

Abstract
Most studies of responses to transcriptional stimuli measure changes in cellular mRNA concentrations. By sequencing nascent RNA instead, it is possible to detect changes in transcription in minutes rather than hours and thereby distinguish primary from secondary responses to regulatory signals. Here, we describe the use of PRO-seq to characterize the immediate transcriptional response in human cells to celastrol, a compound derived from traditional Chinese medicine that has potent anti-inflammatory, tumor-inhibitory, and obesity-controlling effects. Celastrol is known to elicit a cellular stress response resembling the response to heat shock, but the transcriptional basis of this response remains unclear. Our analysis of PRO-seq data for K562 cells reveals dramatic transcriptional effects soon after celastrol treatment at a broad collection of both coding and noncoding transcription units. This transcriptional response occurred in two major waves, one within 10 min, and a second 40–60 min after treatment. Transcriptional activity was generally repressed by celastrol, but one distinct group of genes, enriched for roles in the heat shock response, displayed strong activation. Using a regression approach, we identified key transcription factors that appear to drive these transcriptional responses, including members of the E2F and RFX families. We also found sequence-based evidence that particular transcription factors drive the activation of enhancers. We observed increased polymerase pausing at both genes and enhancers, suggesting that pause release may be widely inhibited during the celastrol response. Our study demonstrates that a careful analysis of PRO-seq time-course data can disentangle key aspects of a complex transcriptional response, and it provides new insights into the activity of a powerful pharmacological agent.
The technique of perturbing cells and then measuring changes in their patterns of gene expression is a reliable and widely used approach for revealing mechanisms of homeostatic regulation. In mammalian cells, a wide variety of stimuli that induce striking changes in transcription are routinely applied, including heat shock, hormones such as estrogen, androgen, and cortisol, lipopolysaccharide, and various drugs. Regardless of the stimulus, transcription is commonly assayed by measuring concentrations of mature mRNA molecules, typically using RNA-seq. This approach is now relatively straightforward and inexpensive and allows for the use of standard analysis tools in detecting many transcriptional responses (Oshlack et al. 2010; Ozsolak and Milos 2011).
Nevertheless, these mRNA-based approaches are fundamentally limited in temporal resolution owing to the substantial lag between changes in transcriptional activity and detectable changes in the level of mRNAs. This lag results in part from the time required for transcription and post-transcriptional processing, and in part because pre-existing mRNAs buffer changes in mRNA concentration. For a typical mammalian gene, significant changes may require hours to detect, making it difficult to distinguish primary responses to a signal from secondary regulatory events. A possible remedy for this limitation is instead to make use of GRO-seq (Core et al. 2008), PRO-seq (Kwak et al. 2013), NET-seq (Churchman and Weissman 2011; Mayer et al. 2015; Nojima et al. 2015), or related methods (Dolken et al. 2008; Rabani et al. 2011, 2014; Li et al. 2016) for assaying nascent RNAs. These assays have the important advantage of directly measuring the production of new RNAs, rather than concentrations of mature mRNAs. As a consequence, they can detect immediate changes in transcriptional activity, and they permit time courses with resolutions on the order of minutes rather than hours (Hah et al. 2011; Danko et al. 2013; Jonkers et al. 2014; Mahat et al. 2016b). An additional benefit of nascent RNA sequencing is that it is effective in detecting unstable noncoding RNAs, including enhancer RNAs (eRNAs), together with protein-coding transcription units (Hah et al. 2011, 2013; Core et al. 2014). As a result, both active regulatory elements (which are generally well marked by eRNAs) and transcriptional responses can be detected using a single assay (Danko et al. 2015).
In this study, we sought to use PRO-seq to characterize the immediate, dynamic transcriptional response to the compound celastrol. Celastrol (also known as tripterine) is a pentacyclic triterpenoid isolated from the root extracts of Tripterygium wilfordii (thunder god vine), which has been used for millennia in traditional Chinese medicine for treatment of fever, joint pain, rheumatoid arthritis, bacterial infection, and other ailments (Westerheide et al. 2004). During the past few decades, celastrol has shown promise as an anti-inflammatory agent in animal models of collagen-induced arthritis, Alzheimer's disease, asthma, systemic lupus erythematosus, and rheumatoid arthritis (Guo et al. 1981; Allison et al. 2001; Xu et al. 2003; Li et al. 2005; Sethi et al. 2007). In addition, celastrol is known to inhibit the proliferation of tumor cells, including those from leukemia, gliomas, prostate, and head/neck cancer (Nagase et al. 2003; Yang et al. 2006; Sethi et al. 2007; Fribley et al. 2015). Recent research has also demonstrated striking obesity-controlling effects in mice (Liu et al. 2015; Ma et al. 2015).
Celastrol is known to activate the mammalian heat shock transcription factor HSF1 and stimulate the heat shock response (Westerheide et al. 2004; Trott et al. 2008) as well as the unfolded protein response (Mu et al. 2008; Fribley et al. 2015). In addition, celastrol activates a battery of antioxidant response genes (Trott et al. 2008). At the same time, celastrol inhibits the activities of other transcription factors, including androgen receptors (Hieronymus et al. 2006), glucocorticoid receptors (Trott et al. 2008), and NF-KB (Sethi et al. 2007). Thus, in several respects, the cellular response to celastrol resembles other well-known stress responses, including, in particular, the response to heat shock. Indeed, this heat shock-like behavior appears to explain, in part, the cytoprotective properties of celastrol. Nevertheless, it remains unclear exactly what distinguishes the celastrol response from heat shock and other stress responses. In particular, little is known about the immediate transcriptional effects or primary targets of celastrol. Thus, an examination using PRO-seq provides an opportunity for a deeper understanding of the specific mechanisms underlying the activity of this potent compound, with potential therapeutic implications.
With these goals in mind, we collected PRO-seq data for K562 cells at tightly spaced time points after treatment with celastrol and analyzed these data using a variety of computational methods. Our analysis sheds new light on the immediate transcriptional response to celastrol at both regulatory elements and target genes. More generally, it demonstrates that time-courses of PRO-seq data together with appropriate bioinformatic analyses can be used to dissect key aspects of a complex transcriptional response.
Results
Celastrol induces broad transcriptional repression and more limited up-regulation
We prepared PRO-seq libraries for K562 cells before celastrol treatment and after 10, 20, 40, 60, and 160 min of celastrol treatment, with two biological replicates per time point (Fig. 1A). To ensure that we could normalize read counts even in the presence of global changes in transcription, we spiked the same number of permeable Drosophila cells into each sample prior to run-on (Booth et al. 2016). Samples were sequenced to a total combined depth of 334.3 M reads, with an average replicate concordance of r2 ≈ 98% (Supplemental Fig. 1). About 0.5 M of these reads (0.1%) were derived from the Drosophila spike in. To obtain gene models appropriate for our cell types and conditions, we developed a probabilistic method, called tuSelector, that considers all GENCODE-annotated isoforms for each gene and identifies the most likely gene model given our PRO-seq data (Supplemental Fig. 2; Supplemental Methods). This step was particularly important for analyses that depend on an accurate transcription start site for each gene. We identified a total of 12,242 protein-coding genes from GENCODE as being actively transcribed in one or more time points (Methods). Of these genes, 75.4% were active across all six time points, 11.7% were active in a single time point, and the remaining 12.9% were active in 2–5 time points. Thus, our PRO-seq data and computational analyses indicate that more than half of all protein-coding genes are transcribed either in the basal condition or during the celastrol response in K562 cells.
Figure 1.
