

Abstract
Immunotherapy adds an exciting new dimension to the treatment of cancer, joining other approaches as a key pillar in the oncotherapeutics armamentarium. Immuno-oncology agents harbor unique mechanisms of antitumor activity by leveraging the host immune system, which may result in response patterns, resistance kinetics, and toxicity profiles that differ from other systemic therapies. These features have led to many discussions on ways to optimally integrate immunotherapy into cancer clinical trials. This overview provides an introduction to the four CCR Focus articles that ensue, with special thoughts paid to clinical trial endpoints, biomarker development and validation, combination strategies, and limitations that arise with increasing use of these agents. In addition, this overview examines design concepts that may be applied to invigorate clinical trials and to maximize their impact in the immuno-oncology era.
INTRODUCTION
In recent decades, practice-altering shifts have transformed the systemic therapy of cancer, including molecular targeting against various oncogenic pathways and genomic sequencing to identify driver aberrations in pursuit of precision medicine. The focus of drug development had primarily been on perturbation of signals that disrupt the growth and spread of cancer cells, however, with the advent of immunotherapy, the focus turned toward harnessing the host immune system to exert anticancer activity. Immunotherapies have unique properties that distinguish them from other systemic therapies, such as their patterns of response, relapse and resistance. Dose-response and dose-toxicity relationships are not typically direct or dose-proportional, as in the case of most cytotoxic chemotherapy and many molecularly targeted agents. Furthermore, immunotherapies have the potential to induce not only sustained, long-term benefits, but also lingering adverse effects. With these features in consideration, three articles in this CCR Focus examine conventional elements of clinical trials – endpoints, biomarkers and combination strategies – in the context of immunotherapy to highlight where standard principles prevail and where innovations are needed (1–3). The limitations and challenges encountered thus far in the design, implementation, and integration of immunotherapy clinical trials are discussed in the final article of this series (4).
Overview of Current Status
The armamentarium that broadly fulfills the definition of immunotherapeutic agents is extensive, including, but not limited to, cancer vaccines, oncolytic viruses, cytokines, adoptive cell transfer, costimulatory molecules, and immune checkpoint inhibitors. The immune checkpoint inhibitors, such as those targeting the cytotoxic T-lymphocyte-associated protein 4 (CTLA4), programmed cell death protein 1 (PD-1) or its ligand (PD-L1), are furthest along in their clinical development path. As such, a retrospective evaluation of the developmental strategies of some immune checkpoint inhibitors might provide insight on gaps that exist in the era of immunotherapeutics.
The first-in-human phase I studies of many anti-CTLA4 anti-PD-1/PD-L1 antibodies, such as ipilimumab (5), pembrolizumab (6), nivolumab (7), durvalumab (8, 9) and atezolizumab (10), all used the 3+3 dose escalation design in patients with advanced solid tumors. The initial studies of PD-1/PD-L1 inhibitors planned expansion cohorts of limited size, but early signs of promising clinical antitumor activity led to substantial increase in the ultimate sample size. All trials rapidly moved to multi-cohort dose expansions in search of early signals of efficacy across different tumor types. In addition to preliminary activity evaluation during the “tail” of phase I trials, many of these agents are also investigated in stand-alone “basket” protocols with multiple cohorts that enroll a variety of histologies and/or enriched patient subsets (e.g. high microsatellite instability [MSI-H] status tumors) at the recommended phase II dose (e.g. KEYNOTE-028 (11)). Methodological issues related to these designs are discussed further in the “Seamless Phase I–II Trial Designs” section below.
Table 1 contains selected clinical trials published in 2016 of two anti-PD-1 antibodies, pembrolizumab and nivolumab; while not comprehensive, it provides a contemporary benchmark of clinical trial design methodologies and selection biomarkers that have been applied. Following the identification of clear signals of antitumor activity, there are two common developmental strategies undertaken in the evaluation of immune checkpoint inhibitors. One approach relies on single-arm or small non-comparative phase II trials (e.g. (12)); others transition seamlessly from phase I trial to multi-cohort expansion phase or basket trial in specific histologies. These studies seek accelerated approval by meeting the US Food and Drug Administration (FDA) criteria for unmet medical need based on a surrogate endpoint, such as objective response rate (ORR). To achieve accelerated regulatory approval, the therapeutic index of the investigational agent must weigh favorably against the standard therapy (if it exists) for the patient population under consideration. The other approach is to directly compare against standard of care therapy through randomized phase II and randomized-controlled phase III trials seeking clinically meaningful benefit in a definitive endpoint such as progression-free survival (PFS) or overall survival (OS), respectively. These randomized comparisons often occur after or near completion of dose expansion in phase I trials to avoid delay; many of these trials aim for large reductions in risk (i.e. hazard ratios of 0.5–0.6). In this CCR Focus, Anagnostou et al. (1). examine the challenges of utilizing the traditional endpoints of ORR, PFS and OS in immuno-oncology clinical trials. Further, as discussed by Mehnert et al. (3), selection biomarkers are not yet universally validated to accurately predict response or resistance to immune checkpoint inhibitors. PD-L1 expression on tumor or immune cells by immunohistochemistry has been the most frequently considered, although this biomarker is confounded by the multiple antibodies and disparate scoring criteria that accompany the various PD-1/PD-L1 inhibitors (13).
