

Abstract
Genome-wide association studies (GWAS) have identified multiple, shared allelic associations with many autoimmune diseases. However, the pathogenic contributions of variants residing in risk loci remain unresolved. The location of the majority of shared disease-associated variants in noncoding regions suggests they contribute to risk of autoimmunity through effects on gene expression in the immune system. In the current study, we test this hypothesis by applying RNA sequencing to CD4+, CD8+, and CD19+ lymphocyte populations isolated from 81 subjects with type 1 diabetes (T1D). We characterize and compare the expression patterns across these cell types for three gene sets: all genes, the set of genes implicated in autoimmune disease risk by GWAS, and the subset of these genes specifically implicated in T1D. We performed RNA sequencing and aligned the reads to both the human reference genome and a catalog of all possible splicing events developed from the genome, thereby providing a comprehensive evaluation of the roles of gene expression and alternative splicing (AS) in autoimmunity. Autoimmune candidate genes displayed greater expression specificity in the three lymphocyte populations relative to other genes, with significantly increased levels of splicing events, particularly those predicted to have substantial effects on protein isoform structure and function (e.g., intron retention, exon skipping). The majority of single-nucleotide polymorphisms within T1D-associated loci were also associated with one or more cis-expression quantitative trait loci (cis-eQTLs) and/or splicing eQTLs. Our findings highlight a substantial, and previously underrecognized, role for AS in the pathogenesis of autoimmune disorders and particularly for T1D.
Type 1 diabetes (T1D) is a disorder of glucose homeostasis that results from T-cell–mediated destruction of the insulin-producing pancreatic beta-cells (Tisch and McDevitt 1996; Delovitch and Singh 1997; Shirangi et al. 2009). Twin concordance rates (∼40% in monozygotic and ∼8% in dizygotic twins) and the aggregation of T1D within families (risk in siblings ∼8%; population prevalence 0.4%) are consistent with a substantial genetic contribution to T1D risk (Barnett et al. 1981; Rich 1990; Redondo et al. 2001; Hyttinen et al. 2003). The human leukocyte antigen (HLA) gene cluster in the major histocompatibility complex (MHC) on Chromosome 6p21 was the first locus to be associated with T1D (Nerup et al. 1974), and as much as half of all the genetic risk for T1D has been attributed to this locus (Todd et al. 1987), with three amino acid positions (HLA-DQβ1 position 57, HLA-DRβ1 positions 13, and 71) accounting for ∼90% of the risk in this region (Hu et al. 2015).
Genome-wide linkage and association studies have identified more than 50 non-MHC loci containing single-nucleotide polymorphisms (SNPs) exhibiting genome-wide significant evidence of association with T1D (Burton et al. 2007; Hakonarson et al. 2007; Barrett et al. 2009; Concannon et al. 2009). The majority of these loci also display associations with additional autoimmune diseases (Onengut-Gumuscu et al. 2015). This suggests that the risk alleles at these SNPs modify genes acting on the immune system rather than on disease-specific target tissues. Fine mapping has revealed that the majority of the potentially causal SNPs are located in noncoding regions (Cortes and Brown 2011). These SNPs are enriched in enhancers, but not promoters, active in immune-relevant cell types: thymus, T cells (CD4+ and CD8+), B cells (CD19+), and stem cells (CD34+), consistent with the increased sharing of susceptibility regions across multiple autoimmune diseases and the expected effects of risk variants on the immune system (Onengut-Gumuscu et al. 2015). In the current study, we build upon these findings, applying RNA sequencing to three of these cell types (CD4+ T cells, CD8+ T cells, and CD19+ B cells) to explore relationships between T1D/autoimmune risk, gene expression, and alternative splicing (AS).
Results
Differential exon usage
A gene was considered expressed if at least one of its exons was detected. There were 47,063 AceView genes expressed (which includes 28,502 AceView “cloud genes,” often-monoexon cDNA clones of unknown biological significance that have not yet been assigned to a known gene) (Thierry-Mieg and Thierry-Mieg 2006), of which 87% were expressed in all three cell types, 6% in at least two cell types, and 7% in only one cell type. Overall, 13% of genes provided evidence for cell-type specificity in expression (Fig. 1A).
Figure 1.

Summary of gene expression and splicing analysis. (A) Genes expressed in CD4+ T cells (red), CD8+ T cells (blue), and CD19+ B cells (green). (B) Proportion of detected exons and splicing events of all genes expressed in at least two cell types in one cell type (red), two cell types (yellow), and all three cell types (blue). The proportion of exons and AS events detected in all three cell types and quantitatively different in at least one cell type is indicated by red hatching. (C) Autoimmune candidate genes expressed in CD4+ T cells, CD8+ T cells, and CD19+ B cells. (D) Proportion of detected exons and splicing events of autoimmune candidate genes expressed in one cell type (red), two cell types (yellow), and all three cell types (blue). (E) T1D candidate genes expressed in CD4+ T cells, CD8+ T cells, and CD19+ B cells. (F) Proportion of detected exons and splicing events of T1D candidate genes expressed in one cell type (red), two cell types (yellow), and all three cell types (blue). (G) cis-eQTL and sQTL in autoimmune disease candidate genes for exons and splicing events detected in all three cell types. The proportion of s/eQTL significant (FDR P < 0.05) in one cell type (red), two cell types (yellow), and all three cell types (blue).
For genes expressed in two cell types, any individual exon that is detected in one cell type but not the other is evidence for alternative exon usage that is most likely to arise from AS. In a comparison of CD4+ and CD8+ T cells, 11% of the 43,314 genes expressed in both cell types had at least one exon detected in only one cell type. In a comparison of T and B cells, the rates of alternative exon usage are higher—17% of approximately 42,000 genes). Altogether, 8077 genes (17% of all genes examined) demonstrated evidence of alternative exon usage.
