

Abstract
We have developed a novel method that enables global subtelomere and haplotype-resolved analysis of telomere lengths at the single-molecule level. An in vitro CRISPR/Cas9 RNA-directed nickase system directs the specific labeling of human (TTAGGG)n DNA tracts in genomes that have also been barcoded using a separate nickase enzyme that recognizes a 7-bp motif genome-wide. High-throughput imaging and analysis of large DNA single molecules from genomes labeled in this fashion using a nanochannel array system permits mapping through subtelomere repeat element (SRE) regions to unique chromosomal DNA while simultaneously measuring the (TTAGGG)n tract length at the end of each large telomere-terminal DNA segment. The methodology also permits subtelomere and haplotype-resolved analyses of SRE organization and variation, providing a window into the population dynamics and potential functions of these complex and structurally variant telomere-adjacent DNA regions. At its current stage of development, the assay can be used to identify and characterize telomere length distributions of 30–35 discrete telomeres simultaneously and accurately. The assay's utility is demonstrated using early versus late passage and senescent human diploid fibroblasts, documenting the anticipated telomere attrition on a global telomere-by-telomere basis as well as identifying subtelomere-specific biases for critically short telomeres. Similarly, we present the first global single-telomere-resolved analyses of two cancer cell lines.
In humans, telomeres are nucleoprotein complexes made up of tandem 5′TTAGGG3′ DNA repeats and associated proteins located at the ends of all 46 chromosomes (Samassekou et al. 2010; Aubert et al. 2012; Vera and Blasco 2012; Montpetit et al. 2014; Blackburn et al. 2015). Telomere (TTAGGG)n tract loss beyond a certain threshold changes the telomere structure (“uncapping”), which causes telomere dysfunction and leads to senescence or apoptosis (Meier et al. 2007; Samassekou et al. 2010; Aubert et al. 2012; Kaul et al. 2012). The senescence threshold is cell line specific for human diploid fibroblasts (HDFs) (Britt-Compton et al. 2006), with the shortest single-telomere length detected at senescence 300 bp (Britt-Compton et al. 2006). In vivo, aberrant senescence or apoptosis can disrupt tissue microenvironments and contribute to aging and cancer. When tumor suppressor pathways are compromised in the presence of dysfunctional telomeres, further telomere attrition occurs and leads to telomere fusions (Capper et al. 2007); genome instability ensues and contributes to the formation of a tumor (Sabatier et al. 2005; Coppe et al. 2010; Davalos et al. 2010; Jaskelioff et al. 2011).
Currently, there are several methods to measure the length of telomere repeats each with their own advantages and disadvantages. Terminal restriction fragment (TRF) estimates the average telomere length of a population of cells with a resolution of 1 kb (Aubert et al. 2012). TRF requires at least 1.5 μg of DNA, can overestimate the telomere length by several kilobases, and is not sensitive to very short telomeres (Samassekou et al. 2010; Montpetit et al. 2014). The quantitative PCR (qPCR) method (Cawthon 2002, 2009) was developed to decrease the amount of DNA required, with only 25–50 ng needed to estimate telomere repeat content of a sample. However, the method actually measures the ratio of total telomere repeat content in a sample relative to a single-copy gene (T/S ratio), which in turn must be related to base pair of telomere using a standard curve of samples whose telomere lengths have been determined by another method (typically TRF analysis), thus providing estimates of average telomere length per sample (Cawthon 2002, 2009; Lynch et al. 2016). Single-telomere length analysis (STELA) (Baird et al. 2003) and quantitative fluorescence in situ hybridization (Q-FISH) (Lansdorp et al. 1996) were developed to detect and measure the length of specific telomeres. STELA can measure single-telomere lengths with a resolution of 0.1 kb and sometimes identifies allelic differences in an individual telomere length (Baird et al. 2003; Samassekou et al. 2010; Aubert et al. 2012; Montpetit et al. 2014). However, it requires unique priming sites in subtelomere regions that are enriched in duplications and is usually limited to subtelomeres XpYp, 2p, 11q, 12q, and 17p (Britt-Compton et al. 2006; Samassekou et al. 2010; Aubert et al. 2012; Montpetit et al. 2014). Q-FISH of telomere repeats (Q-FISH) requires only 15–20 metaphase cells per sample and measures telomeres on all individual chromosome arms. This method is also able to identify chromosome ends without detectable repeats (<0.5 kb), as well as chromosome fusion occurrences (Aubert et al. 2012). A major disadvantage of Q-FISH is that it is limited in the analysis of cells currently in metaphase and is unable to measure telomeres in terminally senescent cells or cells that are no longer able to divide (Aubert et al. 2012; Montpetit et al. 2014). With the current methods to measure telomere length, it is impossible to efficiently acquire global subtelomere and haplotype-resolved telomere length data.
Normal human telomere lengths vary between chromosomes within the same cell and even homologous chromosome arms. The mechanisms regulating subtelomere-specific and haplotype-specific telomere lengths in humans are understudied and poorly understood, primarily because of technical limitations in obtaining these data globally. Although the relative telomere lengths of single chromosome arms within individuals appear to vary considerably with genetic background (including specific subtelomere haplotypes) (Baird et al. 2003; Britt-Compton et al. 2006), there have been some chromosome arm–specific telomere length trends documented. For example, telomeres on chromosome arms 17p, 19p, and 20q have been identified as some of the shortest, whereas 1p, 3p, 2p, and 4q contain telomeres among the longest (Martens et al. 1998; Perner et al. 2003; Britt-Compton et al. 2006; Samassekou et al. 2009, 2010).
Studies measuring average telomere lengths have established a relationship between shorter average telomere lengths and increasing age, age-related diseases, and mortality (Blackburn et al. 2015). However, there have been numerous studies suggesting that the shortest individual telomeres in cells mediate the biological effects associated with short average telomere lengths via telomere dysfunction (Aubert et al. 2012); the shortest telomere or a small number of short telomeres in cells rather than the average telomere length is critical for chromosome stability and cell viability (Samassekou et al. 2010; Vera and Blasco 2012). The identity and frequency of specific critically short telomeres may thus be the most useful telomere biomarker for aging and age-related diseases, including cancer (Vera and Blasco 2012; Roger et al. 2013; Lin et al. 2014; Simpson et al. 2015). Gaining a better understanding of telomere length regulation in healthy humans, including factors involved in single telomere-specific regulation of (TTAGGG)n tract length and stability, may lead to more precise telomere biomarkers and better approaches to defining the role of telomeres in diseases (Blackburn et al. 2015). However, with the current methods to measure telomere length, it is impossible to acquire such global haplotype-resolved telomere length data.
To address this need, we have developed a method that simultaneously measures individual telomere (TTAGGG)n tract lengths and identifies their physically linked subtelomeric DNA. This assay can also be used to identify chromosome ends lacking detectable telomere sequences, characterize novel subtelomeric structural variants and haplotypes, and discover previously uncharacterized subtelomere regions of human genomes. Here, we describe this technology and its application to the analysis of single-telomere lengths in two cancer cell lines and to single-telomere length changes in the IMR90 cellular aging model.
