

Abstract
Background
Following myocardial infarction (MI), peri‐infarct myocardial edema formation further impairs cardiac function. Extracellular RNA (eRNA) released from injured cells strongly increases vascular permeability. This study aimed to assess the role of eRNA in MI‐induced cardiac edema formation, infarct size, cardiac function, and survival after acute MI and to evaluate the therapeutic potential of ribonuclease 1 (RNase‐1) treatment as an eRNA‐degrading intervention.
Methods and Results
C57BL/6J mice were subjected to MI by permanent ligation of the left anterior descending coronary artery. Plasma eRNA levels were significantly increased compared with those in controls starting from 30 minutes after ligation. Systemic application of RNase‐1, but not DNase, significantly reduced myocardial edema formation 24 hours after ligation compared with controls. Consequently, eRNA degradation by RNase‐1 significantly improved the perfusion of collateral arteries in the border zone of the infarcted myocardium 24 hours after ligation of the left anterior descending coronary artery, as detected by micro–computed tomography imaging. Although there was no significant difference in the area at risk, the area of vital myocardium was markedly larger in mice treated with RNase‐1 compared with controls, as detected by Evans blue and 2,3,5‐triphenyltetrazolium chloride staining. The increase in viable myocardium was associated with significantly preserved left ventricular function, as assessed by echocardiography. Moreover, RNase‐1 significantly improved 8‐week survival following MI.
Conclusions
eRNA is an unrecognized permeability factor in vivo, associated with myocardial edema formation after acute MI. RNase‐1 counteracts eRNA‐induced edema formation and preserves perfusion of the infarction border zone, reducing infarct size and protecting cardiac function after MI.
Keywords: edema, extracellular RNA, myocardial infarction, RNase‐1
Subject Categories: Coronary Circulation, Ischemia, Myocardial Biology
Introduction
The heart is one of the organs most sensitive to increased microvascular permeability and interstitial edema formation.1 Importantly, cardiac function is already significantly compromised once the interstitial fluid volume is moderately increased.2 Myocardial edema formation represents a major clinical problem and leads to vessel collapse, stunning of the myocardium, induction of arrhythmias, and impaired cardiac function. Edema occurs in response to ischemia/reperfusion, inflammation, organ transplant rejection, or cardioplegic arrest during surgical interventions such as coronary artery bypass grafting.3 Following myocardial infarction (MI), vascular permeability (VP) across the microvascular endothelium is increased, resulting in persistent postischemic vasogenic edema.2, 4 Myocardial tissue in the border zone of the MI is partially perfused by collateral vessels, preventing necrosis of these acutely undersupplied myocardial regions. When peri‐infarct tissue edema impairs collateral and capillary flow, it further aggravates the effects of the initial hypoxia, contributing to the final extent of tissue injury throughout the ventricular myocardium.5 Even though prevention of edema formation has long been recognized as an attractive approach for the treatment of acute MI, no effective therapeutic strategy is currently available.6
Cell injury resulting from MI leads to the release of intracellular components into the circulation, including RNA.7 As previously demonstrated by our group, the released RNA (mostly ribosomal RNA), designated extracellular RNA (eRNA), is not inert but has specific biochemical activities particularly linked to cardiovascular processes.7, 8, 9, 10 As a procoagulant factor, the negatively charged eRNA activates the contact‐phase pathway of blood coagulation, contributing to thrombus formation.7, 11 Specifically, eRNA promotes the binding of vascular endothelial growth factor (VEGF) to neuropilin 1, which leads to phosphorylation of VEGF‐R2 and disarrangement of vascular endothelial cadherins at cell–cell borders. Activation of these pathways, in turn, results in the release of ribonuclease 1 (RNase‐1) and von Willebrand factor from Weibel‐Palade bodies,12 possibly as a negative feedback loop, to limit the effects of eRNA, as previously published by our group. Importantly, extracellular DNA does not increase the permeability across microvascular endothelial cells via a VEGF‐dependent mechanism.9, 12, 13
Following up on these observations in the current study, we aimed to evaluate the effects of systemic application of pancreatic‐type RNase‐1, a member of the RNase A superfamily that represents the predominant isoform of extracellular RNases, in a mouse model of acute MI to test the therapeutic potential of RNase application on myocardial edema formation, microvascular perfusion, myocardial viability, contractility, and survival.
Methods
Animal Procedures
All procedures involving animals were approved by the local governmental animal care committee (GI 20/10‐Nr.61/2008) and complied with directive 2010/63/EU of the European Parliament. MI was induced in mice, as previously described.13 Male C57BL/6J mice aged 10 to 12 weeks were used, except for survival analysis, for which 2‐year‐old animals were used. In brief, mice were anaesthetized by intraperitoneal injection of ketamine and xylazine and endotracheally ventilated with a small rodent ventilator, whereby 1.5% isoflurane was used for maintenance of anesthesia during surgery.
The surgical procedure included a left‐side thoracotomy followed by incision of the pericardium. MI was induced by permanent ligation of the left anterior descending coronary artery (LAD) with an 8‐0 silk suture at the site of its emergence from under the left atrium, and the incision was closed with a 6‐0 silk suture. ECG (Cardiofax; Nihon‐Koden) was used to document MI by ST‐segment elevation. Analgesia after surgery was performed with buprenorphine (0.01 mg/kg body weight).