Applying the same analyses to genes located in chromosomal regions associated with any autoimmune disease or specifically with T1D revealed a much higher rate of alternative exon usage than that observed when all genes were considered. While the majority of these genes (96% of 1854) were expressed in all three cell types (Fig. 1C), 21% of the 1660 genes expressed in CD4+ and CD8+ T cells had at least one exon detected in only one cell type. Alternative exon usage between T cells and B cells was even higher, with ∼33% of 1637 genes detected in both CD4+ T cells and CD19+ B cells, and 32% of 1642 genes detected in both CD8+ T cells and CD19+ B cells. In total, 37% of the 1690 expressed autoimmune genes demonstrated evidence of alternative exon usage, significantly higher than the 17% observed for all genes. T1D candidate genes were similar to the set of all autoimmune candidate genes (Fig. 1E).
Differential exon expression
Of the 163,713 exons from 33,318 genes detected in all three cell types, 76.5% were differentially expressed (DE; FDR P < 0.05) between at least two of the three cell types (Fig. 1B; Supplemental Fig. S1).
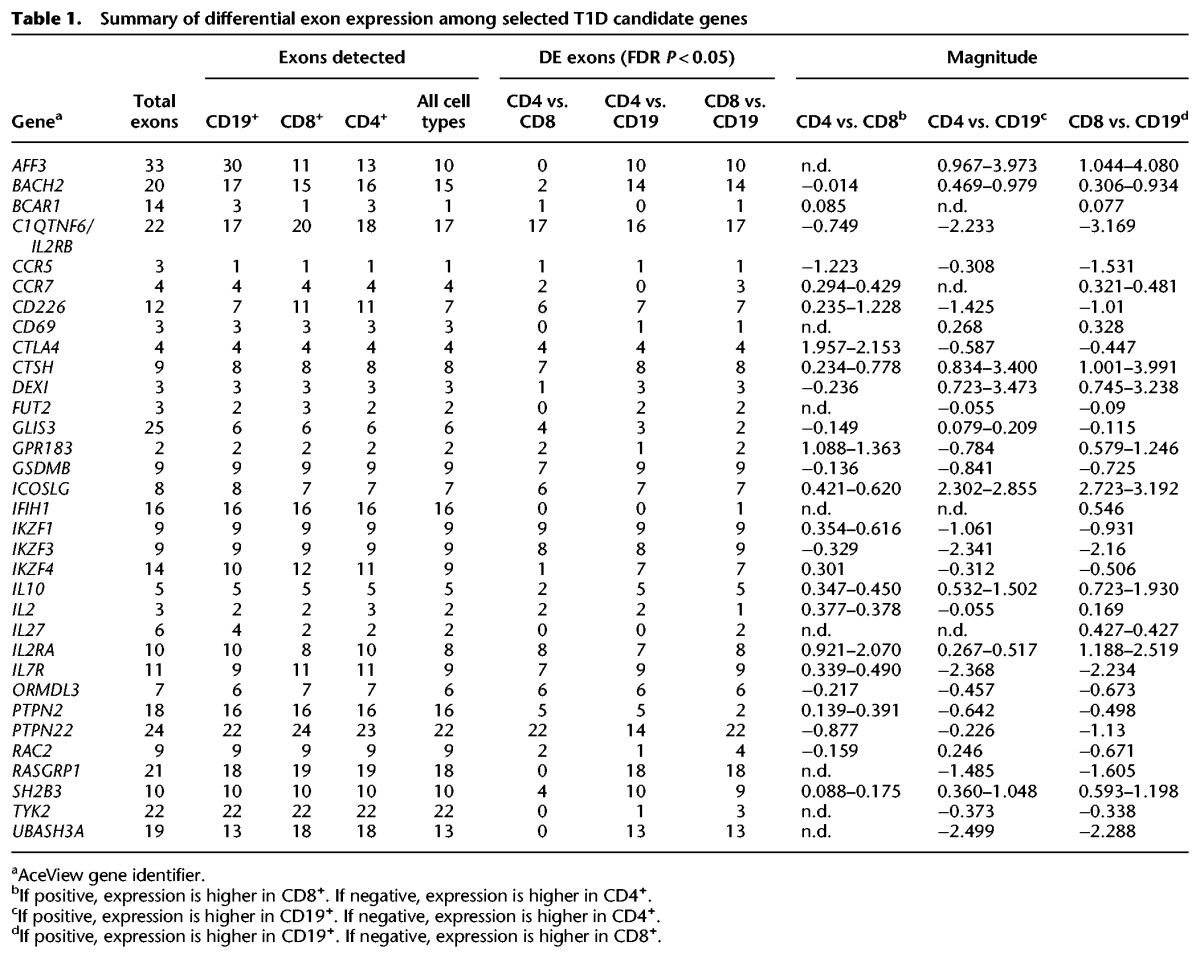
A summary of the differences in exon expression for a subset of T1D candidate genes (Onengut-Gumuscu et al, 2015) is provided in Table 1, and the complete results of the differential exon expression analysis is provided in Supplemental Table S1 with the autoimmune candidate genes indicated in column 9 and the T1D candidate genes indicated in column 10.
Table 1.
Summary of differential exon expression among selected T1D candidate genes
Differential detection of splicing events
Comparing T cells and B cells revealed that ∼25% of genes demonstrated alternative junction event detection (24% and 25% of genes detected in both cell types for comparisons of CD4+ T cells and CD8+ T cells, respectively, to CD19+ B cells). Consistent with the comparisons of alternative exon usage, the rate of alternative junction detection was higher between T and B cells than between T cell types (17% of genes detected in both CD4+ T cells and CD8+ T cell types) (Supplemental Fig. S2).
In total, 15,024 genes expressed in the three cell types studied had evidence of AS (32% of 47,063 genes), where a gene is counted if it had at least one differentially detected exon or at least one differentially detected splicing event. Restricting consideration to only autoimmune or T1D candidate genes resulted in a dramatic enrichment in AS; 76% of expressed autoimmune candidate genes (1690 genes) and 72% of T1D candidate genes (405 genes) had evidence of alternative isoform production (P < 0.0001) (Fig. 1B,D,F; Supplemental Fig. S3). The complete results of the splicing event analysis are provided in Supplemental Table S2, with the autoimmune genes indicated in column 3 and the T1D genes indicated in column 4.