Results
Figure 1 shows an overview of the single-molecule telomere length assay. For our telomere/subtelomere colabeling strategy to work, the genome-wide sequence motif-dependent nick-labeling (Jo et al. 2007; Xiao et al. 2007; Das et al. 2010) must differentiate individual subtelomeres, and the telomere labeling must be specific and proportional to (TTAGGG)n tract lengths. Nt.BspQI, which has been used widely to nick-label (GCTCTTC) sites genome-wide for large single-molecule-based whole-genome map assemblies, was selected initially for sequence motif mapping (Fig. 1; Mak et al. 2016). For telomere-specific labeling, we developed a CRISPR/Cas9 nickase (Cas9n) (Ran et al. 2013)-based protocol directed to telomere (TTAGGG)n tracts by a synthetic guide RNA containing the sequence UUAGGGUUAGGGUUAGGGUU, which is complementary to the 3′ to 5′ telomere DNA repeat strand and followed by the protospacer adjacent motif (PAM) NGG (Zhang et al. 2015). Optimal telomere labeling conditions were defined empirically, with the conditions found to provide maximal overall telomere fluorescent intensity used for all subsequent experiments. We developed two-color and three-color labeling schemes for our single-molecule telomere length assay (Fig. 1). In both schemes, the same gRNA is used to nick-label the telomeric repeats, while the subtelomeric regions are globally nick-labeled using Nt.BspQI to target the GCTCTTC motif. In the two-color scheme, the Cas9n and Nt.BspQI are incubated with the human genomic DNA and simultaneously nick their respective regions. All nicks are labeled with green fluorescently labeled nucleotides using Taq DNA polymerase. In the three-color scheme, Nt.BspQI is first incubated with the human genomic DNA and nicks the GCTCTTC motif. These nicks are labeled with green fluorescently labeled nucleotides using Taq DNA polymerase. The unincorporated green fluorescent nucleotides are removed with shrimp alkaline phosphatase (SAP). The labeled DNA molecules are then nicked with Cas9n and labeled with red fluorescently labeled nucleotides using Taq DNA polymerase. The labeled DNA molecules are then optically imaged in a high-throughput manner using nanochannel arrays (Lam et al. 2012). De novo assembly is performed using the unique subtelomeric Nt.BspQI patterns, and the assemblies are then mapped to subtelomeric reference sequences (human hg38). The individual telomere-terminal DNA molecules thus assembled are used for calculating the telomere length. This allows for the simultaneous identification of the chromosome arms and quantitation of the linked single-telomere lengths.
Figure 1.

Schematic description of the single-molecule telomere length assay. In both telomere labeling schemes, the Cas9 D10A (Cas9n) directed by the telomere gRNA nicks the telomere repeats (TTAGGG)n and Nt.BspQI globally nicks the GCTCTTC motif. In the two-color scheme, all nicks are labeled with green fluorescently labeled nucleotides. In the three-color scheme, the Nt.BspQI-generated nicks are labeled with green fluorescently labeled nucleotides, and the telomeres are labeled with red fluorescently labeled nucleotides. All samples are linearized in the nanochannels and optically imaged. De novo assembly is performed, and the telomere label intensities are used for calculating the telomere length.
Subtelomeric repeat element (SRE) regions positioned immediately adjacent to telomere (TTAGGG)n tracts have complicated efforts to distinguish and identify single chromosome ends in human genomes, effectively precluding global single-telomere analysis at the molecular level. These regions contain highly similar (90%–99.9% sequence identity) segmental duplications, large structural variations, and reference sequence gaps and misassemblies (Stong et al. 2014). While most SRE regions are 40–150 kb in size, long SRE regions of up to ∼300 kb have been identified in a few subtelomeres, including 1p, 8p, and 5q. Motif-dependent nick-labeling of large genomic DNA molecules followed by assembly of mapped single large DNA molecules can effectively distinguish SRE regions and permit connection of chromosome ends harboring the largest SRE regions with specific subtelomeres. Figure 2A demonstrates this for the 1p, 5q, and 8p subtelomeric regions of the HG11381 genome. These subtelomeric regions share some SREs but differ in others, as shown in the paralogy maps of these subtelomere reference sequences (Fig. 2A, top, “paralogy map”; Stong et al. 2014). Following motif-dependent nick-labeling of the HG11381 genome, the 1p, 5q, and 8p subtelomeres were de novo assembled into unique consensus maps from the mixture of single long DNA molecules (Fig. 2A, bottom). 1p and 5q share similar duplicons up to 200 kb from the telomere according to the paralogy map, which is supported in part by the same sequence motif patterns from 55–200 kb (blue bar). However, the sequence motif patterns are clearly distinguishable between 1p and 5q from 0–55 kb, even though this region contains several similar duplicons. As indicated with the red bars in Figure 2A consensus maps, 5q and 8p share very similar duplication structure and organization until 130 kb from the telomere, where the sequence motif patterns start to differ. Based on the above differences, only a 150-kb region is needed to differentiate these three highly similar subtelomeric regions. This is true for all the other subtelomeric chromosome arms sharing similar SREs; furthermore, definitive localization to subtelomere-specific one-copy DNA (gray line segments in the paralogy maps) is typically achieved using large DNA molecules in this size range. This genome mapping method with long DNA molecules provides a unique tool to track and identify individual SRE variants within families of SREs and to connect each chromosome end with a specific subtelomere.
Figure 2.

Global sequence motif-dependent nick-labeling distinguishes subtelomeres and identifies subtelomeric structural variants. (A) The subtelomere repeat elements of 1p, 5q, and 8p are shown as colored blocks, with each color indicating a paralogy block of segmentally duplicated subtelomeric repeat elements (SREs) as defined previously (Stong et al. 2014). The telomeres are designated as a red T. The consensus maps of nick-labeled single molecules after de novo assembly and alignment with the hg38 reference are shown as light blue bars; the dark blue vertical ticks within these bars indicate the Nt.BspQI nick-label sites. The single molecules that comprise the respective consensus map are the yellow lines shown directly below each map. The labels on the single molecules that align with the hg38 reference are dark green, and those that do not align are light blue. The dark blue and red bars designate the overlapping regions between the chromosome arms. (B) The major haplotypes of Chromosome 6p detected from eight individuals. The hg38 6p subtelomere reference sequence is shown as a light blue bar; the dark blue vertical ticks within these bars indicate the in silico Nt.BspQI nick-label sites. The black line segment designates a telomere-adjacent gap in the hg38 reference sequence. Each haplotype is represented as a distinct consensus map of single molecules from the designated genome (yellow line) aligned to the hg38 6p reference sequence. The labels on these consensus maps that align with the hg38 reference are dark green, and those that do not align are light blue. (C) The two distinct haplotypes of the cancer genome UMUC3 for Chromosome 6p.
The Nt.BspQI nickase nick-labeling method can also identify structurally variant haplotypes in highly variable subtelomeric regions. Taking advantage of a related project constructing hundreds of human genome sequence motif maps with Nt.BspQI nick-labeling (GCTCTTC) (Mak et al. 2016), we analyzed and compared assembled subtelomeric regions over 100 genomes to determine whether the Nt.BspQI nick-labeling can distinguish subtelomeric identities and structural variant haplotypes. Figure 2B shows eight major variant haplotypes of Chromosome 6p detected in this large data set. The consensus maps each contain labels that do not align to the hg38 reference and differ from one another (light blue lines). Figure 2C identifies two distinct 6p haplotypes found in the cancer genome of UMUC3.
