

Abstract
Approximately, 50% of all cutaneous melanomas harbor activating BRAF V600 mutations, among these 10–30% carry the V600K mutation. Clinically, patients with V600K tumors experience distant metastases sooner and have an increased risk of relapse and shorter survival than patients with V600E tumors. Despite clinical and other histopathological differences between these BRAF tumor subtypes, little is known about them at the genomic level.
Herein, we systematically compared BRAF V600E and V600K skin cutaneous melanoma (SKCM) samples from the Cancer Genome Atlas (TCGA) for differential protein, gene, and microRNA expression genome-wide using the Mann-Whitney U-test.
Our analyses showed that elements of energy-metabolism and protein-translation pathways were up-regulated and that pro-apoptotic pathways were down-regulated in V600K tumors compared to V600E tumors. We found that c-Kit protein and KIT gene expressions were significantly higher in V600K tumors than in V600E tumors, concurrent with significant down-regulation of several KIT-targeting microRNAs (mir) including mir-222 in V600K tumors, suggesting KIT and mir-222 might key genomic contributors to observed clinical differences.
The relationship that we uncovered among KIT/c-Kit expression, mir-222 expression, and growth and pro-survival signals in V600 tumors is intriguing. We believe that the observed clinical aggressiveness of V600K tumors compared to V600E tumors may be attributable to the increased energy-metabolism, protein-translation and pro-survival signals compared to V600E tumors. If confirmed using larger numbers of V600K tumors, our results may prove useful for designing clinical management and targeted chemotherapeutical interventions for BRAF V600K positive melanomas. Lastly, small sample size in V600K tumors is a major limitation of our study.
Keywords: Melanoma, BRAF, V600E, V600K, c-Kit, mir-222
INTRODUCTION
BRAF is a serine/threonine protein kinase that activates the MAP kinase/ERK-signaling pathway [1]. Approximately, 40–60% of cutaneous melanomas harbor activating BRAF mutations [2]. The majority of the BRAF mutations constitute a substitution of the valine residue at position 600 by a glutamate (V600E) through mutation of a single nucleotide GTG to GAG. Another prevalent BRAF mutation at the same residue is V600K mutation in which the valine residue is replaced by a lysine through two nucleotide substitutions (GTG to AAG). V600K mutation occurs in 10 to 30% of all BRAF V600 melanomas [2–6]. Other less common BRAF mutations at V600 residue include V600R and V600D [1].
Patients with V600K and those with V600E mutation seem to have some distinctive clinical features. Patients with V600K tumors appear to be older (over 50) males, and the tumors often occur in the head and neck area (prone to sun damage) [4, 7–13]. Pathologically, V600K tumors appear to be thicker and are more mitotically active than V600E tumors [8]. Clinically, patients with V600K tumors have an increased risk for brain and lung metastases and are at a siginficantly increased risk of relapse and have a shorter time from diagnosis to metastasis than those with V600E tumors [4, 8–11, 13]. Despite those differences, a large clinical trial showed that V600K tumors were sensitive to vemurafenib, a BRAF inhibitor, and that patients with V600K or V600E tumors, when treated with vemurafenib, had similar overall survival and progression-free survival outcomes [10].
Although the clinical and histopathological differences between V600K and V600E tumors are well documented, little is known about their genomic differences. In this study, we systematically compared protein expression, RNA-seq gene expression, and miRNA-seq microRNA expression between BRAF V600K and BRAF V600E melanoma tumor samples from TCGA. In addition, we analyzed somatic mutation, copy number alteration, and clinical data between those two BRAF tumor subtypes. We aimed to identify signaling pathway that might contribute to the observed clinical and histopathological differences between the two subtypes of BRAF melanoma. Our analysis revealed an intriguing relationship among KIT gene expression and c-Kit protein expression, mir-222 expression, and growth and pro-survival signals in V600 tumors. Although the number of V600K tumor samples available for our analyses was small, our results may provide useful clues for clinical management and targeted chemotherapeutical interventions for the two BRAF melanoma subtypes.
METHODS
We used the online DAVID Bioinformatics Resources 6.7 [14] for gene ontology analysis. We carried out all differential expression analyses (protein, gene and microRNA) between V600E and V600K tumors in Matlab using the Mann–Whitney U test (two-sided) [15]. For our survival analysis, we fit the multivariate Cox proportional hazards model using SAS software (SAS Institute Inc., Cary, NC, USA).
Data
We downloaded all genomic data from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/). We downloaded additional information on the four melanoma subtypes: BRAF, NF1, RAS, and triple wild type from the TCGA cutaneous melanoma publication [2]. Information on the subtypes included clinical, mutation, copy number variation, and other related characteristics. For RNA-seq data, we log2-transformed the normalized read counts (rsem.genes.normalized_results) using [log2(count+1)] but carried out no further normalization. For the RPPA (reverse phase protein array) protein expression data, we used normalized data from TCGA without further normalization or transformation; negative values arose from the normalization by TCGA (http://bioinformatics.mdanderson.org/main/TCPA:Overview).
TCGA provided RNA-seq data on ~24,000 genes for 124 BRAF V600E tumor samples and 17 V600K tumor samples. TCGA sequenced 1,046 micro-RNAs in 121 V600E and 17 V600K tumor samples. TCGA profiled protein expression in up to 91 BRAF V600E tumors and 17 BRAF V600K tumors. Not all proteins were analyzed in all samples; among the 276 proteins or protein modifications TCGA studied, only 103 were analyzed in four or more V600K tumor samples.
Survival
As described in the TCGA publication [2], an accurate stage of disease at the time of the TCGA biospecimen procurement was often unavailable. The available pathology data such as primary tumor staging information were dated to the patient’s initial melanoma diagnosis, not to the time of tumor procurement (i.e., entry into the TCGA database). Often, there was a time-lag (mean: 1,272.9 days; median: 432 days for BRAF-mutation patients) between initial melanoma diagnosis and TCGA biospecimen procurement. TCGA’s analysis of melanoma survival [2] used the patient’s survival from the time of TCGA biospecimen procurement until death or loss to follow-up. Our analysis of melanoma survival used the patient’s survival from diagnosis until death or loss to follow-up. Our survival analysis allowed for right censoring to account for loss to follow-up and left truncation to account for the feature that patients could only enter the study if they survived from diagnosis until TCGA specimen procurement. Of the 124 subjects with V600E tumors and 17 with V600K tumors, TCGA had matched clinical data on 106 and 13, respectively. Among these 119 patients with clinical records, 48 were dead and 71 remained alive at last contact (i.e., were censored for survival analysis).