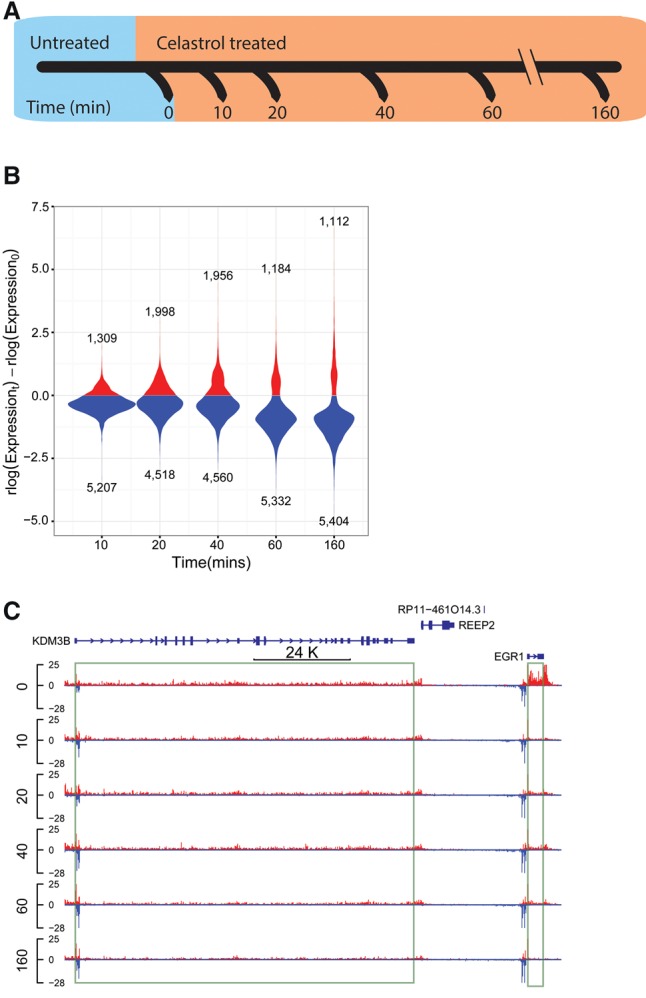
Characterizing the dynamic transcriptional response to celastrol using PRO-seq. (A) PRO-seq was applied to K562 cells collected before celastrol treatment (untreated/0 min) and at 10, 20, 40, 60, and 160 min after celastrol treatment. Two biological replicates were performed for each time point. (B) Distribution of log expression ratios (treated vs. untreated) for each time point (rlog is a regularized log2 estimate obtained from DESeq2). Only genes classified as differentially expressed (DE) throughout the time course are represented. Notice that most DE genes (FDR ≤ 0.01) are down-regulated upon celastrol treatment. (C) A UCSC Genome Browser display showing raw PRO-seq data for two differentially expressed genes, EGR1 and KDM3B. EGR1 is rapidly and strongly repressed (immediate decrease of ∼80%), whereas KDM3B is more gradually repressed, losing ∼50% of its expression by 160 min.
The genes that are differentially transcribed in response to celastrol were of particular interest for further analysis. To measure transcriptional activity specific to each time point, we used counts of PRO-seq reads mapping to the first ∼16 kb of each gene body, omitting the first 500 bp to avoid the effects of promoter-proximal pausing. Because RNA polymerase travels at an average rate of ∼2 kb/min (Singh and Padgett 2009; Danko et al. 2013; Jonkers et al. 2014; Veloso et al. 2014) and our time points are separated by at least 10 min, this strategy conservatively considers new transcription only, yet maintains sufficient statistical power for downstream analysis (see Methods). By applying DESeq2 (Love et al. 2014) to these 16-kb read counts, we identified 6516 (56%) of the active genes as being differentially expressed (DE) relative to the untreated condition (FDR ≤ 0.01). Interestingly, ∼80% of these DE genes were down-regulated. Many genes showed rapid and dramatic down-regulation, with decreases in expression by half or more at 3.5% of DE genes within 10 min, at 7.8% of DE genes within 20 min, and at 48.1% of DE genes within 160 min (Fig. 1B,C). In contrast, many fewer genes showed substantial increases in expression; for example, only 0.03%, 1.9%, and 7.7% of DE genes had doubled in expression after 10, 20, and 160 min, respectively. Nevertheless, extreme up- and down-regulation were both rare, with <1% of DE genes showing increases and <1% showing decreases in transcription by factors of eight or more. These observations are reminiscent of findings for the heat shock response, which have included general decreases in transcription together with up-regulation of selected stress-response elements (Hieda et al. 2004; Mahat et al. 2016b), but the effect of celastrol is somewhat less dramatic. We conclude that celastrol broadly inhibits transcription within minutes after administration but also rapidly activates a set of genes that may be important for continued cellular viability.
Celastrol activates heat shock more strongly and directly than it activates the unfolded protein response
Celastrol has been reported to activate stress response pathways such as the heat shock and unfolded protein responses (Westerheide et al. 2004; Mu et al. 2008; Trott et al. 2008; Fribley et al. 2015). To see whether these effects were detectable at the transcriptional level immediately after treatment with celastrol, we examined our PRO-seq data at genes activated by heat shock factor protein 1 (HSF1) and genes involved in the three branches of the unfolded protein response (UPR), corresponding to activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (ERN1), and endoplasmic reticulum kinase (EIF2AK3) (Fig. 2A). Because the initial stages of the UPR and heat shock response are nontranscriptional, we looked for downstream activity of the first group of transcription factors activated in each pathway, using targets reported in the Reactome pathway database (Fabregat et al. 2016). Most direct targets of HSF1 were up-regulated within 160 min (Fig. 2B). Genes encoding chaperone protein HSPH1 and proteinase inhibitor CBP1 were among the HSF1 targets showing the strongest initial response, with the gene encoding HSPH1 almost quadrupling its expression in 10 min and that for SERPINH1 increasing more than eightfold in 160 min. Most direct targets of the main UPR TFs (ATF4, ATF6, and XBP1), however, were not strongly induced during our time course. There were some exceptions to this general rule, such as genes encoding transcription factor ATF3, chaperone HSPA5, and apoptosis inhibitor DNAJB9, which more than doubled in expression. It is possible that these targets are activated earlier than other targets, perhaps by other TFs. In any case, our observations suggest that celastrol induces a pronounced, rapid transcriptional response in the heat shock pathway and has a much less pronounced transcriptional effect on the UPR, although some targets of the UPR are activated.
Figure 2.

Induction of cellular stress responses by celastrol. (A) Illustration showing key aspects of the unfolded protein response (UPR) and heat shock response (HSR), both of which have been reported to be induced by celastrol (Mu et al. 2008; Trott et al. 2008). Expected transcriptional targets are shown inside the nucleus, with targets of HSF1, the key transcription factor (TF) activated in the HSR, in red, and targets of the TFs associated with the three major branches of the UPR—XBP1, ATF4, and ATF6—in green, purple, and blue, respectively. Asterisks indicate genes that were differentially expressed in our experiments with FDR ≤ 0.01. (B) PRO-seq-based log fold changes in expression in K562 cells after 160 min of treatment by celastrol for numerous known targets of the same four TFs: XBP1, ATF4, ATF6, and HSF1. Genes labeled are the same as those listed in A. Only targets of HSF1 display strong up-regulation.
Celastrol produces distinct temporal patterns of transcriptional response
Our PRO-seq data for closely spaced time points enabled us to examine the temporal patterns of transcriptional response to celastrol treatment across the genome. To group our ∼6500 DE genes by shared transcriptional trajectories across the five time points following celastrol treatment, we used the autoregressive clustering algorithm, EMMIX-WIRE, which considers both expression values at each time point and the correlation of these values over time (Wang et al. 2012). EMMIX-WIRE identified four clusters of DE genes showing distinct patterns of transcription (see Methods; Fig. 3A). Only one of these clusters (cluster #1) displayed dramatic and sustained up-regulation. In contrast, cluster #2 showed rapid and pronounced down-regulation, cluster #3 showed delayed down-regulation, and cluster #4 displayed moderate, continuous down-regulation. Interestingly, the expression patterns for these clusters suggested that the transcriptional response to celastrol occurs largely in two distinct waves: one within the first 10 min, and a second between 40 and 60 min after treatment. It is possible, however, that additional waves occur but are undetectable at the resolution of our time points. These findings were robust to the number of clusters selected, with similar overall behavior for five-, six-, and seven-cluster models (Supplemental Figs. 4–6).
Figure 3.

Clusters of genes showing distinct temporal patterns of response to celastrol. (A) Differentially expressed genes (FDR ≤ 0.01) clustered by time series of log2 fold change (LFC) in expression relative to the untreated condition (0 min). Each gene is represented by a blue line, and the red lines indicate the mean expression per time point per cluster. Below each cluster is a summary of the enriched terms in the Reactome ontology (FDR ≤ 0.01) (see Supplemental Material for details; Fabregat et al. 2016). (B) ChIP-seq data from Vihervaara et al. (2013) describing binding of HSF1 in K562 cells under normal (left) and heat shock (right) conditions, stratified by our cluster assignments. Each line represents an average over all genes in the cluster in the region of the TSS, with lighter-colored bands representing 95% confidence intervals obtained by bootstrap sampling. Notice that cluster #1 is unique in showing a strong enrichment for heat shock-induced binding of HSF1.
