affinity IgG antibodies, so loss of GC B and Tfh cells in Btk-deficient K/BxN indicates lack of support for production of anti-GPI autoantibodies. As GPI is an important autoantigen in both K/BxN and human rheumatoid arthritis (36), we next determined relative serum levels of anti-GPI autoantibody and found striking reduction in Btk-deficient K/BxN (Fig 4e, top, p=0.0004). In contrast, total IgG is only slightly decreased (Fig 4e, bottom, p=0.0146), which may reflect the loss of autoantibodies.
Imaging shows decreased macrophage infiltration in the paws of Btk-deficient K/BxNs
Innate cell contributions to arthritis are well known, so we next used whole-body fluorescent imaging to assess activated macrophage recruitment to inflamed synovia of K/BxN mice. This technique utilizes a fluorescent probe that binds FolRβ, an activation marker on macrophages, and allows sequential, noninvasive, evaluation of mice as arthritis develops (32). From weaning to 7 weeks of age, Btk-sufficient and Btk-deficient K/BxN mice were imaged and fluorescence in each paw measured. Fig 5a shows a representative Btk-sufficient and -deficient K/BxN mouse at 1 and 4 weeks post-wean dates. Fig 5b shows fluorescence of Btk-sufficient and Btk-deficient combined paws from week 1 to week 4 post weaning. This method indicates that significantly more activated macrophages were recruited to the paws in Btk-sufficient mice (p=0.0143).
Figure 5. FolRβ imaging shows increased levels of activated macrophages in the paws of Btk-sufficient K/BxN compared to Btk-deficient counterparts.
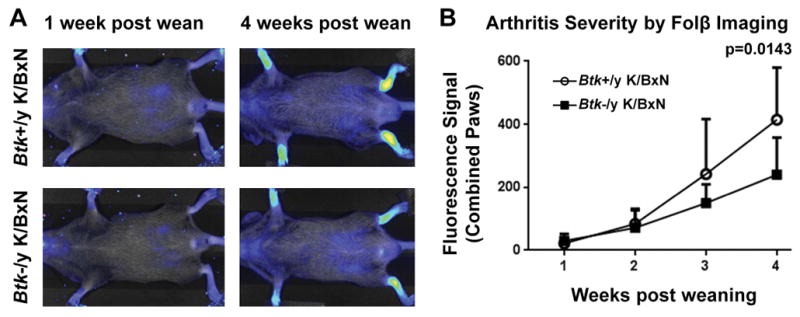
(A) Representative images of a Btk-sufficient (top) and Btk-deficient (bottom) K/BxN at 1 week post weaning and 4 weeks post weaning. Fluorescence in the paws was measured for quantification. (B) Macrophage infiltration into the paw is measured by FolRβ imaging for Btk-sufficient (circles, n=3) and –deficient (squares, n=4) Mean shown ± SD. p=0.0143 between genotypes, as calculated by a 2-way AVOVA with Sidak correction.