RESULTS and DISCUSSION
Survival comparison between BRAF subtypes
An apparent trend of shorter overall survival for patients with V600K tumors than for those with V600E tumors (Figure 1) was not statistically significant (hazard ratio=1.4, 95% confidence interval [0.62, 3.25], p=0.39), similar to other published findings. Nonetheless, this tendency to shorter survival in V600K subjects is consistent with literature findings that V600K tumors tend to have less favorable outcomes than V600E tumors [4, 8–11, 13].
Figure 1.

Fitted survival probabilities as function of days after diagnosis based on Cox regression model for two SKCM subtypes: BRAF V600K and V600E. Circles indicate censored patients. The plots are based on 13 subjects with V600K tumors and 106 with V600E tumors. The apparent difference between survival curves was not statistically significant (p=0.39).
Overall gene, microRNA and protein expression comparisons between BRAF subtypes
Among the ~24,000 genes with RNA-seq gene expression levels from TCGA, we found 1,675 differentially expressed with p<0.05 between BRAF subtypes; among these 593 were up-regulated and 1,082 were down-regulated in BRAF V600K tumors compared to V600E tumors.
Gene ontology (GO) analysis applied to the top 250 (as ordered by p-values for differential expression) genes up-regulated in V600K tumors compared to V600E tumors showed that the up-regulated genes were over-represented in metabolic processes (Table 1A), consistent with up-regulation of proliferation signals in those tumors from protein expression analysis. Among the top 250 genes down-regulated in V600K tumors compared to V600E tumors, GO analysis showed an over-representation in embryonic development pathways (Table 1B).
Table 1.
Enriched GO terms (GOTERM_BP_ALL) for the top 250 up-regulated (A) and 250 down-regulated (B) genes in V600K tumors compared to V600E tumors
| (A) Top 250 up-regulated genes in V600K tumors | ||
|---|---|---|
|
| ||
| GO Term | No. genes in pathway | Benjamini-Hochberg procedure adjusted p-value |
|
| ||
| Cofactor metabolic process | 20 | 9.8E-04 |
| Coenzyme metabolic process | 16 | 1.4E-02 |
| Carboxylic acid metabolic process | 27 | 1.1E-02 |
| Oxoacid metabolic process | 27 | 9.4E-03 |
| Organic acid metabolic process | 28 | 1.2E-02 |
| (B) Top 250 down-regulated genes in V600K tumors | ||
|---|---|---|
|
| ||
| GO Term | No. genes in pathway | Benjamini-Hochberg procedure adjusted p-value |
|
| ||
| Anatomical structure morphogenesis | 54 | 3.7E-03 |
| Embryonic skeletal system development | 10 | 9.7E-03 |
| Embryonic skeletal system morphogenesis | 9 | 6.5E-03 |
| morphogenesis of an epithelium | 19 | 8.1E-03 |
| Embryonic organ morphogenesis | 15 | 7.0E-03 |
Among the 103 proteins for which data were available for at least four V600K tumor samples, 14 were differentially expressed with p<0.05 between BRAF subtypes; among these 7 were up-regulated and 7 were down-regulated in BRAF V600K tumors compared to V600E tumors (Table 2, Supplementary Figure S1A and S1B).
Table 2.
Differentially expressed proteins between V600E and V600K tumors
| Protein | V600E tumors | V600K tumors | p-value | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| median | mean | No. | median | mean | No. | ||
|
| |||||||
| up-regulated in V600K tumors | |||||||
|
| |||||||
| eEF2-R-C | −0.196 | −0.250 | 53 | 0.308 | 0.316 | 9 | 0.003 |
| TFRC-R-V | 0.060 | 0.095 | 91 | 0.877 | 0.807 | 13 | 0.019 |
| ERK2-R-E | −0.023 | 0.001 | 53 | 0.193 | 0.213 | 9 | 0.031 |
| AMPK_alpha-R-C | −0.008 | 0.003 | 91 | 0.228 | 0.174 | 13 | 0.031 |
| c-Kit-R-V | 0.030 | 0.224 | 91 | 0.990 | 0.969 | 13 | 0.032 |
| PCNA-M-C | −0.091 | −0.028 | 53 | 0.081 | 0.195 | 9 | 0.040 |
| S6-R-C | 0.041 | 0.022 | 38 | 0.327 | 0.368 | 4 | 0.042 |
| Tuberin-R-C | −0.202 | −0.366 | 38 | 0.560 | 0.239 | 4 | 0.057 |
|
| |||||||
| down-regulated in V600K tumors | |||||||
|
| |||||||
| HER3_pY1289-R-C | 0.021 | 0.044 | 91 | −0.066 | −0.053 | 13 | 0.004 |
| c-Met_pY1235-R-V | 0.038 | 0.056 | 53 | −0.027 | −0.049 | 9 | 0.017 |
| Rictor_pT1135-R-V | 0.042 | 0.062 | 91 | −0.137 | −0.115 | 13 | 0.018 |
| IGFBP2-R-V | 0.456 | 0.604 | 91 | −0.337 | −0.062 | 13 | 0.024 |
| NF2-R-C | 0.003 | 0.037 | 91 | −0.236 | −0.178 | 13 | 0.028 |
| Caspase-7_cleavedD198-R-C | 0.395 | 0.491 | 91 | −0.594 | −0.242 | 13 | 0.031 |
| Bak-R-E | −0.077 | −0.134 | 38 | −0.392 | −0.454 | 7 | 0.035 |
| Bid-R-C | 0.018 | 0.090 | 91 | −0.090 | −0.043 | 13 | 0.054 |
The negative values were resulted from normalization by TCGA (www.mdanderson.org/). No further normalization/standardization was carried out for data shown here.
Among the significantly down-regulated proteins in V600K tumors were Bak and caspase-7 (Figure 2). Bak belongs to the BCL2 protein family and acts as anti- or pro-apoptotic regulator. Caspase-7 is a member of the cysteine-aspartic acid protease (caspase) family that plays a central role in the execution-phase of cell apoptosis. Another member of the pro-apoptotic family of proteins, Bid, was also down-regulated in V600K samples (Table 2). Bid is also a member of the BCL-2 family of cell death regulators. Taken together, these results suggest that the pro-apoptotic signaling pathways might be down-regulated to promote cell survival and growth in V600K tumors
Figure 2.