Combining AS and differential expression
While the majority of the expressed genes are DE between the three cell types assayed (33,755 of 47,063 expressed genes, 72%) (Table 2), almost all of the genes with evidence of AS are also considered DE (14,056 of 15,024 alternatively spliced genes, 94%) (Table 2). Among both the autoimmune and T1D candidate genes, 98% of the genes considered alternatively spliced are also DE between the three cell types (1259 of 1280 alternatively spliced autoimmune genes) and 95% of all expressed autoimmune genes were DE (1603 of 1690 genes) (Table 2).
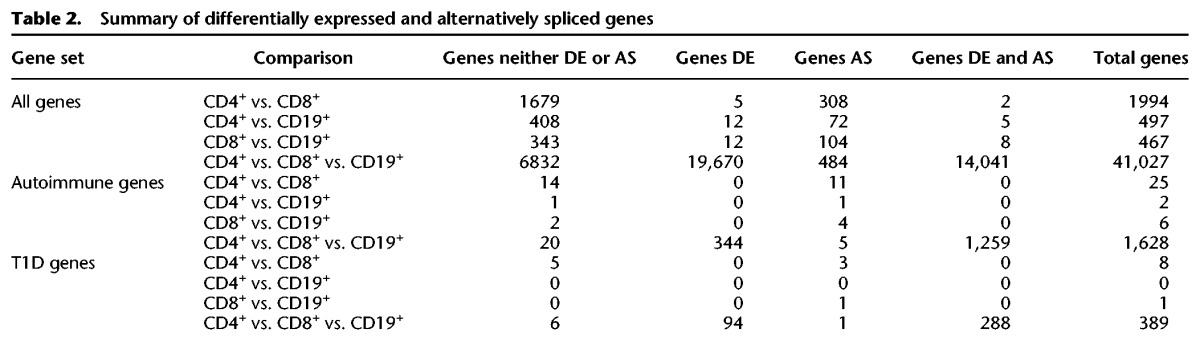
Table 2.
Summary of differentially expressed and alternatively spliced genes
Identification of lymphocyte cis-eQTLs and sQTLs
ImmunoChip SNPs were tested for association with the expression of exons and splicing events from genes in these regions. SNPs were included in the analysis if they could be assigned to one or more of the 1452 genes annotated as a T1D or (other) autoimmune disease candidate that had at least one expressed splicing event type (exon, junction, or IR event) in at least one cell type. There were 7871 significant cis-expression quantitative trait loci (cis-eQTLs) detected at 637 genes (FDR P < 0.05) (Fig. 1G; Supplemental Fig. S4; Supplemental Table S3). While most of the genes were expressed in all three cell types assayed, the majority of the cis-eQTLs were specific to a single cell type (5358 of 7168 cis-eQTLs; 75%) (Fig. 1G; Supplemental Table S4).
The significant cis-eQTLs included 4456 that were associated with splicing events (sQTLs) affecting 427 genes. These sQTLs were further categorized by splicing event type. Similar degrees of cell-type specificity were observed, although putative IR events detected in all three cell types were much more likely to have significant associated sQTLs than other splicing events (P < 0.001) (Supplemental Fig. S4; Supplemental Table S4). About half of the T1D genes with SNP coverage had at least one significant cis-eQTL (156 of 320 genes, 49%) (Supplemental Table S5).
Intersection of T1D-associated SNPs and lymphocyte cis-eQTLs
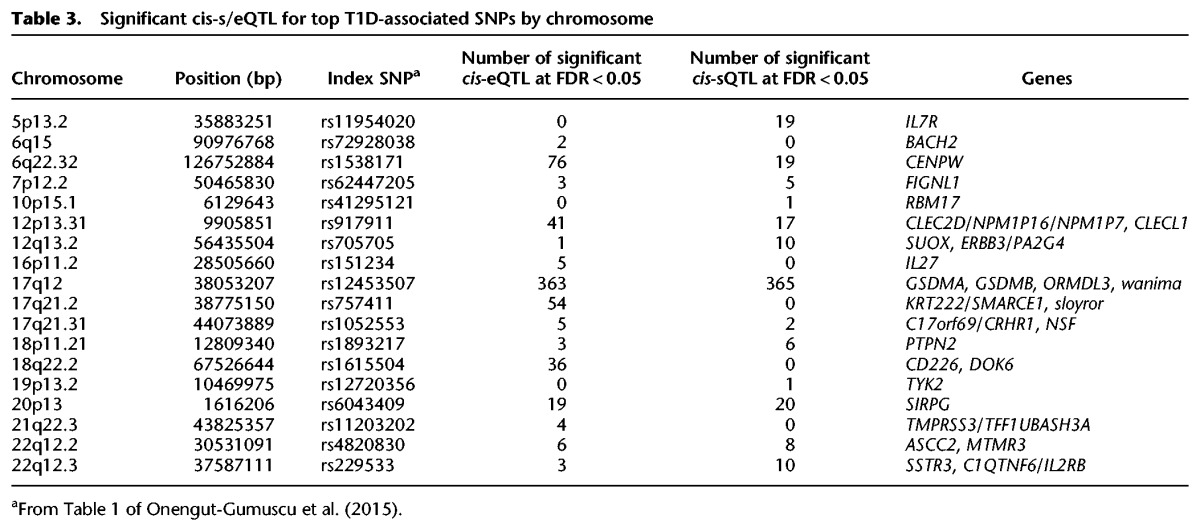
GWAS and subsequent fine mapping on ImmunoChip (Onengut-Gumuscu et al. 2015) has identified significant associations with T1D in 51 distinct chromosomal regions in the genome. To assess the possible functional impact of these associations, the most significant T1D-associated SNP in each region was intersected with the set of significant cis-eQTLs. Three regions were excluded due to missing data for the T1D risk defining SNPs (rs689 on Chromosome 11, INS; rs2611215 on Chromosome 4, CPE/TLL1, and rs35667974 on Chromosome 2, IFIH1). At 41 of the remaining 48 regions, alleles of significant T1D-associated SNPs were correlated (r2 > 0.8) with alleles of one or more cis-eQTLs. Of these 41 SNPs, 18 were correlated (r2 > 0.8) with one or more sQTLs affecting a total of 29 genes (Table 3; Supplemental Tables S6, S7).
Table 3.