Following previously established dose‐finding protocols and reports on different in vivo models from our group and others,14, 15, 16, 17 pancreatic‐type RNase‐1 (50 and 100 μg/kg) or DNase (100 μg/kg) diluted in 50 μL 0.9% saline was injected intravenously via the tail vein 30 minutes, 3 hours, and 6 hours after ligation of the LAD; 50 µL of 0.9% saline was used as a control. Sham‐operated mice were subjected to the same experimental procedure except for the ligation of the LAD. For analysis of the wet/dry weight ratio and the extent of MI and for immunohistochemistry, mice were euthanatized 24 hours after induction of MI. Hearts were harvested and perfused with 0.9% saline with a secured needle via the aortic stump until the remaining blood was rinsed out. A diagram showing the study protocol up to 24 hours and survival after MI is shown in Figure 1A and 1B, respectively.
Figure 1.
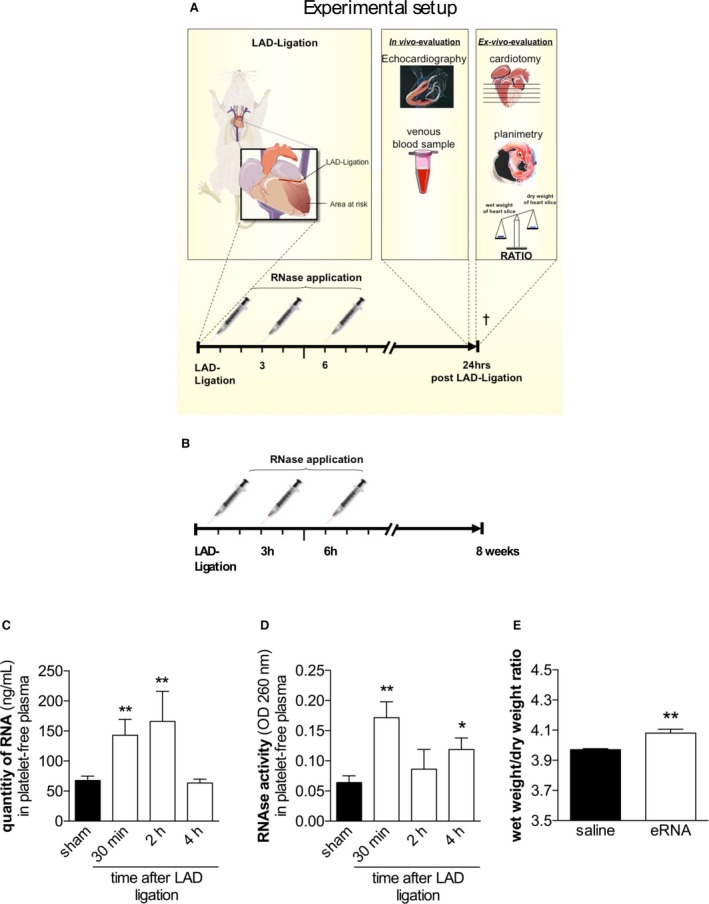
eRNA and intrinsic RNase activity in mice after the induction of myocardial infarction. A and B, A diagram of the study protocol of short‐term (A) and long‐term (B) assessments. C, Quantification of eRNA in platelet‐free plasma samples of mice 24 hours after ligation of the LAD (**P<0.01 vs sham‐operated mice; n=7). D, RNase activity in platelet‐free plasma following ligation of the LAD (*P<0.05, **P<0.01 vs sham‐operated mice; n=7). E, Wet/dry ratios of mouse hearts following intravenous application of external eRNA (15 μg/kg) or saline (*P<0.05, n=7). eRNA indicates extracellular RNA; LAD, left anterior descending coronary artery; OD, optical density; RNase, ribonuclease.
The sample size of each group was determined by power analysis. Depending on the calculation, 5 to 7 mice per group were used.
Calculation of Edema Formation Using the Wet/Dry Weight Ratio
From the harvested hearts, the atria were dissected and ventricles were cut into five 0.8‐ to 1.0‐mm slices. Wet weight was measured immediately after cutting. The tissue was stored in a drying chamber for 48 hours and weighed again. The difference in wet to dry weight was considered the tissue water content and expressed as the wet/dry weight ratio.18 The percentage of H2O was calculated as follows: (1) (wet weight−dry weight)/wet weight×100To quantify edema formation in the area at risk (AAR) and the remote myocardium, the hearts were harvested after perfusion with Evan's blue, cut into five 0.8‐ to 1.0‐mm slices, and the ischemic (pale) myocardium was dissected from the perfused (blue) myocardium in each slice. The wet/dry weight ratio of the pooled ischemic versus perfused myocardial samples was quantified, as described in Equation 1.