Boxplots of expression levels of selected differentially expressed proteins between V600K and V600E tumors. The protein expression data were taken from TCGA RPPA analysis without further standardization and transformation.
Among the proteins up-regulated in V600K tumors compared to V600E tumors, S6 (RPS6) and eEF2 are involved in mRNA translation and ribosomal biogenesis. S6 is a down-stream target of the mTOR/S6K signaling pathway involved in growth factors and mitogen-induced protein translation [16–18]. eEF2 is an essential factor for protein synthesis. Besides eEF2, eEF2K and phosphorylated 4E-BP1 (4E-BP1_pT37_T46) were up-regulated in V600K tumors compared to V600E tumors, although those changes in protein levels were not statistically significant (data not shown). eEF2K is a conserved protein kinase in the calmodulin-mediated signaling pathway that links activation of cell surface receptors to cell division and is involved in the regulation of protein synthesis. It phosphorylates eukaryotic elongation factor 2 (EEF2) and thus inhibits the EEF2 function. eEF2K is expressed at high levels in certain cancers, where it may act to help cell survival, e.g., during nutrient starvation [19]. AMPK is an upstream signaling molecule of the mTOR pathway providing a link between energy and mTOR pathways [16–18]. Tuberin (TSC2), also up-regulated in V600K tumors compared to V600E tumors (Figure 2), forms a complex with TSC1, and the complex serves to control various cellular functions as an important upstream hub for the mTOR pathway [16–18]. Those results suggest that the mTOR pathway may be differentially affected between V600K and V600E tumors. Lastly, the phosphorylated mTOR protein (mTOR_pS2448) level was also elevated in V600K BRAF tumors compared to V600E BRAF tumors, although the elevation did not reach statistical significance (Figure 3). The other proteins that were up-regulated in V600K tumors compared to V600E tumors include TFRC and PCNA. TFRC is a cell surface receptor necessary for cellular iron uptake by the process of receptor-mediated endocytosis. PCNA is involved in DNA replication. It is not clear why those two proteins were up-regulated in V600K tumors compared to V600E tumors. Finally, c-Kit, a receptor tyrosine kinase that plays a multifunctional role in melanogenesis, cell growth, migration and survival [20, 21], was also up-regulated in V600K tumors.
Figure 3.
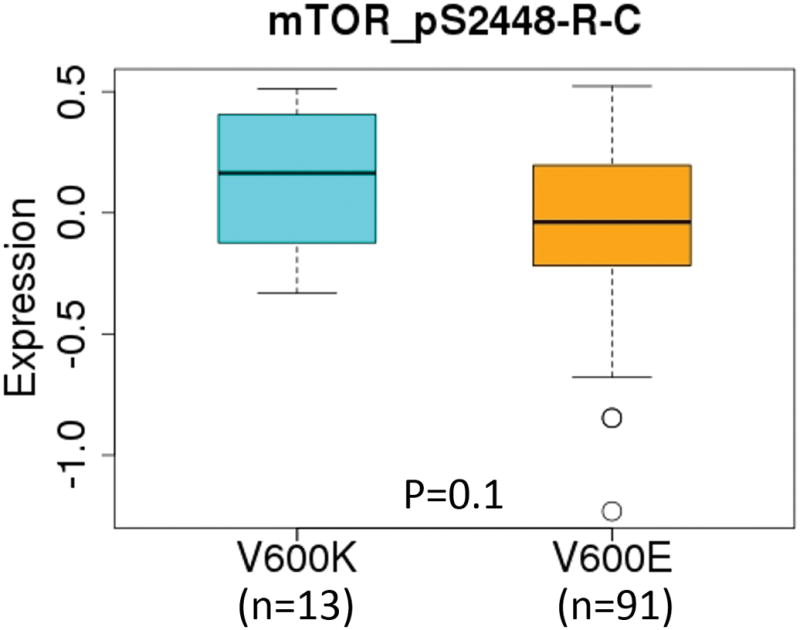
Boxplots of expression levels of mTOR_pS2448-R-C protein between V600K and V600E tumors. The protein expression data were taken from TCGA RPPA analysis without further standardization and transformation.
Among the 1,046 microRNAs analyzed by TCGA, 106 were differentially expressed with p<0.05 between V600E and V600K SKCM tumors; among these 29 were up-regulated and 77 were down-regulated in V600K tumors compared to V600E tumors. Interestingly, several of the most significantly down-regulated microRNAs (Table 3) in V600K tumors compared to V600E tumors. Several of them, such as mir-222[22] (Figure 4), mir-34b/c [23], mir-19b-2 [24], and mir-148a, are either known or predicted to target the KIT gene (see below) (Supplementary Figure S2). Up-regulated miRNAs in V600K tumors compared to V600E tumors include mir-330 and let7g, both of which appear to have antiproliferative effects [25].
Table 3.
Top 15 differentially down-regulated microRNAs in V600K tumors compared to V600E tumors. A total of 1,046 microRNAs were analyzed by TCGA.
| miRNA | p-value | Predicted¶ or known to regulate c-Kit expression |
|---|---|---|
|
| ||
| hsa-mir-222 | 0.0004 | Known |
| hsa-mir-125b-1 | 0.0012 | No |
| hsa-mir-34b | 0.0017 | Predicted |
| hsa-mir-34c | 0.0023 | Known |
| hsa-mir-10b | 0.0032 | No |
| hsa-mir-664 | 0.0032 | No |
| hsa-mir-1271 | 0.0055 | No |
| hsa-mir-708 | 0.0056 | No |
| hsa-mir-100 | 0.0058 | No |
| hsa-mir-653 | 0.0077 | No |
| hsa-mir-19b-2 | 0.0084 | Known |
| hsa-mir-196b | 0.0093 | No |
| hsa-mir-148a | 0.0097 | Known |
| hsa-mir-10a | 0.0117 | No |
| hsa-mir-574 | 0.0120 | No |
based on prediction by microRNA.org. See text for references.
Figure 4.
