

Abstract
Background:
Plants have been a major source of inspiration in developing novel drug compounds in the treatment of various diseases that afflict human beings worldwide. Ruta angustifolia L. Pers known locally as Garuda has been conventionally used for various medicinal purposes such as in the treatment of cancer.
Objective:
A dihydrofuranocoumarin named chalepin, which was isolated from the chloroform extract of the plant, was tested on its ability to inhibit molecular pathways of human lung carcinoma (A549) cells.
Materials and Methods:
Cell cycle analysis and caspase 8 activation were conducted using a flow cytometer, and protein expressions in molecular pathways were determined using Western blot technique.
Results:
Cell cycle analysis showed that cell cycle was arrested at the S phase. Further studies using Western blotting technique showed that cell cycle-related proteins such as cyclins, cyclin-dependent kinases (CDKs), and inhibitors of CDKs correspond to a cell cycle arrest at the S phase. Chalepin also showed inhibition in the expression of inhibitors of apoptosis proteins. Nuclear factor-kappa B (NF-κB) pathway, signal transducer and activation of transcription 3 (STAT-3), cyclooxygenase-2, and c-myc were also downregulated upon treatment with chalepin. Chalepin was found to induce extrinsic apoptotic pathway. Death receptors 4 and 5 showed a dramatic upregulation at 24 h. Analysis of activation of caspase 8 with the flow cytometer showed an increase in activity in a dose- and time-dependent manner. Activation of caspase 8 induced cleavage of BH3-interacting domain death agonist, which initiated a mitochondrial-dependent or -independent apoptosis.
Conclusion:
Chalepin causes S phase cell cycle arrest, NF-κB pathway inhibition, and STAT-3 inhibition, induces extrinsic apoptotic pathway, and could be an excellent chemotherapeutic agent.
SUMMARY
This study reports the capacity of an isolated bioactive compound known as chalepin to suppress the nuclear factor kappa-light-chain-enhancer of activated B cells pathway, signal transducer and activation of transcription 3, and extrinsic apoptotic pathway and also its ability to arrest cell cycle in S phase. This compound was from the leaves of Ruta angustifolia L. Pers. It provides new insight on the ability of this plant in suppressing certain cancers, especially the nonsmall cell lung carcinoma according to this study.
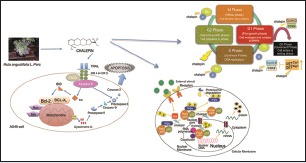
Abbreviations used: °C: Degree Celsius, ANOVA: Analysis of variance, ATCC: American Type Culture Collection, BCL-2: B-Cell CLL/Lymphoma 2, Bcl-xL: B-cell lymphoma extra-large, BH3: Bcl-2 homology 3, BID: BH3-interacting domain death agonist, BIR: Baculovirus inhibitor of apoptosis protein repeat, Caspases: Cysteinyl aspartate-specific proteases, CDK: Cyclin-dependent kinase, CO2: Carbon dioxide, CST: Cell signaling technologies, DISC: Death-inducing signaling complex, DMSO: Dimethyl sulfoxide, DNA: Deoxyribonucleic acid, DR4: Death receptor 4, DR5: Death receptor 5, E1a: Adenovirus early region 1A, ECL: Enhanced chemiluminescence, EDTA: Ethylenediaminetetraacetic acid, ELISA: Enzyme-linked immunosorbent assay, etc.: Etcetera, FADD: Fas-associated protein with death domain, FBS: Fetal bovine serum, FITC: Fluorescein isothiocyanate, G1: Gap 1, G2: Gap 2, HPLC: High-performance liquid chromatography, HRP: Horseradish peroxidase, IAPs: Inhibitor of apoptosis proteins, IC50: Inhibitory concentration at half maximal inhibitory, IKK-α: Inhibitor of nuclear factor kappa-B kinase subunit alpha, IKK-β: Inhibitor of nuclear factor kappa-B kinase subunit beta, IKK-γ: Inhibitor of nuclear factor kappa-B kinase subunit gamma, IKK: IκB kinase, IkBα: Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha, m: Meter, M: Mitotic, mm: Millimeter, mRNA: Messenger ribonucleic acid, NaCl: Sodium chloride, NaVO4: Sodium orthovanadate, NEMO: NF-Kappa-B essential modulator, NF-κB: Nuclear factor kappa-light chain-enhancer of activated B cells, NSCLC: Nonsmall cell lung carcinoma, PBS: Phosphate buffered saline, PGE2: Prostaglandin E2, PI: Propidium iodide, PMSF: Phenylmethylsulfonyl fluoride, pRB: Phosphorylated retinoblastoma, R. angustifolia: Ruta angustifolia L. Pers, Rb: Retinoblastoma, rpm: Rotation per minute, RPMI: Roswell Park Memorial Institute, S phase: Synthesis phase, SD: Standard deviation, SDS-PAGE: Sodium dodecyl sulfate-polyacrylamide gel electrophoresis, Smac: Second mitochondria-derived activator of caspase, SPSS: Statistical Package for the Social Sciences, STAT3: Signal transducer and activation of transcription 3, tBID: Truncated BID, TNF: Tumor necrosis factor, TRADD: Tumor necrosis factor receptor type-1 associated death domain, TRAIL: TNF-related apoptosis- inducing ligand, USA: United States of America, v/v: Volume over volume.
Keywords: Apoptosis, cell cycle, chalepin, nuclear factor kappa-light-chain-enhancer of activated B cells, Ruta angustifolia L. Pers, signal transducer and activation of transcription 3
INTRODUCTION
Lung cancer is a primary malignant tumor killer worldwide. Although the main cause of lung cancer was found to be from carcinogens of tobacco smoke and environment, the mechanism of lung carcinogenesis remains unclear.[1] The major cancer killer worldwide in both sexes is nonsmall cell lung cancer (NSCLC), accounting for >1.2 million deaths each year. The standard therapies that are available currently rarely cure the disease, and the overall 5-year survival rate is only 15% because NSCLC is usually a systemic disease at the time of appearance.[2] In Malaysia, lung cancer is the third most common cancer with 2100 Malaysians diagnosed each year (National Cancer Society Malaysia, 2015).
Plants have been a main source of medicinal compound around the world. Over 60% of cancer therapeutics that are available in the market or at preclinical trial stage are from natural products. Taxol from the Pacific yew tree, vinblastine and vincristine from the Madagascar periwinkle, aspirin from the willow tree, digitalis from foxglove, and artemisinin from wormwood are some of the prominent examples. Natural products from terrestrial and aquatic sources continue to serve as the foundations, from which synthetic compounds could be derived.[3]
Ruta angustifolia L. Pers belonging to the largest families of plant kingdom Rutaceae is a variant of the common rue and it shares the same scientific name with the common rue, Ruta angustifolia var. angustifolia. These plants are commonly used as traditional medicine. R. angustifolia is frequently known as Sadal or Garuda in Malaysia, Godong minggu or Inggu in Indonesia, and Luru in Vietnam. However, it is originated from the Mediterranean region. R. angustifolia has been conventionally used for various medicinal and culinary purposes. It was initially introduced in India and later to Southeast Asia. It is commonly cultivated as potted plants in Malaysia. It is also common in the Indian community in Malaysia to tie it on newborn's hands to avoid insect bites due to its strong smell. It can grow up to 1.5 m in height and has woody stems. It normally grows in mountainous area with a height of 1000 m above sea level. The leaves are compound and light green in color with a width of 2–6 mm and a length of 8–20 mm. The fruits are small and oval in shape, while the seeds are small, black-colored and are in a kidney shape. Its essential oil is commonly used for the manufacturing of perfume and cosmetics.