Each of these clusters was enriched for genes with a distinct biological function, according to Reactome, a carefully curated database of gene-pathway relationships (Fig. 3A). To identify these enrichments, we labeled genes with their associated pathways from Reactome, then used permutation testing to find pathways overrepresented in each of the four clusters relative to the other three. Cluster #1 is enriched for genes responsible for the HSF1 response, including the HSPA family (Supplemental Fig. 7). Consistent with this observation, genes in this cluster have been shown, by ChIP-seq, to bind by HSF1 under heat shock conditions in K562 cells (Fig. 3B; Vihervaara et al. 2013). Cluster #2 is enriched for genes involved in ribosomal assembly, translational initiation, and peptide elongation. Cluster #3 is enriched for pathways that enable DNA replication (e.g., MCM family) and cell-cycle progression (e.g., CDK family) (Supplemental Fig. 10). The delayed down-regulation of these genes may occur as the cell is preparing to enter replicative arrest and, potentially, senescence, consistent with observations that celastrol induces cell-cycle arrest and potentiates apoptosis (Kannaiyan et al. 2011; Feng et al. 2013; Fribley et al. 2015). This transcriptional pattern is also consistent with our observation that celastrol-treated cells failed to replicate and mostly died within three days (Supplemental Fig. 11). Finally, cluster #4 contains essential elements of the RNA splicing machinery (e.g., CD2BP2, CLP1, and the SRSF kinase family) (Supplemental Fig. 11). Down-regulation of this cluster is consistent with previous reports that splicing is inhibited under heat shock (Shalgi et al. 2013). Similar patterns of enrichment were observed with five-, six-, and seven-cluster models. Overall, these results demonstrate that the components of a complex, multilayered transcriptional response can be disentangled to a degree by identifying groups of genes that display distinct temporal patterns of gene expression.
Several key transcription factors are associated with the celastrol response
Can the distinct transcriptional responses in these clusters of genes be traced to particular transcription factors? To address this question, we used linear regression to explain the estimated transcription levels at each time point based on the TFs that apparently bind in the promoter region of each gene (Fig. 4A). We used two orthogonal sources of information about TF binding: (1) ChIP-seq peaks for untreated K562 cells (Dunham et al. 2012); and (2) scores from DeepBind, a machine-learning method that is trained on a combination of ChIP-seq and in-vitro data and predicts TF-specific binding affinity based on the DNA sequence alone (Alipanahi et al. 2015). In both cases, we considered the interval between 500 bp upstream of and 200 bp downstream from the transcription start site of each active gene. Our regression model included a coefficient for each TF at each time point. A positive estimate of this coefficient indicated that increased affinity for a TF was associated with increased expression at a given time point, whereas a negative estimate indicated that increased affinity for a TF was associated with decreased expression at that time point.
Figure 4.

The celastrol response appears to be influenced by various transcription factors. (A) A schematic representation of the regression model used to estimate the impact of TF binding. The predicted expression level for each gene is a linear function of a gene-specific term, a time point-specific term, and a sum of per-time TF-specific effects weighted by the estimated relative TF binding affinity in the promoter regions of differentially expressed genes (from DeepBind or ChIP-seq). Note that the TF binding affinity is estimated from the untreated state and is invariant across time. (B) The estimated TF- and time-specific coefficients from the DeepBind-based regression model. Error bars correspond to the 95% CI estimates for each coefficient. Each gene (e.g., HSF1) is labeled by its DeepBind motif (e.g., D00470.005) in the legend. Only TFs with at least one significant time point (FDR ≤ 0.01) and an absolute effect size in the 90th percentile or above are shown. A positive weight for a TF at a given time point indicates that genes at which that TF is predicted to be bound showed increased expression relative to those without it. Negative weights indicate decreased expression. The three separate plots represent manually selected clusters of TFs associated with distinct temporal patterns. The time point-specific TF coefficients explain ∼11% of the residual variance not explained by gene-specific or time point-specific terms.
Between the two TF binding data sets, we identified >20 TFs as being significantly associated with changes in gene expression and having a large effect size (Fig. 4B; Supplemental Fig. 13). Of these TFs, E2F4 stood out as showing a particularly pronounced impact on expression in both data sets. E2F4 is associated with incrementally increased expression between 0 and 60 min, and with decreased expression thereafter, similar to the expression pattern of genes in cluster #3, which are associated with cell-cycle control. This observation is consistent with reports that E2F4 is an activator in some contexts but primarily acts as a repressor responsible for maintaining G2 arrest (Polager and Ginsberg 2003; Lee et al. 2011). In addition, we found that the dimerizing TFs MYC (from ChIP-seq data) and MAX (from DeepBind predictions) were both associated with an immediate increase in gene expression, followed by decreased expression within 20 min (Fig. 4). This delayed decrease in expression of MYC- and MAX-bound genes could result from the known disruption of MYC-MAX dimerization by celastrol (Westerheide et al. 2004; Wang et al. 2015). Finally, genes predicted to be bound by SRF also displayed elevated gene expression after 40 min. SRF has been previously associated with early and transient induction of cytoskeletal genes in response to heat stress in murine embryonic fibroblasts (Mahat et al. 2016b).
We also found that the paralogous TFs RFX1 (from ChIP-seq) and RFX5 (from DeepBind) were both associated with increases in expression. Both of these TFs have been implicated in regulating the expression of immunity-related human leukocyte antigen (HLA) genes, and both have context-specific transcriptional repression and activation mechanisms (Katan et al. 1997; Villard et al. 2000; Xu et al. 2006), so it is possible that they contribute to celastrol's anti-inflammatory effects. However, RFX1 was not tested with DeepBind and its motif is quite similar to that of RFX5, so it is impossible to know from our data whether one or both of these TFs are important in the celastrol response (notably, they do have different binding patterns in vivo in the untreated condition) (Supplemental Fig. 13B,C). Nevertheless, our regression framework is useful in providing a list of candidate TFs whose binding preferences correlate with aspects of the celastrol response.
Increased polymerase pausing is broadly associated with transcriptional repression
Promoter-proximal pausing of RNA polymerase is a rate-limiting and independently regulated step in productive transcription (Andrulis et al. 2000; Wu et al. 2003). Notably, the peaks of paused RNA polymerase at DE genes doubled in height during our time course (Fig. 5A; Supplemental Fig. 14). Accordingly, we found that the “log pause index,” or log2 ratio of average read depth at the pause peak to that in the proximal gene body, increased by more than 1 (corresponding to a fold-change of more than 2 in the pause index) in DE genes by 160 min (Fig. 5B). Together, these observations indicate that most DE genes undergo increased pausing after celastrol treatment, suggesting that pause release may be widely inhibited during the celastrol response.
Figure 5.

Increased promoter-proximal pausing is associated with transcriptional repression in the response to celastrol. (A) Mean PRO-seq signal at promoters for all active genes, grouped by time point and oriented with respect to the direction of transcription of the gene. The x-axis represents distance to the center between the divergent transcription start sites (see Methods). Intervals around each line represent 95% confidence intervals obtained by bootstrap sampling. Notice the general increase in the height of the pause peaks with time. (B) The distribution of changes in the log fold index with respect to the untreated condition (ΔLPI) (see Methods) for all active genes at each time point. The notch corresponds to median ±1.58×IQR/√n, roughly a 95% confidence interval of the median. (C) The distribution of ΔLPI for all DE genes (FDR ≤ 0.01) by cluster and time point. Notice that all clusters show an increase in the pause index with time, except for cluster #1. Error bars indicate the 25th and 75th percentiles of the data.
To see if particular expression patterns were associated with changes in pausing, we separately examined the changes in log pause index during the time course for each of our six gene expression clusters. Interestingly, we found that pausing increased in all clusters with the exception of cluster #1 (Fig. 5C), the only strongly up-regulated cluster (see Fig. 3A), where pausing decreased for the majority of genes but increased or remained unchanged for a significant fraction of them (Supplemental Fig. 14B). Thus, changes in the log pause index are generally negatively correlated with changes in expression across clusters. This observation suggests that decreases in the rate of release of paused Pol II to productive elongation could contribute to increased pausing and, hence, to down-regulation of transcription, while the absence of such an effect (in cluster #1) might permit up-regulation of transcription (Zeitlinger et al. 2007; Mahat et al. 2016b). As cluster #1 is strongly associated with the HSF1 response, this finding is consistent with previous reports that HSF1 regulates transcription by increasing the rate of release of paused RNA polymerase into productive elongation (Mahat et al. 2016b). Nevertheless, it is also possible that the inverse correlation between pausing and expression is a consequence of “mass action” of available Pol II (see Discussion).