Btk-deficiency is not protective against development of serum transfer arthritis
To directly determine contributions of innate cell intrinsic BTK-signaling to the development of arthritis, we bypassed adaptive immune requirements by transferring K/BxN serum into Btk-sufficient or Btk-deficient NOD mice. Assessment of recipients by clinical score showed that serum transfer arthritis (STA) was not reduced by Btk-deficiency, with Btk-sufficient NOD reaching a clinical score of 4.29±0.756 and –deficient NODs reaching 4.57±0.976 on day 6 (genotype factor p=0.2671) (Fig 6a). In addition, FolRβ imaging of activated macrophages showed no difference between Btk-sufficient and –deficient recipient mice (p=0.5807), indicating that loss of macrophage-intrinsic BTK-mediated signaling did not significantly affect activation and recruitment by transferred autoantibodies (Fig 6b). This finding contrasted previous studies showing that BTK-inhibitors prevent arthritis in serum transfer models, including one that used K/BxN serum (19, 20, 37). We therefore assessed the effects of BTK-deficiency on innate cell numbers and function in spleens from Btk-sufficient and Btk-deficient NOD that served as recipients for these studies. Cell subsets were determined by flow cytometry, using the same criteria as in Figure 2, quantified by percent (Fig 6c, left) and total cell number (Figure 6c, right). We found that the percentages and numbers of splenic neutrophils were significantly increased in Btk-deficient mice (p=0.0063, p=0.0211). Btk-deficiency also resulted in a significant increase in percentage of DCs (p=0.0061), and a trend of higher DC numbers (p=0.1540). In addition, Btk-deficient DCs expressed significantly more CD11b than –sufficient controls (data not shown). Macrophage numbers did not differ, so we explored the effects of BTK-deficiency on their function by generating bone marrow-derived macrophages (BMDMs) and testing their ability to produce TNFα in response to stimulation via FcγRs and TLR4. As shown in Fig 6d, Btk-deficient BMDMs treated with K/BxN serum were able to produce TNFα above baseline (unstimulated=7.103±3.737, stimulated=27.605±4.705, p=0.0297), although the amount of TNFα trended lower than that of Btk-sufficient BMDM (43.182±11.819, p=0.0992). In addition, both Btk-deficient and Btk-sufficient BMDCs responded robustly to LPS, and did not differ in their ability to produce large amounts of TNFα (Btk-sufficient=7838±176.5, Btk-deficient=6819±866.4, p=0.1744) (Fig 6d, right), Thus, Btk-deficiency in this model increases neutrophil numbers and causes a slight trend downward in TNFα production by macrophages in response to K/BxN that is not sufficient to protect mice against STA. Importantly, these imaging studies also suggest that BTK-deficiency does not interfere significantly with the ability of activated macrophages to invade target tissues in response to autoantibodies.
Figure 6. Btk-deficiency is not protective in the serum-transfer model of arthritis.

(A) Btk-sufficient (circles, n=7) and Btk-deficient (squares, n=7) NOD mice were IP injected twice (day 0 and 2) with 200μL of pooled K/BxN sera. Clinical scores over 12 days post-injection are shown. (B) FolRβ imaging was performed on Btk-sufficient (n=3) and –deficient (n=3) NOD mice on day 0, 2, 7 and 10. Values are Mean ± SD. (C) Percentages (left) and total numbers (right) of Btk-sufficient (circles, n=3) and Btk-deficient (squares, n=3) age-matched male NOD. Cell subsets were gated as in Fig 2c. (D) Response of Btk-sufficient (circles, n=3) and Btk-deficient (squares, n=3) NOD BMDMs to incubation overnight with no stimulus, 1/20 K/BxN serum, or LPS. *p≤0.05, **p≤0.01, or as listed on graph, calculated by multi-way ANOVA with Sidak correction (A, D left), Holm-Sidak method of multiple T tests (C) or by unpaired T test with Welch’s correction (D, right).
Discussion
BTK is a promising therapeutic target in autoimmune arthritis, but its mechanisms of action in this disease have not been well-defined. This report presents the first detailed investigation of the role of BTK using genetic deletion in both spontaneous autoimmune and immune complex-mediated models of arthritis. These data demonstrate that Btk-deficiency in the K/BxN model significantly inhibits development of spontaneous arthritis, which depends upon both innate and adaptive immunity (Fig 1). To understand the mechanisms responsible, we undertook a detailed study of immune cells in this model. K/BxN B cells are extremely sensitive to BTK loss, suffering a 74% reduction in numbers (Figs 2 and 3), and T cells show mild reductions, despite the fact that they do not express BTK (Fig 2). Dendritic cell numbers are also somewhat reduced, while other components of the innate system, particularly macrophages, are not (Fig 2). BTK-deficiency strongly inhibits GC development, with large reductions in GC B cells and milder effects on GC Tfh, resulting in loss of anti-GPI autoantibodies that initiate autoimmune arthritis (Fig 4). Therefore, BTK is clearly implicated as supporting adaptive immune drivers of autoimmune arthritis.