Defects in apoptosis are an important factor in progression of cancer apart from role that is played by the proto-oncogenes in activating cancer cell proliferation. Many deregulated oncoproteins that drive cell division also trigger apoptosis (e.g. Myc, E1a, and Cyclin-D1). Mutations generally happen in proto-oncogenes, which would promote tumor cell growth when mutated, and at tumor suppressor genes, which could hinder the inhibition of cell cycle progression and thus encouraging abnormal cell growth.[4] Disruption in cell cycle control mechanism can be resulted from environmental insults, which causes DNA damage. Cell cycle checkpoints control cell growth and development and regulate the intricate network of interaction. Mutations occurring at these checkpoints were found to be present in all types of cancer.[4] Nuclear factor-kappa B (NF-κB) is a common transcription factor. Inflammatory stimuli that are normally environmental pollutants, pro-oxidants, carcinogens, stress, and growth factors usually activate the response of translocation of this transcription factor from cytoplasm to the nucleus. In the nucleus, it would bind to the DNA and initiates the gene transcription. This activation was found to be resulting in the production of various gene products that regulates important mechanisms such as apoptosis, angiogenesis, metastasis, invasion, and inflammation.[5]
R. angustifolia contains a number of bioactive compounds, which has high potential for use in the treatment of various diseases. Some of the constituents of R. angustifolia include arborinine, graveoline, psoralen, bergapten, chalepensin, chalepin, kokusaginine, moskachan (A, B, C, and D), rutamarin, and neophytadiene. Among these compounds, arborinine was reported to exhibit anticancer activity by showing cytotoxicity against A549 lung carcinoma, HeLa cervical carcinoma, KB nasopharyngeal cancer cells, MCF7 breast cancer cells, and A431 skin cancer cells.[6,7,8] Arborinine inhibited the proliferation and induced apoptosis on splenocytes and thymocytes when stimulated with different mitogens.[9] Bergapten, also known as 5-methoxypsoralen, is a compound which is prevalent in R. angustifolia was commonly used to treat various skin ailments such as psoriasis, vitiligo, and multiple sclerosis due to its capacity to intercalate and photo-crosslink DNA.[10,11] The ability of bergapten to halt the proliferation of bladder and melanoma cancer cells and interrupting DNA synthesis in Ehrlich ascites tumor cells was also discovered.[11,12] Graveoline exhibited antibacterial, antitumor, and spasmolytic properties. It was also found to induce apoptosis and autophagy.[13,14,15] Kokusaginine which is one of the compounds present in R. angustifolia was reported to possess antimalarial and antifungal activities.[14,16] It was also found to exhibit inhibitory activity against Schistosoma mansoni miracidia which may lead to schistosomiasis, which is a tropical disease in Africa.[17] Recently, there was a report which showed kokusaginine's potential as a good inhibitor against hepatitis C virus.[18] It was also reported to possess cytotoxicity against A2780 ovarian carcinoma cells, HeLa cervical cancer cells, and KB nasopharyngeal cancer cells.[7,19] Moskachan D, isolated from R. angustifolia, was reported to have good inhibition against human lung carcinoma cells.[20] Graveoline which is the most abundantly available compound in R. angustifolia[20] exhibited antibacterial, antitumor, antifungal, and spasmolytic properties.[13,14] Graveoline also induced cell death through apoptosis and autophagy in A375 skin melanoma cells.[15] Drugs which have apoptotic and autophagic properties are effective for the treatment of cancer.[15] Neophytadiene, which was also a compound found and isolated from R. angustifolia,[20] was reported to have antipyretic, analgesic, anti-inflammatory, antimicrobial, and antioxidant activity.[21] Rutamarin, a compound reported in R. angustifolia, was discovered to possess cytotoxicity against lung, colon, breast, and liver carcinomas.[22] This shows that rutamarin has potential as a chemotherapeutic agent. Another study has also shown the potential of rutamarin in glucose homeostasis and improving insulin sensitivity in mice. Rutamarin was found to be having potential to treat Type 2 diabetes.[23] Methoxsalen was also used in the treatment of vitiligo as it could assist in repigmentation of the skin with the help of sunlight.[24] Chalepensin showed cytotoxicity against colon (HT29), lung (A549), breast (MCF7), kidney (A498), and pancreatic (PACA-2) cancer cell lines.[20,25] This shows that it could be a potential chemotherapeutic agent. Another recent report showed that chalepensin has antimicrobial effect against Streptococcus mutans, a microbe which causes dental caries.[26] Chalepin [Figure 1], a dihydrofuranocoumarin, is one of the bioactive compounds isolated from R. angustifolia. Previous studies have revealed that chalepin possesses anticoagulative potential and is able to induce necrotic cell death on hepatic cells;[27] besides that, chalepin was found to inhibit mitochondrial respiration in rat liver mitochondria.[28] A study in 2014 revealed that chalepin isolated from the leaves of R. angustifolia has the potential to inhibit hepatitis C virus replication.[18] It was also reported that chalepin stimulated insulin release in rat insulinoma cell line and mild antimicrobial activity.[29] Chalepin was also found to exhibit good cytotoxicity against Huh7.5 hepatoma cell line.[18] Another recent study revealed that chalepin possesses excellent cytotoxicity with an IC50 value of 8.69 ± 2.43 μg/ml (27.64 μM) and induced apoptosis toward human lung carcinoma (A549) cells.[20] This has initiated further investigation to gain further insight on the underlying mechanisms responsible for the induction of apoptotic cell death in cancer cells.
Figure 1.
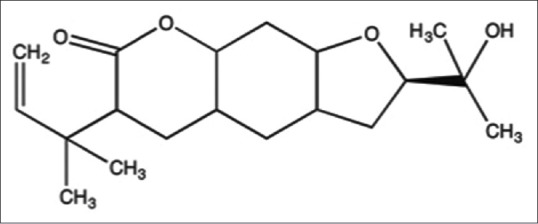
Chemical structure of chalepin
MATERIALS AND METHODS
Cell culture, compounds, and reagents
Human lung carcinoma (A549) was purchased from American Type Culture Collection (ATCC, USA). It was cultured in Roswell Park Memorial Institute (RPMI) media supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin/streptomycin (100 μg/ml), and 1% amphotericin B (100 μg/ml). The cells were maintained in a humidified incubator (5% CO2 in air at temperature of 37°C). Chalepin was isolated from the leaves of R. angustifolia using a high-performance liquid chromatography as previously described.[20] It was dissolved in dimethyl sulfoxide (DMSO) for further analyses.
Antibodies and chemicals
Antibodies used in this research were primary antibodies monoclonal cyclin D1, monoclonal cyclin E1, monoclonal cyclin-dependent kinases (CDK2), monoclonal CDK4, monoclonal p21, polyclonal p21, polyclonal CIAP1, monoclonal CIAP2, monoclonal Mcl-1, monoclonal cyclooxygenase-2 (COX-2), monoclonal c-myc, monoclonal p65, monoclonal phosphorylated p65, polyclonal IKβα, polyclonal phosphorylated Ikβα, polyclonal inhibitor of nuclear factor kappa-B kinase subunit alpha (IKK-α), polyclonal IKK-β, polyclonal signal transducer and activation of transcription 3 (STAT-3), polyclonal phosphorylated STAT-3 (pSTAT-3), monoclonal death receptor 4 (DR4), polyclonal DR5, polyclonal Bid, monoclonal retinoblastoma (Rb), and polyclonal phosphorylated Rb (pRb). which were all purchased from cell signaling technologies. Bradford reagent used for protein estimation was purchased from Bio-Rad.
Cell culture
The human cell line A549 (lung cancer) was cultured as monolayer in RPMI 1640 growth media. The cell was purchased from the American Tissue Culture Collection (ATCC, USA). All the media were supplemented with 10% v/v FBS, 100 μg/mL penicillin/streptomycin, and 50 μg/mL amphotericin B. The cells were cultured in a 5% CO2 incubator at 37°C.
Cell cycle analysis
After 48 and 72 h of treatments with chalepin, adherent and floating cells were collected, centrifuged, and fixed in 70% ethanol overnight. All samples were centrifuged to remove ethanol, and the cell pellets were washed with cold phosphate-buffered saline (PBS). The cells were subsequently resuspended in 300 μL of staining solution containing concentrations of 50 μg/mL propidium iodide, 50 μg/mL RNase, 0.1% Triton X-100, 1 mg/mL sodium citrate, and distilled water. Following 30 min incubation at room temperature, the cells were analyzed with a flow cytometer and data analysis was performed by Modfit LT software (Verity Software House, US).