Heat shock induces a similar but more pronounced transcriptional response than celastrol
Celastrol is known to mimic heat shock in many respects (Westerheide et al. 2004), but it remains unclear how similar the transcriptional responses to these two stimuli are. To address this question more directly, we obtained PRO-seq data for heat shock-treated K562 cells from a recently published study (Vihervaara et al. 2017) and processed it identically to our celastrol data. We focused on comparing the heat shock data for 0 and 30 min (the only time points available) with our celastrol data for 0 and 40 min, additionally considering our 60- and 160-min time points for some analyses (Supplemental Fig. 15A,B). In general, heat shock induced a broader response than celastrol treatment, with twice as many genes differentially expressed after 30 min of heat shock (4604) vs. after 40 min of celastrol treatment (2302). Of the 1301 genes that were up-regulated in response to either treatment, 21% were shared between the heat shock and celastrol responses, and of the 4230 that were down-regulated in response to either treatment, 25% were shared (Fig. 6). As with celastrol treatment, the pause index significantly increased after 30 min of heat shock. Taken together, these results suggest that there are many commonalities between the early transcriptional responses to the heat shock and celastrol treatments but also many differences.
Figure 6.
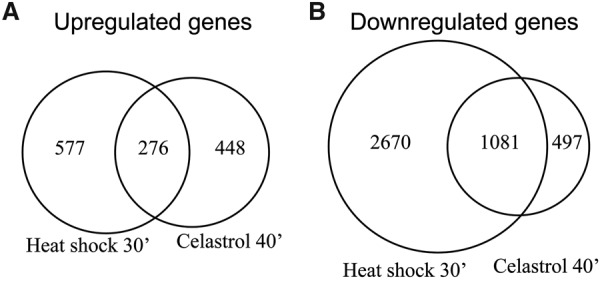
Celastrol down-regulates most of the same genes as heat shock but up-regulates many different genes. (A) Venn diagram of genes that are up-regulated after 30 min of heat shock vs. after 40 min of celastrol treatment. (B) Venn diagram of genes that are down-regulated after 30 min of heat shock vs. after 40 min of celastrol treatment.
We then sought to characterize the pathways underlying major differences between the celastrol and heat shock responses. Using Reactome, we tested for functional enrichments among shared and nonshared DE genes, separately considering up- and down-regulated genes. We found that genes down-regulated only in the celastrol response were enriched for mitochondrial energy production and translation of mitochondrial genes (Supplemental Fig. 16A). In contrast, down-regulated genes specific to the heat shock response were enriched for MAP kinase signaling and cell-cycle progression (Supplemental Fig. 16B), whereas down-regulated genes that were shared in both responses were strongly enriched for ribosomal formation and translation (Supplemental Fig. 16C). For the up-regulated genes, the heat shock-only genes were enriched for GPCR signaling, and the shared genes were dominated by heat shock response pathways in agreement with the analyses discussed above (Supplemental Fig. 17A,B); no pathways were significantly enriched in the celastrol-only response.
Finally, we searched for pathways whose genes tended to change expression in opposite directions in the heat shock and celastrol responses. We compared the 40, 60, and 160-min time points for celastrol to the 30-min heat shock time point. One pathway, cholesterol biosynthesis, emerged from this analysis as down-regulated in celastrol at both 60 and 160 min but up-regulated in heat shock (Supplemental Fig. 18A). This observation is consistent with previous findings for mammalian cells that heat shock increases activity of the MVA pathway, a key cholesterol biosynthesis pathway (Shack et al. 1999). The central regulatory enzyme in the MVA pathway, HMGCR, is clearly up-regulated in heat shock and down-regulated at 60 min in celastrol. We investigated whether sterol response element binding factor 1 (SREBF1), an important TF for cholesterol biosynthesis genes (Brown and Goldstein 1997), was a potential mechanism for decreased genic expression in the celastrol response by asking if genes bound by SREBF1 in untreated cells showed decreased expression relative to those not bound across the celastrol time-course (Supplemental Fig. 18B). We found that genes that were strongly bound by SREBF1 in untreated cells, in comparison to unbound genes, went from being more highly expressed in the untreated condition to having similar mean expression at 60 min of treatment and lower mean expression at 160 min of treatment. These results demonstrate that, despite having many similar effects, celastrol and heat shock have opposite effects on the expression of genes involved in cholesterol biosynthesis.
Enhancers show similar functional associations and pausing patterns to genes
Previous studies have shown that putative enhancers are divergently transcribed, producing nascent RNAs that can be detected via PRO-seq (De Santa et al. 2010; Core et al. 2014; Danko et al. 2015). Using dREG (Danko et al. 2015), which predicts divergent transcription start sites (dTSSs) from stranded GRO/PRO-seq data, we identified 25,891 apparent dTSSs from our PRO-seq data, pooling calls across time points. Based on the distance from nearest annotated genic TSSs, we classified 7334 of these dTSSs as likely transcribed enhancers, 15,941 as likely promoters, and the remaining 2616 as ambiguous. For validation, we examined ChIP-seq data from ENCODE for untreated K562 cells and found, as expected, that enhancer and promoter classes were both strongly enriched for acetylation of histone H3 at lysine 27 (H3K27ac), and that the promoter class was more strongly enriched for RNA polymerase and trimethylation of histone H3 at lysine 4 (H3K4me3) (Fig. 7A). The enhancer class also showed moderate enrichment for monomethylation of histone H3 at lysine 4 (H3K4me1). These observations confirm that PRO-seq serves as an efficient single-assay approach for characterizing both transcribed enhancers and protein-coding genes in our system (Danko et al. 2015).
Figure 7.

Response to celastrol at predicted transcribed enhancers. (A) Divergent transcription start sites (dTSS) that were classified by distance-based rules as likely enhancers or promoters show distinct patterns of histone marks in untreated K562 cells. Shown are H4K4me1 (enriched at enhancers), H3K4me3 (enriched at promoters), and H3K27ac (enriched at both). PRO-seq read counts are shown for comparison. The x-axis is oriented by direction of transcription of nearest gene. (B) Gene Ontology (GO) biological processes associated with differentially expressed enhancers using GREAT (McLean et al. 2010). Bar plot represents −log10 P-values for enrichment, with numerical fold enrichments indicated at right. (C) Metaplot of PRO-seq signal at all enhancers, centered on the dTSS, per time point. Units of PRO-seq signal are average numbers of reads per 10-bp bin. Intervals around each line represent a 95% confidence interval obtained by bootstrap sampling. (D) The distribution of DeepBind scores for HSF2 and JUND for transcriptionally activated and unchanged sets of enhancers. The notch corresponds to the median ±1.58×IQR/√n, roughly a 95% confidence interval of the median.
To better understand the role of the noncoding regulatory elements in the celastrol response, we further examined 1479 (∼20%) of the 7334 dTSS-based enhancers that were classified as differentially transcribed. We attempted to find functional enrichments for potential target genes of these differentially transcribed enhancers using the Genome Regions Enrichment of Annotations Tool (GREAT, ver. 3.0) (see Methods), which associates candidate regulatory elements with likely target genes according to distance-based rules and then tests those genes for functional enrichments (McLean et al. 2010). GREAT identified enrichments for processes relating to apoptosis, translational regulation, and responses to various environmental stresses (Fig. 7B), in general agreement with our analysis of DE genes. We also found that our set of putative enhancers displayed an accumulation of paused polymerase after celastrol treatment (Fig. 7C; Supplemental Fig. 19). Although the functional significance of pausing at enhancers is unknown, this observation suggests that global shifts in pause levels at genic TSSs are also reflected at enhancers.