Measurement of Infarct Size
Before harvesting of the hearts, retrograde intra‐aortical injection of 1 mL of 0.5% Evans blue dye in PBS was carried out, and the ventricles were sliced into 5 sections from the apex to the valvular level with a razor blade, resulting in 0.8‐ to 1.0‐mm slices. The AAR was quantified via computer‐aided planimetry, using a dissection microscope equipped with a digital camera, by measuring the area of unstained myocardium. To investigate viable myocardium within the AAR, ex vivo staining of the heart slices with 2,3,5‐triphenyltetrazolium chloride (TTC 1%) was performed. The stained slices were used for computer‐aided planimetry from both sides. Within the AAR, TTC‐stained myocardium represents viable tissue, whereas nonstained areas indicate necrosis.19
Immunohistochemistry
For immunohistochemistry, hearts were embedded in Tissue‐Tek OCT (Sakura Finetek) embedding medium, snap‐frozen, and stored at −80°C until sectioning. For this purpose, the ventricle was cut in 6‐μm serial sections, and 10 cross‐sections from regular intervals within the border zone of the AAR were examined. The apoptotic cells were quantified by terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling (TUNEL) according to the supplier's instructions (In Situ Cell Death Detection Kit; Roche). Nuclei were stained with nuclear DAPI (4′,6‐diamidino‐2‐phenylindole). Images were captured and processed using an immunofluorescence microscope equipped with appropriate filter blocks.
Echocardiography
Transthoracic echocardiography was performed in anesthetized (ketamine/xylazine) mice 24 hours after ligation (ATL HDI 5000; Philips) with a scan head of 17.5 MHz (8 mice per group). Two‐dimensional M‐mode images were obtained from the short‐axis view at the mid–papillary muscle level in the greatest left ventricular diameter. Fractional shortening was measured 3 times for each animal before harvesting of heart tissues for further investigation, and the average was taken into consideration for the analysis of cardiac function. A single researcher blinded to treatment allocation performed the analysis.
Micro–Computed Tomography
For ex vivo micro–computed tomography (micro‐CT) analysis, the aorta was cannulated in situ, and heparinized saline (10 mL of 0.9% sodium chloride with 1000 IU heparin) was injected in a retrograde manner until the venous effluent from the right atrium no longer contained blood. A plumbiferous radiopaque polymer (Microfil MV‐122; Flow Tech) was then injected into the left ventricle in situ until it emerged from the inferior caval vein. After polymerization of the compound, the heart was removed and scanned using the SkyScan 1173 micro‐CT system (Bruker Micro‐CT). The tube voltage was set to 40 kVp, the tube current was 200 μA, and the exposure time was 2 seconds per frame. Specimens were scanned over 240° with rotation steps of 0.25° and an isotropic voxel side length of 6 μm. A 4‐fold frame averaging was carried out for image noise reduction. Cubic volumes of interest of 0.11 mm3 were obtained from standardized locations of the left ventricle (lateral wall, apex), the septum, and the right ventricle. Image analysis and morphometry of the microvasculature were carried out using the CT Analyzer Software (CTAn; Bruker Micro‐CT).
Analysis of Venous Blood Samples
Total RNA of murine blood samples was quantified in platelet‐free plasma using a GeneQuant photometer (Amersham Pharmacia). RNAse activity was measured using the Quant‐iT RNA Assay Kit (Invitrogen).
Statistical Analysis
Data were analyzed with Prism 6.01 (GraphPad Software). One‐way ANOVA followed by the Holm‐Sidak multiple comparisons method was used to compare >2 groups. Two‐way ANOVA followed by the Holm‐Sidak multiple comparison method was used to compare groups with >1 factor to test an interaction between the factors. The mean of every group in the above‐mentioned analyses was compared with the mean of every other group. The Student t test was chosen to compare 2 groups with normally distributed values. Against a background of small sample sizes, normality was visually checked. Survival was presented as a Kaplan–Meier curve. The log‐rank (Mantel‐Cox) test was used to compare the 2 curves. All data are presented as the mean±SEM. P<0.05 was considered statistically significant for all comparisons.
Results
Plasma eRNA Increases After LAD Ligation
The level of circulating eRNA in platelet‐free plasma was significantly increased 30 minutes after ligation of the LAD compared with levels in plasma from sham‐operated mice (67.64±7.23 versus 142.8±26.61 ng/mL; P=0.007; n=7). The circulating eRNA content peaked at 166.1±49.93 ng/mL 2 hours after ligation of the LAD (P=0.004). However, at 4 hours after MI, the eRNA content returned to the basal level detected in sham‐operated controls (63.47±6.37 ng/mL; P=0.858; Figure 1C).
The activity of extracellular circulating RNases was significantly increased in plasma samples 30 minutes after ligation of the LAD compared with that in samples from sham‐operated mice (optical density: 260 nm; 0.064±0.01 versus 0.0172±0.026; P=0.002; n=7). Although the RNAse activity decreased to the basal level 2 hours after ligation of the LAD, a second peak in RNAse activity was detected 4 hours following experimental MI (optical density: 260 nm; 0.119±0.019; P=0.039; n=7; Figure 1D).
To test the effects of circulating eRNA on endothelial permeability and cardiac edema formation in vivo, we administered exogenous eRNA at a concentration that was comparable to plasma concentrations detected after cardiac ischemia/reperfusion20 or after acute MI, as assessed in this study, via tail vein injection (15 μg/kg). At 4 hours after administration, the mice were euthanatized, and the hearts were weighed immediately after excision and 48 hours later, following storage in a drying chamber at 50°C. Compared with saline control treatment, the application of eRNA significantly increased the wet/dry weight ratio, indicating that an increase in eRNA indeed results in cardiac edema formation (3.97±0.007 versus 4.08±0.025; P=0.003; n=5; Figure 1E). Moreover, eRNA led to increased water content in the lungs and liver compared with saline control treatment (data not shown).