Caspase-8 activity
Caspases play an important role in cell death. Caspase activity assays were performed according to the instructions in the manual of the Caspases-8 Staining Kit (CaspILLUME). Cells were seeded at a density of 1 × 106 cells per culture dish (60 mm). After being subjected to treatment of chalepin at various concentrations (18 μg/ml [57.3 mM], 27 μg/ml [86.0 mM], 36 μg/ml [114.6 mM], and 45 μg/ml [143.3 mM]) for 48 and 72 h, the cells were detached using accutase, washed, and resuspended in PBS. Next, 300 mL of each sample was aliquoted into centrifuge tubes after which 1 mL of fluorescent substrate (FITC-IETD-FMK) was added to each tube and incubated for 1 h at 37°C incubator and 5% CO2. At the end of the incubation period, the cells were centrifuged at 3000 rpm for 5 min and the supernatant was removed. After that, the cells were resuspended in 0.5 mL wash buffer and centrifuged at 3000 rpm for 5 min. This step was repeated before performing the analysis with flow cytometer (Accuri C6) and a minimum of 10,000 events were acquired for each replicate. Untreated cells in 0.5% DMSO served as the control.
Western blot analysis
For this analysis, 1 × 106 of A549 cells were plated per tissue culture dish. Chalepin-treated whole-cell extracts were lysed in lysis buffer (20 mM Tris (pH 7.4), 250 mM natrium chloride, 2 mM ethylenediaminetetraacetic acid (pH 8.0), 0.1% Triton X-100, 0.01 μg/ml aprotinin, 0.005 μg/ml leupeptin, 0.4 mM phenylmethylsulfonyl fluoride, and 4 mM sodium orthovanadate). Lysates were then spun at 15,000 rpm for 10 min to remove insoluble material and resolved on a 10.0% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel. After electrophoresis, the proteins were electrotransferred to a nitrocellulose membrane, blocked with Blocking One (Nacalai Tesque, Inc.), and probed with various antibodies (1:1000) overnight at 4°C. The blot was washed, exposed to horseradish peroxidase-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence (enhanced chemiluminescence, Advansta).
Preparation of nuclear and cytoplasm extract
Nuclear extract and cytoplasmic fractions from A549 cells were prepared to determine the expression of proteins involved in the NF-κB pathway using kit from Active Motif. For preparation of nuclear extracts, the cells were first collected in ice-cold PBS. The cell suspension is centrifuged at 2000 × g at 4°C and pellet were resuspended in hypotonic buffer for 15 min on ice. A volume of 25 μl of detergent was added for every 100 μl of hypotonic buffer, and it was vortexed for 10 s at the highest speed. The lysate was then centrifuged at 14,000 ×g for 30 s in a microcentrifuge tube which has been precooled. The supernatant which was the cytoplasmic fraction was transferred into a prechilled microcentrifuge tube and stored in −80°C until further use. After collection of the cytoplasmic fraction, the pellet was used for the nuclear fraction collection. The nuclei were lysed using the complete lysis buffer provided in the kit, and the nuclear proteins were solubilized in detergent-free lysis buffer in the presence of the protease inhibitor cocktail. The suspension was incubated for 30 min on ice on a rocking platform set at 150 rpm. It was then vortexed for 30 s at the highest setting and next, centrifuged for 10 min at 14,000 ×g in a microcentrifuge tube which has been precooled at 4°C. The supernatant which was the nuclear fraction was transferred into another prechilled microcentrifuge tube and the aliquot was stored at −80°C until further use.
Statistical analysis
All the values reported are shown as mean ± standard error of the mean, and all the experiments were conducted at least twice using sample triplicates. Figures from morphological studies, flow cytometry plots, and Western blot analyses are representative of the experiment replicates. Comparison between control and treated groups was performed using one-way analysis of variance with post hoc Tukey test (*P < 0.05 considered statistically significant). Statistical analyses were performed using SPSS 20.0 software (IBM, USA) and calculations were done using the Microsoft Excel. Quantification of the bands in the Western blot analysis was done using the ImageJ software (developed by Wayne Rasband).
RESULTS
Cell cycle analysis using the flow cytometry revealed that chalepin inhibited cell cycle at the S phase. Cell cycle analysis segregates cells based on its DNA content. The DNA was stained/labeled and measured to determine the phase in which the cell is located. There was a significant accumulation of cells in the S phase after treatment of chalepin for 48 and 72 h [Figure 2]. The accumulation was 27.73% at 48 h and 25.38% at 72 h after treatment with 45 μg/ml of chalepin. For the control (untreated) cells, the number of cells at the S-phase was only about 4.02%. Accumulation of cells at a certain phase indicates that the cell cycle is arrested at that particular point. As concentration of chalepin increases, the number of cells in S phase increased. From the cell cycle analysis, it is apparent that chalepin arrests A549 cells at the S phase which is the synthesis phase where DNA replication occurs.
Figure 2.

Cell cycle analysis of A549 cells treated with chalepin at 48 h and 72 h as compared to untreated control. The data expressed as mean ± standard deviation of three replicates. Asterisks indicate significantly different value from control (*P < 0.05)
This preliminary result was further studied by determining the expression of the cell cycle-related proteins upon the treatment of chalepin [Figure 3].
Figure 3.
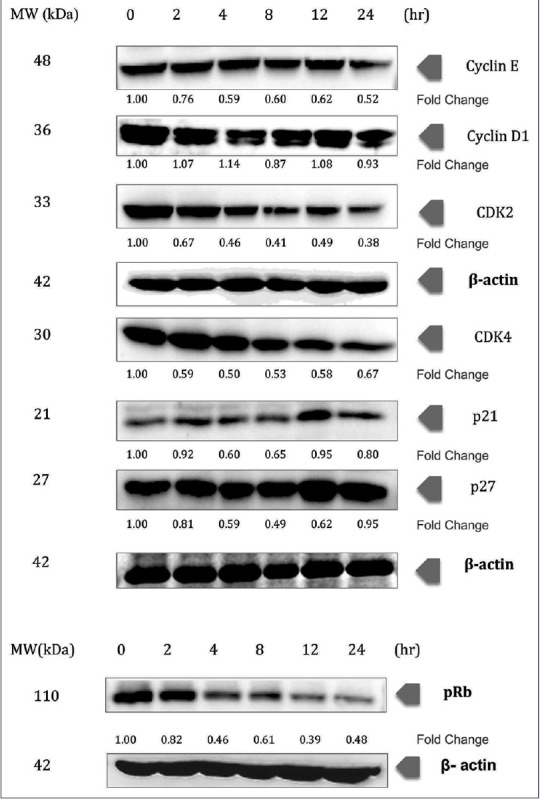
Chalepin induces changes expression in cell cycle-related proteins in a dose-dependent manner. A549 cells (2 × 106 mL−1) were treated with 36 μg/ml of chalepin at various time points, after which the whole-cell extracts were prepared and 30 μg of protein was resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, electrotransferred onto nitrocellulose membranes, and probed for the cyclin E, cyclin D1, CDK2, CDK4, p21, p27, and phosphorylated retinoblastoma proteins. The experiments were conducted in triplicates
The effect of chalepin on antiapoptotic proteins such as cIAP-1, cIAP-2, and MCL-1 was evaluated. Western blot results showed that the antiapoptotic gene products were downregulated with a decrease of up to 0.79-, 0.56-, and 0.59-fold for the proteins cIAP-1, cIAP-2, and MCL-1, respectively, at 24 h incubation of chalepin. Inhibitor of apoptosis proteins (IAPs) is commonly involved in the regulation of caspases and immune signaling [Figure 4].
Figure 4.