After confirming that global sequence motif labeling can differentiate subtelomeres, we optimized the CRISPR/Cas9 telomeric labeling using a circular fosmid containing an 800-bp tract of telomeric repeats cloned from the human chromosome arm 8q. There was no fluorescent labeling of the telomeric repeats (TTAGGG) without the presence of either Cas9n or gRNA. The fluorescent labeling of telomeric repeats was detected only when both Cas9n and gRNA were present in the reaction (data not shown). We then combined the Cas9n (TTAGGG) and Nt.BspQI (GCTCTTC) to nick-label the telomere and the adjacent subtelomeric DNA of the fosmid at the same time. After linearization of fosmid DNA with the enzyme NotI, the Nt.BspQI labeling pattern of the fosmid matched the reference sequence, and the telomeric labeling was always at the end of the molecules (Supplemental Fig. S1A), indicating the telomeric labeling.
There are two possible methods to measure the telomere repeat lengths from fluorescent labels: based on the contour of telomeric labeling and the intensity of telomeric labeling. The longer the telomere, the more pixels it will occupy. However, the ends of DNA molecules tend to fold back onto themselves which affects the length measurements. More importantly, even with a single-point emitter, several pixels collect photons due to photon scattering. By use of the contour method, it is impossible to differentiate and resolve a telomere length <1 kb as all telomeres <1 kb will occupy the same number of pixels. The intensity method uses the total intensity of the telomeric labeling. We reasoned, the longer the telomere, the more frequently Cas9n will nick the (TTAGGG)n tract and fluorescent nucleotides become incorporated. The total intensity should therefore be proportional to the telomere length. We used fosmids with known telomere length to quantify the system and normalize the intensity measurement for estimating single-telomere lengths (Supplemental Fig. S1), and then extrapolated the intensities/base pair (TTAGGG)n tract from these standards to estimate base pair in genomic (TTAGGG)n tracts on single molecules based upon the measured fluorescent intensities.
The initial results of the two colabeling strategies for the single-molecule telomere length assay are shown in Figure 3. In the first application, we tested the three-color scheme on human genomic DNA sample GM11832. The results for Chromosomes 3p and 12p are shown in Figure 3, A and B, respectively. The telomeres are clearly labeled in red at the end of each molecule. The three-color scheme easily differentiates subtelomeres and labels genomic (TTAGGG)n tracts at the ends of the mapped molecules but is a time-consuming assay that involves two separate nick-labeling reactions with an intermediate step to remove the free fluorescently labeled nucleotides. This additional step along with frequent pipetting tend to break the DNA into smaller fragments, resulting in fewer long DNA fragments ideal for distinguishing the larger SRE regions. We hypothesized that the two-color scheme could also differentiate subtelomeres and label (TTAGGG)n tracts at the ends of long mapped subtelomere fragments while at the same time maintaining longer DNA molecules. This scheme was first tested with the IMR90 cell line, and indeed, this was the case. The single molecules produced from both color schemes generated the same consensus maps that were used to identify single telomeres and haplotypes, as well as labeled (TTAGGG)n tracts at the ends of the mapped molecules. The consensus maps of both haplotypes as well as examples of single molecules for Chromosome 2q from IMR90-PD17 are shown in Figure 3C. The ends of the molecules were labeled in green. As expected, when Cas9 was not used on the same sample, the ends of the molecules with the sample pattern were not labeled (Supplemental Fig. S2). The two-color scheme generates longer DNA molecules for mapping, has a simpler experimental procedure, and involves much less imaging time with two-thirds of the data storage requirements.
Figure 3.

Colabeling telomere (TTAGGG)n tracts using gRNA-directed CRISPR/Cas9 nickase and subtelomeres using global motif-dependent nickase Nt.BspQI. The hg38 subtelomere reference sequences are shown as light blue bars; the dark blue vertical ticks within these bars indicate in silico Nt.BspQI nick-label sites. The black line segments designate telomere-adjacent gaps in the hg38 reference sequence. Individual single-molecule maps were de novo assembled into the consensus maps (yellow lines), which were then aligned with the hg38 reference (light blue bars). The green lines on the consensus maps are Nt.BspQI (GCTCTTC) sites that align to the corresponding reference site. Those that do not align with the reference are designated with light blue lines. Representative examples of raw images of single molecules that were used to create the consensus maps are shown. (A,B) Three-color consensus maps and single molecules for Chromosomes 3p and 12p of sample GM11832. The DNA backbone is blue, labeled subtelomeric Nt.BspQI sites are green, and the telomere (TTAGGG)n tracts are red (C) Two-color consensus maps and representative images of single molecules of both haplotypes for Chromosome 2q of sample IMR90-PD17. The DNA backbone is blue, and both the labeled subtelomeric Nt.BspQI sites and the telomere (TTAGGG)n tracts are green.
Next, we applied the two-color labeling scheme to identify individual telomere lengths in the IMR90 fibroblast aging model cell line at early passage, late passage, and senescence, as well as the UMUC3 bladder cancer and the LNCaP prostate cancer cell lines. Typical raw imaging results used for determination of the average single-telomere lengths, in this case for Chromosome 8q, are shown at the top of Figure 4. The consensus maps aligned well to the hg38 human reference. It is visually obvious by the intense end telomere labels that IMR90-PD17 has the relatively longest telomeres. At population doubling of 45, the telomeres get shorter. Interestingly, when the IMR90 cells were senescent, a fraction of the 8q telomeres were very short, whereas others were still relatively long. The individual single-molecule telomere length distribution for 8q is plotted for each cell culture sample at the bottom of Figure 4B, where each spot is a single-telomere length measurement and the mean size is indicated by the horizontal bar for each sample. The broad size ranges of single telomere measurements in a given sample is similar to what is found for the PCR-based STELA single-telomere length distributions for bulk cell cultures (Baird et al. 2003). These reflect in part the mixed clonal subpopulations of IMR90 cells doubling at different rates (Britt-Compton et al. 2006) within the average cell division rate designated by the population doubling number of the cell culture overall, along with mixed clone-specific telomere attrition rates affected by human telomere rapid deletion (TRD) events and telomere end-processing, among other potential factors (Britt-Compton et al. 2006). The relatively large standard deviation associated with each average single-telomere length in the IMR90 samples thus reflects primarily single cell-dependent biological variation rather than measurement error; coefficients of variation (CVs) calculated for average single-telomere lengths in technical replicates using DNA from the IMR90-PD17 cell culture (inter-assay, identical samples processed and analyzed weeks to months apart) were 14.6%, which is in the ballpark of CVs reported for published inter-assay telomere measurements using Q-FISH, Flow-FISH, and qPCR (O'Sullivan et al. 2004; Gutierrez-Rodrigues et al. 2014; Martin-Ruiz et al. 2015). Our method is still new, and further improvements in reproducibility are likely to come with refinements to the protocol and wider experience with the methodology. Interestingly, the two telomerase-positive cancer cell lines have a tighter and shorter distribution of single-telomere lengths at 8q compared with the IMR90 samples, consistent with the shorter and more homogenous overall average telomere lengths for these cell lines (Table 1).
Figure 4.