Finally, we sought to determine which TFs influenced activity at enhancers. Because sparse data at enhancers resulted in noisier estimates of transcriptionally engaged RNA polymerase than at genes, we focused in this case on a relatively small group of 480 enhancers that showed little activity at 0 min but greatly increased activity by 160 min (see Methods). We compared DeepBind scores for these activated enhancers with those for non-DE enhancers that had similar absolute expression levels and found six TFs whose motifs had significantly elevated scores for sequence elements in the activated enhancers: HSF1/2, JUND, FOSL2, MAFK, STAT3, and THRA (Fig. 7D; Supplemental Fig. 20). Of these TFs, HSF2 was also associated with increased expression in genes, while JUND and FOSL2 are subunits of AP-1, a TF previously found to regulate cellular growth and senescence (Shaulian and Karin 2001). Because these TFs were identified simply based on their sequence preferences, TFs with similar motifs are also potential regulators. For example, it is possible that JUN, whose expression increases over the time-course and which is known to be activated by HSF1 (Sawai et al. 2013), is actually responsible for the apparent association with JUND, which does not appear to be activated. We also cannot effectively distinguish between HSF1 and HSF2 binding here. In addition, since this analysis was limited to TFs that increased transcription at enhancers, it is unsurprising that it did not identify TFs associated with decreased expression in the genic response, such as MAX. Despite these caveats, these results demonstrate that PRO-seq can be used to detect transiently activated enhancers and identify candidate TFs that may drive the enhancer response.
Discussion
This study represents the first genome-wide assessment of the immediate transcriptional effects of celastrol, including transcribed regulatory elements as well as genes, shedding light on some of the possible primary targets and mechanisms of action of this potent therapeutic compound. We find that celastrol treatment results in pervasive transcriptional down-regulation, with nearly half of expressed genes being down-regulated within 160 min. A much smaller group of genes, roughly 10% of those expressed, are up-regulated during the same time interval. By analyzing the sequences nearby transcription units, we were able to identify several transcription factors whose binding patterns partially explain these transcriptional responses. We also observed a clear impact from celastrol on polymerase pausing at both genes and enhancers, which is negatively correlated with changes in transcriptional activity. While there are limits to what can be learned from PRO-seq data alone, we have shown that when these data are collected at relatively high temporal resolution and analyzed together with other data for the untreated condition, they can provide valuable insights into a multifaceted, multistage cellular response to a transcriptional stimulus.
We find that celastrol treatment generally induces a similar response to heat shock, consistent with previous reports (Westerheide et al. 2004; Trott et al. 2008). Heat shock has also been observed to induce widespread down-regulation in mammalian cells (Hieda et al. 2004; Mahat et al. 2016b; Vihervaara et al. 2017). Moreover, many of the same genes that are up-regulated upon celastrol treatment are also bound by HSF1 after heat shock or participate in heat shock pathways. In addition, sequences associated with HSF1 binding are associated with increased gene expression, according to our regression analysis. Finally, our direct comparison showed that both treatments lead to up-regulation of genes involved in the heat shock response and down-regulation of genes involved in ribosomal formation and translation.
Together, these findings suggest that HSF1 is activated soon after celastrol treatment, whereupon it activates a large group of genes. These observations suggest that, in part, the transcriptional response to celastrol may simply be a general cellular stress response. Cellular stress responses are known to affect a broad range of cellular functions, including cell-cycle arrest, transcription of molecular chaperones, activation of DNA damage repair pathways, removal of irretrievably damaged macromolecules, and apoptosis upon severe damage (de Nadal et al. 2011), and they are relevant in many diseases, including cancer (Bi et al. 2005), proteotoxic diseases (Mu et al. 2008), and autoimmune disorders (Todd et al. 2008). Previous studies have investigated cellular stress responses at the delayed transcriptional (Teves and Henikoff 2011; Mahat et al. 2016b), post-transcriptional (Gardner 2008), translational (Shalgi et al. 2013), and post-translational levels (Urano et al. 2000; Golebiowski et al. 2009). Together with similar studies of heat shock (Mahat et al. 2016b), our study helps to illuminate specific features of the early transcriptional response to stress.
Nevertheless, we observed several differences between the heat shock and celastrol responses. The most striking difference was that genes associated with the cholesterol synthesis pathway are activated by heat shock but repressed by celastrol. This observation is consistent with reports that celastrol decreases endogenous cholesterol in mice and reduces obesity (Liu et al. 2015; Ma et al. 2015; Zhang et al. 2016). Further analysis suggested that loss of SREBF1 binding may be responsible for the difference in the celastrol response. Given that SREBFs are activated via proteolytic cleavage from an inter-membrane protein when sterols are scarce (Wang et al. 1994), it is possible that celastrol inhibits cholesterol synthesis by inhibiting SREBF1 cleavage and release. Alternatively, or in addition, celastrol could inhibit cholesterol synthesis by decreasing the stability of the SCAP-SREBF complex (Zhang et al. 2009; Kuan et al. 2017).
Another apparent difference between the heat shock and celastrol responses has to do with the kinetics at genes that appear to be regulated by SRF, a transcription factor associated with HSF1/2-independent up-regulation after heat shock. We observed an enrichment for SRF binding sites in the core promoters of genes that displayed elevated expression after 40 min of celastrol treatment. In contrast, previous findings for heat shock (Mahat et al. 2016b) have indicated that SRF-mediated up-regulation occurs much more rapidly, as early as 2.5 min after treatment. Finally, our analysis suggests that the loss of binding by MYC-MAX may be responsible, in part, for the broad transcriptional repression we observe within 20 min of celastrol treatment. To our knowledge, MYC-MAX inhibition has not been reported to be important in the heat shock response. The finding for celastrol is supported by previous studies showing that celastrol directly inhibits MYC-MAX functionality (Wang et al. 2015). Since MYC-MAX is a strong transcriptional activator and is bound at over 6000 promoters of active genes in K562, its inhibition may be an important contributing factor to widespread down-regulation after celastrol treatment (Amati et al. 1992, 1993).
A major strength of our experimental approach is that it allows us to observe transient as well as sustained transcriptional responses. For example, we found that E2F4, a transcriptional repressor, was quickly down-regulated after celastrol treatment, reducing in expression by half within 20 min. Despite loss of this repressor, target genes of E2F4 were increasingly down-regulated (rather than up-regulated, as expected) after 60 min (Lee et al. 2011). These observations suggest that the apparent increased repression activity of E2F4 during the celastrol response may have a nontranscriptional basis. Interestingly, previous studies have shown that celastrol inhibits CDK4 and that CDK4 overexpression disrupts E2F4 DNA-binding ability (Scimè et al. 2008; Peng et al. 2010). Thus, it is possible that celastrol increases the DNA binding of E2F4 to DNA, which in turn could contribute to cell-cycle arrest (Polager and Ginsberg 2003; Lee et al. 2011).
Another advantage of our densely sampled PRO-seq time-course is that it allows us to measure changes in promoter-proximal RNA polymerase pausing. We observed that pause indices increased by more than twofold at differentially expressed genes during our time-course. We also found that increased pausing was associated with decreased transcription in genes, as previously reported for heat shock conditions (Mahat et al. 2016b), although we did not observe a converse association between decreased pausing and up-regulation of genes. A possible mechanism that could contribute to this increased pausing in down-regulated genes is the celastrol-induced disruption of the MYC-MAX complex, which has been shown to recruit P-TEFb, which in turn broadly facilitates pause release (Kanazawa et al. 2003). This mechanism could, in principle, affect down-regulated genes only, for example, if up-regulated genes recruit P-TEFb independently of MYC-MAX (e.g., through the activity of HSF1) (Lis et al. 2000).
While it is possible that increased pausing causes decreased expression by limiting productive elongation and therefore reducing transcription levels, an alternative possibility is that increased pausing is a consequence of “mass action”—that is, decreased transcriptional activity across many genes results in increased availability of free Pol II, some of which ends up being loaded on promoters and coming to rest at pause sites (Mahat et al. 2016b). In other words, the negative correlation between pausing and expression could be explained by causality in either direction, or perhaps in both directions. Additional experiments will be needed to establish the causal basis of these correlations. In any case, our observations suggest that changes in pausing are widespread and broadly associated with transcriptional repression and therefore may play an important role in the celastrol response.