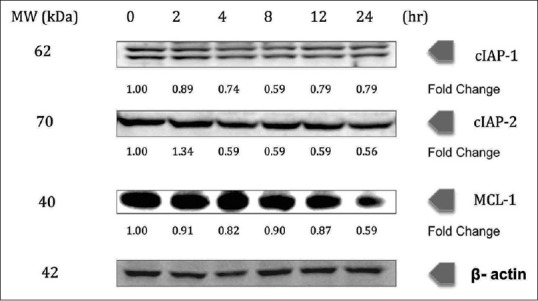
Chalepin inhibits the expression of antiapoptotic gene products, cIAP-1, cIAP-2, and MCl-1 in a dose-dependent manner. A549 cells (2 × 106 mL−1) were treated with 36 μg/ml of chalepin at various time points, after which the whole-cell extracts were prepared and 30 μg of protein was resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, electrotransferred onto nitrocellulose membranes, and probed for cIAP-1, c-IAP-2, and MCl-1. The experiments were conducted in triplicates
COX-2 was downregulated upon treatment of chalepin toward A549 cells at 24 h treatment incubation time, with a fold change of 0.56 at 24 h incubation [Figure 5]. Treatment of chalepin resulted in downregulation of c-myc at earlier time points such as 2 and 4 h with a fold change up to 0.55 at 4 h, some upregulation at 8 and 12 h, and further downregulation at 24 h incubation time (0.72-fold) [Figure 5].
Figure 5.
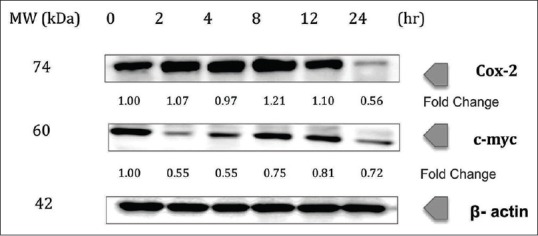
Chalepin inhibits the expression of cyclooxygenase-2 and c-myc in a dose-dependent manner A549 cells (2 × 106 mL−1) were treated with 36 μg/ml of chalepin at various time points, after which the whole-cell extracts were prepared and 30 μg of protein was resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, electrotransferred onto nitrocellulose membranes, and probed for cyclooxygenase-2 and c-myc. The experiments were conducted in triplicates
Our results also showed that the Iκβα phosphorylation was downregulated upon treatment of chalepin (0.62-fold at 6 h) [Figure 6]. Degradation of Iκβα however showed an inconsistent result, with upregulation (1.49-fold at 1 h) and slight downregulation (0.92-fold at 6 h) in the protein expression. The inhibition was also through modulation of the p65 subunit of NF-κB where the phosphorylation of p65 and the translocation of the p65 subunit to nucleus were inhibited. Results obtained indicated an upregulation and then a downregulation (0.57-fold) at the 6 h incubation time in the p65 subunit in the nuclear fraction. This shows that there is an inhibition in the translocation of p65 into the nucleus. Phosphorylation of p65 in the nucleus was inhibited as there was a downregulation in the expression of the phosphorylated p65 upon treatment of chalepin (0.58-fold at 6 h). Overall, the results indicate that the NF-κB pathway is suppressed, especially at earlier incubation time.
Figure 6.

Chalepin suppresses nuclear factor-kappa B pathway in a time-dependent manner. A549 cells (2 × 106 mL−1) were treated with 36 μg/ml of chalepin at various time points (0, 1, 2, 4 and 6 h), after which the cell nuclear extracts were prepared and 30 μg of protein was resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, electrotransferred onto nitrocellulose membranes, and probed for phosphorylated p65, p65, phosphorylated IKβα, and IKβα. The experiments were conducted in triplicates
In our study, treatment of chalepin suppressed the pSTAT3, thus indicating an inactivation of the transcription factor [Figure 7]. The pSTAT3 was downregulated to 0.59-fold at 6 h treatment of chalepin. Chalepin therefore could inhibit the progression of A549 cells. The expression of STAT3 protein remains almost constant.
Figure 7.

Chalepin inhibits constitutive signal transducer and activation of transcription 3 phosphorylation in A549 cell line in a time-dependent manner. A549 cells (2 × 106 mL−1) were treated with 36 μg/ml of chalepin at various time points, after which the whole-cell extracts were prepared and 30 μg of protein was resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, electrotransferred onto nitrocellulose membranes, and probed for phosphorylated signal transducer and activation of transcription 3 and signal transducer and activation of transcription 3. The experiments were conducted in triplicates
Caspase 8 is associated with the activation of the extrinsic apoptotic pathway. Cells with activated caspase 8 would move to the right side (V1-R) of the density plot [Figure 8]. The results show that there is a remarkable increase in the caspase 8 activity of cells treated with chalepin as compared to the control. It was observed that after 48 h of incubation, there is a 5-fold increase in the activation of caspase 9 from 4.90% ± 0.35% in untreated control to 26.10% ± 0.21% in cells treated with 45 μg/ml of chalepin. As for the 72 h incubation period, there was an increase from 6.00% ± 0.15% for untreated control cells to 51.7% ± 0.35% for treated cells with 45 μg/ml of chalepin which corresponds to an 8.61-fold increase. These results show that chalepin-treated cells undergo activation of caspase 8.
Figure 8.

Caspase 8 activity of A549 cells treated with chalepin and incubated for 48 h and 72 h. (a) Flow cytometric analysis of apoptotic (V1-R) and nonapoptotic populations (V1-L) for active caspase 9 activity at 48 h incubation (b) Flow cytometric analysis of apoptotic (V1-R) and nonapoptotic populations (V1-L) for active caspase 8 activities at 72 h incubation (c) Fold increase of A549 cells treated with chalepin at different incubation times. The data expressed as mean ± standard deviation of three replicates. Asterisks indicate significantly different value from control (*P < 0.05)
DR4 and DR5 showed an upregulation at 24 h incubation time, with fold changes of 2.17 and 2.84, respectively, upon treatment with chalepin in the Western blot study. Interestingly, there was an initial downregulation in the expression and then a significant upregulation at the 24 h incubation time [Figure 9], whereas BH3-interacting domain death agonist (BID) showed downregulation in expression and showed the most downregulation at 12 h with 0.47-fold change [Figure 10].
Figure 9.
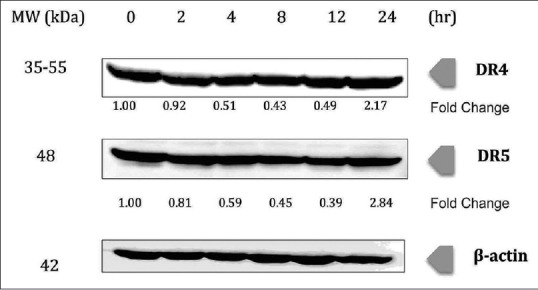
Chalepin increases expression level of death receptors in A549 cell line in a time-dependent manner. A549 cells (2 × 106 mL−1) were treated with 36 μg/ml of chalepin at various time points, after which the whole-cell extracts were prepared and 30 μg of protein was resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, electrotransferred onto nitrocellulose membranes, and probed for DR4 and DR5. The experiments were conducted in triplicates
Figure 10.
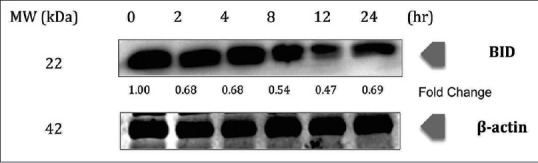
Chalepin promotes cleavage of BH3-interacting domain death agonist in A549 cell line in a time-dependent manner. A549 cells (2 × 106 mL−1) were treated with 36 μg/ml of chalepin at various time points, after which the whole-cell extracts were prepared and 30 μg of protein was resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, electrotransferred onto nitrocellulose membranes, and probed for BH3-interacting domain death agonist protein. The experiments were conducted in triplicates
DISCUSSION
There is a strong correlation between cell cycle and cancer. Cell cycle is machinery that controls cell proliferation and cancer is a disease, which is caused by inappropriate cell proliferation. Several fiascos in the cell cycle's normal signal delivery, which tells the cell to adhere, differentiate, or die that causes the cancer cells to proliferate uncontrollably.[30] The cell cycle comprises a series of complex molecular and biochemical signaling pathways. It has four phases, which include G1 phase, S phase, G2 phase, and M phase. There are several checkpoints that are important control points that regulate the cell cycle to proceed further or to halt and undergo apoptosis.