Significant cis-s/eQTL for top T1D-associated SNPs by chromosome
An example of a novel T1D-associated cis-eQTL detected in the current study is the association between alleles at rs1893592 and expression of the gene UBASH3A. The minor “C” allele at rs1893592 is associated with a reduced risk of several different autoimmune diseases (Trynka et al. 2011; Okada et al. 2014), and other SNPs in the region have been associated with T1D (Concannon et al. 2008; Barrett et al. 2009). The “C” allele of rs1893592 replaces the more common purine residue at the +3 position of the splice donor sequence following exon 10 of UBASH3A with a less commonly used pyrimidine (Exome Aggregation Consortium et al. 2015). Increased retention of introns 10 and 11 of the major UBASH3A transcript (introns 11 and 12 of the UBASH3A gene) was observed in carriers of the “C” allele of rs1893592 in both CD4+ (Fig. 2) and CD8+ T cells. The “C” allele at rs1893592 was also associated with elevated expression of eight exons and three splice junctions in CD4+ T cells, suggesting that rs1893592 may also alter overall expression of the UBASH3A gene (Fig. 2). While the retention of introns 10 and 11 were apparent in individuals homozygous for the minor allele of rs1893592 (and in heterozygotes to a lesser extent), additional subject-specific splicing dysregulation was also evident, such as the retention of additional introns (Supplemental Fig. S5). A similar example illustrating the effect of rs1217414 on PTPN22 expression and splicing is presented in Supplemental Figure S6.
Figure 2.
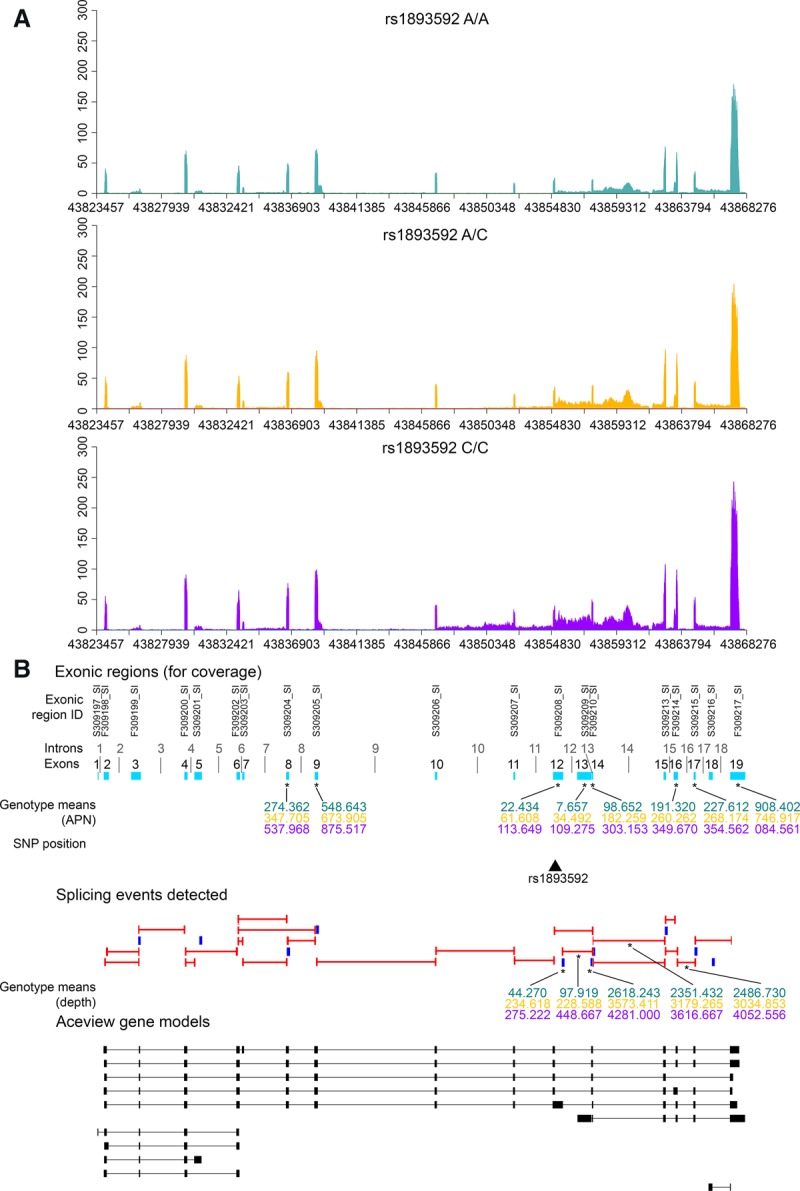
UBASH3A gene expression by rs1893592 genotypes in CD4+ T cells. (A) Distribution of sequencing coverage across UBASH3A for each genotype of rs1893592: A/A (teal), A/C (gold), C/C (purple). (B) Splicing events detected in UBASH3A and UBASH3A gene models from AceView annotations. Detected junctions are indicated by red bars and putative IR events by blue blocks. AceView gene models for UBASH3A are indicated by black blocks and bars, and cyan blocks represent the collapsed gene model for UBASH3A. Splicing events and exons associated with rs1893592 are denoted by an asterisk (*), with mean coverage for the A/A (teal), A/C (gold), and C/C (purple) genotypes. Exons (cyan blocks in collapsed gene models) are numbered sequentially by start and stop positions 5′ to 3′ on the + strand.
Discussion
Variation in the regulation of gene expression is an important mechanism for determining cellular programming and phenotypes. Dysregulation of this process has been implicated in the molecular etiology of many human diseases (Stanford et al. 2000; Cartegni et al. 2006; Sellier et al. 2010; Arnold et al. 2013; Sevcik et al. 2013; Qiu et al. 2014; Danan-Gotthold et al. 2015; Liu et al. 2015; Wen et al. 2015). Dysregulation can arise through effects on synthesis or degradation of transcripts resulting in over- or underexpression of protein products, or through effects on splicing. In the case of autoimmune diseases in general and T1D specifically, a few individual examples of AS at risk loci have been described (Ueda et al. 2003; Kralovicova et al. 2006; Onengut-Gumuscu et al. 2006; Kralovicova and Vorechovsky 2010; Gerold et al. 2011; Ge et al. 2016), and a broader role for AS in disease risk has been hypothesized (Juan-Mateu et al. 2015). These considerations, as well as our prior observation of an association between T1D risk loci and enhancers active in lymphocytes, prompted our examination of gene expression in CD4+ T cells, CD8+ T cells, and CD19+ B cells. As we were interested in determining whether the alleles associated with risk are also associated with changes in expression and splicing, we employed a case-only study design to enrich for these risk alleles and thereby potentially increase our power to detect these associations.