Application of RNase‐1 Prevents Myocardial Edema Formation in Mice
Following induction of MI (Figure 2A), RNase‐1, DNase (for comparison and to exclude unspecific effects of RNase treatment), or saline was administered intravenously at 30 minutes, 3 hours, and 6 hours after ligation of the LAD. At 24 hours after the treatments, mice were euthanatized and the heart wet/dry weight ratios were determined. Treatment with 50 μg/kg RNase‐1 significantly reduced myocardial water content in infarcted mouse hearts compared with the control group (wet/dry ratio 3.56±0.44 versus 4.14±0.88; P=0.039; n=6) (Figure 2B). In contrast, application of DNase (100 μg/kg) did not alter myocardial edema formation (wet/dry ratio 3.99±0.43; P=0.718 versus RNase‐1; n=6). An increase in the dose of RNase‐1 to 100 μg/kg showed no additional benefit (wet/dry ratio 3.69±0.41; P=0.875 versus 50 μg/kg RNase‐1; n=6), indicating that a dose of 50 μg/kg is sufficient to exert maximum effects. Moreover, further analysis of the separate compartments revealed that RNase treatment (50 μg/kg) resulted in a significant reduction of the water content of the perfused remote myocardium, probably in the direct peri‐infarct region (wet/dry weight ratio 3.53±0.16 versus 4.45±0.19; P=0.0059; n=6; Figure 2D), and of the nonperfused AAR (wet/dry weight ratio 3.54±0.13 versus 4.33±0.21; P=0.015; n=6; Figure 2E).
Figure 2.
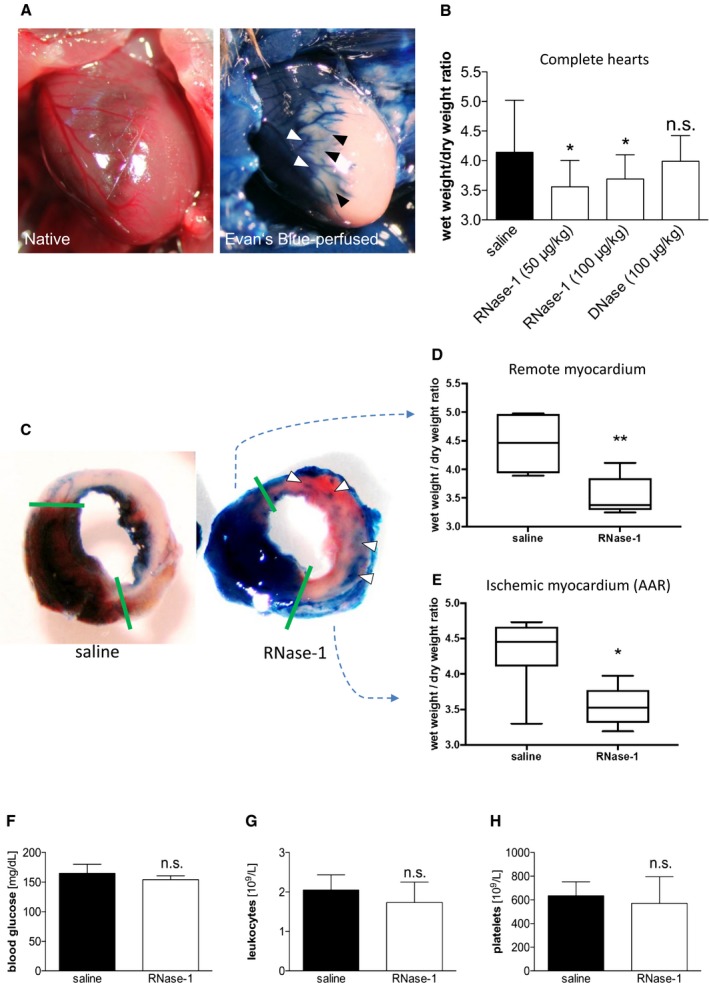
RNase‐1 application reduces myocardial edema formation following MI. A, Representative image of a native edematous murine heart 24 hours after ligation of the LAD (left panel). Evans blue perfusion delineates the AAR of the LAD perfusion bed (unstained; right panel). B, Wet/dry ratios of mice 24 hours after induction of MI following intravenous administration of RNase‐1, DNase, or saline (*P<0.05, n=7). C, Representative images of murine hearts stained with Evans blue (perfused myocardium) and TTC (viable myocardium) 24 hours after ligation of the LAD treated with saline only (left) or RNase‐1 (50 μg RNase‐1/kg; right). Arrows indicate perfused arteries (blue) in the AAR. D and E, Separate wet/dry weight ratios of the perfused remote myocardium (D) and the ischemic myocardium/AAR (E) 24 hours after induction of MI following intravenous administration of RNase‐1 or saline (*P<0.05, **P<0.005, n=6). Treatment of ternary RNase‐1 injection had no significant impact on total concentration of glucose (F) and total amount of leukocytes (G) or platelets (H) in venous blood samples of mice 24 hours following LAD ligation (P=n.s., n=7). AAR indicates area at risk; LAD, left anterior descending coronary artery; MI, myocardial infarction; n.s., not significant; RNase‐1, ribonuclease 1; TTC, 2,3,5‐triphenyltetrazolium chloride.