Cells use a complex set of enzymes called kinases to control various steps in the cell cycle. CDKs are a specific family of enzymes that use signals to switch on cell cycle mechanisms. These kinases require association with a second subunit, which is the regulatory protein, known as cyclin. It is transiently expressed at the appropriate period of the cell cycle to be activated. Cyclin subunit complexes with its partner CDK to create an active complex.[30] When functioning properly, the cell cycle regulatory proteins (cyclins) act as tumor suppressors by controlling cell growth and inducing the death of damaged cells. Genetic mutations which can lead to the absence of one or more of the regulatory proteins at the cell cycle checkpoints can result in the “molecular switch” being turned permanently on, permitting uncontrolled multiplication of the cell, leading to carcinogenesis or tumor development. Throughout the G1 phase, Rb is underphosphorylated. It is phosphorylated just before S phase and remains phosphorylated until late mitosis.[31] Cyclin E complexes with CDK2 to regulate the G1/S phase transition, whereas cyclin D complexes with CDK4 to regulate the G1 phase.[32] These CDK-cyclin complexes are negatively regulated by endogenous CDK inhibitors such as p21Cip and p27Kip or degradation of cyclin. CDK4/cyclin D complex, which initiates the progression of cell cycle through the G1 phase,[33] is inhibited by p21cip1, while CDK2/cyclin E complex, which initiates the progression of cell cycle to the S-phase,[33] is inhibited by p27Kip1. This inhibition would result in hypo-pRb proteins that would result in inhibition of E2F transcription factor into nucleus that would subsequently activate the transcription of cell cycle related genes. p21Cip1 arrests cells in both G1 and G2 cell cycle phases; p27Kip1 blocks the G1/S phase transition.[34]
The regulatory subunit, i.e., cyclin D and E, showed a downregulation, 0.52- and 0.93-fold, respectively, at 24 h, when treated with chalepin at concentration 36 μg/μl (114.6 μM). CDK2 and CDK4, which complex with cyclin E and D, respectively, showed a dramatic decrease in expression. It showed a decrease of up to 0.38-fold (24 h) and 0.50-fold (4 h), respectively. This shows that the complex formation that is required for progression of cell cycle through G1 and S phase is inhibited. This could also be the reason why there was some accumulation of cells observed in the G1 phase in the flow cytometry cell cycle analysis. The inhibitors of CDKs (p21 and p27) showed slight downregulation; however, it was upregulated (to 0.80 and 0.95, respectively) at higher incubation time. pRb protein showed hypophosphorylation. The expression of pRb showed 0.48-fold at 24 h. This is an indication that E2F gene is not activated and transcription of genes required for cell cycle progress is halted as pRb protein is essential for this downstream pathway. This result correlates with the S-phase arrest.
Western blot was conducted at a time point of 2, 4, 8, 12, and 24 h for whole-cell extract-related analysis of proteins. A time point up to 24 h was chosen due to the reason that cell death (apoptosis) is initiated approximately at 24 h. The study of the expression of proteins when cell death is occurring would result in inaccurate results. The time point where cell death initiates was studied using the trypan blue dye exclusion assay. The results show that at 24 h, about 91.17% ± 1.53% of viable cells were observed. The percentage of viable cells dropped sharply to 39.67% ± 1.89% at 48 h and to 12.0% ± 1.57% at 72 h. Hence, the time point of up to 24 h has been selected for the study of the protein expression.
IAPs are highly conserved in viruses and mammals and are also known as Baculovirus IAP Repeat (BIR) domain-containing proteins that are a class of protein characterized by the presence of BIR domain. IAPs are Zn2+ ion coordinating protein–protein interaction domain. IAPs are overexpressed in a number of tumors and are known to regulate carcinogenesis at various stages. c-IAP1 and c-IAP2 have been suggested as proto-oncogenes by various genetic studies.[35] c-IAP1 and c-IAP2 bind and inhibit caspase 3 and 7.[36] Rudimental cancer cells fail to respond to treatment because the malignant cells fail to respond to chemotherapy, radiation, or the immune surveillance by endogenous cytotoxic T-cells and natural killer cells. The failure of cell death results from failure of the cells to undergo apoptosis and initiate caspase activation. IAPs, which are the antiapoptotic proteins, play a role in blocking cell death, by inhibiting the downstream caspase activation pathways.[36] When c-IAP1 and c-IAP2 are absent in the death receptor complex, the binding of death ligands to their receptors leads to the formation of the death-inducing signaling complex, in which adaptor proteins (Fas-associated death domain [FADD] and/or tumor necrosis factor [TNF] receptor type-1 associated death domain) bind with their death domain and induce the recruitment and activation of the initiator caspases, caspase-8 or -10.[37] A new therapeutic approach is to develop small molecule drugs that mimic second mitochondria-derived activator of caspase (Smac), a pro-apoptotic mitochondrial protein that inhibits IAPs. Smac mimetics also known as the IAP antagonists suppresses IAPs to induce cancer cell death. The unique action of Smac mimetics could enhance therapeutic activity of many existing cancer therapies.[37]
In our research, it is evident that treatment of chalepin on A549 cells inhibits the expression of c-IAP1 and c-IAP2, 0.79- and 0.56-fold, respectively [Figure 4]. Modulation of IAPs results in chalepin being similar to a Smac mimetic. The mode of action for commonly available drugs for the treatment of NSCLC such as doxorubicin, erlotinib, gemcitabine, paclitaxel, and vinorelbine is through its ability to act as a Smac mimetic.[38] Ability of chalepin to also act as a Smac mimetic is an interesting observation, which may promise a solution in the treatment of lung cancer.
The antiapoptotic protein MCL-1 functions as a key regulator of cancer cell survival and a known resistance factor for small molecule B-cell CLL/lymphoma (BCL-2) family inhibitors making it an attractive therapeutic target.[39] However, to directly inhibit MCL-1 requires the disruption of high-affinity protein–protein interactions, and therefore, designing small molecules potent enough to inhibit MCL-1 in cells has proven extremely challenging.[39] MCL-1 has been proven to mediate multiple types of tumor. MCL-1 gene locus was amplified in a variety of tumor types including NSCLC.[39] A549 cells treated with chalepin showed downregulation in the expression of MCL-1 protein with the protein suppression of up to 0.59-fold [Figure 4]. This result indicates that chalepin might be able to act as MCL-1-specific BH3 mimetics. Members of the BCL-2 family such as BCL-2, BCL-XL, and MCL-1 are antiapoptotic proteins and are overexpressed in many cancers. These proteins act to bind and sequester proapoptotic proteins such as Bax and Bak. Targeting the BCL-2 family proteins promises a good chemotherapeutic passage. Small molecules that have the capacity to mimic the BH3 domain of BH3-only BCL-2 family members could serve as inhibitor. BH3 mimetics are generally short peptides or organic molecules. The latter are indeed antagonists of the prosurvival members.[40] MCL-1 inhibition was found to cause tumor regression and cell death in various experiments. MCL-1 inhibition could be executed through various approaches such as indirect CDK inhibitors, small molecules, and BH3 peptides, which could directly bind to MCL-1 to antagonize its activity. However, BH3 mimetics are considered to be the best type of MCL-1 inhibitor. High binding affinity is difficult to achieve, which may be due to the nature of the protein/protein interaction and the conformational rigidity of the hydrophobic binding groove of MCL-1.[40] To determine the mode of action of chalepin in inhibition of MCL-1, further studies need to be conducted.