Table 1.
Selected Immune Checkpoint Inhibitor Studies Published in 2016
| Agent | Phase | N Enrolled | Tumor Type | Design | Biomarkers (Selection) | Endpoints | Reference |
|---|---|---|---|---|---|---|---|
| Late Phase (Randomized) | |||||||
| Pembrolizumab Keynote-024 | III | 305 | NSCLC | Randomized 1:1 Pembrolizumab vs investigator’s choice chemotherapy |
PD-L1 ≥50% TPS | PFS Target HR 0.55 |
Reck et al. (41) |
| Pembrolizumab Keynote-021 | II | 123 | NSCLC | Randomized 1:1 Pembrolizumab plus chemotherapy vs chemotherapy |
Stratified by PD-L1 (<1% vs ≥1%) | ORR by RECIST 1.1 (30% difference in ORR) Target HR 0.5 (PFS is secondary endpoint) |
Langer et al.(42) |
| Pembrolizumab Keynote-010 | II/III | 1034 | NSCLC | Randomized 1:1:1 Pembrolizumab (2 doses): chemotherapy |
PD-L1 ≥1% TPS Stratified by PD-L1 (TPS <50% vs ≥50%) |
OS and PFS Target HR 0.6 |
Herbst et al.(43) |
| Nivolumab Checkmate-141 | III | 361 | SCCHN | Randomized 2:1 Nivolumab vs investigator’s choice systemic therapy |
Not selected by PD-L1 | OS Target HR 0.667 |
Ferris et al.(44) |
| Nivolumab Checkmate-064 | II | 140 | MM | Randomized 1:1 Nivolumab followed by ipilumumab or vice versa |
Not selected by PD-L1 | Treatment related grade 3-5 adverse events till week 25 (end of induction) | Weber et al.(45) |
| Early Phase | |||||||
| Pembrolizumab Keynote-012 | Ib | 32 | TNBC | Part of a multi-cohort study | PD-L1 in stroma or ≥1% of tumor cells | ORR by RECIST 1.1 P0=0.2, P1=0.45 |
Nanda et al.(46) |
| Pembrolizumab | II | 36 | MM or NSCLC with brain metastases | Single arm phase II, 2 parallel cohorts | Not selected by PD-L1 | Brain metastasis response by modified RECIST | Goldberg et al.(47) |
| Pembrolizumab Keynote-012 | Ib | 60 | SCCHN | Part of a multi-cohort study | PD-L1 in stroma or ≥1% of tumor cells | ORR by RECIST 1.1 P0=0.1, p1=0.35 |
Seiwert et al.(48) |
| Pembrolizumab | II | 26 | MCC | Single arm phase II study | Not selected by PD-L1 or by MCPyV status | ORR by RECIST 1.1 P0=0.05, P1=0.25 |
Nghiem et al.(49) |
| Pembrolizumab Keynote-013 | II | 31 | HL | Single arm phase II study | Not selected by PD-L1 | CRR by IHP criteria P0=0.1, P1=0.3 |
Armand et al.(50) |
| Pembrolizumab Keynote-012 | Ib | 39 | Gastric | Part of a multi-cohort study | PD-L1 in stroma or ≥1% of tumor cells | ORR by RECIST 1.1 |
Muro et al.(51) |
| Pembrolizumab Keynote-001 | I | 655 | MM | Expansion cohort of phase I study | Not selected by PD-L1 | ORR by RECIST 1.1 | Ribas et al.(52) |
| Nivolumab Checkmate-032 | I/II | 86 | UC | Single arm phase II study | Not selected by PD-L1 | ORR by RECIST 1.1 P0=0.1, P1=0.25 |
Sharma et al.(53) |
| Nivolumab Checkmate-012 | Ib | 52 | NSCLC | Part of a multi-cohort study | Not selected by PD-L1 | Safety and tolerability | Gettinger et al. (54) |
| Nivolumab Checkmate-205 | II | 80 | HL | Single arm phase II study part of a multi-cohort study | Not selected by PD-L1 | ORR by IHP criteria | Younes et al.(55) |
| Nivolumab | Ib | 81 | HM | Phase I dose escalation study | Not selected by PD-L1 | Safety and tolerability | Lesokhin et al.(56) |
| Nivolumab Checkmate-032 | I/II | 216 | SCLC | Multiple cohorts with nivolumab alone or nivolumab plus ipilimumab | Not selected by PD-L1 | ORR by RECIST 1.1 | Antonia et al.(57) |
CRR = complete response rate
HL = Hodgkin lymphoma
HM = hematological malignancies
HR = hazard ratio
ICI = immune checkpoint inhibitor
IHP = International Harmonization Project
MCC = merkel cell carcinoma
MCPyV = merkel-cell polyomavirus
MM = malignant melanoma
NSCLC = non-small-cell lung cancer
ORR = objective response rate
PFS = progression-free survival
RECIST = Response Evaluation Criteria in Solid Tumours
SCCHN = squamous cell carcinoma of the head and neck
SCLC = small-cell lung cancer
TNBC = triple negative breast cancer
TPS = tumor proportion score
UC = urothelial cancer
Identification of Gaps in Immunotherapy Clinical Trials
The articles in this CCR Focus provide a critical appraisal of the gaps that exist in the current clinical trial landscape for immunotherapies and suggest ways that the drug development paths for this class of agents can be improved and modernized.