The cellular blood composition and plasma glucose levels can influence edema formation; however, we did not detect significant changes in plasma glucose, leukocytes, or platelets following RNase treatment compared with saline‐treated mice (Figure 2F through 2H).
Application of RNase‐1 Preserves Coronary Blood Flow
Reduced coronary and capillary flow are among the most detrimental sequelae of myocardial edema formation. We assessed myocardial perfusion using contrast agent–based micro‐CT angiography 24 hours after LAD ligation (Figure 3A). Compared with saline, RNase‐1 application (50 μg/kg) at 30 minutes, 3 hours, and 6 hours following LAD ligation significantly increased collateral perfusion, as shown by the increased accumulation of contrast agent within the ischemic myocardium (Figure 3A and 3B). Moreover, the density of perfused vessels in the border zone of the AAR was significantly enhanced following RNase‐1 treatment (0.23±0.22% versus 2.15±1.92% of total area; P<0.001; Figure 3C). Collateral perfusion was quantified by determining the average vessel diameter per area in the AAR following treatment with RNase‐1 or saline, respectively. The data revealed a significant increase in large perfused collateral vessels in the infarct border zone in the RNase‐1–treated group (structure linear density 0.11±0.09 versus 0.73±0.62 mm−1; P<0.001; Figure 3D). Importantly, these vessels were part of a functionally active microvascular network in which the perfusion was significantly preserved in response to RNase‐1 treatment (connectivity density 27.47±27.03 versus 96.68±93.21 mm−3; n=22; P=0.027; Figure 3E). There were no significant differences between the RNase‐1‐ and saline‐treated animals with regard to structure separation (0.27±0.05 versus 0.26±0.08 mm; n=22; P=0.699; Figure 3F) and thickness of the perfused collaterals (0.018±0.004 versus 0.021±0.01 mm; n=22; P=0.268; Figure 3G), indicating that no vascular remodeling or angiogenesis occurred during this short period in RNase‐1–treated hearts but that the connective vessels are preexisting mature collaterals.
Figure 3.
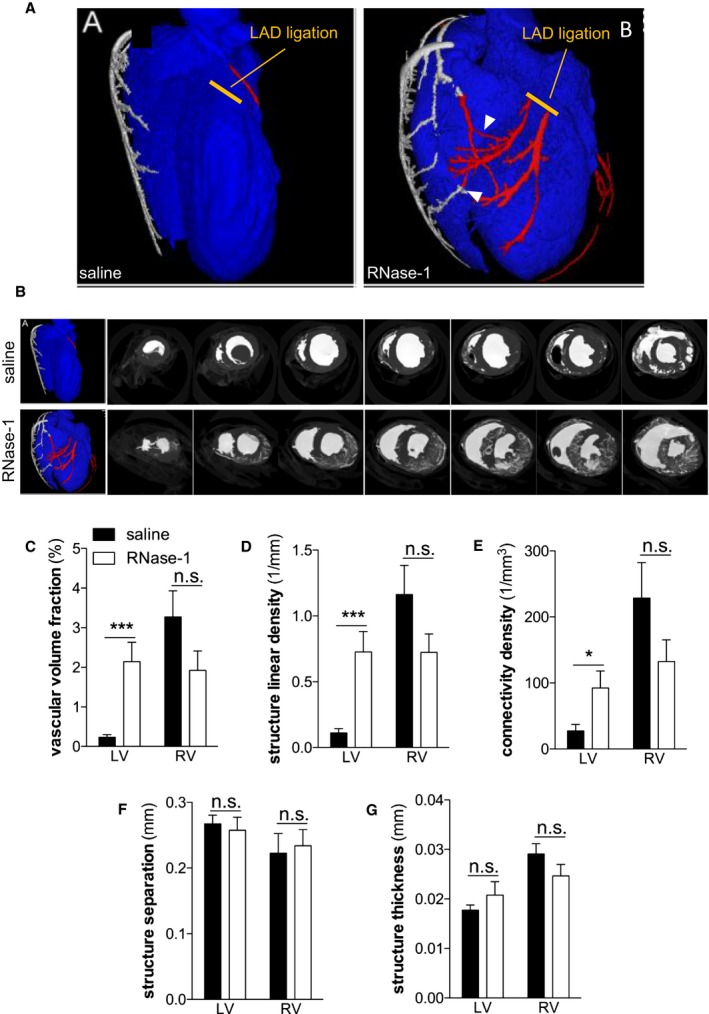
RNase‐1 application preserves coronary blood flow 24 hours after ligation of the LAD. A and B, Representative micro–computed tomography images of hearts 24 hours after ligation of the LAD and after treatment of the mice with saline (upper and left panels, respectively) or RNase‐1 (50 μg RNase‐1/kg; lower and right panels, respectively). Arrowheads indicate collateral connections. C, Density of contrast agent calculated in coronary arteries 24 hours after ligation of the LAD, indicating vascular volume structure. D, Structure linear density measured as the average amount of vessel diameters per area. E, Connectivity density of myocardial vessels. F, Structure separation measured as the average distance between vessels. G, Structure thickness measured as the average thickness of the vessel wall (*P<0.05, *** P<0.001, n=5). LAD indicates left anterior descending coronary artery; LV, left ventricle, n.s., not significant; RNase‐1, ribonuclease 1; RV, right ventricle.