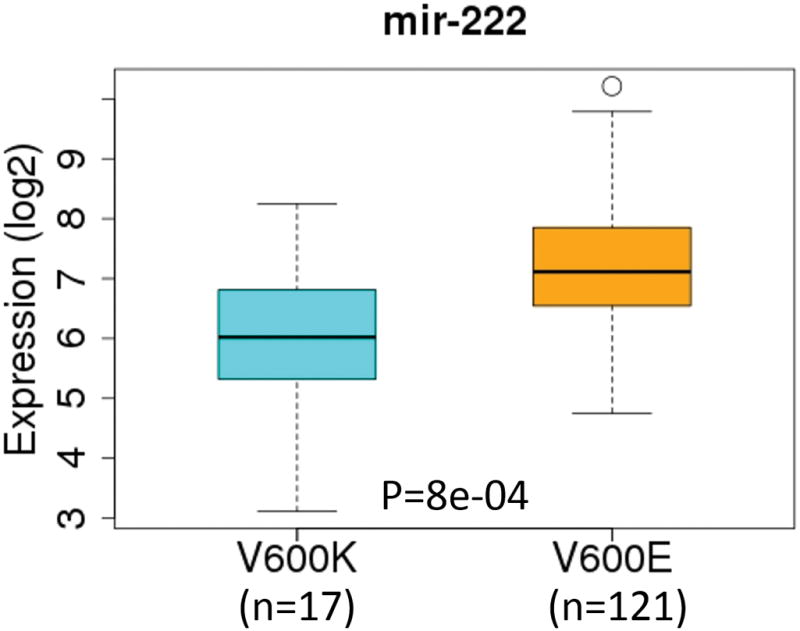
Boxplots of mir-222 expression levels across two SKCM BRAF tumor subtypes defined by TCGA melanoma working group. The numbers of microRNA-seq samples for V600K and V600E subtypes were 17 and 121, respectively.
Preliminary synthesis of gene, protein, and microRNA expression findings
Our gene expression analysis suggested the up-regulation of metabolic processes and down-regulation of embryonic development pathways in V600K tumors compared to V600E tumors. Our protein expression analysis suggested that pro-apoptotic pathways were down-regulated, possibly contributing to enhanced cell survival and growth in V600K tumors compared to V600E tumors. In addition, our protein expression analysis indicated that the mTOR signaling pathway may be differentially affected between the two BRAF subtypes. Our analyses to this point, however, failed to uncover a single regulatory or signaling feature that might underpin or connect these differentially expressed pathways.
Perhaps the most striking feature to emerge from our differential expression analyses was the fact that c-Kit was significantly up-regulated in V600K tumors while several micro-RNAs known or predicted to target KIT were significantly down-regulated. Both KIT gene expression and protein expression were up-regulated in V600K tumors compared to V600E tumors (p=0.03) (Figure 5). In addition, using 352 matched SKCM protein-expression (RPPA) and gene-expression (RNA-seq) samples, we found a strong correlation in expression levels between c-Kit and KIT (Spearman ρ=0.86, and p<0.001). The coordinated up-regulation of c-Kit protein level and KIT gene expression and concomitantly down-regulation of several KIT-targeting microRNAs suggested to us that the c-Kit signaling pathway is possibly an important feature distinguishing between V600E and V600K BRAF tumors. Those observations prompted us to focus our downstream analysis on KIT/c-Kit and its signaling pathways.
Figure 5.
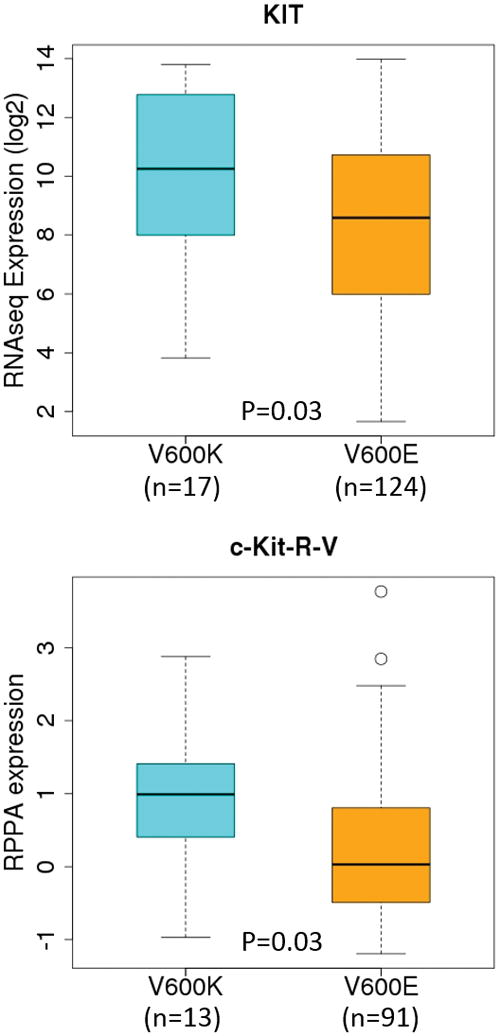
Boxplots of c-Kit protein (top) and KIT gene (bottom) expression across the two SKCM mutation subtypes (BRAF V600E and V600K) defined by TCGA melanoma working group. The numbers samples for V600K and V600E, respectively, were 13 and 91 for RPPA; and 17 and 124 for RNA-seq. Both RNA-seq and RPPA (protein expression) data were normalized by TCGA; we subsequently applied a log2-transformation to the RNA-seq data before analysis.
Additional findings related to mir-222 and the c-Kit signaling pathway
In the TCGA data, mir-222 expression was negatively correlated with that of KIT in all 450 SKCM samples (Spearman ρ = −0.27, p= 7.0E-09) and 14 V600K samples (Spearman ρ =−0.32, p=0.26) (Figure 6). Up-regulation of mir-222 results in dramatic loss of KIT transcript and c-Kit protein by targeting the 3′-untranslated region (UTR) of KIT in papillary thyroid carcinoma [26], gastrointestinal stromal tumors [27, 28], erythroleukemic cells [29], prostate cancer [30], acute myelogenous leukemia [31], cutaneous melanoma [32] and human umbilical vein endothelial cells (HUVECs) [33].
Figure 6.