Anagnostou and colleagues (1) reviewed nuances associated with applying traditional efficacy and toxicity endpoints to immuno-oncology agents, given potential differences in response and resistance kinetics from other systemic therapies. The emergence of various response criteria to meet the specific effects of these agents (e.g. irRC (14), irRECIST (15) and iRECIST (16)) has added further complexity to the field. Standardization of universally accepted tumor immune response criteria is of top priority. Likewise, the description and attribution of immune-mediated adverse events arising from auto-immunity developing in host tissue need to be appropriately documented, including the need for intervention (e.g. corticosteroids), as well as the timing of onset and resolution. As for efficacy evaluation, Kaplan-Meier plots for PFS and OS in many immuno-oncology trials have a distinctive configuration, characterized by a delay in clinical effect and a non-zero tail representing long-term survivors (17). Landmark survival estimates and non-proportional hazard models have been proposed as more appropriate for reporting clinical outcomes from immunotherapy. Health-related quality of life and patient-reported outcome evaluations are underrepresented in immuno-oncology research and increased attention should be paid to these patient-based endpoints.
Mehnert and colleagues (3) proposed key recommendations related to immuno-oncology biomarker research, highlighting the complex and dynamic characteristics of many biomarker candidates being interrogated for their ability to predict for response, resistance, or toxicity to immunotherapy. The need for high quality pre-analytics such as biospecimen acquisition, standardized assays, and clinical annotation is emphasized, which resonates with the mandate of the US National Cancer Institute’s new program under the Cancer Moonshot directive called the Partnership for Accelerating Cancer Therapies (PACT) (18). Strategic incorporation of biomarker endpoints into early and late stage immuno-oncology clinical trials must consider their scientific value to the development of these agents and, importantly, the perspectives of patients under study.
Day and colleagues (2) focused on one of the most challenging areas in immuno-oncology drug development – creating a rational framework to design and assess combination therapy. Priority should be given to combinations that have the strongest scientific evidence for additivity or synergy and a favorable therapeutic index, although interspecies differences limit the utility of most nonclinical models to nominate the most appropriate drug schedules and sequences to enter clinical testing. Many clinical trials evaluating immunotherapy-based doublets or even triplets with an anti-PD-1/PD-L1 backbone are ongoing or being planned; the vast majority of these emerge from empiricism or minimal scientific justifications. Lessons learned from the nivolumab and ipiliumab combination are examined to further inform patient selection, dose and schedule optimization, and toxicity management.
Baik and colleagues (4) offered a thoughtful interpretation of the limitations and challenges in immuno-oncology clinical trials. Insufficient representation of patient subsets including those with autoimmune disease, virally initiated diseases, etc. in immunotherapy trials negatively affects the generalization of results to these individuals. Unless clinical trials are inclusive of these patient populations or are specifically designed to enroll such patients, their access to promising immuno-oncology compounds will be restricted. Furthermore, with the regulatory approval of anti-PD-1/PD-L1 antibodies in multiple indications, compounded by the large number of ongoing clinical trials incorporating these agents alone or in combination, it has become increasingly difficult to identify research participants who are naïve to PD-1/PD-L1 inhibitors. Other unaddressed questions, such as the optimal length of therapy of immune checkpoint inhibitors in patients whose tumors demonstrate response, tracking of effects on subsequent therapies in those who discontinued such drugs, and documentation of late toxicities, are also discussed.
Checklists for Early and Late Phase Immunotherapy Trials
The rapid entry of new immuno-oncology compounds into the clinic has led to an exponential increase in the number of early and late phase trials, a phenomenon that is likely unsustainable due to limited patient, infrastructure, and financial resources. Despite the broad activity observed with PD-1/PD-L1 inhibitors in many tumor types (Figure 1), there are clear instances that such agents exert minimal antitumor activity, as in the case of pembrolizumab in microsatellite stable colorectal cancer or castrate resistant prostate cancer (7). There is an urgent need for efficient and nimble clinical trials to seamlessly advance the most promising drugs or drug combinations while stopping futile efforts in a decisive and reasonable timeframe. Table 2 provides checklists of important items that should be considered during the conduct of early and late phase immunotherapy clinical trials. Figure 2 provides an outline of the structure of the clinical developmental pathway for immune-oncology agents.