Single-telomere length analysis results for 8q. (Top) The hg38 subtelomere reference sequence for 8q is shown as a light blue bar; the dark blue vertical ticks within these bars indicate in silico Nt.BspQI nick-label sites. The black line segment designates a telomere-adjacent gap in the hg38 reference sequence. The green lines on the consensus maps are Nt.BspQI (GCTCTTC) sites that align to the corresponding reference site. Those that do not align with the reference are designated with light blue lines. Representative examples of raw images of single molecules from two-color labeling experiments for each of the samples that were used to create the consensus map are shown. The human diploid fibroblast cell line IMR90 was sampled at population doubling 17 (IMR90-PD17), at PD45 (IMR90-PD45), and at senescence (IMR90-senescence; PD61). The immortal telomerase-positive cancer cell lines UMUC3 (bladder cancer) and LNCaP (prostate cancer) were also sampled. (Bottom) Telomere (TTAGGG)n tract length results for 8q from each of these samples. Each single-molecule telomere length measurement for 8q is represented by a dot, and the average telomere length for 8q in each sample is shown as a horizontal line.
Table 1.
Longest and shortest average single-telomere lengths
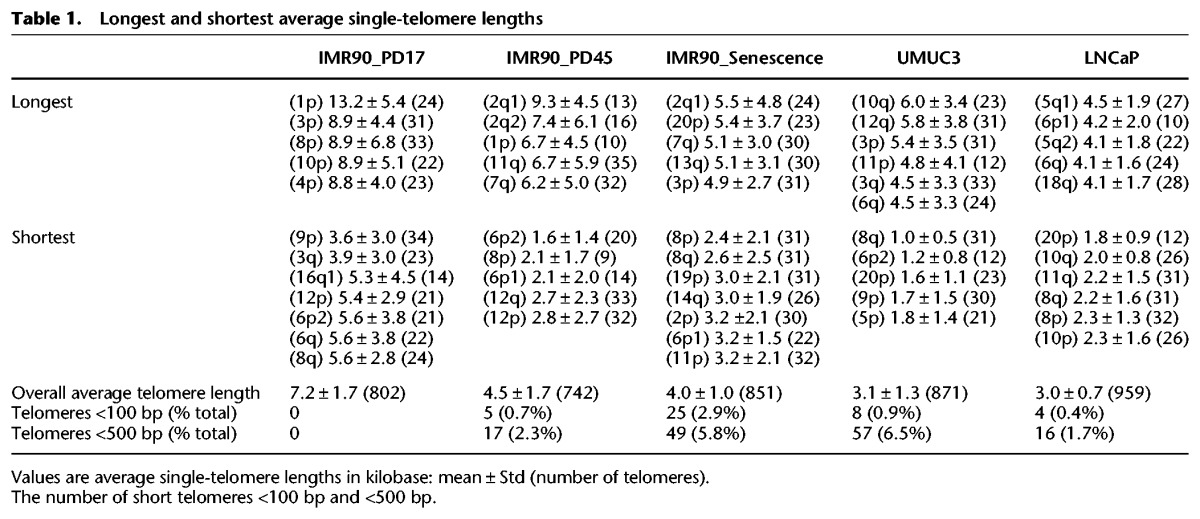
Globally, we could measure 36 out of 46 telomeres with about 30 molecules per telomere and detect telomere tracts <100 bp (Supplemental Table S1). We did not observe any mapped, apparently terminal, large DNA molecules totally lacking telomere signal, which may indicate that “telomere-free ends” documented by Q-FISH studies of metaphase chromosomes may in fact retain extremely short telomeres that are below the detection limit for Q-FISH but are still detectable using our technique. A full set of raw single-molecule images are shown in Supplemental Figure S3. The chromosome arms 13p, 14p, 15p, 21p, and 22p could not be identified and measured due to the lack of reference sequences for acrocentric short-arm telomeres. The telomeres of XpYp were also not measured because there were no contigs that spanned the telomere adjacent sequences of the XpYp subtelomere reference, possibly due to many gaps in the hg38 reference and a large highly variable region containing minisatellite repeats near this telomere (Inglehearn and Cooke 1990). Lastly, 16p, 17p, 19q, and 22q failed the assembly with most samples because of inverted nick pair (INP) sites. These are where two closely spaced nicking enzymes sites are found on opposite strands, which causes double-strand breaks in molecules to be mapped, that preclude the assembly on long continuous contigs.
Several interesting features and trends can be ascertained from the average single-telomere lengths measured in the IMR90 samples. First, there is a large difference between the longest and the shortest average single-telomere lengths in each of the IMR90 samples, with this difference decreasing as the cell culture progresses toward cellular senescence (13.2 kb vs. 3.6 kb at PD17, 9.3 kb vs. 2.4 kb at PD45, and 5.5 kb vs. 2.4 kb at senescence) (Table 1). The overall average telomere lengths for each sample (obtained by combining all of the single-telomere lengths) are reduced from 7.2 kb in PD17 to 4.5 kb in PD45 and to 4.0 kb at senescence in the IMR90 fibroblasts. Interestingly, no telomeres approaching a dysfunctional length (<500 bp) or expected to be well below the threshold for telomere dysfunction (<100 bp) were detected in the PD17 sample, whereas these begin to appear by PD45. From PD45 to senescence, the overall average telomere length decreases slightly (4.5 kb to 4.0 kb), but the number of telomeres <100 bp increases fivefold (Table 1).
Second, the average single-telomere length trends do not all follow the same trajectory with increasing IMR90 passage number. While most of the average single-telomere lengths decrease to about one-half of their initial value from PD17 to PD45 (including 1p, 3p, 4p, 5p, 6p1, 6p2, 8p, 11p, 12p, 1q, 5q, 12q, 14q, 15q, 21q), others do not decrease significantly from PD17 to PD45 (2p, 8q, 9q, and 11q), and one group remains relatively constant from PD17 through senescence (9p, 20p, 2q haplotype1, and 17q). Third, in almost all cases (with the exception of 2p) the average single-telomere lengths of senescent IMR90 fibroblasts (PD61) do not differ statistically from PD45 fibroblasts, even though the frequency of very short telomeres (<100 bp) increases fivefold. Finally, identities of very short (<100 bp) telomeres are biased in senescent IMR90, with about one-third of these telomeres on 8q and one-eighth on 14q (Table 2).
Table 2.