Application of RNase‐1 Reduces Infarct Size and Apoptosis Within the Border Zone After MI
To analyze the infarct size and the content of viable myocardium after ligation of the LAD, the mouse hearts were perfused with Evans blue in vivo and cut into slices that were incubated with TTC ex vivo (Figure 4A). Planimetry of TTC‐stained AAR indicated that significantly more viable myocardium was preserved in mice treated with RNase‐1 (50 μg/kg) than in the saline‐treated control group 24 hours after induction of MI (vital area/AAR 0.112±0.009 versus 0.344±0.03; P<0.0001; n=5; Figure 4C). Consequently, the nonvital myocardium within the AAR, identified by a lack of staining with Evans blue or TTC, was significantly reduced after RNase‐1 compared with control treatment (infarcted area/AAR 0.888±0.009 versus 0.656±0.014; P<0.0001; n=5; Figure 4D).
Figure 4.
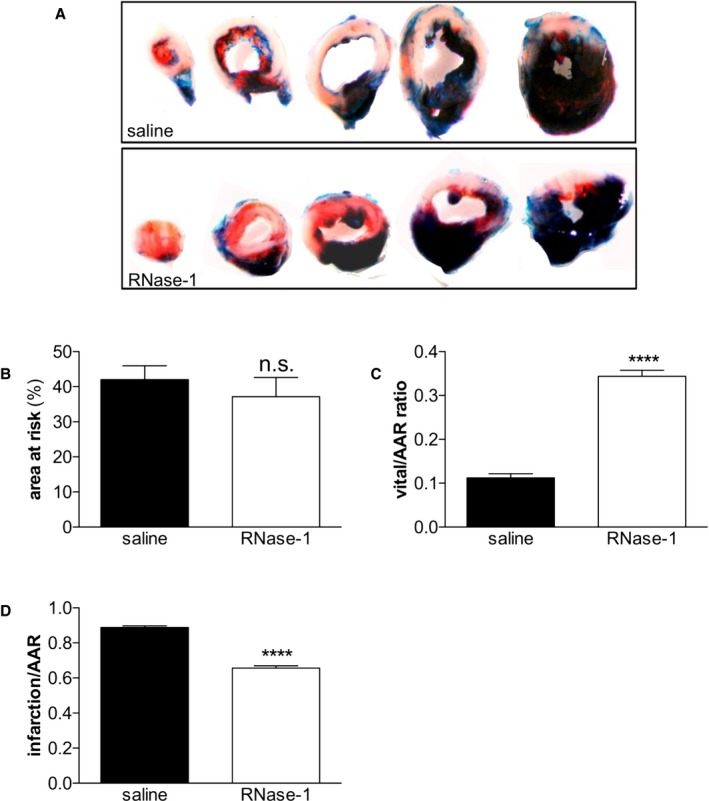
Application of RNase‐1 reduces infarct size 24 hours following ligation of the LAD. A, Representative images of murine hearts stained with Evans blue and TTC 24 hours after ligation of the LAD treated with RNase‐1 (50 μg RNase‐1/kg; lower panel) or saline (upper panel). B, LAD ligation resulted in comparable AAR size (P=ns, n=5). C, Quantification of vital myocardium normalized to AAR. D, Quantification of infarct size normalized to AAR after treatment with RNase‐1 or saline 24 hours after ligation of the LAD (**** P<0.0001, n=5). AAR indicates area at risk; LAD, left anterior descending artery; n.s., not significant; RNase‐1, ribonuclease 1; TTC, 2,3,5‐triphenyltetrazolium chloride.
To correlate myocardial perfusion with cellular markers of apoptosis, we performed TUNEL staining on cross‐sections of infarcted hearts (Figure 5A). At 24 hours after ligation of the LAD, we detected a significant reduction in TUNEL‐positive cells within the AAR border zone in mice treated with RNase‐1 (50 μg/kg) compared with that in controls (0.462±0.031 versus 0.338±0.015 TUNEL‐positive/total cells; P<0.001; n=4; Figure 5B).
Figure 5.