Figure 1.
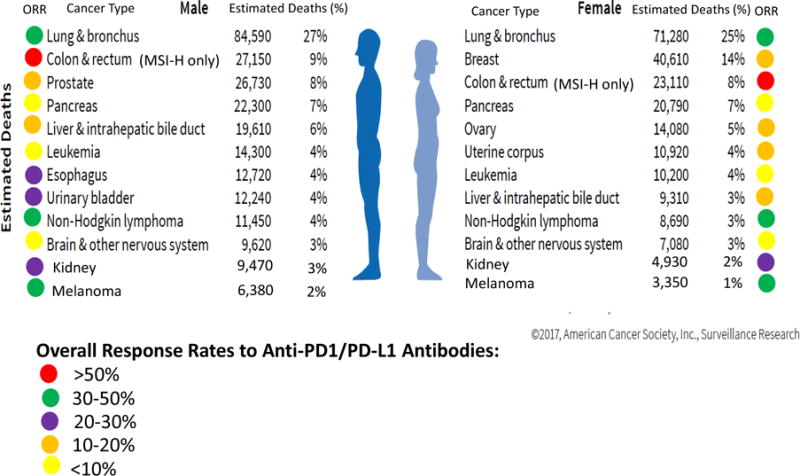
Objective Response Rate to Anti-PD1/PD-L1 Antibodies in the Most Lethal Cancers. Estimated deaths data from the American Cancer Society (58).
Table 2.
Important Steps for Consideration in the Conduct of Early and Late Phase Immunotherapy Trials
| Early Phase Trials: |
|---|
|
|
|
| Late Phase Trials: |
|
|
|
DLT = dose-limiting toxicity
PFS = progression-free survival
OS = overall survival RECIST = Response Evaluation Criteria in Solid Tumors
RP2D = recommended phase II dose
Figure 2.
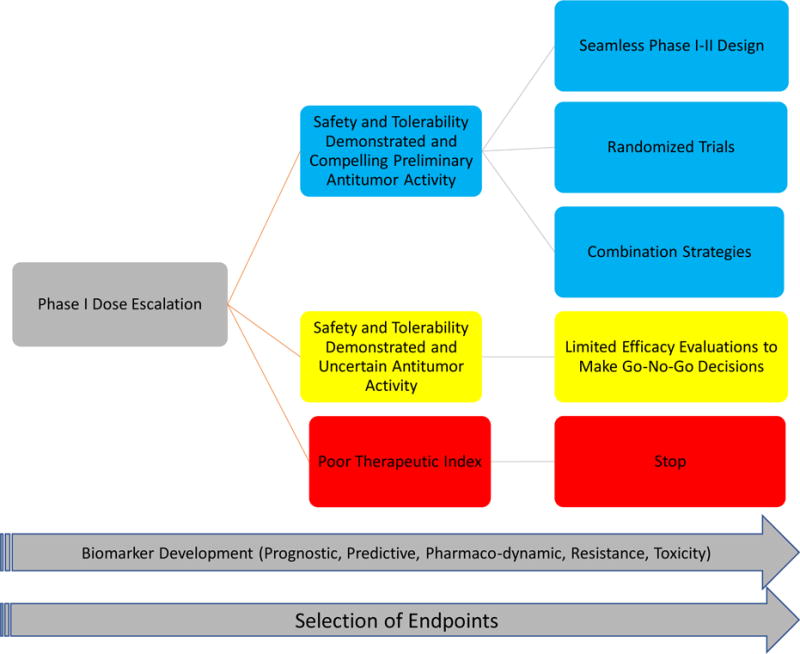
Clinical Developmental Pathway for Immuno-Oncology Agents
Areas for Innovation and Impact in the Design of Immunotherapy Clinical Trials
Innovations in clinical trial designs to increase operational efficiency and optimize patient outcomes are needed as immunotherapy becomes integrated as a key therapeutic pillar in oncology; some examples are described below.
Evaluation of Tumor Growth Modulation
While tumor shrinkage remains one of the most validated biomarkers of antitumor activity, many drugs with primarily growth inhibitory potential, including some immuno-oncology agents, demonstrate only cytostatic effects when given to patients with heavily pretreated and highly resistant cancers in early phase clinical trials. This may be particularly relevant in cases of patients previously exposed to PD-1/PD-L1 inhibitors who have primary or acquired resistance to these agents and subsequently enter clinical trials evaluating immuno-oncology combinations (e.g. the combination of a co-stimulatory molecule and a PD-1/PD-L1 inhibitor). While objective responses may occur in some cases, the preliminary efficacy of such combinations in the absence of tumor shrinkage may be based on the achievement of clinically meaningful disease stabilization, which remains an elusive endpoint in a heterogeneous early phase trial patient population with variable tumor biology. Some experts consider prolonged stable disease of at least 6 months or longer as evidence of clinical benefit, while others have attempted to use patients as their own controls by comparing tumor growth rate or PFS on current regimen versus the same parameter on immediate prior regimen (19, 20). The application of these measures of cytostasis as surrogates of efficacy requires systematic validation in clinical trials and demonstration of reproducibility. There are also ongoing efforts to develop novel radiomics biomarkers of response or resistance to immune checkpoint inhibitors by using advanced image processing techniques to extract quantitative texture and geometric features from CT or MRI scans and subject them to machine learning algorithms (21). The ability to distinguish pseudo-progression (i.e. an uncommon phenomenon observed with some immuno-oncology agents associated with the appearance of initial tumor growth and/or development of new lesions, presumably due to immune cell infiltration into tumor, followed by subsequent tumor regression) from true progression early, to avoid exposing the latter group of patients to ineffective treatment, is challenging to understand and predict, and one of the highest research priorities.