COX-2 is formed by a cell in response to the presence of its substrate and not usually expressed in normal tissue. COX-2 is overexpressed in neoplasms and premalignant lesions. Inhibitors of COX-2 have been investigated as chemotherapeutic agents for chemoprevention. Preclinical data suggest that inhibitors of COX-2 may have the tendency to protect against colon, breast, lung, esophageal, and oral tumors.[41] COX-2 overexpression is often associated with an increased production of prostaglandin E2 (PGE2) which is a major by-product of COX-2 that is involved in cell death, cell proliferation, and tumor invasion in many types of cancer. PGE2 affects through membrane receptors such as EP1, EP2, EP3, and EP4, which activates different types of signaling pathways. NSCLC is associated with Ras mutation, which leads to upregulation of COX-2 that result in increased PGE2 production. PGE2 increases cell proliferation in A549 cell line and this activation is associated with activation of Ras pathway through EP4 receptor. PGE2 acts by affecting the release of amphiregulin, which is the most abundant ligand in A549 cells. Several signaling pathways are associated with tumor progression, which is linked to PGE2, and this is why inhibition of COX-2 is a good strategy in fighting cancer.[42] In our study, COX-2 was downregulated upon treatment of chalepin toward A549 cells at 24 h treatment time (0.56-fold) [Figure 5]. However, the study on the expression of the protein after 24 h was not undertaken as the cells starts to undergo cell death after 24 h, according to the trypan blue dye exclusion assay results. This indicates that chalepin could inhibit the production of PGE2 and activation of Ras pathway thus inhibiting cell proliferation.
c-Myc is an oncogene which regulates cellular growth and metabolism. In cancer cells, metabolic changes need to take place to support the increased demand of nucleic acids, lipids, and proteins that are necessary for rapid cellular proliferation. In these situations, c-myc is often overexpressed.[43] The c-myc gene was amplified in many human cancers, including breast cancer, lung cancer, and colon cancer.[44] According to a study, c-myc was found to induce metastasis in NSCLC.[45] In our study, treatment of chalepin resulted in downregulation of c-myc at earlier time points such as 2 and 4 h (0.55-fold change). There was an increase in the expression at 8 and 12 h and a decrease in the expression at 24 h (0.72 fold) [Figure 5]. The study of the expression later than 24 h was not done due to the reason where the cell death commences after the 24 h time point. Since there was a inconsistency in the expression of c-myc, this particular gene product, which is responsible for cellular growth and metabolism, could be inhibited by chalepin. As c-myc was found to promote metastasis in A549 cells,[45] chalepin could be an agent that inhibits metastasis.
NF-κB is an omnipresent transcription factor. It consists of a few subunits such as p50, p65, and Iκβα, which reside in the cytoplasm. When there are external stimuli such as inflammation, environmental pollutions, prooxidants, carcinogens, stress, and growth factors, these subunits are activated. External stimuli generally activate the IKK complex. There are a few kinases, which assist in this activation, which is known as IKK. The regulatory step in this pathway requires activation of IKK complex that has high molecular weight. The catalysis of IKK is generally carried out by three tightly associated IKK subunits. IKK-α and IKK-β serve as the catalytic subunits of the kinase. IKK-γ or commonly known as NEMO serves as the regulatory subunit.[46] The activation of IKK initiates upon phosphorylation at Ser177 and Ser181 in the activation loop of IKK-β (Ser176 and Ser180 in IKK-α), which causes conformational changes, resulting in kinase activation. The increase in the activity of kinase will result in the phosphorylation of the NF-κB inhibitor, which is Iκβα that would result in dissociation of the inhibitor from NF-κB and ubiquitination of the Iκβα. The ubiquitination would then recruit proteasome, which would degrade the Iκβα, an inhibitor.[47] Serine phosphorylation at various sites of the p65 subunit has been shown to be important in initiating transcription.[48] The free NF-κB would then move to nucleus to initiate transcription of DNA to mRNA and then further translate to proteins which would subsequently respond to the initial external stimuli by causing changes in the cell. This activation has been known to affect various gene products, which regulates inflammation, cancer, angiogenesis, metastasis, apoptosis, and chemoresistance.[5]
Chalepin was found to suppress the NF-κB activation, which is commonly induced by various carcinogens and inflammatory agents. In this study, chalepin was found to act through direct interaction with the p65 subunit of NF-κB pathway and also through its ability to inhibit phosphorylation of the inhibitor of NF-κB, i.e., Iκβα, thus causes the pathway to be in an inactive state. The pathway was inhibited through suppression of Iκβα phosphorylation and degradation.
Signal transducer and activator of transcription 3 or STAT3 is overexpressed in many human tumors. The STAT3 family of transcription factor controls various physiological processes such as immunity, metabolism, development, and differentiation. STAT3 is often abnormally expressed in cancer.[49] STAT3 has been a promising target for the development of novel cancer drugs. Several studies have shown that modulation of constitutive STAT3 has a critical role in the tumor progression.[50] pSTAT3 contributes to an important role in cell proliferation and survival of tumor cells in various types of cancer including head and neck cancer, multiple myeloma, lymphomas, and leukemia.[51] Treatment of chalepin suppressed the pSTAT3 (0.59-fold change) thus indicating an inactivation of the transcription factor [Figure 7]. Chalepin therefore could inhibit the progression of A549 cells. Chalepin is a good candidate for the modulation of STAT3. However, further research needs to be conducted. Chalepin was found to be able to suppress expression of several STAT3 regulated genes such as cyclin D1 which is responsible for cell proliferation, several antiapoptotic genes such as Mcl-1 and c-Myc, immune suppression, and inflammation-related genes, e.g., COX-2 and p21, which is associated with cell survival and metastasis. With this results, it is noteworthy that chalepin is not specific to NF-κB pathway as it can also affect pSTAT3. Hence, it is a multitargeted agent that can be used against various malignancies as well.
Apoptosis can be initiated through two major interconnected pathways. The first one involves the activation of the TNF family of death receptors, also known as extrinsic pathway. The second involves the release of cytochrome c from mitochondria, also known as intrinsic apoptosis pathway. Caspase 8 exists in an inactive proenzyme form and is converted to the active form upon recruitment to the cytoplasmic domain of death receptors. TNF-related apoptosis-inducing ligand or commonly known as TRAIL, when binding with DR4 or DR5, results in the recruitment of an adaptor protein FADD. The activated caspase 8 initiates apoptosis through two different mechanisms: (i) directly activates caspase 3 to stimulate apoptosis and (ii) by cleaving BID, i.e., the BH3-interacting death domain which acts as a precursor in activating the intrinsic apoptosis pathway.[52] DR4 and DR5 showed an upregulation at 24 h incubation time upon treatment with chalepin. The upregulation was 2.17-fold and 2.84-fold change for DR4 and DR5, respectively. Interestingly, there was an initial downregulation in the expression and then a significant upregulation at the 24 h incubation time [Figure 9]. Activation of caspase 8, which was observed through flow cytometry analysis [Figure 8], further confirms that the extrinsic pathway is activated upon treatment of chalepin. Caspase 8 could activate mitochondrial apoptosis pathway/intrinsic apoptosis pathway, which is mediated by caspase 8 substrate BID. Caspase 8 cleaves BID to truncated BID (tBID) and it translocates to the mitochondria to interact with members of the Bcl-2 family to promote cytochrome c release. Release of cytochrome c would then activate caspase 9 and then caspase 3 to initiate apoptosis.[53] Western blot analysis showed the downregulation of the protein BID, 0.69-fold change at 24 h [Figure 10]. This indicated the cleavage of BID to tBID which would initiate mitochondria to release cytochrome c and activate apoptosis through the mitochondrial pathway. A recent report showed that chalepin activated caspase 9 and caspase 3 which confirmed the occurrence of the downstream intrinsic apoptosis pathway.[20]
If we were to analyze the mechanism of action of chalepin as a whole, we can conclude that chalepin targets multiple pathways which are involved in tumorigenesis. Chalepin can be described as possessing various mechanism of action. Chalepin has been predominantly found to cause cell death A549 human lung carcinoma through the intrinsic apoptotic pathway from a previous report.[20] Our study further confirms that chalepin not only acts through the intrinsic apoptotic pathway, and it also inhibits the death receptor-induced apoptotic pathway. Chalepin also acts as to inhibit the pSTAT3 which is essential for the activation of transcription in cell. This makes chalepin as a potentially effective suppressor of tumor cell survival, angiogenesis, and proliferation. In addition, chalepin showed suppression toward the NF-κB pathway via inhibition in the phosphorylated Iκβα and degradation of Iκβα. To add to this, chalepin also inhibited the phosphorylation of p65 and translocation of this protein to the nucleus to initiate transcription. Chalepin also downregulated the expression of NF-κB regulated antiapoptotic (c-IAP1, c-IAP2, XIAP, survivin, Bcl-2, and Bcl-xL) and proliferative (cyclin D1, cyclin E, COX-2, and c-myc) gene products.[20] These results suggest that chalepin may be assisted by NF-κB pathway to play its role in causing cell death.