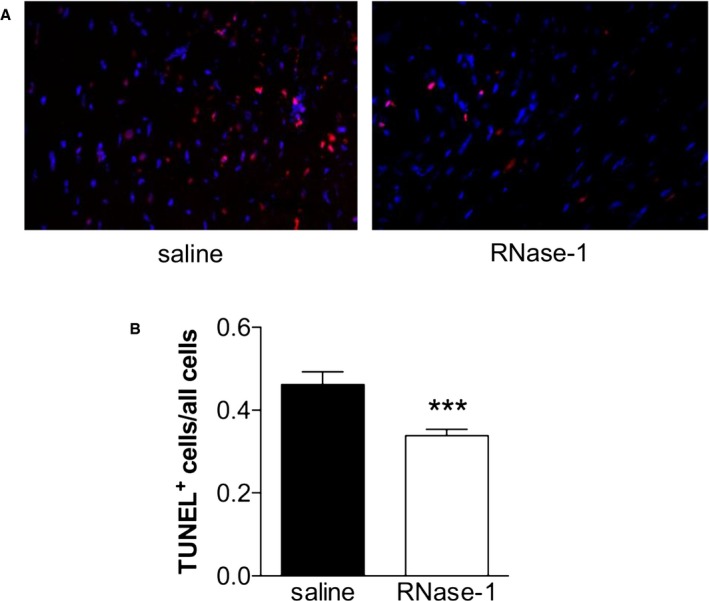
Application of RNase‐1 reduces apoptosis 24 hours following ligation of the LAD. A, Representative images of TUNEL‐stained cross‐sections within the peri‐infarction zone 24 hours after induction of myocardial infarction (TUNEL red, DAPI blue) treated with saline (left panel) or RNase‐1 (50 μg RNase‐1/kg; right panel). B, The amount of apoptotic cells in the border zone of the AAR was normalized to all cells in the panel (*** P<0.001, n=4). AAR indicates area at risk; DAPI, 4′,6‐diamidino‐2‐phenylindole; LAD, left anterior descending coronary artery; RNase‐1, ribonuclease 1; TUNEL, terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling.
Application of RNase‐1 Preserves Myocardial Function Following Acute MI
To assess the functional aspects of RNase‐1 treatment after induction of MI, high‐frequency transthoracic echocardiography was performed 24 hours after ligation of the LAD (Figure 6A) to analyze fractional shortening of the anterolateral wall of the left ventricle. Mice treated with RNase‐1 showed a significantly preserved contractility of the left ventricle compared with animals treated with saline only (fractional shortening 22.34±5.72 at 50 μg/kg RNase‐1 versus 13.33±2.29 with saline; P=0.013; n=4; Figure 6A and 6B).
Figure 6.
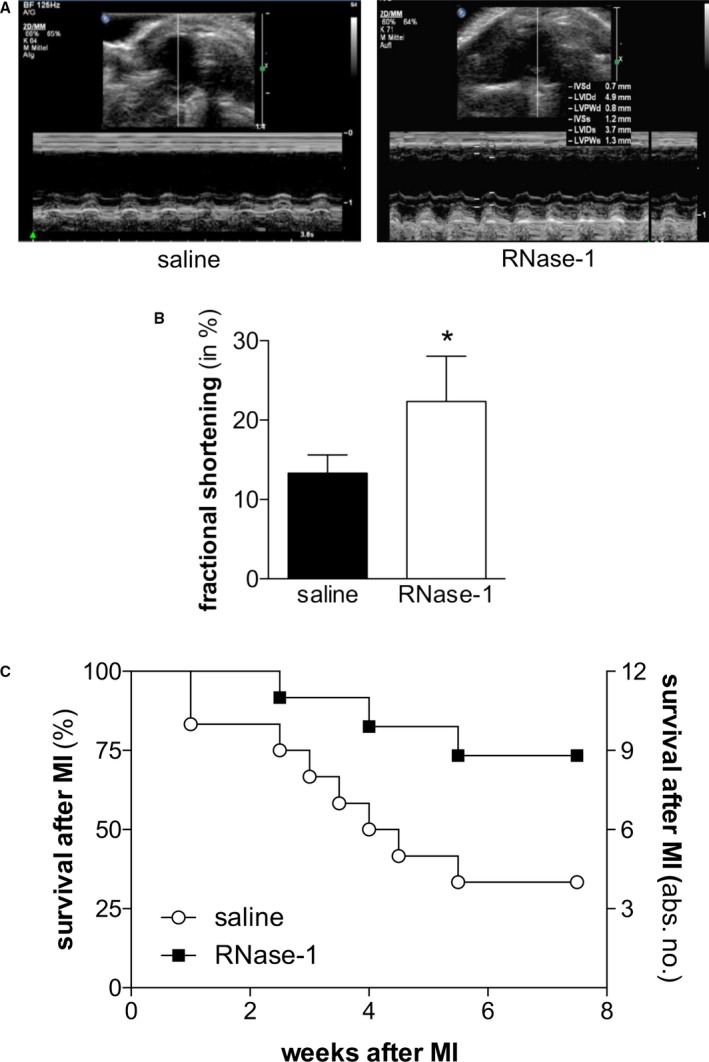
Application of RNase‐1 after MI preserves myocardial function. A, Representative images of echocardiography (Philips 17.5‐MHz scan head) 24 hours after ligation of the LAD and after treatment with saline (left panel) or RNase‐1 (right panel). B, Fractional shortening as a marker of ventricular wall motility was assessed 24 hours after induction of MI and after treatment with RNase‐1 (50 μg RNase‐1/kg) or saline (*P<0.05, n=8). C, Kaplan–Meier curve showing the survival of 2‐year‐old mice following ligation of the LAD and following treatment with RNase‐1 (50 μg RNase‐1/kg) or saline (*P<0.05, n=12). LAD indicates left anterior descending coronary artery; MI, myocardial infarction; RNase‐1, ribonuclease 1.