The effect of BTK-deficiency on B cells in this model is profound, much more so than in any previous study using non-transgenic B cells. BTK is a well-defined cytosolic component of the signalosome that propagates signals from the BCR (6, 7, 38–42). Btk-deficiency reduces B cell numbers by 50% in C57BL/6 mice and by 18% in NOD mice (4, 7). Therefore, the extreme 74% reduction in overall B cell numbers in Btk-deficient K/BxN is striking. Analysis of K/BxN B cell subsets shows increased percentages of transitional cells in Btk-deficient mice, indicating a block at both T1 and T2 stages. While the T2 block is classically found in other models, the T1 block is usually far less pronounced (4, 7). Furthermore, unlike Btk-deficient C57BL/6 or NOD, B cells fail to accumulate at blocked stages in Btk-deficient K/BxN. This suggests significant cell loss at transitional stages, with further loss at the mature FO stage, resulting in very low FO numbers. This pattern is similar to transgenic models of autoimmunity, including an anti-insulin B cell model we reported to have 95% reduction in the absence of BTK (15, 43). Equally striking is the profound reduction of the MZ compartment, which begins even at the pMZ stage. Though in most models the MZ develops independently of BTK, we recently published that NOD MZ B cells rely in part on BTK signals (44). Even in NOD mice, however, loss of BTK causes a block at the pMZ stage, with an increase in numbers, and only partial reduction of MZ B cells (4). Again, this unusual reliance of the MZ compartment in Btk-deficient K/BxN mice mirrors anti-insulin B cells (15). The only other endogenous B cells known to rely so heavily on BTK-signaling are autoimmune-prone subsets such as B1a and anergic An1 (15). Further work is needed to determine the mechanism underlying this unusual pattern of B cell reduction in Btk-deficient K/BxN mice.
Germinal centers are critical to immune responses. GC B cells are primary responders in infection or autoimmunity, proliferating, undergoing somatic hypermutation and class switch to IgG, then transforming into antibody-producing plasma cells. BTK is known to contribute to GC formation (13, 45), so reduction of GC B cells in Btk-deficient K/BxN is unsurprising. BTK is not present in T cells, and indeed, those that reached the Tfh and GC Tfh stages did not exhibit loss of activation marker or transcription factor expression, as shown by quantification of BCL6, CD44, and ICOS (Fig 4). However, GC Tfh and B cells are reciprocally dependent. T-B interactions at multiple stages are necessary for Tfh development, including cognate interactions at the T-B border and non-cognate interactions that facilitate Tfh motility and follicular migration (34). Therefore we conclude that lack of available GC B cells in Btk-deficient mice removed cellular stimuli necessary for proper TfH development. GC failure in turn blocked development of anti-GPI autoantibodies (Fig. 4), consistent with loss of autoantibodies in other Btk-deficient models (4, 14, 46). Anti-GPI antibody is preferentially targeted, as BTK-deficiency resulted in an 83% decrease in anti-GPI IgG, but only 16% in total IgG. This reflects similar findings in models of lupus and T1D, and further supports the conclusion that loss of BTK profoundly affects autoreactive B cells (4, 14, 47).
In contrast to previous reports using pharmacologic inhibition (17, 19, 20), BTK-contributions to innate mediators of arthritis are not apparent in this genetically deficient model. While FolRβ imaging shows reduced synovial macrophage infiltration in the spontaneous model (Fig 5), this is likely secondary to reduced autoantibodies, since there is no difference in clinical score or FolRβ outcomes when autoantibodies are supplied exogenously in the serum transfer model (Fig. 6). The most obvious way to interpret this difference in outcomes is to attribute the efficacy of BTK-inhibitors to off-target effects. Most recent studies regarding the role of BTK in autoimmune arthritis have focused on its role in FcγR stimulated phagocytosis and cytokine production by macrophages (10, 12, 16, 17, 19, 20, 48) and have relied solely on BTK inhibitors, rather than genetic deletion, to make their conclusions. However, kinase-specific inhibition is difficult. For example, ibrutinib also binds many other kinases, including Tec, Jak3 and, importantly, the T cell signaling protein ITK, with known effects on T cell function (21). LFM-A13, used in well-cited macrophage studies, also interacts with Tec (10, 49). Our studies indicate that BTK-deficiency may blunt, but does not eliminate, macrophage inflammatory responses as measured by TNFα production by BMDCs in response to K/BxN serum autoantibodies (Fig 6d). The fact that macrophages also respond very dramatically to TLR4 stimulation, regardless of BTK status, further supports the idea that BTK may play only a minor role in macrophage driven inflammation. Our findings are the first to use BTK-deficiency, rather than a small molecular inhibitor, to study the role of this protein in innate cell contributions to arthritis, and demonstrate the need for further studies.