Very short telomeres in senescent and cancer cell lines
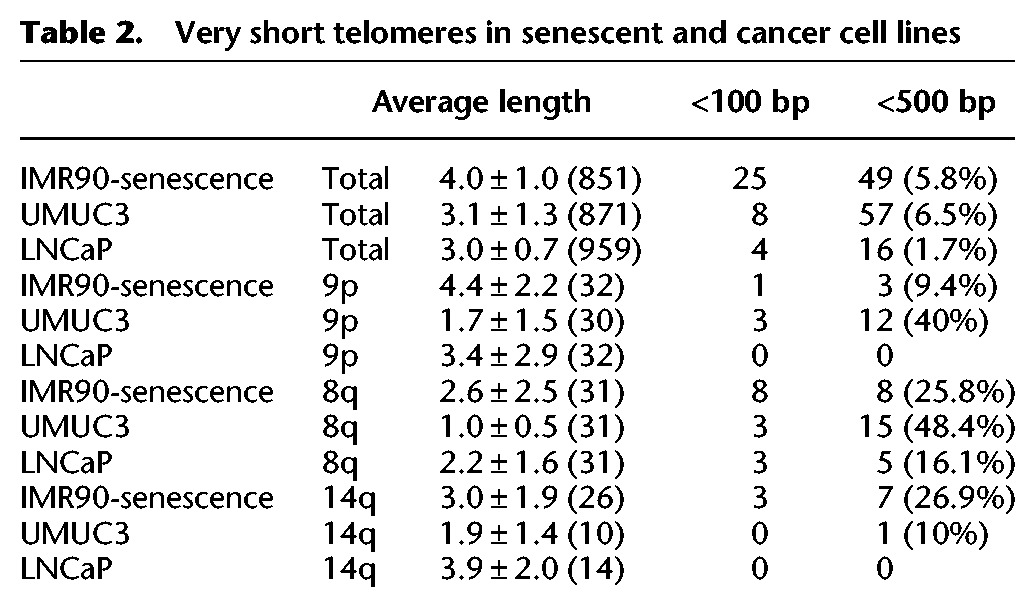
The UMUC3 bladder cancer and the LNCaP prostate cancer cell lines each have shorter telomere lengths with more uniform single-telomere length distributions compared to IMR90, as expected from prior telomere length analysis of these cell lines (Kageyama et al. 1997; Xu and Blackburn 2007). UMUC3 exhibited a relatively large number of very short telomeres, with 57 telomeres <500 bp. Forty-seven percent of these 57 shorter telomeres are on 9p (12 copies) and 8q (15 copies). These results are consistent with the published STELA results for this cell line, which designated these short telomeres “t-stumps” (Xu and Blackburn 2007). Overall, the 8q arm has the largest number of shorter telomeres for both IMR90 at senescence and UMUC3 cell line (Table 2). The 9p arm of UMUC3 also has a large number of short telomeres, which is not the case for IMR90 at senescence. On the other hand, the 14q arm of IMR90 at senescence has a greater number of short telomeres than the 14q arm of UMUC3 cell line (Table 2). The LNCaP cancer cell line has an overall average telomere length similar to that of the UMUC3 cancer cell line. However, there are not a large number of telomeres <500 bp. Even in this case, the 8q arm still accounts for most telomeres <100 bp (three out of four) and telomeres <500 bp (five out of 16).
Taken together, these results from our first single-telomere analyses reveal intriguing trends and apparently dynamic subtelomere-specific regulation of single-telomere lengths, while establishing the basic feasibility of the technology. For some chromosome arms with structurally variant subtelomeres, the resolution can be extended to individual haplotypes. For example, the haplotypes for the 2q arm of the IMR90 cell line at different population doublings have similar telomere lengths. On the other hand, one 6p subtelomere haplotype has significantly longer telomeres than the other haplotype for both IMR90 and UMUC3 cell lines (P < 0.01). These results are consistent with prior STELA analyses of several selected individual telomeres, where single-telomere (TTAGGG)n tract regulation and stability were found to be haplotype specific and dependent on cis-factors (Britt-Compton et al. 2006).
In addition to its utility for quantifying single-telomere lengths on large molecules linked to specific known telomeres, the specific fluorescent tagging of telomeres is a very powerful tool to identify previously uncharacterized subtelomeres. This makes it particularly valuable for improving the quality of subtelomeric reference sequences and for identifying new structurally variant subtelomeres. For example, the subtelomeric region 0–500 kb of Chromosome 1p arm is missing in all of the genomes we mapped. However, we noticed that a 1p DNA-containing contig mapping 600 kb from the 1p telomere in hg38 contained either intense telomere gRNA-directed CRISPR/Cas9n-dependent labeling green end labels (two-color labeling scheme) or intense red labels (three-color labeling scheme) in all of the genomes analyzed (Fig. 5A). We concluded that our experiments had identified the true 1p telomere and that 0–600 kb of hg38 is incorrect and should be removed from the reference assembly.
Figure 5.

Telomere labeling identifies incorrectly represented and uncharacterized subtelomeres. (A) The 0.55- to 0.8-megabase region of the hg38 subtelomere reference sequence for 1p of UMUC3 is shown as a light blue bar; the dark blue vertical ticks within these bars indicate in silico Nt.BspQI nick-label sites. The hg38 reference sequence continuing toward the putative 1p telomere is not shown. The green lines on the single-molecule maps are Nt.BspQI (GCTCTTC) sites. Representative examples of raw images of several of these single molecules from two-color labeling experiments are shown. The single DNA molecules used to form the consensus map for Chromosome 1p each align with the 0.6-Mb region of the hg38 reference, and all contain intense telomere end labels. (B) Five telomere-containing consensus maps that could not be aligned with the hg38 reference sequence. Red bar designates an SRE located on 4p, 4q, and several acrocentric short-arm subtelomeres (Youngman et al. 1992).
In addition, five consensus maps that could not be mapped to the hg38 reference were found to have a telomere label at one end (Fig. 5B). Consensus maps 1, 2, and 3 share a 40-kb conserved pattern, marked as the red bars in Figure 5B. This pattern is similar to the pattern of Chromosome 4p of hg38 from 25 kb to 65 kb and corresponds to a segmental duplication that is shared by the subtelomeres of 4p, 4q, and many acrocentric short-arm subtelomeres (Youngman et al. 1992). Taken together with the telomeric labels at the end of DNA molecules, it is strongly suggested these three consensus maps belong to the five unknown acrocentric chromosome ends 13p,14p, 15p, 21p, and 22p. Interestingly, they all have 7-kb repetitive sequence units following this 40-kb conserved pattern. Consensus map 1 has the longest 7-kb repeat region (>140 kb), which makes the sequence assembly impossible with current sequencing technologies. Based on these patterns, we are able to locate more unaligned consensus maps with the same patterns from other genomes, with some >700 kb long. The other two unaligned consensus maps with telomeric labels (Fig. 5B, bottom) reflect yet to be identified subtelomere alleles that cannot be incorporated into the current reference sequence on the basis of their nickase mapping patterns.
Discussion
Here, we have developed novel methods that facilitate global subtelomere-specific analysis of human lengths at the single-molecule level. One of the key technology advances is to use the CRISPR/Cas9 genome editing system to covalently tag the telomeric repeats with fluorescent dyes. There have been reports using an EGFP-linked, deactivated dCas9 protein to label telomeres for imaging (Chen et al. 2013). While useful for some purposes (e.g., in vivo imaging), this approach is not appropriate for our application using nanochannel analysis for two reasons. First, the EGFP-dCas9 is noncovalently linked to the telomere target sequence, and some fraction will not remain bound during the nanochannel array analysis procedure, making accurate quantitation difficult. Our method covalently incorporates fluorescently labeled nucleotides into the target telomere sequence and is therefore a far more stable interaction than the EGFP-dCas9. Second, the EGFP-dCas9 would introduce DNA-bound protein to the final sample, which would make it more difficult to load the sample into the nanochannels.