The RNA-seq studies described here allow us to draw several broad conclusions about lymphocyte differentiation and the genes implicated in autoimmunity: (1) The three cell types studied do not differ substantially in the genes they express but rather in the relative levels of expression and alternative exon usage of these genes, suggesting that, among lymphocytes, cell types are defined more by differential expression and AS of a common set of genes than by expression of distinct sets of genes; (2) among autoimmune candidate genes, there is enrichment for differential expression and AS; and (3) a number of the T1D/autoimmune candidate SNPs are associated with sQTLs for genes within those regions providing a potential mechanistic link between these disease associations and the function of the gene.
Annotating splicing events
Existing methods to estimate transcript isoform expression (Martin and Wang 2011; Steijger et al. 2013) are sensitive to structural properties like guanine-cytosine content and repeat regions that can lead to false positives with regards to alternative isoform usage (Love et al. 2015). Examining gene expression in terms of splicing events allows for the direct measurement of individual splicing events rather than relying on an estimate of isoform abundance and avoids statistical problems associated with the assignment of reads to multiple transcripts. Annotations for each splicing event are directly accessible enabling the examination of global shifts in gene expression and identification of novel splicing events. Although the hg19 genome was used in this study due to the availability of the more accurate AceView gene models (SEQC/MAQCIII Consortium 2014), our analysis of splicing events would not likely be substantially different if the same annotations were available for the more-recent hg38 genome build.
Distinct AS patterns in T1D and autoimmune candidate genes
Many of the same exons and splicing events were detected, but differentially expressed, in all three cell types: CD4+ T cells, CD8+ T cells, and CD19+ B cells. This differential expression is consistent with a previous multi-immune cell expression study, HaemAtlas (Watkins et al. 2009). Exons in autoimmune disease and T1D candidate genes were more likely to be differentially expressed and alternatively spliced than the expressed exons of other genes. These findings suggest that autoimmune genes are enriched for cell-type–specific functions and encourages both further exploration of their expression and splicing in specific subtypes of the broad lymphocyte classes studied here and functional studies focusing on putative protein isoforms arising through AS.
Among different splicing event types, some, such as IR or the inclusion of previously nonannotated junctions, are more likely to result in the production of novel protein isoforms. Putative IR and unannotated junctions were observed in all three cell types examined, and each accounted for ∼18% of detected splicing events. These splicing event types displayed the greatest lymphocyte subset specificity, with a higher proportion of events expressed in a single cell type compared with all other splicing event types. The higher level of lymphocyte subset specificity for these events may reflect a frequent mechanism whereby the products of these genes contribute to autoimmunity. Some caution is warranted in this interpretation since the findings may be inadvertently biased by focusing on a selection of genes and/or the use of only T1D cases in this study. In addition, putative IR events may represent a mechanism other than actual retention of introns, such as unprocessed mRNA or novel donor sites.
cis-sQTLs and the role of splicing in autoimmunity
Several functional studies of individual T1D candidate genes have identified AS as an underlying mechanism. The soluble protein isoform of CTLA4, resulting from the exclusion of exon 3, is reported to reduce risk for T1D by potentiating regulatory T-cell function (Clark et al. 2007). The disease-associated SNP rs3087243 (also referred to as CT60) is associated with decreased mRNA expression of soluble CTLA4 in CD4+/CD25+ regulatory T cells (Lewis et al. 2003). In the PTPN22 gene, rs56048322 increases the production of alternative transcripts, resulting in either the retention of intron 18 or skipping of exon 18 (Onengut-Gumuscu et al. 2006; Ge et al. 2016). Based on these precedents, we hypothesized that SNPs within or flanking autoimmune-associated genes might similarly regulate gene expression via effects on splicing.
We found that 637 of the 1197 autoimmune candidate genes with both detectable expression and SNP coverage in our study had at least one cis-sQTL. To the best of our knowledge, this is the first study examining the differences in AS between multiple lymphocyte populations and the first to directly associate genomic variation at autoimmune disease genes with these differences. While we have used gene expression data from lymphocytes from T1D patients, the shared genetic risk between autoimmune diseases suggests that dysregulation in these lymphocyte subsets likely has implications for other autoimmune diseases.
We observed a striking level of cell-type specificity for cis-s/eQTLs in our analysis. Over 75% of the significant s/eQTLs for autoimmune genes in our study were confined to a single subset of lymphocytes. This is in contrast to other genome-wide, multitissue eQTL studies where the proportion of reported cis-eQTLs that are specific to a single tissue is, in some cases, substantially lower (Nica et al. 2011; Flutre et al. 2013). It has been previously reported by the Genotype-Tissue Expression (GTEx) project that >50% of all eQTLs, and ∼45% of eQTL from the same set of genes we tested, are shared among tissues (The GTEx Consortium 2015). In our study, we assayed three cell types that are transcriptionally similar and are likely more phenotypically similar than in the prior cited studies.
In summary, our findings suggest that splicing is affected by common genetic variants to an unexpectedly large degree in lymphocytes and that genes implicated in autoimmunity in general and T1D specifically are enriched for such variants. Given the potential for variation in the frequency of splicing events to alter the functions of the affected genes, the phenotypic consequences of genetic regulation of splicing and its relevance to autoimmune disease risk warrant further investigation.
Methods
Subject ascertainment
The Type 1 Diabetes Genetics Consortium (T1DGC) ascertained families with parents having one affected child (trios) and two or more affected children (affected sib pairs [ASPs]) with T1D as previously described (Concannon et al. 2009; Hilner et al. 2010). Index cases (probands) from 83 families, consisting of 44 male and 39 female subjects (mean age at ascertainment, 32 ± 8 yr), were selected for these studies. Demographics are provided in Table 4. All subjects self-reported European ancestry; this was confirmed with an analysis of population structure as previously reported (Onengut-Gumuscu et al. 2015).