Seamless Phase I–II Trial Designs
Many studies seek to evaluate both the appropriate dose and schedule, and to identify histologies that appear to respond to these agents. Protocols begin with dose finding and then enroll patients across multiple histologies (22); sometimes they include a safety run-in for each cancer subtype due to context-dependent toxicity. The goal of these designs is to increase efficiency by allowing the study to add different cancers via protocol amendments instead of developing new protocols. By combining dose finding with subsequent assessment of activity in disease-specific cohorts, these master protocols progress seamlessly from phase I to phase II. The rapid pace at which immuno-oncology agents are entering the clinical research arena has sometimes meant that disease-specific cohorts receive treatment without the protocol specifying a statistical design. Even though these “expansion” cohorts are part of a phase I study, in fact, the cohort is providing evidence of the agent’s activity. These protocols should include appropriate statistical designs to protect patients in expansion cohorts, either through safety stopping boundaries after the initial dose escalation or futility boundaries to avoid treatment with ineffective therapies. Further research into efficient study designs that balance the desire for rapid drug development against the need for generation of rigorous scientific evidence is a high priority.
Adaptive Designs in Combination Trials
Most clinical studies in cancer are adaptive in some sense, including study designs with group-sequential boundaries guiding decisions relating to interim analysis results, futility analyses, dose-escalation rules, adaptive enrichment, and adaptive randomization. Several high-profile clinical trials in cancer have incorporated adaptive randomization by allowing the probabilities governing treatment assignments to change in favor of better performing treatment regimens (23). Examples of such trials are the BATTLE studies (24, 25) and I-SPY-2 (26, 27), the latter has “graduated” two agents to further study (28, 29). Both studies use relatively quickly available endpoints (i.e. 8-week disease control and pathologic complete response in the neo-adjuvant setting, respectively) to inform activity assessment. Studies evaluating immuno-oncology agents may require longer follow-up of patients, since clinical response is not always seen immediately. Immune-related biomarkers that can show efficacy-related activity earlier than clinical endpoints may emerge as intermediate endpoints, allowing adaptive randomization to be more useful for studies of immuno-oncology agents. Although such adaptations may well accelerate the development of effective treatment regimens, careful thought must go into the study’s design to weigh potential benefits against possible problems (30, 31). Studies that adapt ongoing randomization to pair sensitive disease subtypes to their more active treatment regimens will likely find greatest use in the phase II setting, where screening for activity takes precedence over performing a confirmatory trial.
Duration of Therapy and Dose Sequencing Trials
As discussed in Baik et al. (4), the optimal duration of therapy is unclear for immuno-oncology agents that are typically delivered by repeated dosing, such as costimulatory molecules and immune checkpoint inhibitors. This uncertainty exists not only in the setting of advanced disease for patients whose tumors achieved sustained objective response or prolonged disease stabilization, but also in the curative scenario whereby such agents are given as adjuvant therapy. As such, clinical trials evaluating short courses of maintenance or adjuvant immunotherapy (e.g. 3–6 months) versus longer courses are needed. In addition, clinical trials specifically designed to ascertain the effect of dosing schedule for immune combinations are lacking, this gap also needs to be corrected. For instance, whether anti-PD-1/PD-L1 antibodies and adoptive cell therapies can be safely combined is unknown; whether they should be administered concurrently or sequentially to achieve the most favorable therapeutic index requires exploration (32). From the clinical trials design perspective, many of these questions pose non-inferiority hypotheses and require large sample sizes to address. Despite the potential impact on patient outcome that the answers to these questions may bring, pharmaceutical companies are generally not financially incentivized to conduct such trials. They will likely rely on the concerted efforts of cooperative groups or other research consortia to complete.