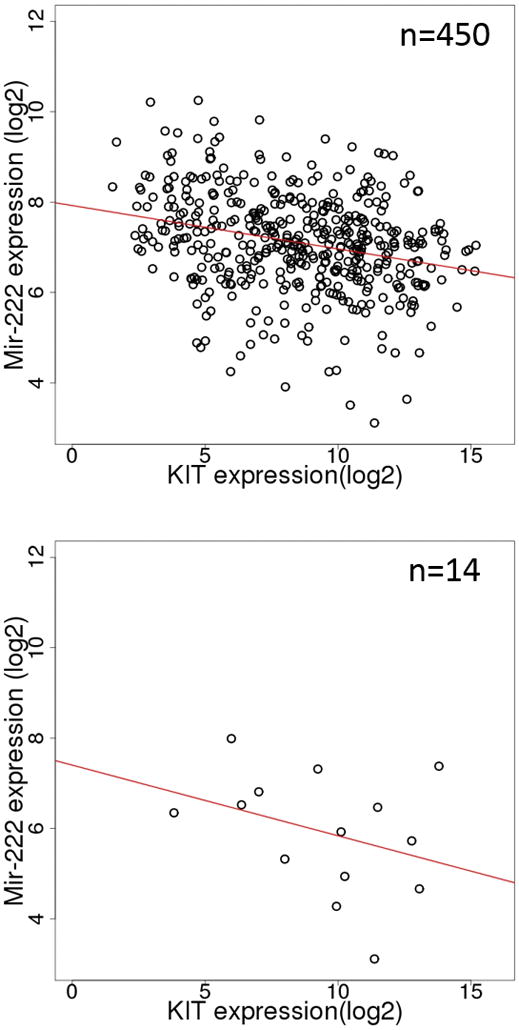
Scatter plots of KIT and mir-222 expression in all SKCM tumors (top) and BRAF V600K only SKCM tumors (bottom). The red lines are the least-squares fits. In this Figure, we first identified matching RNA-seq and miRNA-seq samples and extracted KIT expression and mir-222 expression respectively.
Besides targeting KIT, mir-222 has been shown to regulate the expression of other genes including BIM [34], DDIT4 [35], ETV1 [28], ICAM1 [36], MMP1 and SOD2 [37], p27 (KIP1) and p57 (KIP2) [22, 38, 39], PTEN and TIMP3 [40], PUMA [41], and TRPS1 [42]. The role of mir-221/222 in cancer has been reviewed in [43].
To examine whether down-regulation of mir-222 is associated with up-regulation of metabolism in other TCGA tumor types, we mapped all TCGA RNASeqV2 samples to all miRNA-seq samples (from HiSeq only). In total, 8,343 samples from 31 unique tumor types were matched between the two platforms (Supplementary Table S1). The number of matched samples in each tumor type ranged from 57 to 1,224. We then computed the pair-wise Spearman-rank correlation between mir-222 and each of the 20,531 genes for each of the 31 tumor types. This resulted in 31 pair-wise correlation coefficients between each gene and mir-222, one for each tumor type. Not surprisingly, GO terms related to metabolism are enriched with the 150 most negatively correlated genes whereas GO terms related to immune response and cell motility are enriched with the 150 most positively correlated genes (data not shown). KIT was among the top 150 most positively correlated genes. When we considered only SKCM tumors, the enrichment of metabolism genes among the 150 most negatively correlated genes persisted. Those results suggest a link between down-regulation of mir-222 and the over-representation of metabolism genes among genes whose expression is up-regulated in V600K tumors, although the exact relationship between the two is unclear.
TCGA had previously identified 18 significantly mutated genes in melanoma [2]; three (GNA11, KIT, and PTEN) showed proportionally higher number of mutations in V600K tumors than in V600E tumors (Table 4A). A closer examination of the mutations in each of the three genes did not find a pattern of association between the mutations and c-Kit expression among the 13 V600K tumors (Table 4B). In fact, the three V600K tumors with the highest c-Kit expression were all wild-type for the three genes (highlighted in Table 4B). We saw no evident association between the elevated expression of c-Kit and any copy number changes in the KIT gene (Table 4B). No differential methylation in KIT promoter was found between V600K and V600E tumors (data not shown).
Table 4A.
Somatic mutation patterns in BRAF V600E and V600K tumors. The three genes (GNA11, KIT, and PTEN) with significant difference between the two subtypes are highlighted
| Mutation | No. V600E tumors with mutations | No. V600K tumors with mutations | No. V600E tumors without mutation | No. V600K tumors without mutation | p-value |
|---|---|---|---|---|---|
|
| |||||
| ARID2_mut | 13 | 4 | 111 | 13 | 0.13 |
| CDK4_mut | 2 | 0 | 122 | 17 | 1.00 |
| CDKN2A_mut | 3 | 0 | 121 | 17 | 1.00 |
| DDX3X_mut | 18 | 1 | 106 | 16 | 0.47 |
| GNA11_mut | 4 | 3 | 120 | 14 | 0.04 |
| GNAQ_mut | 0 | 0 | 124 | 17 | 1.00 |
| IDH1_mut | 3 | 1 | 121 | 16 | 0.41 |
| KIT_mut | 3 | 3 | 121 | 14 | 0.02 |
| KRAS_mut | 0 | 0 | 124 | 17 | 1.00 |
| MAP2K1_mut | 0 | 0 | 124 | 17 | 1.00 |
| NF1_mut | 0 | 0 | 124 | 17 | 1.00 |
| NRAS_mut | 0 | 0 | 124 | 17 | 1.00 |
| PPP6C_mut | 7 | 1 | 117 | 16 | 1.00 |
| PTEN_mut | 5 | 4 | 119 | 13 | 0.01 |
| RAC1_mut | 16 | 2 | 108 | 15 | 1.00 |
| RB1_mut | 4 | 0 | 120 | 17 | 1.00 |
| TERT_MUT | 12 | 3 | 112 | 14 | 0.39 |
| TP53_mut | 3 | 0 | 121 | 17 | 1.00 |
Data taken from the Supplementary Table 1 of TCGA publication [2]. p-value was computed using a two-side Fisher exact test.
Table 4B.