For the first time, large numbers of individual telomere lengths can be tracked efficiently in the context of total genomic DNA, as well as cis-factors influencing their length regulation and stabilities potentially evaluated. This new experimental capability may lead to much more complete and powerful analyses of genetic and epigenetic factors influencing telomere elongation, attrition, processing, and stability than those previously feasible using average telomere lengths. The methodology also connects subtelomere and haplotype specificity with individual SRE organization and variation, providing a window into the dynamics and potential functions of these complex and structurally variant telomere-adjacent DNA regions in telomere regulation and genome biology.
The results from our initial analyses of single-telomere lengths in an aging fibroblast cell line model and in a pair of cancer cell lines provide proof of principle for the efficacy of the methods, yielding data consistent with previous studies of these cell lines as well as new insights into their subtelomere-dependent telomere length dynamics. The overall average telomere length we measured from our single-telomere length data for senescent IMR90 cells (4.0 kb) (Table 1) was very close to the average of the four STELA-measured single telomeres measured in bulk IMR90 senescent cultures previously (4.1 kb) (Britt-Compton et al. 2006). The overall telomere attrition rate of IMR90 with population doublings is also very similar to what is expected (96 bp per doubling between PD17 and PD45) (Karlseder et al. 2002; Britt-Compton et al. 2006); as with IMR90 telomere attrition measured using STELA (Britt-Compton et al. 2006), we saw differences in subtelomere-specific telomere attrition rates. However, in our analysis of many more single telomeres, the arm-specific (and sometimes haplotype-specific) attrition rate differences were often much more pronounced than those seen previously. Importantly, the distribution of very short telomeres likely to be dysfunctional was biased, suggesting particular subtelomere-specific vulnerabilities to telomere dysfunction; in the case of IMR90, they are enriched at 8q and 14q (Table 2).
We unexpectedly observed a subset of single telomeres whose average single-telomere lengths changed very little between PD17 and PD45, and the average single-telomere lengths of most telomeres stayed roughly the same or even increased slightly from PD45 to senescence of the bulk IMR90 cultures. These dynamics may be attributable to several possible factors. First, subtelomere-specific and allele-specific cis-regulation of telomere length and stability has been documented previously (Baird et al. 2003; Britt-Compton et al. 2006). Second, changes in the sizes of IMR90 clonal subpopulations may occur as the bulk culture expands and especially as it approaches senescence. STELA analysis of clonal IMR90 cultures identified very large differences in subtelomere specific attrition rates between individual clones (from <50 bp/PD to >2000 bp/PD) (Britt-Compton et al. 2006). IMR90 cultures propagated to senescence using standard culture conditions, such as those we have used here, have different global expression profiles than those propagated under low oxygen conditions (Lackner et al. 2014), suggesting that oxidative stress may impact the clonal subpopulations contributing to the bulk senescing cultures. Future experiments measuring global single-telomere lengths following IMR90 culture of clonal populations grown in varying oxygen tensions are required to clarify these issues.
The two telomerase-positive immortal cancer cell lines analyzed here have very distinct single-telomere dynamics from the IMR90 senescing fibroblast model. Both the bladder cancer cell line UMUC3 and the prostate cancer cell line LNCaP have much narrower distributions of single-telomere lengths than IMR90, perhaps reflecting their respective clonal derivations from somatic cells with already very short single-telomere distributions. UMUC3 in particular has a relatively high percentage of very short telomeres <500 bp in length (Table 1). Identified previously as T-stumps (Xu and Blackburn 2007), it has been proposed that in the setting of active telomerase and compromised checkpoints typical of immortal cancer cells, these very short telomeres are the minimal telomere (TTAGGG)n tract that can still form a TRF1- and TRF2-containing capping complex and, furthermore, that telomerase is required to maintain/protect this complex and prevent telomere fusion and end-joining reactions that would otherwise kill the cancer cell (Xu and Blackburn 2007). The subtelomere-specific biases are especially evident in UMUC3, where almost half of the very short telomeres detected are present at either 8q or 9p (Table 2).
The single-telomere resolved (TTAGGG)n length data sets acquirable using our current methods are still incomplete. While only 36 of 46 subtelomeric regions and their associated telomere lengths were successfully mapped and analyzed, we were able to identify several additional consensus maps with telomere labeling. These consensus maps likely include the acrocentric p arms (13p, 14p, 15p, 21p, and 22p) and the XpYp arm; the hg38 human reference sequence lacks complete information for these subtelomeres, which are required to connect to the consensus maps using global nickase-dependent patterns. Experiments are underway to confirm the origins of these subtelomere consensus maps using gRNA-directed CRISPR/Cas9 nickase labeling of known subtelomere sequences from these regions paired with telomere-specific labeling. The other four subtelomeres and their associated telomeres (16p, 17p, 19q, and 22q) were not mapped due to double-strand DNA breaks caused by INP sites of the nicking enzyme used, Nt.BspQI. New England Biolabs recently developed a new nicking enzyme, Nb.BssSI. We are currently testing this nicking enzyme, alone and in combination, to detect the remaining telomeres. We also have the option of marking these individual telomeres using gRNA-directed CRISPR/Cas9 nickase labeling (McCaffrey et al. 2016).
One particular strength of this telomere length–measuring method is its potential ability to differentiate haplotype-specific telomere lengths. Current haplotype linked telomere length measurements are largely limited to subtelomeres with large structural variants detectable using global nickase-dependent labeling patterns (e.g., Fig. 2B). We have recently shown that CRISPR/Cas9 labeling can differentiate alleles with single-nucleotide polymorphism (SNPs) in the PAM protospacer regions of specific gRNAs that can be used to direct allele-specific nick-labeling (data not shown). Thus, while the scope of the mapping is currently limited to 36 subtelomeres and structural variants, with these additional refinements the technology has the potential to resolve all subtelomeres and subtelomere haplotypes. The combination of CRISPR/Cas9-specific sequence tagging with long-range single-molecule mapping may find other important applications in other target genomic regions.
Methods
Cell preparation
UMUC3 cells, a human urinary bladder carcinoma cell line, were obtained from ATCC and cultured in Eagle's minimum essential medium (EMEM) containing Earle's salts, NEAA, and L-glutamine (2 mM; ATCC) with 10% fetal bovine serum (FBS; Corning). LNCaP cells, a human prostate cancer cell line, were obtained from ATCC and cultured in RPMI 1640 media containing 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4500 mg/L glucose, and 1500 mg/L sodium bicarbonate (ATCC) supplemented with 10% FBS (Corning). IMR-90 cells, a human fetal lung fibroblast cell line, were obtained from Coriell Cell Repository and maintained in EMEM containing Earle's salts, NEAA, and L-glutamine (2 mM; ATCC) supplemented with 15% FBS (Corning). UMUC3 and LNCaP cells were passaged using 0.25% trypsin-EDTA (Gibco), and IMR-90 cells were passaged using 0.05% Trypsin-EDTA (Gibco).
High-molecular-weight DNA extraction
Mammalian cells were embedded in gel plugs, and high-molecular-weight DNA was purified as described in a commercial large DNA purification kit (BioRad no. 170-3592). Plugs were incubated with lysis buffer and proteinase K for 4 h at 50°C. The plugs were washed and then solubilized with GELase (Epicentre). The purified DNA was subjected to 2.5 h of drop-dialysis. It was quantified using Quant-iTdsDNA assay kit (Life Technology), and the quality was assessed using pulsed-field gel electrophoresis (Schwartz and Cantor 1984).