Table 4.
Subject demographics

ImmunoChip SNP genotyping and definition of candidate genes
All subjects were genotyped with the ImmunoChip, a custom Illumina genotyping array containing SNPs from 186 chromosomal regions selected based on genome-wide significance for association with at least one of 12 different autoimmune diseases, including T1D (Cortes and Brown 2011; Onengut-Gumuscu et al. 2015). Sample and SNP quality control procedures were conducted as part of the T1DGC fine-mapping project (Onengut-Gumuscu et al. 2015; NCBI dbGaP accession phs000911.v1.p1).
Three categories of genes were defined for comparison of gene expression: “T1D candidate genes” representing 432 genes from 51 T1D-associated genomic regions cataloged in Immunobase (http://www.immunobase.org/), “autoimmune genes” representing the 1774 candidate genes for autoimmunity (including T1D) cataloged in ImmunoBase, and “nonautoimmune genes” representing all other protein coding genes excluding those previously defined as “T1D candidate genes” or “autoimmune genes.”
Sample preparation and RNA sequencing
Peripheral blood mononuclear cells (PBMCs) were fractionated by positive selection on antibody-coated magnetic beads into CD4+ T-cell, CD8+ T-cell, and CD19+ B-cell populations. Purities of the resulting populations (>90%) were confirmed by flow cytometry. Sample preparation metadata, including live cell counts, cell purity, and time before freezing, were recorded. RNA was purified, libraries prepared, and sequencing (50 million reads/sample) performed using the Illumina HiSeq 2000 platform (HudsonAlpha Genome Services Laboratory). Sufficient RNA could not be obtained for one subject, who was excluded from subsequent analyses. Samples were prepared and sequenced in three pools with paired-end 50-bp reads (Supplemental File S1). After quality control assessments of the sequencing data, an additional five individual cell samples were excluded on the basis of low coverage (Supplemental File S2). Thus, 81 subjects were included in analysis, with sequencing data from all three cell types from 79 study subjects.
Annotation of splicing events
The catalog of all possible splicing events was created using the genomic features file (GFF) from AceView (2010 release) (Thierry-Mieg and Thierry-Mieg 2006) gene models for the hg19 human genome build (https://www.ncbi.nlm.nih.gov/ieb/research/acembly/). Possible exon–exon junctions were generated from all logical combinations of exon pairs within a gene from 5′-to-3′. Junctions were centered (based on the read length of 50 bp) such that the junction site was no >12 bp from the center of a read. (Supplemental Fig. S7). Overlapping exons were grouped into an exonic region. The longest 5′ most exon in each region was used as a baseline to classify alternative donor and acceptor events. (Supplemental Fig. S7). There were 678,664 exons representing 349,564 exonic regions. The minimum start and maximum end positions were used to estimate expression (Graze et al. 2012). Exon–exon junctions were classified as “exon skipping” if at least one exon occurred between the exons of a junction. For each exon-skipping junction, a list of skipped exons was created. Exons within each exonic region were classified as having an alternative donor or acceptor site if their genomic start (acceptor site) or stop (donor site) differed from that of the reference exon, and the corresponding junction were also annotated. A list of exon–exon junctions from the transcripts in the GFF file was used to determine if a junction was annotated to a known transcript. The classifications of exon-skipping, alternative donor, and alternative acceptor are not mutually exclusive. In each exonic region, putative IR events from the 3′-most exon were generated by extending the donor site sequence into the neighboring intron. There were 6,390,703 possible exon–exon junctions and 232,249 IR events. A total of 292,753 junctions were annotated to at least one known transcript. Junctions were classified as exon-skipping (5,837,064), alternative donor (1,240,520), alternative acceptor (1,257,096), and alternative donor and alternative acceptor (903,513).
Gene expression analysis
RNA sequencing data were aligned to the splicing catalog reference sequences using the Bowtie algorithm (version 0.12.9) (Langmead et al. 2009) to ensure that no gaps were introduced in junction alignments. Reads that did not align to splicing events were mapped to the complete human genome (GRCh37/hg19 version, release 73) using the BWA-MEM algorithm (version 0.7.9) (Li 2013; https://github.com/McIntyre-Lab/papers/tree/master/newman_t1d_cases_2017). Exon coverage was quantified within an exonic region as average depth per nucleotide (APN), and detection was defined as an APN > 0 for at least half of all samples per cell type. Junctions were quantified as the number of reads aligned to an event, and detection was defined as having at least 10 aligned reads in at least half of all samples per cell type; as junctions are typically much shorter than exons, this was used to ensure that only junctions that were definitely detected were examined. Several transformations of the coverage data were considered to normalize the expression data, and upper-quartile normalization and log2 transformation were selected due to better performance of the residuals (Bullard et al. 2010; Dillies et al. 2013).
Several potential covariates were examined, including live cell count, cell purity, time before freezing, and other sample preparation parameters. None of these parameters accounted for significant variation. Ten latent (unmeasured) confounders were estimated from the gene expression data (Stegle et al. 2012). Models were fit adding all latent confounders; however, no improvement over the model with Factor 2 alone was observed (Supplemental File S3). Factor 2 was included in all subsequent analyses.
The mixed effects model, Yijkn = μ + ci + sj + cisj + pk + vin + εijkn, was fit separately for each exon/junction where Yijkn is the normalized expression, i is the cell type (i = CD4+, CD8+, CD19+), j is subject sex (j = male, female), k is the pool samples were prepared and sequenced in (k = 1, 2, 3), and n is the individual subject (n = 1, 2, …, 81). Variables cell type (c), sex (s), pool (p), and latent factor estimate (v) were fixed effects, and pool was considered as a random effect. The residual ε were assumed to be distributed N(0, σn), and degrees of freedom were adjusted using Kenward–Roger approximations (Kenward and Roger 1997). Individual comparisons of the different cell types were calculated in a pairwise fashion (CD4+ vs. CD8+; CD4+ vs. CD19+; CD8+ vs. CD19+). A false-discovery rate (FDR) of 0.05 was considered to be significant (Benjamini and Hochberg 1995; Verhoeven et al. 2005). Other FDR thresholds (FDR < 0.1, FDR < 0.2) were considered, and the results were qualitatively similar to that of FDR < 0.05.