GNA11, KIT, and PTEN mutations and KIT copy number alterations in V600K tumors in relation to c-Kit protein expression.
| Name | Mutation | Copy No. Alteration | c-Kit expression | ||
|---|---|---|---|---|---|
|
| |||||
| GNA11 | KIT | PTEN | KIT_cnv | ||
|
| |||||
| TCGA-BF-A3DL-01 | wt | wt | wt | 0 | 2.6437 |
| TCGA-BF-A3DM-01 | wt | wt | H92Y | 0 | −0.3460 |
| TCGA-BF-A5EQ-01 | wt | wt | wt | 0 | 2.7293 |
| TCGA-DA-A1I0-06 | wt | F182_splice | wt | 0 | 0.9899 |
| TCGA-DA-A3F8-06 | wt | Y200S | R301C;H92Y | −1 | 0.4071 |
| TCGA-EB-A3XB-01 | wt | wt | R301C | 1 | 0.5543 |
| TCGA-EE-A183-06 | wt | wt | wt | 0 | n/a |
| TCGA-EE-A20H-06 | wt | wt | wt | 0 | n/a |
| TCGA-EE-A29E-06 | wt | R311* | wt | 0 | 1.4094 |
| TCGA-EE-A2GB-06 | wt | wt | wt | 1 | n/a |
| TCGA-EE-A2GT-06 | G8S | wt | wt | −1 | 1.2554 |
| TCGA-EE-A2ME-06 | wt | wt | wt | 0 | n/a |
| TCGA-ER-A19J-06 | wt | wt | wt | 0 | −0.9046 |
| TCGA-ER-A19K-01 | wt | wt | wt | 0 | 2.8772 |
| TCGA-ER-A42L-06 | R132C | wt | wt | −1 | 1.1497 |
| TCGA-FS-A1ZC-06 | wt | wt | R301C | 0 | −0.9696 |
| TCGA-IH-A3EA-01 | R132C | wt | wt | 0 | 0.7967 |
‘n/a’ denotes no data from TCGA. Mutation and copy number alteration data are from [2]. The three tumors with the highest c-Kit expression are highlighted.
In conclusion, our analyses of multiple genomic data suggest that both c-Kit protein and KIT gene expression levels were significantly higher in V600K tumors than in V600E tumors. Concurrently, mir-222 expression was significantly down-regulated in V600K tumors compared to V600E tumors. We also found that those differential changes concomitantly correlated with increased proliferation and pro-survival signals/pathways in V600K tumors compared to V600E tumors, providing a plausible link between the observed clinical phenotypes and genomics between the two subtype BRAF tumors.
Discussion
An increasing number of studies have demonstrated that BRAF V600K and V600E tumors are not identical. V600K tumors are more aggressive in that patients with those tumors have a significantly higher risk for relapse and a shorter time from diagnosis to metastasis compared to V600E tumors. Despite of the clinical and histopathological differences, little is known about the underlying mechanisms at the genomic level.
Structurally, both V600E and V600K mutations are located in the activation segment helix 1 inside the kinase domain and are predicted to perturb the structure of the helix, thus, promoting dimerization of the protein [44]. The exact structural basis for the clinical and histopathological differences between the two BRAF tumor subtypes is, however, not entirely clear.
Our computational analysis of multiple genomic data from TCGA suggested that V600K tumors had higher c-Kit protein and KIT gene expression levels than V600E tumors. We also found evidence of down-regulation of pro-apoptotic proteins and up-regulation of metabolism in V600K tumors compared to V600E tumors. Proteins involved in mRNA translation and ribosomal biogenesis were also up-regulated in V600K tumors compared to V600E tumors, suggesting that the mTOR pathway [16–18] may be further activated in V600K tumors. Several proteins involved in DNA repair such as ATM, CHEK2, and PCNA, also had higher expression in V600K tumors compared to V600E tumors (Supplementary Figure S1A).
c-Kit is a receptor tyrosine kinase that plays a multifunctional role in melanogenesis, cell growth, migration and survival [20, 21]. Activation of c-Kit either by the stem cell factor (SCF, a ligand) or oncogenic mutation activates the phosphatidylinositol 3-kiase (PI3K) signaling pathway as the dominant effector for cell proliferation and the mitogen-activated protein kinase (MAPK) cascade as an ancillary survival pathway [45, 46]. Although c-Kit has an important role in normal melanocyte differentiation, growing evidence also suggests that it plays a role in melanoma progression [21].
What caused the upregulation c-Kit and KIT expression in V600K tumors is unclear. We could find no association with somatic mutation, copy number alteration, or KIT promoter methylation data. KIT has been shown to be transcriptionally regulated by MITF [47, 48]. However, MITF gene expression did not differ between V600K and V600E tumors (data not shown).
We found that mir-222 expression was significantly down-regulated in V600K tumors compared to V600E tumors. Mir-222 is known to regulate c-Kit protein and KIT gene expression. Thus, it is possible that up-regulation of c-Kit resulted from down-regulation of mir-222. If so, what caused the down-regulation of mir-222 in V600K tumors? No differential methylation changes (TCGA methylation450 data, not shown) between V600K and V600E tumors were found in the promoter of human mir-222 gene. Mir-222 expression has been shown to be regulated transcriptionally by several transcription factors. The promyelocytic leukemia zinc finger (PLZF) transcription factor represses mir-222 expression in cutaneous melanoma [22]. PLZF (ZBTB16) expression was not differential between V600K and V600E subtypes, however (data not shown). Other transcription factors known to regulate mir-222 expression include AML1 (RUXN1) [31], AP-1 [40], FOSL1 [42] and NF-κB and c-Jun [49]. Although FOSL2 and RUNX3 were significantly down-regulated in V600K tumors compared to V600E tumors, whether either of the two played a role in the differential regulation of mir-222 expression is unclear. Hypoxia has been shown to down-regulate mir-222 expression [50], raising the question whether hypoxia in V600K tumors is more severe than in V600E tumors. Besides c-Kit, mir-222 is also known to target at least four tumor suppressors (PTEN, p27, p57, and TIMP3), suggesting that having the “right” dosage of mir-222 may be critical, if its expression level was manipulated for potential therapeutic benefit.
We suggest that up-regulation of c-Kit protein and KIT gene expression, down-regulation of mir-222, and increased metabolic signals may be intertwined. We believe that enhanced growth and pro-survival signals in V600K tumors compared to V600E tumors may be in part responsible for the observed clinical aggressiveness of V600K tumors.
Lastly, our observations/conclusions were built on a small number of V600K tumors (13–19) vs a more substantial number of V600E tumors (91-124). The sample sizes for some of the differentially expressed proteins identified in V600K tumors were smaller (4–13). Small sample size in V600K tumors is a major limitation of our study. If we had a larger sample of that subtype, our results could change. It is important that our results are further validated in an independent study. Furthermore, underlying differences between V600E and V600K tumors are likely complex; c-Kit/mir-222 are likely not the only players.