Clinical Trials that Target Large Differences in Effect Size
Many contemporary randomized clinical trials evaluating immune checkpoint inhibitors have set a reasonably high efficacy bar seeking large reductions in risk (i.e. hazard ratios of 0.5–0.6) for PFS or OS, which translate to doubling or near doubling of these time-based parameters (Table 1). Small incremental gains are not affordable in the context of scarce resources. Unless there are system-wide solutions to the prohibitively high prices of cancer drugs including immunotherapy (33), their access to many patients will remain limited. As such, it is critical to design clinical trials that demonstrate substantial benefits via clinically meaningful efficacy endpoints, which may vary by patient population and disease status, while also considering the treatment’s toxicity profile and patients’ health-related quality of life. For instance, in 2016 the US FDA provided accelerated approval for nivolumab in combination with ipilimumab for the treatment of patients with unresectable or metastatic malignant melanoma based on PFS (CHECKMATE-067 (34): 11.5 months with the combination, versus 2.9 months with ipilimumab alone and 6.9 months with nivolumab alone). Approval was given despite a higher rate of grade 3 or 4 treatment-related adverse events observed with the doublet than either agent given alone. As the OS data from this study become available (35), continued approval for this indication may depend on confirmation of a sustained benefit that is reflected by relevant reductions in death rates (36). The enrichment for biomarker-positive subgroups is a logical approach to achieve large effect size in clinical trials (3), but this strategy is currently challenged by the lack of validated predictive biomarkers in immunotherapy.
Clinical Trials at Minimal Residual Disease Stage to Increase Cure Rates
While there continues to be tremendous hope that immunotherapy offers cures to some patients with advanced disease, the present reality is that only a small proportion qualify as long-term survivors. Clinical trials targeting patients with minimal residual disease after definitive therapy but are at high risk for relapse have the greatest potential to improve cancer cure rates. EORTC 18071 evaluated adjuvant ipilimumab in patients with stage III resected melanoma and demonstrated a 5-year rate of recurrence-free survival of 40.8% in the ipilimumab group versus 30.3% in the placebo group (hazard ratio 0.76; 95% C.I. 0.64–0.89) (37). Similar studies with other immuno-oncology agents are ongoing (e.g., KEYNOTE-054 (38); CHECKMATE-238 (39)). Likewise, the Canadian Cancer Trials Group BR.31 trial evaluates the role of adjuvant durvalumab in patients with completed resected stage IB, II and IIIA non-small cell lung cancer (NCT02273375). These patient populations also offer the opportunity to examine the role of circulating tumor and immune biomarkers (i.e. circulating tumor DNA, exosomes, cytokines) that may further refine patient selection and enable monitoring of therapeutic resistance.
Conclusion
Several national and international initiatives seek to leverage approaches in immune-oncology, including the NCI Cancer Moonshot, which received a boost when the U.S. Congress passed the 21st Century Cures Act, creating the Blue-Ribbon Panel working group to define strategic initiatives in clinical cancer research (40). This initiative bolsters cancer discovery to accelerate treatment and cures over the next 7 years and provides $1.8 billion in funding. Working through the Foundation of the National Institutes of Health (FNIH), PACT is actively exploring public-private partnerships with government, academe, and the pharmaceutical industry to broaden the expansion of immunotherapy and combination therapy research in biomarkers and treatment. The NCI has embarked on major initiatives in precision medicine using genomics, proteomics, and transcriptomics that can be directly applied to immune-oncology as predictive biomarkers are identified for response, resistance, pseudo-progression, and progression. Evidence-driven biomarker development will enable the precise selection of patients whose tumors are most likely to respond to immunotherapy and combination clinical trials, and to segment successful treatment of cancer patients. Efficient and effective novel clinical trial designs that are purposely suited for immunotherapy should also accelerate drug development in general.
Acknowledgments
Grant support
NCI UM1-CA186644 (Siu LL), NCI-UM1-CA186691 (Rosner GL)
Footnotes
Disclosures
Siu LL: Consultancy/Advisory Board: AstraZeneca/MedImmune, Boehringer Ingelheim, Celgene, Merck, Pfizer. Research Funding: Abraxis BioScience, AstraZeneca/MedImmune, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Genentech/Roche, GlaxoSmithKline, Merck, Novartis, Pfizer.
Ivy SP: No relationships to disclose
Dixon EL: No relationships to disclose
Gravell AE: No relationships to disclose
Reeves S: No relationships to disclose
Rosner GL: Equity Ownership: Johnson & Johnson. Other (served on independent data monitoring board for a study): Novartis
References
- 1.Anagnostou V, Yarchoan M, Hansen A, Sharon E, Collyar D, Chow L, et al. Immuno-oncology Trial Endpoints: Capturing Clinically Meaningful Activity. Clin Cancer Res. 2017;23:xx–xx. doi: 10.1158/1078-0432.CCR-16-3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Day D, Monjazeb A, Sharon E, Ivy P, Rubin E, Rosner G, et al. From Famine to Feast: Developing Early Phase Combination Immunotherapy Trials Wisely. Clin Cancer Res. 2017;23:xx–xx. doi: 10.1158/1078-0432.CCR-16-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehnert J, Monjazeb A, Beerthuijzen J, Collyar D, Rubinstein L, Harris L. The Challenge for Development of Valuable Immuno-Oncology Biomarkers. Clin Cancer Res. 2017;23:xx–xx. doi: 10.1158/1078-0432.CCR-16-3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baik C, Rubin E, Forde P, Mehnert J, Collyar D, Butler M. Limitations and Challenges in Immuno-Oncology Clinical Trials. Clin Cancer Res. 2017;23:xx–xx. doi: 10.1158/1078-0432.CCR-16-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maker AV, Phan GQ, Attia P, Yang JC, Sherry RM, Topalian SL, et al. Tumor Regression and Autoimmunity in Patients Treated With Cytotoxic T Lymphocyte–Associated Antigen 4 Blockade and Interleukin 2: A Phase I/II Study. Annals of Surgical Oncology. 2005;12:1005–16. doi: 10.1245/ASO.2005.03.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:4286–93. doi: 10.1158/1078-0432.CCR-14-2607. [DOI] [PubMed] [Google Scholar]
- 7.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MedImmune LLC. A Phase 1/2 Study to Evaluate MEDI4736. 2012 [cited 2017 Apr 7]; Available from: https://clinicaltrials.gov/ct2/show/NCT01693562.