Overall, macrophage and other innate cell numbers were mostly stable, with the exception of dendritic cells, which were decreased in Btk-deficient K/BxNs. This is not the case in Btk-deficient non-diabetic NODs (Fig 6c), suggesting that changes in dendritic cell numbers are not due to a developmental block. Rather, the ongoing immune reaction in BTK-sufficient K/BxN most likely drives expansion of DCs needed to facilitate antigen-presentation in the T cell zone. Alternatively, loss of BTK-signaling from innate receptors in DCs may contribute indirectly to failure of adaptive responses. Interestingly, previous studies using C57BL/6 mice have shown that Btk-deficient DCs have reduced IL10 production, and exhibit increased T cell stimulatory activity (50). Thus, the role of BTK in DC contributions to autoimmune arthritis requires additional investigation, and would benefit from studies using DC-targeted deletion in the future. Of note, the increase in neutrophil numbers found in Btk-deficient NOD recipients of K/BxN serum do not necessarily reflect increased functional contributions to inflammation in that model. Btk-deficient neutrophils in xid and C57BL/6 mouse models showed decreased E-selectin mediated recruitment (51) and decreased granules per cell (52). Interestingly, this contrasts neutrophils from human XLA patients that have showed no defects in effector function (53), and even had increased production of reactive oxygen species (54). Future studies are necessary to resolve these conflicts in the literature.
This study is the first to rigorously define contributions of BTK to autoimmune arthritis. The findings support development of BTK-inhibitors for RA but demonstrate the need to more completely understand their effects on the immune system. Furthermore, sensitivity of B cells to BTK inhibition could potentially be used as an indicator of autoreactivity to predict response to treatment as we move towards a personalized approach to treatment of RA.
Acknowledgments
This work was supported by the Department of Veterans’ Affairs (TSB) and by National Institutes of Health Grants: National Institute of Arthritis and Musculoskeletal and Skin Diseases R01 AR049010 (LC), National Institute of Diabetes and Digestive and Kidney Diseases R01 DK084246 (PLK), and National Institute of Heart, Lung and Blood R01 HL116358 (TSB) and T32HL069765 (LEN), as well as by Juvenile Diabetes Research Foundation Grant 3-2013-121(RHB). Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). Histology was performed by the Translational Pathology Shared Resource, supported by the Mouse Metabolic & Phenotyping Center National Institutes of Health Grant 5U24 DK059637 from the National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Kendall is the inventor of a Utility Patent Application: Methods for delaying or preventing the onset of type 1 diabetes, which regards the use of BTK-inhibitors for treatment of autoimmune diabetes.