Medicinal plants have important role in large group of the world's population. Currently, the recognition and development of medicinal plants are on increase in developing nations as well as industrialized nations.[54] Asian and African countries particularly have infused herbal medicine into their primary healthcare. It remains as an important element in their medicinal systems.[55] There is an estimation that medicines derived from plant constituents contribute to 25% of the total modern medicinal drugs.[54] R. angustifolia which has been well documented for its ethnopharmacological property, especially in Asian region, proves to possess valuable medicinal property, especially its chemical constituent, chalepin, which was proven by this scientific research to possess chemotherapeutic property which is beneficial in combating cancer.
CONCLUSION
This is the first report on the effect of chalepin isolated from R. angustifolia L. Pers against A549 cell line on the mechanistic pathway of cell death and modulation in the cell cycle of A549 cells. Cell cycle arrest was found to occur at the S phase with downregulation in cyclin D, cyclin E, CDK2, and CDK4 and also with slight upregulation in the inhibitors of CDKs, i.e., p21 and p27. A downregulation was observed in the pRb protein. Chalepin showed suppression of the NF-κB pathway with downregulation in the pIκβα and Iκβα degradation and inhibition in the phosphorylation and translocation of the p65 protein subunit. Besides that, extrinsic apoptosis pathway was triggered upon treatment of chalepin on the A549 cells. Caspase 8, which is a key initiator kinase in the extrinsic apoptosis pathway, was activated upon treatment of chalepin. Besides that, DR4 and DR5 were upregulated and BID cleavage further strengthened the activation of the extrinsic apoptosis pathway by chalepin. pSTAT3 was inhibited upon introduction of chalepin. Chalepin was found to inhibit several STAT3-regulated genes such as c-myc, COX-2, Mcl-1, proteins. STAT3 and pSTAT3 were also downregulated. c-IAP1 and c-IAP2, which are antiapoptotic proteins, were found to be downregulated after treatment of chalepin. It can therefore be concluded that chalepin induces apoptosis and suppresses pSTAT3, NF-κB pathway, and extrinsic apoptosis pathway through BID cleavage. This compound could be an excellent candidate to be considered as a chemotherapeutic agent.
Financial support and sponsorship
We would also like to acknowledge the Ministry of Higher Education of Malaysia and the University of Malaya for financial assistance to support this study, which was received through the HIR MOHE F000002-21001 Grant and IPPP PG200/2014B Grant.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
Jaime Stella Moses Richardson would like to acknowledge Ministry of Higher Education for providing scholarship under the MyPhD Scheme in the MyBrain15 program. We would also like to acknowledge the Ministry of Higher Education of Malaysia and the University of Malaya for financial assistance to support this study, which was received through the HIR MOHE F000002-21001 Grant and IPPP PG200/2014B Grant. We would like to acknowledge Dr. Gautam Sethi from NUS, Singapore, for giving his valuable comments on this work.
REFERENCES
- 1.Chen W, Li Z, Bai L, Lin Y. NF-kappaB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front Biosci (Landmark Ed) 2011;16:1172–85. doi: 10.2741/3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brambilla E, Gazdar A. Pathogenesis of lung cancer signalling pathways: Roadmap for therapies. Eur Respir J. 2009;33:1485–97. doi: 10.1183/09031936.00014009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fadlalla K, Watson A, Yehualaeshet T, Turner T, Samuel T. Ruta graveolens extract induces DNA damage pathways and blocks Akt activation to inhibit cancer cell proliferation and survival. Anticancer Res. 2011;31:233–41. [PMC free article] [PubMed] [Google Scholar]
- 4.Foster I. Cancer: A cell cycle defect. Radiography. 2008;14:144–9. [Google Scholar]
- 5.Sethi G, Ahn KS, Aggarwal BB. Targeting nuclear factor-kappa B activation pathway by thymoquinone: Role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol Cancer Res. 2008;6:1059–70. doi: 10.1158/1541-7786.MCR-07-2088. [DOI] [PubMed] [Google Scholar]
- 6.Ngoumfo RM, Jouda JB, Mouafo FT, Komguem J, Mbazoa CD, Shiao TC, et al. In vitro cytotoxic activity of isolated acridones alkaloids from Zanthoxylum leprieurii Guill. et Perr. Bioorg Med Chem. 2010;18:3601–5. doi: 10.1016/j.bmc.2010.03.040. [DOI] [PubMed] [Google Scholar]
- 7.Wu TS, Shi LS, Wang JJ, Iou SC, Chang HC, Chen YP, et al. Cytotoxic and antiplatelet aggregation principles of Ruta graveolens. J Chin Chem Soc. 2003;50:171–8. [Google Scholar]
- 8.Réthy B, Zupkó I, Minorics R, Hohmann J, Ocsovszki I, Falkay G. Investigation of cytotoxic activity on human cancer cell lines of arborinine and furanoacridones isolated from Ruta graveolens. Planta Med. 2007;73:41–8. doi: 10.1055/s-2006-951747. [DOI] [PubMed] [Google Scholar]
- 9.Roseghini R, Moreira P, Vale V, Pinheiro AM, Costa JF, Bittencourt T, et al. Different effects of arborinine alkaloid obtained from Brazilian Erthela baihensis on spleen and thymus cells stimulated in vitro with different mitogens. Immunopharmacol Immunotoxicol. 2006;28:361–76. doi: 10.1080/08923970600809579. [DOI] [PubMed] [Google Scholar]
- 10.Orlita A, Sidwa-Gorycka M, Kumirska J, Malinski E, Siedlecka EM, Gajdus J, et al. Identification of Ruta graveolens L. metabolites accumulated in the presence of abiotic elicitors. Biotechnol Prog. 2008;24:128–33. doi: 10.1021/bp070261d. [DOI] [PubMed] [Google Scholar]
- 11.Panno ML, Giordano F, Palma MG, Bartella V, Rago V, Maggiolini M, et al. Evidence that bergapten, independently of its photoactivation, enhances p53 gene expression and induces apoptosis in human breast cancer cells. Curr Cancer Drug Targets. 2009;9:469–81. doi: 10.2174/156800909788486786. [DOI] [PubMed] [Google Scholar]
- 12.Lee YM, Wu TH, Chen SF, Chung JG. Effect of 5-methoxypsoralen (5-MOP) on cell apoptosis and cell cycle in human hepatocellular carcinoma cell line. Toxicol In Vitro. 2003;17:279–87. doi: 10.1016/s0887-2333(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 13.An ZY, Yan YY, Peng D, Ou TM, Tan JH, Huang SL, et al. Synthesis and evaluation of graveoline and graveolinine derivatives with potent anti-angiogenesis activities. Eur J Med Chem. 2010;45:3895–903. doi: 10.1016/j.ejmech.2010.05.043. [DOI] [PubMed] [Google Scholar]
- 14.Oliva A, Meepagala KM, Wedge DE, Harries D, Hale AL, Aliotta G, et al. Natural fungicides from Ruta graveolens L. leaves, including a new quinolone alkaloid. J Agric Food Chem. 2003;51:890–6. doi: 10.1021/jf0259361. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S, Bishayee K, Khuda-Bukhsh AR. Graveoline isolated from ethanolic extract of Ruta graveolens triggers apoptosis and autophagy in skin melanoma cells: A novel apoptosis-independent autophagic signaling pathway. Phytother Res. 2014;28:1153–62. doi: 10.1002/ptr.5107. [DOI] [PubMed] [Google Scholar]
- 16.Wansi JD, Hussain H, Tcho AT, Kouam SF, Specht S, Sarite SR, et al. Antiplasmodial activities of furoquinoline alkaloids from Teclea afzelii. Phytother Res. 2010;24:775–7. doi: 10.1002/ptr.2894. [DOI] [PubMed] [Google Scholar]
- 17.Njogu MK, Matasyoh JC, Kibor AC. Isolation of four furoquinoline alkaloids from Teclea nobilis and their activity against Schistosoma mansoni miracidia. J Biomed Pharm Res. 2014;3:87–93. [Google Scholar]