Methods
Celastrol treatment
K562 cells were cultured at 37°C in RPMI media (Gibco) containing 10% FBS (Gibco), Pen Strep (Gibco), and 2 mM L-glutamine (Gibco). Biological replicate cell cultures were prepared as follows: after thawing K562 cells and seeding a fresh culture, cells were split into two separate flasks, which would remain separated through six passages and expansions until treatment and collection for preparation of PRO-seq libraries. Cells from each expanded replicate were seeded onto six 30-mL dishes (one for each time point) at a density of 5 × 105 cells/mL and then incubated for an additional doubling cycle (∼20 h). For treatments, fresh celastrol was dissolved in DMSO at a final concentration of 20 mM. Celastrol-treated samples received celastrol (Sigma) at a final concentration of 3 µM, whereas untreated (0-min) samples received an equivalent volume of DMSO. Cells remained in culture dishes in the incubator during the time-course. Time-course treatments were carried out in reverse order so that all samples would be collected at the same time (starting with 160-min time point and ending with the untreated).
Cell permeablization and PRO-seq
Samples were then prepared for precision run-on reactions by subjecting cells to permeablizing conditions. Briefly, cultures were spun down and resuspended in ice cold 1×PBS. Samples were spun again and washed in 5 mL wash buffer (10 mM Tris-Cl, pH 7.5, 10 mM KCl, 150 mM sucrose, 5 mM MgCl2, 0.5 mM CaCl2, 0.5 mM DTT 1× protease inhibitor cocktail [Roche], and 20 U RNase inhibitor [SUPERase In, Invitrogen]). Cell pellets were then resuspended in permeabilization buffer (10 mM Tris-Cl, pH 7.5, 10 mM KCl, 250 mM sucrose, 5 mM MgCl2, 1 mM EGTA, 0.05% Tween-20, 0.1% NP40, 0.5 mM DTT, 1× protease inhibitor cocktail [Roche], and 20 U RNase inhibitor [SUPERase In, Invitrogen]) and left on ice for 5 min. Cells were checked for penetration by trypan blue to assess permeability (∼99% permeable). Cells were then washed two times in 5 mL wash buffer before being resuspended in 200 µL storage buffer (50mM Tris-Cl, pH 8.3, 40% glycerol, 5 mM MgCl2, 0.1 mM EDTA, and 0.5 mM DTT). A 1:50 dilution was prepared using 2 µL of each sample and used to take OD600 measurements. All samples were then diluted to an equal density (OD600 = 0.181) in a final volume of 110 µL of storage buffer. Then, 5 × 104 pre-permeabilized S2 cells were spiked in to each cell count-normalized sample before flash-freezing the permeabilized cells and storing them at −80°C.
Stored permeable cells with spike-ins were thawed on ice, and each sample was subjected to the precision run-on protocol (Mahat et al. 2016a). Run-on reactions incorporated only biotinylated NTPs with no unmodified NTPs. All libraries were subjected to nine cycles of PCR amplification before size selection and gel purification.
Cell counting
Cells were either treated with 3 µM celastrol, DMSO, or left untreated for 4 d. Live/dead cells were determined based on trypan blue staining. Counts were measured with an automatic cell counter (Bio-Rad).
Read mapping
All filtered reads were removed from each fastq file, then cutadapt (v1.9.1) was run with the following options:
cutadapt -a TGGAATTCTCGGGTGCCAAGG -m 15 to remove the Illumina adapters and discard all remaining reads that were <15 bp in length. All reads were then trimmed to 34 bp in length using fastx_trimmer (v0.0.13.2) to avoid biasing read mapping away from gene promoters. The trimmed reads were then aligned to the hg19 and bdgp6 genomes using the STAR aligner (v2.4.0i) (Dobin et al. 2013). Reads aligning to hg19 and bdgp6 were then separated, and bigwigs were created by converting each read to a single count at its 5′ end. While human assembly hg19 was used for read mapping, we do not expect the use of the more recent hg38/GRCh38 would have an appreciable impact on our results, as the major differences between these assemblies concern alternative haplotypes and centromeric regions.
Detection and resolution of dTSSs
dREG was run on each sample as described previously (Danko et al. 2015), producing a set of genomic intervals corresponding to predicted divergent transcription starts sites. These initial dREG calls had fairly coarse resolution, ranging in size from several hundred to thousands of bases. We therefore applied a heuristic scanning method to identify one or more higher-resolution dTSSs within each dREG call. Briefly, this method involved sliding a window along a dREG interval and considering the relative read counts among three subintervals: a peak, a flank, and center. To identify pairs of divergent peaks, the test was applied simultaneously to each strand in a strand-specific manner, and the results were combined. Specifically, for a scan initiated at base i, the center was defined as the interval [i, i + 110), the shoulder as [i − 50, i), and the flank as [i − 250, i − 150). Three one-sided binomial tests were performed, testing that there are fewer reads in the center than the flank, the center than the shoulder, and the flank than the shoulder. The sum of the resulting six negative log P-values (three for each strand) then became the per-base score. The best scoring window in a dREG region was taken as a dTSS. In addition, up to two other dTSSs were called if their score exceeded 20.
Classifications of dTSSs
dTSSs were classified as either enhancers or promoters based on their relative distance from the set of all TSSs annotated in GENCODE v19. To classify each dTSS, the following rules were applied: (1) if the dTSS was >1 kb, and at most 1 Mb, away from the nearest annotated promoter, it was classified as an enhancer; (2) if the dTSS was within 200 bp of an annotated promoter, or it was within 1 kb of an annotated promoter and it was the closest dTSS to the promoter, it was classified as a promoter; (3) if the dTSS was between 600 bp and 1 kb away from the nearest annotated promoter and not the closest dTSS to the promoter, it was classified as an enhancer; (4) otherwise, the dTSS was classified as unknown.
Selection of active transcripts in K562 cells
Selection of transcripts was performed by a new program, called TuSelector. First, a list of potential transcripts was obtained from GENCODE v19. The genic regions and data were partitioned into 100-bp intervals. For each gene, a set of coarse-grained overlapping transcript models was created, where for each transcript model and interval, the interval was assigned to the transcript model if and only if it overlapped the corresponding annotated transcribed region by more than 50% at the nucleotide level. Next, the PRO-seq read counts in each 100-bp interval were summarized by a 1 if there were reads aligned to the interval or a 0 otherwise. TuSelector computed a likelihood for each of the possible coarse-grained transcript models at a given gene, as follows:
where T is the transcript model, the products range across genomic intervals i, xi is the summary of the data in interval i (0 or 1), δ(i, T) is an indicator function that takes value 1 when interval i is included in T and 0 otherwise, X is a Bernoulli random variable, and θt and θu are the parameters for this random variable in the transcribed and untranscribed states, respectively. P(θt) is assumed to be uniform over the interval (0.3, 1), and P(θu) is assumed to be uniform over the interval (0.01, 0.03). In practice, we discretized θu into segments of size 0.01 and θt into segments of size 0.05 and approximated the integrals with finite sums. Finally, in addition to the annotated transcripts, we considered a competing model representing a completely untranscribed gene.
TuSelector was run separately for each replicate and time point and potentially produced discordant transcript calls across these runs. Therefore, we selected at most one “consensus” transcript model per gene for use in further analysis, as follows. To be considered a consensus call, TuSelector had to identify the same transcript model at least 80% of the time with at least 50% of replicate pairs both having the same transcript call. Two transcript models were considered “the same” if their endpoints differed by less than 500 bp. If no transcript model met these criteria, the gene was not considered in further analysis.
Estimating expression and detecting differentially expressed genes
For all active, protein-coding transcripts, reads were taken from up to the first 16 kb of the gene, excluding the first 500 bp to avoid an influence from promoter-proximal pausing. This strategy allowed us to focus on the most recent transcription at each time point and avoid averaging over long time intervals. The maximum interval of 16 kb was based on a minimum interval between time points of 10 min and an average polymerase transcription rate of ∼2 kb/min, minus a few kilobases of “padding.” Any genes that were shorter than 700 bp were removed from the analysis. A size factor for each sample was obtained by taking the number of spike-in reads for that sample divided by the median number of spike-in reads per sample. To estimate expression of transcriptional enhancers, reads were taken from 310 bases (assuming a 110-base spacing between dTSSs as reported by Core et al. 2014, plus 100 bp to either side) centered on the dTSS. Both sets of read counts were fed jointly into DESeq2, and enhancers and genes were subsequently separated for further analysis. An enhancer or gene was called as DE with an FDR ≤ 0.01 using a likelihood ratio test.