References
- 1.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. The New England journal of medicine. 2011;365(23):2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 2.Donahue KE, Gartlehner G, Jonas DE, Lux LJ, Thieda P, Jonas BL, et al. Systematic review: comparative effectiveness and harms of disease-modifying medications for rheumatoid arthritis. Annals of internal medicine. 2008;148(2):124–34. doi: 10.7326/0003-4819-148-2-200801150-00192. [DOI] [PubMed] [Google Scholar]
- 3.Smolen JS, Aletaha D. Rheumatoid arthritis therapy reappraisal: strategies, opportunities and challenges. Nature reviews Rheumatology. 2015;11(5):276–89. doi: 10.1038/nrrheum.2015.8. [DOI] [PubMed] [Google Scholar]
- 4.Kendall PL, Moore DJ, Hulbert C, Hoek KL, Khan WN, Thomas JW. Reduced diabetes in btk-deficient nonobese diabetic mice and restoration of diabetes with provision of an anti-insulin IgH chain transgene. J Immunol. 2009;183(10):6403–12. doi: 10.4049/jimmunol.0900367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antony P, Petro JB, Carlesso G, Shinners NP, Lowe J, Khan WN. B cell receptor directs the activation of NFAT and NF-kappaB via distinct molecular mechanisms. Experimental cell research. 2003;291(1):11–24. doi: 10.1016/s0014-4827(03)00338-0. [DOI] [PubMed] [Google Scholar]
- 6.Khan WN. Regulation of B lymphocyte development and activation by Bruton’s tyrosine kinase. Immunologic research. 2001;23(2–3):147–56. doi: 10.1385/IR:23:2-3:147. [DOI] [PubMed] [Google Scholar]
- 7.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3(3):283–99. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 8.Petro JB, Khan WN. Phospholipase C-gamma 2 couples Bruton’s tyrosine kinase to the NF-kappaB signaling pathway in B lymphocytes. The Journal of biological chemistry. 2001;276(3):1715–9. doi: 10.1074/jbc.M009137200. [DOI] [PubMed] [Google Scholar]
- 9.Petro JB, Rahman SM, Ballard DW, Khan WN. Bruton’s tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. The Journal of experimental medicine. 2000;191(10):1745–54. doi: 10.1084/jem.191.10.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jongstra-Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol. 2008;181(1):288–98. doi: 10.4049/jimmunol.181.1.288. [DOI] [PubMed] [Google Scholar]
- 11.Paracha RZ, Ali A, Ahmad J, Hussain R, Niazi U, Muhammad SA. Structural evaluation of BTK and PKCdelta mediated phosphorylation of MAL at positions Tyr86 and Tyr106. Computational biology and chemistry. 2014;51:22–35. doi: 10.1016/j.compbiolchem.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Hartkamp LM, Fine JS, van Es IE, Tang MW, Smith M, Woods J, et al. Btk inhibition suppresses agonist-induced human macrophage activation and inflammatory gene expression in RA synovial tissue explants. Annals of the rheumatic diseases. 2015;74(8):1603–11. doi: 10.1136/annrheumdis-2013-204143. [DOI] [PubMed] [Google Scholar]
- 13.Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood. 2012;119(16):3744–56. doi: 10.1182/blood-2011-12-397919. [DOI] [PubMed] [Google Scholar]
- 14.Whyburn LR, Halcomb KE, Contreras CM, Lowell CA, Witte ON, Satterthwaite AB. Reduced dosage of Bruton’s tyrosine kinase uncouples B cell hyperresponsiveness from autoimmunity in lyn−/− mice. J Immunol. 2003;171(4):1850–8. doi: 10.4049/jimmunol.171.4.1850. [DOI] [PubMed] [Google Scholar]
- 15.Bonami RH, Sullivan AM, Case JB, Steinberg HE, Hoek KL, Khan WN, et al. Bruton’s tyrosine kinase promotes persistence of mature anti-insulin B cells. J Immunol. 2014;192(4):1459–70. doi: 10.4049/jimmunol.1300125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. The Journal of pharmacology and experimental therapeutics. 2013;346(2):219–28. doi: 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- 17.Xu D, Kim Y, Postelnek J, Vu MD, Hu DQ, Liao C, et al. RN486, a selective Bruton’s tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. The Journal of pharmacology and experimental therapeutics. 2012;341(1):90–103. doi: 10.1124/jpet.111.187740. [DOI] [PubMed] [Google Scholar]
- 18.Robak T, Robak E. Tyrosine kinase inhibitors as potential drugs for B-cell lymphoid malignancies and autoimmune disorders. Expert opinion on investigational drugs. 2012;21(7):921–47. doi: 10.1517/13543784.2012.685650. [DOI] [PubMed] [Google Scholar]