- 18.Wahyuni TS, Widyawaruyanti A, Lusida MI, Fuad A, Soetjipto Fuchino H, et al. Inhibition of hepatitis C virus replication by chalepin and pseudane IX isolated from Ruta angustifolia leaves. Fitoterapia. 2014;99:276–83. doi: 10.1016/j.fitote.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Prakash Chaturvedula VS, Schilling JK, Miller JS, Andriantsiferana R, Rasamison VE, Kingston DG. New cytotoxic alkaloids from the wood of Vepris punctata from the Madagascar rainforest. J Nat Prod. 2003;66:532–4. doi: 10.1021/np020578h. [DOI] [PubMed] [Google Scholar]
- 20.Richardson JS, Sethi G, Lee GS, Malek SN. Chalepin: Isolated from Ruta angustifolia L. Pers induces mitochondrial mediated apoptosis in lung carcinoma cells. BMC Complement Altern Med. 2016;16:389. doi: 10.1186/s12906-016-1368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raman BV, La S, Saradhi MP, Rao BN, Khrisna A, Sudhakar M, et al. Antibacterial, antioxidant activity and GC-MS analysis of Eupatorium odoratum. Asian J Pharm Clin Res. 2012;5:99–106. [Google Scholar]
- 22.Yang QY, Tian XY, Fang WS. Bioactive coumarins from Boenninghausenia sessilicarpa. J Asian Nat Prod Res. 2007;9:59–65. doi: 10.1080/10286020500382397. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Zhang H, Yao XG, Shen H, Chen J, Li C, et al. (+)-Rutamarin as a dual inducer of both GLUT4 translocation and expression efficiently ameliorates glucose homeostasis in insulin-resistant mice. PLoS One. 2012;7:e31811. doi: 10.1371/journal.pone.0031811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elliott JA., Jr Methoxsalen in the treatment of vitiligo;an appraisal of the permanency of the repigmentation. AMA Arch Derm. 1959;79:237–41. doi: 10.1001/archderm.1959.01560140099014. [DOI] [PubMed] [Google Scholar]
- 25.Anaya AL, Macías-Rubalcava M, Cruz-Ortega R, García-Santana C, Sánchez-Monterrubio PN, Hernández-Bautista BE, et al. Allelochemicals from Stauranthus perforatus a Rutaceous tree of the Yucatan Peninsula, Mexico. Phytochemistry. 2005;66:487–94. doi: 10.1016/j.phytochem.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Flores R, Gloria-Garza MA, de la Garza-Ramos MA, Quintanilla-Licea R, Tamez-Guerra P. Antimicrobial effect of chalepensin against Streptococcus mutans. J Med Plant Res. 2016;10:631–4. [Google Scholar]
- 27.Emerole G, Thabrew MI, Anosa V, Okorie DA. Structure-activity relationship in the toxicity of some naturally occurring coumarins-chalepin, imperatorin and oxypeucedanine. Toxicology. 1981;20:71–80. doi: 10.1016/0300-483x(81)90107-4. [DOI] [PubMed] [Google Scholar]
- 28.Olorunsogo OO, Emerole GO, Malomo SO, Thabrew MI. Inhibition of mitochondrial respiration by chalepin, a naturally occurring furocoumarin. Toxicol Lett. 1983;15:323–7. doi: 10.1016/0378-4274(83)90151-0. [DOI] [PubMed] [Google Scholar]
- 29.Adebajo AC, Iwalewa EO, Obuotor EM, Ibikunle GF, Omisore NO, Adewunmi CO, et al. Pharmacological properties of the extract and some isolated compounds of Clausena lansium stem bark: Anti-trichomonal, antidiabetic, anti-inflammatory, hepatoprotective and antioxidant effects. J Ethnopharmacol. 2009;122:10–9. doi: 10.1016/j.jep.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 30.Collins K, Jacks T, Pavletich NP. The cell cycle and cancer. Proc Natl Acad Sci U S A. 1997;94:2776–8. doi: 10.1073/pnas.94.7.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–82. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 32.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–49. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dickson MA, Schwartz GK. Development of cell-cycle inhibitors for cancer therapy. Curr Oncol. 2009;16:36–43. doi: 10.3747/co.v16i2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muñoz-Alonso MJ, Acosta JC, Richard C, Delgado MD, Sedivy J, León J. p21Cip1 and p27Kip1 induce distinct cell cycle effects and differentiation programs in myeloid leukemia cells. J Biol Chem. 2005;280:18120–9. doi: 10.1074/jbc.M500758200. [DOI] [PubMed] [Google Scholar]
- 35.Oberoi-Khanuja TK, Murali A, Rajalingam K. IAPs on the move: Role of inhibitors of apoptosis proteins in cell migration. Cell Death Dis. 2013;4:e784. doi: 10.1038/cddis.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schimmer AD. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res. 2004;64:7183–90. doi: 10.1158/0008-5472.CAN-04-1918. [DOI] [PubMed] [Google Scholar]
- 37.McKinlay M.A. DDW; 2011. SMAC Mimetics: a new class of targeted agents that activate apoptotic cell death and block pro-survival signalling in cancer cells; p. 37. [Google Scholar]
- 38.Owens TW, Gilmore AP, Streuli CH, Foster FM. Inhibitor of apoptosis proteins: Promising targets for cancer therapy. J Carcinog Mutagen. 2013;(Suppl 14):pii: S14-004. doi: 10.4172/2157-2518.S14-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Besbes S, Billard C. First MCL-1-selective BH3 mimetics as potential therapeutics for targeted treatment of cancer. Cell Death Dis. 2015;6:e1810. doi: 10.1038/cddis.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dang CT, Shapiro CL, Hudis CA. Potential role of selective COX-2 inhibitors in cancer management. Oncology (Williston Park) 2002;16(5 Suppl 4):30–6. [PubMed] [Google Scholar]
- 42.Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol. 2010;2010:215158. doi: 10.1155/2010/215158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18:5546–53. doi: 10.1158/1078-0432.CCR-12-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rapp UR, Korn C, Ceteci F, Karreman C, Luetkenhaus K, Serafin V, et al. MYC is a metastasis gene for non-small-cell lung cancer. PLoS One. 2009;4:e6029. doi: 10.1371/journal.pone.0006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karin M. How NF-kappaB is activated: The role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–74. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 48.Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on serine 536 defines an IkBa-independent NF-κB pathway. J Biol Chem. 2005;280:34538–47. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- 49.Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, et al. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845:136–54. doi: 10.1016/j.bbcan.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 50.Yue P, Turkson J. Targeting STAT3 in cancer: How successful are we? Expert Opin Investig Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sethi G, Chatterjee S, Rajendran P, Li F, Shanmugam MK, Wong KF, et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol Cancer. 2014;13:66. doi: 10.1186/1476-4598-13-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rogalska A, Gajek A, Marczak A. Epothilone B induces extrinsic pathway of apoptosis in human SKOV-3 ovarian cancer cells. Toxicol In Vitro. 2014;28:675–83. doi: 10.1016/j.tiv.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Gnesutta N, Minden A. Death receptor-induced activation of initiator caspase 8 is antagonized by serine/threonine kinase PAK4. Mol Cell Biol. 2003;23:7838–48. doi: 10.1128/MCB.23.21.7838-7848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain S, Dwivedi J, Jain PK, Satpathy S, Patra A. Medicinal plants for treatment of cancer: A brief review. Pharmacogn J. 2016;8:87–102. [Google Scholar]
- 55.Cock I. The safe usage of herbal medicines: Counter-indications, cross-reactivity and toxicity. Pharmacogn Commun. 2015;5:2–38. [Google Scholar]