Supplementary Material
Acknowledgments
Source of funding: This research was supported by Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (ES101765).
We thank Xuting Wang and Grace Kissling for comments and suggestions and Gordon Flake for discussions. This research was supported by Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (ES101765).
Footnotes
Disclosure of potential conflicts of interest: No potential conflicts of interest were disclosed.
References
- 1.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stadelmeyer E, Heitzer E, Resel M, Cerroni L, Wolf P, Dandachi N. The BRAF V600K mutation is more frequent than the BRAF V600E mutation in melanoma in situ of lentigo maligna type. J Invest Dermatol. 2014;134(2):548–550. doi: 10.1038/jid.2013.338. [DOI] [PubMed] [Google Scholar]
- 4.Bucheit AD, Syklawer E, Jakob JA, Bassett RL, Jr, Curry JL, Gershenwald JE, Kim KB, Hwu P, Lazar AJ, Davies MA. Clinical characteristics and outcomes with specific BRAF and NRAS mutations in patients with metastatic melanoma. Cancer. 2013;119(21):3821–3829. doi: 10.1002/cncr.28306. [DOI] [PubMed] [Google Scholar]
- 5.Menzies AM, Haydu LE, Visintin L, Carlino MS, Howle JR, Thompson JF, Kefford RF, Scolyer RA, Long GV. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res. 2012;18(12):3242–3249. doi: 10.1158/1078-0432.CCR-12-0052. [DOI] [PubMed] [Google Scholar]
- 6.Rubinstein JC, Sznol M, Pavlick AC, Ariyan S, Cheng E, Bacchiocchi A, Kluger HM, Narayan D, Halaban R. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med. 2010;8:67. doi: 10.1186/1479-5876-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mar VJ, Wong SQ, Li J, Scolyer RA, McLean C, Papenfuss AT, Tothill RW, Kakavand H, Mann GJ, Thompson JF, et al. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histologic and molecular signatures of UV damage. Clin Cancer Res. 2013;19(17):4589–4598. doi: 10.1158/1078-0432.CCR-13-0398. [DOI] [PubMed] [Google Scholar]
- 8.Mar VJ, Liu W, Devitt B, Wong SQ, Dobrovic A, McArthur GA, Wolfe R, Kelly JW. The role of BRAF mutations in primary melanoma growth rate and survival. Br J Dermatol. 2015;173(1):76–82. doi: 10.1111/bjd.13756. [DOI] [PubMed] [Google Scholar]
- 9.Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–1246. doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- 10.McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–332. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Osta H, Falchook G, Tsimberidou A, Hong D, Naing A, Kim K, Wen S, Janku F, Kurzrock R. BRAF mutations in advanced cancers: clinical characteristics and outcomes. PLoS One. 2011;6(10):e25806. doi: 10.1371/journal.pone.0025806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ponti G, Manfredini M, Tomasi A, Pellacani G. Distinctive clinical and dermoscopic features of BRAF V600K mutated melanomas. Br J Dermatol. 2015;172(5):1438–1440. doi: 10.1111/bjd.13484. [DOI] [PubMed] [Google Scholar]
- 13.Jewell R, Chambers P, Harland M, Laye J, Conway C, Mitra A, Elliott F, Cook MG, Boon A, Newton-Bishop J. Clinicopathologic features of V600E and V600K melanoma--letter. Clin Cancer Res. 2012;18(24):6792. doi: 10.1158/1078-0432.CCR-12-2974. author’s reply p 6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 15.Mann HB, Whitney DR. On a Test of Whether One of 2 Random Variables Is Stochastically Larger Than the Other. Annals of Mathematical Statistics. 1947;18(1):50–60. [Google Scholar]
- 16.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 17.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hocker TL, Singh MK, Tsao H. Melanoma genetics and therapeutic approaches in the 21st century: moving from the benchside to the bedside. J Invest Dermatol. 2008;128(11):2575–2595. doi: 10.1038/jid.2008.226. [DOI] [PubMed] [Google Scholar]
- 19.De Gassart A, Martinon F. Translating the anticancer properties of eEF2K. Cell Cycle. 2017;16(4):299–300. doi: 10.1080/15384101.2016.1254974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galli SJ, Tsai M, Wershil BK. The c-kit receptor, stem cell factor, and mast cells. What each is teaching us about the others. Am J Pathol. 1993;142(4):965–974. [PMC free article] [PubMed] [Google Scholar]
- 21.Lennartsson J, Ronnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev. 2012;92(4):1619–1649. doi: 10.1152/physrev.00046.2011. [DOI] [PubMed] [Google Scholar]
- 22.Felicetti F, Errico MC, Bottero L, Segnalini P, Stoppacciaro A, Biffoni M, Felli N, Mattia G, Petrini M, Colombo MP, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008;68(8):2745–2754. doi: 10.1158/0008-5472.CAN-07-2538. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Dong K, Gao P, Long M, Lin F, Weng Y, Ouyang Y, Ren J, Zhang H. microRNA-34a sensitizes lung cancer cell lines to DDP treatment independent of p53 status. Cancer Biother Radiopharm. 2013;28(1):45–50. doi: 10.1089/cbr.2012.1218. [DOI] [PubMed] [Google Scholar]
- 24.Xie R, Lin X, Du T, Xu K, Shen H, Wei F, Hao W, Lin T, Lin X, Qin Y, et al. Targeted Disruption of miR-17-92 Impairs Mouse Spermatogenesis by Activating mTOR Signaling Pathway. Medicine (Baltimore) 2016;95(7):e2713. doi: 10.1097/MD.0000000000002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho WC. MicroRNAs in cancer - from research to therapy. Biochim Biophys Acta. 2010;1805(2):209–217. doi: 10.1016/j.bbcan.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 26.He H, Jazdzewski K, Li W, Liyanarachchi S, Nagy R, Volinia S, Calin GA, Liu CG, Franssila K, Suster S, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2005;102(52):19075–19080. doi: 10.1073/pnas.0509603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koelz M, Lense J, Wrba F, Scheffler M, Dienes HP, Odenthal M. Down-regulation of miR-221 and miR-222 correlates with pronounced Kit expression in gastrointestinal stromal tumors. Int J Oncol. 2011;38(2):503–511. doi: 10.3892/ijo.2010.857. [DOI] [PubMed] [Google Scholar]