eQTL analysis
Of the 164,643 ImmunoChip SNPs passing quality control, 55,032 were excluded on the basis of minor allele frequency (MAF) < 5%. Linkage disequilibrium (LD)–based filtering, as implemented in PLINK (Purcell et al. 2007; Chang et al. 2015), was performed to exclude strongly correlated SNPs (r2 > 0.9) within a 50-SNP window. After LD filtering, a total of 48,650 SNPs remained. eQTL analyses were restricted to cis effects. To determine the window size around each gene for selecting SNPs to test as cis-eQTL, LD was calculated between the index SNP for each T1D-associated chromosomal region (Onengut-Gumuscu et al. 2015) and all SNPs within a 2-Mb window. Regions of LD with the index SNP, representing the region with the most likely causal variant, ranged from 0–186 kb, with a median range of 5.2 kb. To have a consistent window size for each gene likely to capture the SNPs contributing to cis-effects, LD-pruned SNPs were assigned to a gene if they were located within a 5-kb window at either end of the gene. For each cell type, tests for association between SNPs in a gene region and the set of detected exons/junctions were conducted using a mixed effects model, Yjkmn = μ + gm + sj + gmsj + pk + vin + εjkmn, fit separately for each SNP–exon/junction pair where Yjkmn is the normalized expression, m is the SNP genotype coded as the number of alternative alleles (m = 0, 1, 2), j is subject sex (j = male, female), k is the pool samples were prepared and sequenced in (k = 1, 2, 3), and n is the individual subject (n = 1, 2, …, 81). Variables SNP genotype (g), sex (s), pool (p), and latent factor estimate (v) were fixed effects, and pool was also considered as a random effect. The residual ε were assumed to be distributed N(0, σn), and degrees of freedom were adjusted using Kenward–Roger approximations (Kenward and Roger 1997). No test for interaction between cell type and genotype was included in the model as there was insufficient statistical power to make a meaningful interpretation of this test. A FDR of 0.05 was considered to be significant (Benjamini and Hochberg 1995; Verhoeven et al. 2005). To clearly distinguish between allelic associations with exon expression and splicing events, “eQTL” is used to refer to all SNPs at which an allelic association with variation in the expression of an exon is observed, while “sQTL” (splicing QTL) refers to the subset of eQTL for which the expressed event is a splice junction or IR event. To put cis-s/eQTLs into the context of T1D susceptibility, the LD between the list of T1D-credible SNPs from Onengut-Gumuscu et al. (2015) and SNPs used in the cis-s/eQTL analysis in the present study was calculated. SNPs tested as cis–s/eQTL were considered to be in LD with T1D-credible SNPs if the pairwise correlation was high (r2 > 0.8).
Software, assembly, and alignment availability
All scripts pertaining in this study are available as in Supplemental Scripts and at the GitHub code repository (https://github.com/McIntyre-Lab/papers/tree/master/newman_t1d_cases_2017).
Data access
The sequencing data from this study have been submitted to the NCBI database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap) under accession number phs001426.v1.p1.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Institutes of Health (DP3DK085678/DK/NIDDK, AI42288/NIAID, GM1022 27/NIGMS) and utilizes resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by NIDDK, National Institute of Allergy and Infectious Diseases, National Human Genome Research Institute, National Institute of Child Health and Human Development, and Juvenile Diabetes Research Foundation International.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.217984.116.
References
- Arnold ES, Ling S-C, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, et al. 2013. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci 110: E736–E745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett AH, Eff C, Leslie RDG, Pyke DA. 1981. Diabetes in identical twins: a study of 200 pairs. Diabetologia 20: 87–93. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al. 2009. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41: 703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300. [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, et al. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. 2006. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet 78: 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TA, Schweitzer AC, Chen TX, Staples MK, Lu G, Wang H, Williams A, Blume JE. 2007. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biology 8: R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon P, Onengut-Gumuscu S, Todd JA, Smyth DJ, Bergholdt R, Akolkar B, Erlich HA, Hilner JE, Julier C, Morahan G, et al. 2008. A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes 57: 2858–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon P, Chen W-M, Julier C, Morahan G, Akolkar B, Erlich HA, Hilner JE, Nerup J, Nierras C, Pociot F, et al. 2009. Genome-wide scan for linkage to type 1 diabetes in 2,496 multiplex families from the type 1 diabetes genetics consortium. Diabetes 58: 1018–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes A, Brown MA. 2011. Promise and pitfalls of the Immunochip. Arthritis Res Ther 13: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. 2015. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res 43: 5130–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delovitch TL, Singh B. 1997. The nonobese diabetic mouse as a model of autoimmune diabetes: Immune dysregulation gets the NOD. Immunity 7: 727–738. [DOI] [PubMed] [Google Scholar]
- Dillies M-A, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, Keime C, Marot G, Castel D, Estelle J, et al. 2013. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform 14: 671–683. [DOI] [PubMed] [Google Scholar]
- Exome Aggregation Consortium, Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O'Donnell-Luria A, Ware J, Hill A, et al. 2015. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 10.1101/030338. [DOI] [PMC free article] [PubMed]
- Flutre T, Wen X, Pritchard J, Stephens M. 2013. A statistical framework for joint eQTL analysis in multiple tissues. PLoS Genet 9: e1003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Onengut-Gumuscu S, Quinlan AR, Mackey AJ, Wright JA, Buckner JH, Habib T, Rich SS, Concannon P. 2016. Targeted deep sequencing in multiple-affected sibships of European ancestry identifies rare deleterious variants in PTPN22 that confer risk for type 1 diabetes. Diabetes 65: 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerold KD, Zheng P, Rainbow DB, Zernecke A, Wicker LS, Kissler S. 2011. The soluble CTLA-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes 60: 1955–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graze RM, Novelo LL, Amin V, Fear JM, Casella G, Nuzhdin SV, McIntyre LM. 2012. Allelic imbalance in Drosophila hybrid heads: exons, isoforms, and evolution. Mol Biol Evol 29: 1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2015. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonarson H, Grant SFA, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Casalunovo T, Taback SP, Frackelton EC, et al. 2007. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 448: 591–594. [DOI] [PubMed] [Google Scholar]
- Hilner JE, Perdue LH, Sides EG, Pierce JJ, Waegner AM, Aldrich A, Loth A, Albret L, Wagenknecht LE, Nierras C, et al. 2010. Designing and implementing sample and data collection for an international genetics study: the Type 1 Diabetes Genetics Consortium (T1DGC). Clin Trials 7: S5–S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Deutsch AJ, Lenz TL, Onengut-Gumuscu S, Han B, Chen W-M, Howson JMM, Todd JA, De Bakker PIW, Rich SS, et al. 2015. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat Genet 47: 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J. 2003. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: a nationwide follow-up study. Diabetes 52: 1052–1055. [DOI] [PubMed] [Google Scholar]
- Juan-Mateu J, Villate O, Eizirik DL. 2015. Mechanisms in endocrinology: alternative splicing: the new frontier in diabetes research. Eur J Endocrinol 174: R225–R238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenward MG, Roger JH. 1997. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 53: 983–997. [PubMed] [Google Scholar]
- Kralovicova J, Vorechovsky I. 2010. Allele-specific recognition of the 3′ splice site of INS intron 1. Hum Genet 128: 383–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovicova J, Gaunt TR, Rodriguez S, Wood PJ, Day INM, Vorechovsky I. 2006. Variants in the human insulin gene that affect pre-mRNA splicing: Is −23HphI a functional single nucleotide polymorphism at IDDM2? Diabetes 55: 260–264. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Green RE, Brenner SE. 2003. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci 100: 189–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997.