- 19.Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nature chemical biology. 2011;7(1):41–50. doi: 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- 20.Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, et al. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis research & therapy. 2011;13(4):R115. doi: 10.1186/ar3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–49. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansson L, Holmdahl R. Genes on the X chromosome affect development of collagen-induced arthritis in mice. Clinical and experimental immunology. 1993;94(3):459–65. doi: 10.1111/j.1365-2249.1993.tb08218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87(5):811–22. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286(5445):1732–5. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 25.Kyburz D, Corr M. The KRN mouse model of inflammatory arthritis. Springer seminars in immunopathology. 2003;25(1):79–90. doi: 10.1007/s00281-003-0131-5. [DOI] [PubMed] [Google Scholar]
- 26.Ji H, Korganow AS, Mangialaio S, Hoglund P, Andre I, Luhder F, et al. Different modes of pathogenesis in T-cell-dependent autoimmunity: clues from two TCR transgenic systems. Immunological reviews. 1999;169:139–46. doi: 10.1111/j.1600-065x.1999.tb01312.x. [DOI] [PubMed] [Google Scholar]
- 27.Block KE, Huang H. The cellular source and target of IL-21 in K/BxN autoimmune arthritis. J Immunol. 2013;191(6):2948–55. doi: 10.4049/jimmunol.1301173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh A, Leng L, Fan J, Gajda M, Brauer R, Fingerle-Rowson G, et al. Macrophage-derived, macrophage migration inhibitory factor (MIF) is necessary to induce disease in the K/BxN serum-induced model of arthritis. Rheumatology international. 2013;33(9):2301–8. doi: 10.1007/s00296-013-2713-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang QQ, Birkett R, Koessler RE, Cuda CM, Haines GK, 3rd, Jin JP, et al. Fas signaling in macrophages promotes chronicity in K/BxN serum-induced arthritis. Arthritis Rheumatol. 2014;66(1):68–77. doi: 10.1002/art.38198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, et al. Arthritis critically dependent on innate immune system players. Immunity. 2002;16(2):157–68. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 31.Kojima F, Kapoor M, Yang L, Fleishaker EL, Ward MR, Monrad SU, et al. Defective generation of a humoral immune response is associated with a reduced incidence and severity of collagen-induced arthritis in microsomal prostaglandin E synthase-1 null mice. J Immunol. 2008;180(12):8361–8. doi: 10.4049/jimmunol.180.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han W, Zaynagetdinov R, Yull FE, Polosukhin VV, Gleaves LA, Tanjore H, et al. Molecular imaging of folate receptor beta-positive macrophages during acute lung inflammation. American journal of respiratory cell and molecular biology. 2015;53(1):50–9. doi: 10.1165/rcmb.2014-0289OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan WN, Sideras P, Rosen FS, Alt FW. The role of Bruton’s tyrosine kinase in B-cell development and function in mice and man. Annals of the New York Academy of Sciences. 1995;764:27–38. doi: 10.1111/j.1749-6632.1995.tb55802.x. [DOI] [PubMed] [Google Scholar]
- 34.Pratama A, Vinuesa CG. Control of TFH cell numbers: why and how? Immunology and cell biology. 2014;92(1):40–8. doi: 10.1038/icb.2013.69. [DOI] [PubMed] [Google Scholar]
- 35.Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. The Journal of experimental medicine. 2015;212(4):539–53. doi: 10.1084/jem.20141380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaller M, Burton DR, Ditzel HJ. Autoantibodies to GPI in rheumatoid arthritis: linkage between an animal model and human disease. Nature immunology. 2001;2(8):746–53. doi: 10.1038/90696. [DOI] [PubMed] [Google Scholar]
- 37.Lou Y, Han X, Kuglstatter A, Kondru RK, Sweeney ZK, Soth M, et al. Structure-based drug design of RN486, a potent and selective Bruton’s tyrosine kinase (BTK) inhibitor, for the treatment of rheumatoid arthritis. Journal of medicinal chemistry. 2015;58(1):512–6. doi: 10.1021/jm500305p. [DOI] [PubMed] [Google Scholar]
- 38.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Molecular immunology. 2004;41(6–7):599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Shinners NP, Carlesso G, Castro I, Hoek KL, Corn RA, Woodland RT, et al. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J Immunol. 2007;179(6):3872–80. doi: 10.4049/jimmunol.179.6.3872. [DOI] [PubMed] [Google Scholar]