- 28.Gits CM, van Kuijk PF, Jonkers MB, Boersma AW, van Ijcken WF, Wozniak A, Sciot R, Rutkowski P, Schoffski P, Taguchi T, et al. MiR-17-92 and miR-221/222 cluster members target KIT and ETV1 in human gastrointestinal stromal tumours. Br J Cancer. 2013;109(6):1625–1635. doi: 10.1038/bjc.2013.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felli N, Fontana L, Pelosi E, Botta R, Bonci D, Facchiano F, Liuzzi F, Lulli V, Morsilli O, Santoro S, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci U S A. 2005;102(50):18081–18086. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spahn M, Kneitz S, Scholz CJ, Stenger N, Rudiger T, Strobel P, Riedmiller H, Kneitz B. Expression of microRNA-221 is progressively reduced in aggressive prostate cancer and metastasis and predicts clinical recurrence. Int J Cancer. 2010;127(2):394–403. doi: 10.1002/ijc.24715. [DOI] [PubMed] [Google Scholar]
- 31.Brioschi M, Fischer J, Cairoli R, Rossetti S, Pezzetti L, Nichelatti M, Turrini M, Corlazzoli F, Scarpati B, Morra E, et al. Down-regulation of microRNAs 222/221 in acute myelogenous leukemia with deranged core-binding factor subunits. Neoplasia. 2010;12(11):866–876. doi: 10.1593/neo.10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Igoucheva O, Alexeev V. MicroRNA-dependent regulation of cKit in cutaneous melanoma. Biochem Biophys Res Commun. 2009;379(3):790–794. doi: 10.1016/j.bbrc.2008.12.152. [DOI] [PubMed] [Google Scholar]
- 33.Poliseno L, Tuccoli A, Mariani L, Evangelista M, Citti L, Woods K, Mercatanti A, Hammond S, Rainaldi G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108(9):3068–3071. doi: 10.1182/blood-2006-01-012369. [DOI] [PubMed] [Google Scholar]
- 34.Terasawa K, Ichimura A, Sato F, Shimizu K, Tsujimoto G. Sustained activation of ERK1/2 by NGF induces microRNA-221 and 222 in PC12 cells. FEBS J. 2009;276(12):3269–3276. doi: 10.1111/j.1742-4658.2009.07041.x. [DOI] [PubMed] [Google Scholar]
- 35.Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM, Dejean A. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(1):264–269. doi: 10.1073/pnas.0907904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueda R, Kohanbash G, Sasaki K, Fujita M, Zhu X, Kastenhuber ER, McDonald HA, Potter DM, Hamilton RL, Lotze MT, et al. Dicer-regulated microRNAs 222 and 339 promote resistance of cancer cells to cytotoxic T-lymphocytes by down-regulation of ICAM-1. Proc Natl Acad Sci U S A. 2009;106(26):10746–10751. doi: 10.1073/pnas.0811817106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Yu J, Jiang L, Wang A, Shi F, Ye H, Zhou X. MicroRNA-222 regulates cell invasion by targeting matrix metalloproteinase 1 (MMP1) and manganese superoxide dismutase 2 (SOD2) in tongue squamous cell carcinoma cell lines. Cancer Genomics Proteomics. 2009;6(3):131–139. [PMC free article] [PubMed] [Google Scholar]
- 38.Fornari F, Gramantieri L, Ferracin M, Veronese A, Sabbioni S, Calin GA, Grazi GL, Giovannini C, Croce CM, Bolondi L, et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27(43):5651–5661. doi: 10.1038/onc.2008.178. [DOI] [PubMed] [Google Scholar]
- 39.Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, Jacob S, Majumder S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem. 2008;283(44):29897–29903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P, et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer cell. 2009;16(6):498–509. doi: 10.1016/j.ccr.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Zhang CZ, Zhang JX, Zhang AL, Shi ZD, Han L, Jia ZF, Yang WD, Wang GX, Jiang T, You YP, et al. MiR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Mol Cancer. 2010;9:229. doi: 10.1186/1476-4598-9-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O’Brien C, Spoerke J, Jhunjhunwala S, Boyd Z, Januario T, et al. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci Signal. 2011;4(177):ra41. doi: 10.1126/scisignal.2001538. [DOI] [PubMed] [Google Scholar]
- 43.Garofalo M, Quintavalle C, Romano G, Croce CM, Condorelli G. miR221/222 in cancer: their role in tumor progression and response to therapy. Curr Mol Med. 2012;12(1):27–33. doi: 10.2174/156652412798376170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, Therrien M. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol. 2015;22(1):37–43. doi: 10.1038/nsmb.2924. [DOI] [PubMed] [Google Scholar]
- 45.Roskoski R., Jr Structure and regulation of Kit protein-tyrosine kinase--the stem cell factor receptor. Biochem Biophys Res Commun. 2005;338(3):1307–1315. doi: 10.1016/j.bbrc.2005.09.150. [DOI] [PubMed] [Google Scholar]
- 46.Carlino MS, Todd JR, Rizos H. Resistance to c-Kit inhibitors in melanoma: insights for future therapies. Oncoscience. 2014;1(6):423–426. doi: 10.18632/oncoscience.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemesath TJ, Price ER, Takemoto C, Badalian T, Fisher DE. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature. 1998;391(6664):298–301. doi: 10.1038/34681. [DOI] [PubMed] [Google Scholar]
- 48.Tsujimura T, Morii E, Nozaki M, Hashimoto K, Moriyama Y, Takebayashi K, Kondo T, Kanakura Y, Kitamura Y. Involvement of transcription factor encoded by the mi locus in the expression of c-kit receptor tyrosine kinase in cultured mast cells of mice. Blood. 1996;88(4):1225–1233. [PubMed] [Google Scholar]
- 49.Galardi S, Mercatelli N, Farace MG, Ciafre SA. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res. 2011;39(9):3892–3902. doi: 10.1093/nar/gkr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Camps C, Saini HK, Mole DR, Choudhry H, Reczko M, Guerra-Assuncao JA, Tian YM, Buffa FM, Harris AL, Hatzigeorgiou AG, et al. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol Cancer. 2014;13:28. doi: 10.1186/1476-4598-13-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.