Clustering differentially expressed genes
Gene expression log-fold changes were computed relative to the untreated (zero-minute) time point using the DESeq-based estimates of absolute expression (rlog values). All DE genes were then fed into the autoregressive clustering program EMMIX-WIRE using default settings. Likelihood values for between two and 10 clusters were computed. We selected four clusters as a value at which the increases in likelihood with the number of clusters began to decline. To check for the robustness of our selection, we repeated our analyses with five and seven clusters and found that they were not highly sensitive to the cluster number.
Computing functional enrichment for gene clusters
Reactome (v52) was used to assign genes to functional categories. Genes that were not annotated in Reactome were removed. The background set for all enrichments was the set of DE genes present in Reactome. Odds ratios were computed per cluster (c) and pathway (p) as
where X represents a count and c and ¬c denote the sets of genes in, and not in, cluster c, respectively, and, similarly, p and ¬p denote the sets of genes in, and not in, pathway p. An empirical null distribution of odds ratios was computed by randomly shuffling the gene assignments to pathways 100,000 times. P-values were then computed from this distribution and the Benjamini-Hochberg procedure was applied to estimate false discovery rates.
Characterizing genic regulation
ChIP-seq data were downloaded from the ENCODE website (https://www.encodeproject.org) in narrowPeak format (optimal idr) on Sept. 30th, 2016. Scores for each gene-TF pair were computed by taking the peaks with the maximum signal that intersected [−200,+500] around the promoter. DeepBind v0.11 was run over [−200,+500] around the promoter with standard settings using all nondeprecated motifs for DNA binding proteins (Alipanahi et al. 2015). DeepBind and ChIP-seq scores were then standardized to control for differences in range. To analyze TFs that may be involved in different regulatory patterns, we linearly regressed genic expression (as estimated by DESeq2) against scores from ChIP-seq or DeepBind with time point-specific coefficients for each TF and a time-independent, gene-specific coefficient to capture the fixed effect of unmodeled regulation (Neph et al. 2012; Alipanahi et al. 2015). In this framework, the expected expression of a given gene i at time j is expressed as
where βi is the gene-specific expression bias term, βj is a time-specific bias term, and βijk is the coefficient for the time point-specific effect of a TF k on gene i. Standard deviations for each coefficient were estimated via 1000 bootstraps. Finally, the list of TFs was filtered to keep those with FDR ≤ 0.01 in at least one time point and with a maximal change between any two coefficients in the 90th percentile. This procedure selected for TFs having an effect that was both statistically significant and of large magnitude. Per-TF F-statistics were also calculated and are available as Supplemental Tables (Supplemental Tables 1, 2).
Identification and analysis of genic pause peaks
To locate pause peaks, we scanned each active transcript (see above) greater than 1 kb in length in the region of the annotated TSS ([TSS − 200, TSS + 200]) taking the number of reads in a 50-bp sliding window, with a sliding increment of 5 bp. The window with the largest number of reads in the untreated condition (0-min time point) was designated as the pause peak. To compute a log2 pause index (LPI), we subtracted the DESeq-estimated log2 read count (the “rlog” value) for the gene body from the equivalent DESeq-estimated log2 read count at the peak. Furthermore, to compute changes in this value over time, we subtracted the LPI for the zero time point from the LPI for each subsequent time point; that is, the change in LPI at time t, denoted ΔLPIt, was given by ΔLPIt = LPIt − LPI0. Notice that normalizing changes in the pause peak by changes in the gene body in this way causes ΔLPIt to increase only if the number of reads in the peak increases by more than the number of reads in the gene body.
Analysis of heat shock data
PRO-seq heat shock gene expression values were computed in an identical manner to the celastrol data with the exception of the size factors, which were obtained directly from a previous analysis (Vihervaara et al. 2013). Gene transcripts were the same as those used for the celastrol data. P-values were (re-)computed using the Wald test instead of the LRT for both the celastrol and heat shock data to allow for a single time point analysis. Differences in the distributions of gene expression within a pathway between HS and celastrol responses were evaluated using the Kolmogorov-Smirnov test.
Estimating expression in enhancers
To prevent contamination from genic transcription, all dTSSs previously annotated as enhancers were extended by 1 kb to either side and removed if any part of the extended enhancer was within 5 kb of a gene body. DESeq was used to estimate the transcription level in the enhancer peaks, tails, and the whole enhancer body. The enhancer peak was defined as ±250 bp from the center of the enhancer, the tail was ±400 to ±1000 bp from the center, and the whole enhancer was 0 to ±1000 bp from the center of the enhancer. Read counts were summed from both strands for each region (i.e., peak = plus strand [0,+250]+ minus strand[−250,0]), and then DESeq2 was used to estimate fold changes. Enhancers were called as strongly activated if they were differentially expressed with FDR ≤ 0.01, rlog(expression at 0 min) ≤ 1, and were in the 90th percentile for fold change between 0 and 160 min. To get the same number of similarly expressed nondifferentially expressed enhancers, we performed rejection sampling on enhancers that were differentially expressed with FDR > 0.5, using their average expression values between 0 and 160 min and probabilities calculated from a kernelized histogram of the activated enhancer's expression at 160 min.
Data access
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE96869.
Supplementary Material
Acknowledgments
We thank other members of the Siepel, Lis, and Danko laboratories for helpful discussions. This research was supported, in part, by US National Institutes of Health (NIH) grants HG007070 (to J.T.L. and A.S.), GM102192 (to A.S.), and HG009309 (to C.G.D.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.222935.117.
References
- Alipanahi B, Delong A, Weirauch MT, Frey BJ. 2015. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 33: 831–838. [DOI] [PubMed] [Google Scholar]
- Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. 2001. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry 25: 1341–1357. [DOI] [PubMed] [Google Scholar]
- Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H. 1992. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 359: 423–426. [DOI] [PubMed] [Google Scholar]
- Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. 1993. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 72: 233–245. [DOI] [PubMed] [Google Scholar]
- Andrulis ED, Guzmán E, Döring P, Werner J, Lis JT. 2000. High-resolution localization of Drosophila Spt5 and Spt6 at heat shock genes in vivo: roles in promoter proximal pausing and transcription elongation. Genes Dev 14: 2635–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al. 2005. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 24: 3470–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth GT, Wang IX, Cheung VG, Lis JT. 2016. Divergence of a conserved elongation factor and transcription regulation in budding and fission yeast. Genome Res 26: 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89: 331–340. [DOI] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. 2011. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. 2014. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46: 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, Kraus WL. 2013. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol Cell 50: 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hyland SL, Core LJ, Martins AL, Waters CT, Lee HW, Cheung VG, Kraus WL, Lis JT, Siepel A. 2015. Identification of active transcriptional regulatory elements from GRO-seq data. Nat Methods 12: 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nadal E, Ammerer G, Posas F. 2011. Controlling gene expression in response to stress. Nat Rev Genet 12: 833–845. [DOI] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei C-L, Natoli G. 2010. A large fraction of extragenic RNA Pol II transcription sites overlap enhancers. PLoS Biol 8: e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, et al. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, et al. 2016. The reactome pathway knowledgebase. Nucleic Acids Res 44: D481–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Zhang D, Fan C, Ma C, Yang W, Meng Y, Wu W, Guan S, Jiang B, Yang M, et al. 2013. ER stress-mediated apoptosis induced by celastrol in cancer cells and important role of glycogen synthase kinase-3β in the signal network. Cell Death Dis 4: e715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribley AM, Miller JR, Brownell AL, Garshott DM, Zeng Q, Reist TE, Narula N, Cai P, Xi Y, Callaghan MU, et al. 2015. Celastrol induces unfolded protein response-dependent cell death in head and neck cancer. Exp Cell Res 330: 412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LB. 2008. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol 28: 3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golebiowski F, Matic I, Tatham MH, Cole C, Yin Y, Nakamura A, Cox J, Barton GJ, Mann M, Hay RT. 2009. System-wide changes to SUMO modifications in response to heat shock. Sci Signal 2: ra24. [DOI] [PubMed] [Google Scholar]