- 9.Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti–Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. Journal of Clinical Oncology. 2016;34:3119–25. doi: 10.1200/JCO.2016.67.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toshihiko D, Piha-Paul SA, Jalal SI, Mai-Dang H, Saraf S, Koshiji M, et al. Updated results for the advanced esophageal carcinoma cohort of the phase Ib KEYNOTE-028 study of pembrolizumab (MK-3475) Journal of Clinical Oncology. 2016;34 :abstr 7. [Google Scholar]
- 12.Hazarika M, Chuk MK, Theoret MR, Mushti S, He K, Weis SL, et al. U.S. FDA Approval Summary: Nivolumab for Treatment of Unresectable or Metastatic Melanoma Following Progression on Ipilimumab. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-0712. [DOI] [PubMed] [Google Scholar]
- 13.Hansen AR, Siu LL. PD-L1 Testing in Cancer: Challenges in Companion Diagnostic Development. JAMA oncology. 2016;2:15–6. doi: 10.1001/jamaoncol.2015.4685. [DOI] [PubMed] [Google Scholar]
- 14.Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 15.Nishino M, Giobbie-Hurder A, Gargano M, Suda M, Ramaiya NH, Hodi FS. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:3936–43. doi: 10.1158/1078-0432.CCR-13-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:e143–e52. doi: 10.1016/S1470-2045(17)30074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen T-T. Statistical issues and challenges in immuno-oncology. Journal for immunotherapy of cancer. 2013;1:18. doi: 10.1186/2051-1426-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.National Cancer Institute. NCI-Related Moonshot Activities. 2017 [cited 2017 Apr 7]; Available from: www.cancer.gov/research/key-initiatives/moonshot-cancer-initiative/milestones/nci-activities.
- 19.Ferté C, Fernandez M, Hollebecque A, Koscielny S, Levy A, Massard C, et al. Tumor growth rate is an early indicator of antitumor drug activity in phase I clinical trials. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:246–52. doi: 10.1158/1078-0432.CCR-13-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodon J, Soria JC, Berger R, Batist G, Tsimberidou A, Bresson C, et al. Challenges in initiating and conducting personalized cancer therapy trials: perspectives from WINTHER, a Worldwide Innovative Network (WIN) Consortium trial. Annals of oncology : official journal of the European Society for Medical Oncology. 2015;26:1791–8. doi: 10.1093/annonc/mdv191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prawira A, Dufort P, Halankar J, Paravasthu DM, Hansen A, Spreafico A, et al. Development of a predictive radiomics signature for response to immune checkpoint inhibitors (ICIs) in patients with recurrent or metastatic squamous cell carcinoma of the head and neck (RM-SCCHN) Annals of Oncology. 2016;27 [Google Scholar]
- 22.Renfro LA, Sargent DJ. Statistical controversies in clinical research: basket trials, umbrella trials, and other master protocols: a review and examples. Annals of oncology : official journal of the European Society for Medical Oncology. 2017;28:34–43. doi: 10.1093/annonc/mdw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berry DA. Adaptive clinical trials in oncology. Nature reviews Clinical oncology. 2011;9:199–207. doi: 10.1038/nrclinonc.2011.165. [DOI] [PubMed] [Google Scholar]
- 24.M.D. Anderson Cancer Center. BATTLE-2 Program: A Biomarker-Integrated Targeted Therapy Study in Previously Treated Patients With Advanced Non-Small Cell Lung Cancer. 2010 [cited 2017 Apr 7]; Available from: https://clinicaltrials.gov/ct2/show/NCT01248247.