- 40.Satterthwaite AB, Lowell CA, Khan WN, Sideras P, Alt FW, Witte ON. Independent and opposing roles for Btk and lyn in B and myeloid signaling pathways. The Journal of experimental medicine. 1998;188(5):833–44. doi: 10.1084/jem.188.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Middendorp S, Dingjan GM, Maas A, Dahlenborg K, Hendriks RW. Function of Bruton’s tyrosine kinase during B cell development is partially independent of its catalytic activity. J Immunol. 2003;171(11):5988–96. doi: 10.4049/jimmunol.171.11.5988. [DOI] [PubMed] [Google Scholar]
- 42.Kersseboom R, Ta VB, Zijlstra AJ, Middendorp S, Jumaa H, van Loo PF, et al. Bruton’s tyrosine kinase and SLP-65 regulate pre-B cell differentiation and the induction of Ig light chain gene rearrangement. J Immunol. 2006;176(8):4543–52. doi: 10.4049/jimmunol.176.8.4543. [DOI] [PubMed] [Google Scholar]
- 43.Middendorp S, Hendriks RW. Cellular maturation defects in Bruton’s tyrosine kinase-deficient immature B cells are amplified by premature B cell receptor expression and reduced by receptor editing. J Immunol. 2004;172(3):1371–9. doi: 10.4049/jimmunol.172.3.1371. [DOI] [PubMed] [Google Scholar]
- 44.Case JB, Bonami RH, Nyhoff LE, Steinberg HE, Sullivan AM, Kendall PL. Bruton’s Tyrosine Kinase Synergizes with Notch2 To Govern Marginal Zone B Cells in Nonobese Diabetic Mice. J Immunol. 2015;195(1):61–70. doi: 10.4049/jimmunol.1400803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vinuesa CG, Sunners Y, Pongracz J, Ball J, Toellner KM, Taylor D, et al. Tracking the response of Xid B cells in vivo: TI-2 antigen induces migration and proliferation but Btk is essential for terminal differentiation. European journal of immunology. 2001;31(5):1340–50. doi: 10.1002/1521-4141(200105)31:5<1340::AID-IMMU1340>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 46.Halcomb KE, Musuka S, Gutierrez T, Wright HL, Satterthwaite AB. Btk regulates localization, in vivo activation, and class switching of anti-DNA B cells. Molecular immunology. 2008;46(2):233–41. doi: 10.1016/j.molimm.2008.08.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steinberg BJ, Smathers PA, Frederiksen K, Steinberg AD. Ability of the xid gene to prevent autoimmunity in (NZB X NZW)F1 mice during the course of their natural history, after polyclonal stimulation, or following immunization with DNA. The Journal of clinical investigation. 1982;70(3):587–97. doi: 10.1172/JCI110651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rankin AL, Seth N, Keegan S, Andreyeva T, Cook TA, Edmonds J, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol. 2013;191(9):4540–50. doi: 10.4049/jimmunol.1301553. [DOI] [PubMed] [Google Scholar]
- 49.Gilbert C, Levasseur S, Desaulniers P, Dusseault AA, Thibault N, Bourgoin SG, et al. Chemotactic factor-induced recruitment and activation of Tec family kinases in human neutrophils. II. Effects of LFM-A13, a specific Btk inhibitor. J Immunol. 2003;170(10):5235–43. doi: 10.4049/jimmunol.170.10.5235. [DOI] [PubMed] [Google Scholar]
- 50.Kawakami Y, Inagaki N, Salek-Ardakani S, Kitaura J, Tanaka H, Nagao K, et al. Regulation of dendritic cell maturation and function by Bruton’s tyrosine kinase via IL-10 and Stat3. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(1):153–8. doi: 10.1073/pnas.0509784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, et al. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood. 2010;115(15):3118–27. doi: 10.1182/blood-2009-11-254185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fiedler K, Sindrilaru A, Terszowski G, Kokai E, Feyerabend TB, Bullinger L, et al. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood. 2011;117(4):1329–39. doi: 10.1182/blood-2010-04-281170. [DOI] [PubMed] [Google Scholar]
- 53.Marron TU, Rohr K, Martinez-Gallo M, Yu J, Cunningham-Rundles C. TLR signaling and effector functions are intact in XLA neutrophils. Clin Immunol. 2010;137(1):74–80. doi: 10.1016/j.clim.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK, et al. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nature immunology. 2012;13(4):369–78. doi: 10.1038/ni.2234. [DOI] [PubMed] [Google Scholar]