- Guo JL, Yuan SX, Wang XC, Xu SX, Li DD. 1981. Tripterygium wilfordii Hook Fin rheumatoid arthritis and ankylosing spondylitis. Preliminary report. Chin Med J (Engl) 94: 405–412. [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. 2011. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145: 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. 2013. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 23: 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieda M, Winstanley H, Maini P, Iborra FJ, Cook PR. 2004. Different populations of RNA polymerase II in living mammalian cells. Chromosome Res 13: 135–144. [DOI] [PubMed] [Google Scholar]
- Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, et al. 2006. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10: 321–330. [DOI] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, Lis JT. 2014. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 3: e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM. 2003. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene 22: 5707–5711. [DOI] [PubMed] [Google Scholar]
- Kannaiyan R, Shanmugam MK, Sethi G. 2011. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett 303: 9–20. [DOI] [PubMed] [Google Scholar]
- Katan Y, Agami R, Shaul Y. 1997. The transcriptional activation and repression domains of RFX1, a context-dependent regulator, can mutually neutralize their activities. Nucleic Acids Res 25: 3621–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan Y-C, Hashidume T, Shibata T, Uchida K, Shimizu M, Inoue J, Sato R. 2017. Heat shock protein 90 modulates lipid homeostasis by regulating the stability and function of sterol regulatory element-binding protein (SREBP) and SREBP cleavage-activating protein. J Biol Chem 292: 3016–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, Lis JT. 2013. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339: 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B-K, Bhinge AA, Iyer VR. 2011. Wide-ranging functions of E2F4 in transcriptional activation and repression revealed by genome-wide analysis. Nucleic Acids Res 39: 3558–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhang Y, Huang X-Y, Sun Y, Jia Y, Li D. 2005. Beneficial effect of tripterine on systemic lupus erythematosus induced by active chromatin in BALB/c mice. Eur J Pharmacol 512: 231–237. [DOI] [PubMed] [Google Scholar]
- Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK. 2016. RNA splicing is a primary link between genetic variation and disease. Science 352: 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis JT, Mason P, Peng J, Price DH, Werner J. 2000. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev 14: 792–803. [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. 2015. Treatment of obesity with celastrol. Cell 161: 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Xu L, Alberobello AT, Gavrilova O, Bagattin A, Skarulis M, Liu J, Finkel T, Mueller E. 2015. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1α transcriptional axis. Cell Metab 22: 695–708. [DOI] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, Lis JT. 2016a. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11: 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Salamanca HH, Duarte FM, Danko CG, Lis JT. 2016b. Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol Cell 62: 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, Churchman LS. 2015. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 161: 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu T-W, Ong DST, Wang Y-J, Balch WE, Yates JR III, Segatori L, Kelly JW. 2008. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 134: 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase M, Oto J, Sugiyama S, Yube K, Takaishi Y, Sakato N. 2003. Apoptosis induction in HL-60 cells and inhibition of topoisomerase II by triterpene celastrol. Biosci Biotechnol Biochem 67: 1883–1887. [DOI] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. 2012. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. 2015. Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161: 526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshlack A, Robinson MD, Young MD. 2010. From RNA-seq reads to differential expression results. Genome Biol 11: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. 2011. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 12: 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng B, Xu L, Cao F, Wei T, Yang C, Uzan G, Zhang D. 2010. HSP90 inhibitor, celastrol, arrests human monocytic leukemia cell U937 at G0/G1 in thiol-containing agents reversible way. Mol Cancer 9: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polager S, Ginsberg D. 2003. E2F mediates sustained G2 arrest and down-regulation of stathmin and AIM-1 expression in response to genotoxic stress. J Biol Chem 278: 1443–1449. [DOI] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. 2011. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol 29: 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Raychowdhury R, Jovanovic M, Rooney M, Stumpo DJ, Pauli A, Hacohen N, Schier AF, Blackshear PJ, Friedman N, et al. 2014. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159: 1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai M, Ishikawa Y, Ota A, Sakurai H. 2013. The proto-oncogene JUN is a target of the heat shock transcription factor HSF1. FEBS J 280: 6672–6680. [DOI] [PubMed] [Google Scholar]
- Scimè A, Li L, Ciavarra G, Whyte P. 2008. Cyclin D1/cdk4 can interact with E2F4/DP1 and disrupts its DNA-binding capacity. J Cell Physiol 214: 568–581. [DOI] [PubMed] [Google Scholar]
- Sethi G, Ahn KS, Pandey MK, Aggarwal BB. 2007. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-κB-regulated gene products and TAK1-mediated NF-κB activation. Blood 109: 2727–2735. [DOI] [PubMed] [Google Scholar]
- Shack S, Gorospe M, Fawcett TW, Hudgins WR, Holbrook NJ. 1999. Activation of the cholesterol pathway and Ras maturation in response to stress. Oncogene 18: 6021–6028. [DOI] [PubMed] [Google Scholar]
- Shalgi R, Hurt JA, Krykbaeva I, Taipale M, Lindquist S, Burge CB. 2013. Widespread regulation of translation by elongation pausing in heat shock. Mol Cell 49: 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, Karin M. 2001. AP-1 in cell proliferation and survival. Oncogene 20: 2390–2400. [DOI] [PubMed] [Google Scholar]
- Singh J, Padgett RA. 2009. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol 16: 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, Henikoff S. 2011. Heat shock reduces stalled RNA polymerase II and nucleosome turnover genome-wide. Genes Dev 25: 2387–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd DJ, Lee A-H, Glimcher LH. 2008. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol 8: 663–674. [DOI] [PubMed] [Google Scholar]
- Trott A, West JD, Klaić L, Westerheide SD, Silverman RB, Morimoto RI, Morano KA. 2008. Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol-targeted molecule. Mol Biol Cell 19: 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. 2000. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666. [DOI] [PubMed] [Google Scholar]
- Veloso A, Kirkconnell KS, Magnuson B, Biewen B, Paulsen MT, Wilson TE, Ljungman M. 2014. Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res 24: 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, Sergelius C, Vasara J, Blom MAH, Elsing AN, Roos-Mattjus P, Sistonen L. 2013. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci 110: E3388–E3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, Mahat DB, Guertin MJ, Chu T, Danko CG, Lis JT, Sistonen L. 2017. Transcriptional response to stress is pre-wired by promoter and enhancer architecture. Nat Commun 8: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villard J, Peretti M, Masternak K, Barras E, Caretti G, Mantovani R, Reith W. 2000. A functionally essential domain of RFX5 mediates activation of major histocompatibility complex class II promoters by promoting cooperative binding between RFX and NF-Y. Mol Cell Biol 20: 3364–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sato R, Brown MS, Hua X, Goldstein JL. 1994. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 77: 53–62. [DOI] [PubMed] [Google Scholar]
- Wang K, Ng SK, McLachlan GJ. 2012. Clustering of time-course gene expression profiles using normal mixture models with autoregressive random effects. BMC Bioinformatics 13: 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Teriete P, Hu A, Raveendra-Panickar D, Pendelton K, Lazo JS, Eiseman J, Holien T, Misund K, Oliynyk G, et al. 2015. Direct inhibition of c-Myc-Max heterodimers by celastrol and celastrol-inspired triterpenoids. Oncotarget 6: 32380–32395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Bosman JD, Mbadugha BNA, Kawahara TLA, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. 2004. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem 279: 56053–56060. [DOI] [PubMed] [Google Scholar]
- Wu C-H, Yamaguchi Y, Benjamin LR, Horvat-Gordon M, Washinsky J, Enerly E, Larsson J, Lambertsson A, Handa H, Gilmour D. 2003. NELF and DSIF cause promoter proximal pausing on the hsp70 promoter in Drosophila. Genes Dev 17: 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wu Z, Xu C, Ren Y, Ge Y. 2003. Observation on serum anti-double stranded DNA antibodies of tripterine in systemic lupus erythematosus of (NZBxW)F1 mice. Ann Rheum Dis 62: 377–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Sengupta PK, Seto E, Smith BD. 2006. Regulatory factor for X-box family proteins differentially interact with histone deacetylases to repress collagen α2(I) gene (COL1A2) expression. J Biol Chem 281: 9260–9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Chen D, Cui QC, Yuan X, Dou QP. 2006. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 66: 4758–4765. [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong J-W, Nechaev S, Adelman K, Levine M, Young RA. 2007. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet 39: 1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. 2009. Characterization of celastrol to inhibit Hsp90 and Cdc37 interaction. J Biol Chem 284: 35381–35389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Geng C, Liu X, Li M, Gao M, Liu X, Fang F, Chang Y. 2016. Celastrol ameliorates liver metabolic damage caused by a high-fat diet through Sirt1. Mol Metab 6: 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.