- 25.Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Tsao A, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer discovery. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clinical pharmacology and therapeutics. 2009;86:97–100. doi: 10.1038/clpt.2009.68. [DOI] [PubMed] [Google Scholar]
- 27.Carey LA, Winer EP. I-SPY 2–Toward More Rapid Progress in Breast Cancer Treatment. The New England journal of medicine. 2016;375:83–4. doi: 10.1056/NEJMe1603691. [DOI] [PubMed] [Google Scholar]
- 28.Park JW, Liu MC, Yee D, Yau C, van ’t Veer LJ, Symmans WF, et al. Adaptive Randomization of Neratinib in Early Breast Cancer. The New England journal of medicine. 2016;375:11–22. doi: 10.1056/NEJMoa1513750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rugo HS, Olopade OI, DeMichele A, Yau C, van ’t Veer LJ, Buxton MB, et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. The New England journal of medicine. 2016;375:23–34. doi: 10.1056/NEJMoa1513749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berry DA. Adaptive clinical trials: the promise and the caution. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:606–9. doi: 10.1200/JCO.2010.32.2685. [DOI] [PubMed] [Google Scholar]
- 31.Thall P, Fox P, Wathen J. Statistical controversies in clinical research: scientific and ethical problems with adaptive randomization in comparative clinical trials. Annals of oncology : official journal of the European Society for Medical Oncology. 2015;26:1621–8. doi: 10.1093/annonc/mdv238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maude SL, Hucks GE, Seif AE, Talekar MK, Teachey DT, Baniewicz D, et al. Pembrolizumab to augment response to CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL) Journal of Clinical Oncology. 2017;35 :abstr 103. [Google Scholar]
- 33.Tefferi A, Kantarjian H, Rajkumar SV, Baker LH, Abkowitz JL, Adamson JW, et al. In Support of a Patient-Driven Initiative and Petition to Lower the High Price of Cancer Drugs. Mayo Clinic proceedings. 2015;90:996–1000. doi: 10.1016/j.mayocp.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larkin J, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, CL C. Overall survival (OS) results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naïve patients with advanced melanoma (CheckMate 067) American Association for Cancer Research. 2017 Abstract CT075. [Google Scholar]
- 36.U.S. Food and Drug Administration. Accelerated approval for Opdivo (nivolumab) Injection. 2016 [cited 2017 Apr 7]; Available from: www.accessdata.fda.gov/drugsatfda_docs/appletter/2016/125554Orig1s007ltr.pdf.
- 37.Eggermont AMM, Chiarion-Sileni V, Grob J-J, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. The New England journal of medicine. 2016;375:1845–55. doi: 10.1056/NEJMoa1611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merck Sharp & Dohme Corp. Study of Pembrolizumab (MK-3475) Versus Placebo After Complete Resection of High-Risk Stage III Melanoma (MK-3475-054/KEYNOTE-054) 2015 [cited 2017 Apr 7]; Available from: https://clinicaltrials.gov/ct2/show/NCT02362594.
- 39.Bristol-Myers Squibb. Efficacy Study of Nivolumab Compared to Ipilimumab in Prevention of Recurrence of Melanoma After Complete Resection of Stage IIIb/c or Stage IV Melanoma (CheckMate 238) 2015 [cited 2017 Apr 7]; Available from: https://clinicaltrials.gov/ct2/show/NCT02388906.
- 40.National Cancer Institute. Cancer Moonshot Blue Ribbon Panel Report. 2016 [cited 2017 Apr 7]; Available from: www.cancer.gov/research/key-initiatives/moonshot-cancer-initiative/blue-ribbon-panel.
- 41.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375:1823–33. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 42.Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17:1497–508. doi: 10.1016/S1470-2045(16)30498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–50. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 44.Ferris RL, Blumenschein G, Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856–67. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL, Jr, Lawrence DP, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol. 2016;17:943–55. doi: 10.1016/S1470-2045(16)30126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol. 2016;34:2460–7. doi: 10.1200/JCO.2015.64.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldberg SB, Gettinger SN, Mahajan A, Chiang AC, Herbst RS, Sznol M, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17:976–83. doi: 10.1016/S1470-2045(16)30053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–65. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 49.Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med. 2016;374:2542–52. doi: 10.1056/NEJMoa1603702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, et al. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J Clin Oncol. 2016 doi: 10.1200/JCO.2016.67.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016;17:717–26. doi: 10.1016/S1470-2045(16)00175-3. [DOI] [PubMed] [Google Scholar]
- 52.Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA. 2016;315:1600–9. doi: 10.1001/jama.2016.4059. [DOI] [PubMed] [Google Scholar]
- 53.Sharma P, Callahan MK, Bono P, Kim J, Spiliopoulou P, Calvo E, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol. 2016;17:1590–8. doi: 10.1016/S1470-2045(16)30496-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, et al. Nivolumab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34:2980–7. doi: 10.1200/JCO.2016.66.9929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Younes A, Santoro A, Shipp M, Zinzani PL, Timmerman JM, Ansell S, et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016;17:1283–94. doi: 10.1016/S1470-2045(16)30167-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol. 2016;34:2698–704. doi: 10.1200/JCO.2015.65.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17:883–95. doi: 10.1016/S1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- 58.American Cancer Society. Cancer Facts & Figures 2017. Atlanta, GA: American Cancer Society; 2017. [Google Scholar]