Guide RNA preparation
The seed sequence of 20 nucleotides complementary to the 3′-5′ strand of the telomere (UUAGGGUUAGGGUUAGGGUU) was designed via a gRNA design tool (Feng Laboratory CRISPR design web tool at http://crispr.mit.edu). This seed sequence was incorporated into the crRNA. The crRNA and the universal tracrRNA were synthesized by GE Dharmacon. The telomere gRNA was created by preincubating the tracrRNA (0.1 nmol) and crRNA (0.1 nmol) on ice for 30 min.
The two-color labeling scheme
The gRNA (2.5 µM) was incubated with 200 ng of Cas9 D10A (LabOmics), 1× NEBuffer 3 (New England BioLabs, NEB), and 1× BSA (NEB) at 37°C for 15 min. The DNA (300 ng) and 5 U of Nt.BspQI (NEB) were added to the mixture and incubated at 37°C for 60 min. The nicked DNA was labeled with 5 U of Taq DNA Polymerase (NEB), 1× green labeling mix (BioNano Genomics), and 1× Thermopol buffer (NEB) at 72°C for 60 min. The nicks were repaired with 20 kU of Taq DNA Ligase (NEB), 1 mM NAD+ (NEB), 100 nM dNTPs, and 1× Thermopol buffer (NEB) at 37°C for 30 min. The small quantity (300 ng) of labeled genomic DNA required for a typical experiment is sufficient to generate at least 60× coverage of a genome, making it feasible to apply this method to small clinical sample sources such as blood.
The three-color labeling scheme
The DNA (300 ng) was first nicked with 5 U of Nt.BspQI (NEB) in 1× NEBuffer 3 (NEB) at 37°C for 2 h. The nicked DNA was then labeled with 5 U of DNA Taq Polymerase (NEB), 100 nM ATTO532-dUTP dAGC, and 1× NEBuffer 3.1 (NEB) at 72°C for 60 min. The sample was treated with 0.3 U of SAP (USB Products) at 37°C for 10 min and then 65°C for 5 min. The gRNA (2.5 µM) was incubated with 200 ng of Cas9 D10A (LabOmics), 1× NEBuffer 3 (NEB), and 1× BSA (NEB) at 37°C for 15 min. The green-labeled sample was then added to the reaction and incubated at 37°C for 1 h. The Cas9 D10A nicks were labeled with 2.5 U of Taq DNA Polymerase (NEB), 1× IrysPrep labeling mix red (BioNano Genomics), and 1× NEBuffer 3.1 (NEB) at 72°C for 60 min. The nicks were repaired with 20 kU of Taq DNA ligase (NEB), 1 mM NAD+ (NEB), 100 nM dNTPs, and 1× NEBuffer 3.1 (NEB) at 37°C for 30 min.
DNA loading and imaging
After nick-labeling with either the two- or three-color schemes, the samples were treated with 6 mAU of QIAGEN Protease at 56°C for 30 min, and the reaction was stopped with 1 µL of IrysPrep stop solution (BioNano Genomics). The DNA backbone was stained with 333 nM YOYO-1 (Invitrogen) and is shown in blue in all figures. The stained samples were loaded and imaged inside the nanochannels following the established protocol (Lam et al. 2012). BioNano Genomics labeling kit and IrysChip were used to generate the nick labeling data. The next generation mapping system from BioNano has dramatically improved the throughput; our custom-made systems are very similar to this new BioNano Genomics system. Each IrysChip contains two nanochannel devices, which can generate at least 60 Gb of data (molecules >150 kb). Normally, 60× coverage (180 Gb) is needed to generate 30 molecules of each chromosome end containing the telomeres. This assay runs 3 d, collecting over 24,000 images. It currently costs $1000 per sample to run whole-genome mapping. The image analysis was done using BioNano commercial software for segmenting and detecting DNA backbone based on the YOYO-1 staining similar to early optical mapping method (Lin et al. 1999), and localizing the green labels by fitting the point-spread functions.
De novo genome map assembly
Single-molecule maps were assembled de novo into consensus maps using software tools developed at BioNano Genomics, specifically Refaligner and Assembler (Mak et al. 2016). Briefly, the assembler is a custom implementation of the overlap-layout-consensus paradigm with a maximum likelihood model. An overlap graph was generated based on pairwise comparison of all molecules as input. Redundant and spurious edges were removed. The assembler outputs the longest path in the graph, and consensus maps were derived. Consensus maps are further refined by mapping single-molecule maps to the consensus maps, and label positions are recalculated. Refined consensus maps are extended by mapping single molecules to the ends of the consensus and calculating label positions beyond the initial maps. After merging of overlapping maps, a final set of consensus maps was output and used for subsequent analysis.
Telomere length analysis
The molecules from the consensus maps, which were mapped to the ends of the individual chromosomes (hg38 reference) were designated as the molecules containing telomere and analyzed. These molecules contain additional labels not found in the reference, which were classified as telomere labels. The integrated fluorescence intensity of these labels was calculated after subtracting the background intensity. The intensity was then converted to base pairs based on standard established using fosmids (Supplemental Fig. S1).
Data access
The BioNano whole-genome assembly data from this study have been submitted to the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA396850. The BioNano supplemental data at this accession refer to IMR90 (SUPPF_0000001237), LNCaP (SUPPF_0000001236), and UMUC3 (SUPPF_0000001235).
Supplementary Material
Acknowledgments
This research is supported in part by US National Institutes of Health (NIH) grants to H.R. and M.X. (R21HG007205 and R21CA177395). Part of the informatics analysis was run on hardware supported by Drexel's University Research Computing Facility.
Author contributions: J.M., J.S., K.L., and E.Y. generated data. J.M., E.Y., S.P., and M.X. analyzed data. J.M., H.R., and M.X. wrote the manuscript. H.R. and M.X. designed and supervised the project.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.222422.117.