- Liu X-Y, Li H-L, Su J-B, Ding F-H, Zhao J-J, Chai F, Li Y-X, Cui S-C, Sun F-Y, Wu Z-Y, et al. 2015. Regulation of RAGE splicing by hnRNP A1 and Tra2β-1 and its potential role in AD pathogenesis. J Neurochem 133: 187–198. [DOI] [PubMed] [Google Scholar]
- Love MI, Hogenesch JB, Irizarry RA. 2015. Modeling of RNA-seq fragment sequence bias reduces systematic errors in transcript abundance estimation. bioRxiv 10.1101/025767. [DOI] [PMC free article] [PubMed]
- Martin JA, Wang Z. 2011. Next-generation transcriptome assembly. Nat Rev Genet 12: 671–682. [DOI] [PubMed] [Google Scholar]
- Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, Ryder LP, Thomsen M, Nielsen LS, Svejgaar A. 1974. HL-A antigens and diabetes mellitus. Lancet 2: 864–866. [DOI] [PubMed] [Google Scholar]
- Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, Travers M, Potter S, Grundberg E, Small K, et al. 2011. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 7: e1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. 2014. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506: 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onengut-Gumuscu S, Buckner JH, Concannon P. 2006. A haplotype-based analysis of the PTPN22 locus in type 1 diabetes. Diabetes 55: 2883–2889. [DOI] [PubMed] [Google Scholar]
- Onengut-Gumuscu S, Chen W-M, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, Farber E, Bonnie JK, Szpak M, Schofield E, et al. 2015. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 47: 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. 2007. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y, Wang W-Y, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, et al. 2014. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest 124: 981–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo MJ, Yu L, Hawa M, Mackenzie T, Pyke DA, Eisenbarth GS, Leslie RDG. 2001. Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 44: 354–362. [DOI] [PubMed] [Google Scholar]
- Rich SS. 1990. Mapping genes in diabetes–genetic epidemiologic perspective. Diabetes 39: 1315–1319. [DOI] [PubMed] [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, et al. 2010. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29: 1248–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEQC/MAQCIII Consortium. 2014. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol 32: 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevcik J, Falk M, Macurek L, Kleiblova P, Lhota F, Hojny J, Stefancikova L, Janatova M, Bartek J, Stribrna J, et al. 2013. Expression of human BRCA1 Δ17–19 alternative splicing variant with a truncated BRCT domain in MCF-7 cells results in impaired assembly of DNA repair complexes and aberrant DNA damage response. Cell Signal 25: 1186–1193. [DOI] [PubMed] [Google Scholar]
- Shirangi TR, Dufour HD, Williams TM, Carroll SB. 2009. Rapid evolution of sex pheromone-producing enzyme expression in Drosophila. PLoS Biol 7: e1000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford PM, Halliday GM, Brooks WS, Kwok JBJ, Storey CE, Creasey H, Morris JGL, Fulham MJ, Schofield PR. 2000. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene - Expansion of the disease phenotype caused by tau gene mutations. Brain 123: 880–893. [DOI] [PubMed] [Google Scholar]
- Stegle O, Parts L, Piipari M, Winn J, Durbin R. 2012. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc 7: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steijger T, Abril JF, Engstrom PG, Kokocinski F, Hubbard TJ, Guigo R, Harrow J, Bertone P, Consortium R. 2013. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods 10: 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry-Mieg D, Thierry-Mieg J. 2006. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol 7: S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisch R, McDevitt H. 1996. Insulin-dependent diabetes mellitus. Cell 85: 291–297. [DOI] [PubMed] [Google Scholar]
- Todd JA, Bell JI, McDevitt HO. 1987. HLA-DQ β gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 329: 599–604. [DOI] [PubMed] [Google Scholar]
- Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, Bakker SF, Bardella MT, Bhaw-Rosun L, Castillejo G, et al. 2011. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet 43: 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, et al. 2003. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423: 506–511. [DOI] [PubMed] [Google Scholar]
- Verhoeven KJF, Simonsen KL, McIntyre LM. 2005. Implementing false discovery rate control: increasing your power. Oikos 108: 643–647. [Google Scholar]
- Watkins NA, Gusnanto A, de Bono B, De S, Miranda-Saavedra D, Hardie DL, Angenent WGJ, Attwood AP, Ellis PD, Erber W, et al. 2009. A HaemAtlas: characterizing gene expression in differentiated human blood cells. Blood 113: E1–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Toomer KH, Chen Z, Cai X. 2015. Genome-wide analysis of alternative transcripts in human breast cancer. Breast Cancer Res Treat 151: 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.