References
- Aubert G, Hills M, Lansdorp PM. 2012. Telomere length measurement-caveats and a critical assessment of the available technologies and tools. Mutat Res 730: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird DM, Rowson J, Wynford-Thomas D, Kipling D. 2003. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat Genet 33: 203–207. [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Epel ES, Lin J. 2015. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science 350: 1193–1198. [DOI] [PubMed] [Google Scholar]
- Britt-Compton B, Rowson J, Locke M, Mackenzie I, Kipling D, Baird DM. 2006. Structural stability and chromosome-specific telomere length is governed by cis-acting determinants in humans. Hum Mol Genet 15: 725–733. [DOI] [PubMed] [Google Scholar]
- Capper R, Britt-Compton B, Tankimanova M, Rowson J, Letsolo B, Man S, Haughton M, Baird DM. 2007. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev 21: 2495–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM. 2002. Telomere measurement by quantitative PCR. Nucleic Acids Res 30: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM. 2009. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res 37: e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li G-W, Park J, Blackburn EH, Weissman JS, Qi LS. 2013. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155: 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Austin MD, Akana MC, Deshpande P, Cao H, Xiao M. 2010. Single molecule linear analysis of DNA in nano-channel labeled with sequence specific fluorescent probes. Nucleic Acids Res 38: e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos AR, Coppe JP, Campisi J, Desprez PY. 2010. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev 29: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Rodrigues F, Santana-Lemos BA, Scheucher PS, Alves-Paiva RM, Calado RT. 2014. Direct comparison of flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans. PLoS One 9: e113747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglehearn CF, Cooke HJ. 1990. A VNTR immediately adjacent to the human pseudoautosomal telomere. Nucleic Acids Res 18: 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, et al. 2011. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo K, Dhingra DM, Odijk T, de Pablo JJ, Graham MD, Runnheim R, Forrest D, Schwartz DC. 2007. A single-molecule barcoding system using nanoslits for DNA analysis. Proc Natl Acad Sci 104: 2673–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Kamata S, Yonese J, Oshima H. 1997. Telomere length and telomerase activity in bladder and prostate cancer cell lines. Int J Urol 4: 407–410. [DOI] [PubMed] [Google Scholar]
- Karlseder J, Smogorzewska A, de Lange T. 2002. Senescence induced by altered telomere state, not telomere loss. Science 295: 2446–2449. [DOI] [PubMed] [Google Scholar]
- Kaul Z, Cesare AJ, Huschtscha LI, Neumann AA, Reddel RR. 2012. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep 13: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner DH, Hayashi MT, Cesare AJ, Karlseder J. 2014. A genomics approach identifies senescence-specific gene expression regulation. Aging Cell 13: 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al. 2012. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol 30: 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdorp PM, Verwoerd NP, vandeRijke FM, Dragowska V, Little MT, Dirks RW, Raap AL, Tanke HJ. 1996. Heterogeneity in telomere length of human chromosomes. Hum Mol Genet 5: 685–691. [DOI] [PubMed] [Google Scholar]
- Lin J, Qi R, Aston C, Jing J, Anantharaman TS, Mishra B, White O, Daly MJ, Minton KW, Venter JC. 1999. Whole-genome shotgun optical mapping of Deinococcus radiodurans. Science 285: 1558–1562. [DOI] [PubMed] [Google Scholar]
- Lin TT, Norris K, Heppel NH, Pratt G, Allan JM, Allsup DJ, Bailey J, Cawkwell L, Hills R, Grimstead JW, et al. 2014. Telomere dysfunction accurately predicts clinical outcome in chronic lymphocytic leukaemia, even in patients with early stage disease. Br J Haematol 167: 214–223. [DOI] [PubMed] [Google Scholar]
- Lynch SM, Peek M, Mitra N, Ravichandran K, Branas C, Spangler E, Zhou W, Paskett ED, Gehlert S, DeGraffinreid C. 2016. Race, ethnicity, psychosocial factors, and telomere length in a multicenter setting. PLoS One 11: e0146723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AC, Lai YY, Lam ET, Kwok TP, Leung AK, Poon A, Mostovoy Y, Hastie AR, Stedman W, Anantharaman T, et al. 2016. Genome-wide structural variation detection by genome mapping on nanochannel arrays. Genetics 202: 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens UM, Zijlmans JM, Poon SS, Dragowska W, Yui J, Chavez EA, Ward RK, Lansdorp PM. 1998. Short telomeres on human chromosome 17p. Nat Genet 18: 76–80. [DOI] [PubMed] [Google Scholar]
- Martin-Ruiz CM, Baird D, Roger L, Boukamp P, Krunic D, Cawthon R, Dokter MM, van der Harst P, Bekaert S, de Meyer T, et al. 2015. Reproducibility of telomere length assessment: an international collaborative study. Int J Epidemiol 44: 1749–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey J, Sibert J, Zhang B, Zhang Y, Hu W, Riethman H, Xiao M. 2016. CRISPR-CAS9 D10A nickase target-specific fluorescent labeling of double strand DNA for whole genome mapping and structural variation analysis. Nucleic Acids Res 44: e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier A, Fiegler H, Munoz P, Ellis P, Rigler D, Langford C, Blasco MA, Carter N, Jackson SP. 2007. Spreading of mammalian DNA-damage response factors studied by ChIP-chip at damaged telomeres. EMBO J 26: 2707–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit AJ, Alhareeri AA, Montpetit M, Starkweather AR, Elmore LW, Filler K, Mohanraj L, Burton CW, Menzies VS, Lyon DE, et al. 2014. Telomere length: a review of methods for measurement. Nurs Res 63: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan JN, Finley JC, Risques RA, Shen WT, Gollahon KA, Moskovitz AH, Gryaznov S, Harley CB, Rabinovitch PS. 2004. Telomere length assessment in tissue sections by quantitative FISH: Image analysis algorithms. Cytometry Part A 58A: 120–131. [DOI] [PubMed] [Google Scholar]
- Perner S, Brüderlein S, Hasel C, Waibel I, Holdenried A, Ciloglu N, Chopurian H, Nielsen KV, Plesch A, Högel J, et al. 2003. Quantifying telomere lengths of human individual chromosome arms by centromere-calibrated fluorescence in situ hybridization and digital imaging. Am J Pathol 163: 1751–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger L, Jones RE, Heppel NH, Williams GT, Sampson JR, Baird DM. 2013. Extensive telomere erosion in the initiation of colorectal adenomas and its association with chromosomal instability. J Natl Cancer Inst 105: 1202–1211. [DOI] [PubMed] [Google Scholar]
- Sabatier L, Ricoul M, Pottier G, Murnane JP. 2005. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol Cancer Res 3: 139–150. [DOI] [PubMed] [Google Scholar]
- Samassekou O, Ntwari A, Hébert J, Yan J. 2009. Individual telomere lengths in chronic myeloid leukemia. Neoplasia 11: 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samassekou O, Gadji M, Drouin R, Yan J. 2010. Sizing the ends: normal length of human telomeres. Ann Anat 192: 284–291. [DOI] [PubMed] [Google Scholar]
- Schwartz DC, Cantor CR. 1984. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell 37: 67–75. [DOI] [PubMed] [Google Scholar]
- Simpson K, Jones RE, Grimstead JW, Hills R, Pepper C, Baird DM. 2015. Telomere fusion threshold identifies a poor prognostic subset of breast cancer patients. Mol Oncol 9: 1186–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stong N, Deng Z, Gupta R, Hu S, Paul S, Weiner AK, Eichler EE, Graves T, Fronick CC, Courtney L, et al. 2014. Subtelomeric CTCF and cohesin binding site organization using improved subtelomere assemblies and a novel annotation pipeline. Genome Res 24: 1039–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera E, Blasco MA. 2012. Beyond average: potential for measurement of short telomeres. Aging 4: 379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M, Phong A, Ha C, Chan T-F, Cai D, Leung L, Wan E, Kistler AL, DeRisi JL, Selvin PR. 2007. Rapid DNA mapping by fluorescent single molecule detection. Nucleic Acids Res 35: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Blackburn EH. 2007. Human cancer cells harbor T-stumps, a distinct class of extremely short telomeres. Mol Cell 28: 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman S, Bates GP, Williams S, McClatchey AI, Baxendale S, Sedlacek Z, Altherr M, Wasmuth JJ, MacDonald ME, Gusella JF, et al. 1992. The telomeric 60 kb of chromosome arm 4p is homologous to telomeric regions on 13p, 15p, 21p, and 22p. Genomics 14: 350–356. [DOI] [PubMed] [Google Scholar]
- Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. 2015. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4: e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.