Application of RNase‐1 Improves Survival Following Acute MI
Finally, the survival rates after MI of aged mice treated with RNase‐1 (50 μg/kg) or saline were compared. Compared with control mice, the survival rate of mice treated with RNase‐1 was significantly increased 8 weeks after MI (73.33% versus 33.33%; n=12; P=0.043; Figure 6C).
Discussion
Edema formation following MI further impairs perfusion of the ischemic region of the heart and of adjacent regions from which collateral vessels could partially compensate the acute lack of blood supply, resulting in a further reduction of cardiac function and expansion of cardiac necrosis.1 Despite promising experimental strategies for targeting myocardial edema formation, convincing therapeutic concepts in the clinical setting are still missing.21 The data presented clearly show that application of RNase‐1 in a murine model of permanent LAD ligation decreases myocardial edema formation and improves myocardial collateral perfusion, preserving viable myocardium within the AAR. In addition, the application of RNase‐1 improved left ventricular function and overall survival of mice following MI. We thus conclude that application of RNase‐1 may represent a natural cardioprotective, feasible, and effective treatment strategy to prevent myocardial edema formation and myocardial damage following acute myocardial ischemia.
Our data indicated that plasma levels of eRNA were elevated between 30 minutes and 2 hours following permanent ligation of the LAD. Concurrently, the intrinsic RNase activity was increased following MI, possibly as a direct response to the release of eRNA following myocardial injury or via eRNA‐induced inflammatory stimuli,22 as eRNA has been reported to induce RNase release from endothelial cells in vitro following inflammatory stimulation. The eRNA was measured in platelet‐free plasma and may be released by different sources including necrotic cells, apoptotic cells, or activated platelets at the sites of injury. In a previous study of cardiac ischemia/reperfusion injury (IRI), we found cardiomyocytes to be the predominant source of eRNA.20 During MI, high amounts of eRNA are released into the circulation and thus may disrupt the intercellular integrity of the endothelial layer, especially in the direct vicinity of the damaged or necrotic myocardium.23
We previously assessed the effect of eRNA on myocardial IRI, a condition that best resembles the condition of patients with acute ST‐segment–elevation MI who undergo immediate reperfusion therapy.20 Given the highly promising results, we aimed in this study to extend these observations to conditions of permanent ischemia that are found after insufficient therapeutic or spontaneous reperfusion and often comprise smaller myocardial areas than acute ST‐segment–elevation MI events. Indeed, edema formation has been shown to occur very early in the ischemic myocardium,24, 25, 26 even though to a smaller extent than after IRI.27 Our data now indicate that this edema formation in the ischemic myocardium and the peri‐infarct region can be reduced by RNase treatment. Despite the fact that the impact of myocardial edema formation is far less well established after permanent occlusion, our data support a prominent and robust effect of an RNA‐depleting therapy also under these conditions. Nevertheless, the specific findings of this study are limited to conditions of permanent ischemia and cannot be transferred to conditions of IRI.
We recently showed that the induction of VP at the endothelium by eRNA is driven by a VEGF‐dependent mechanism.12 Specifically, eRNA induces the mobilization of extracellularly bound VEGF, leading to the activation of its specific tyrosine kinase receptor VEGF‐R2.28 Even though VEGF is a key player in angiogenesis and thus contributes to the restoration of infarcted or stunned myocardial tissue in the long term, the early effects of VEGF on VP are clearly detrimental. Consequently, short‐term targeting of eRNA as an activator of early VEGF signaling should not influence the positive long‐term effects of VEGF.
Our micro‐CT data confirmed the existence of fully functional collateral arteries in healthy mouse hearts. Furthermore, we demonstrated a clear correlation between myocardial edema formation with collapse of these collaterals and reduced perfusion in the border zone of and directly in the AAR after LAD ligation. The collapse of these collaterals, in turn, aggravates local hypoxia and leads to increased cell death within the AAR, further deteriorating ventricular function and increasing mortality in mice after MI. Importantly, RNase‐1 application targeted the process of edema formation at a very early stage and thus might prevent subsequent negative effects on perfusion of the infarct border zone. Importantly, we did not detect deleterious side effects after application of this natural cardioprotective enzyme.
RNase‐1 application also reduced the number of apoptotic cells in the border zone of the AAR; however, as described recently, these cells most likely do not represent cardiomyocytes because apoptotic cell death was demonstrated not to significantly affect either the number of viable cardiomyocytes or cardiac function under ischemic conditions.29 Consequently, further studies must define the extent to which apoptosis of noncardiomyocyte cardiac cells contributes to the aggravation of ischemic damage.
In a previous study of the in vivo effects of RNase‐1, it was shown that exogenously administered RNase‐1 strongly reduced vasogenic edema formation and ischemic lesion volume in a rat stroke model.12 Importantly, maximal beneficial effects of RNase‐1 with regard to prevention of edema formation and neuroprotection were achieved with a dose of 50 μg RNase‐1/kg in rats16; therefore, this concentration was used in the present study. Our data confirmed this dose to be effective for the prevention of cardiac edema formation after acute MI in mice, whereas a further increase in the dose to 100 μg RNase‐1/kg did not result in further improvement of the protective effects on edema formation (Figure 2B) or cardiac function (data not shown).
In addition to changes in VP, edema formation depends on oncotic pressure.30 Even though it is very unlikely that protein applied at an amount as low as the one used in this study (50 μg RNase‐1/kg mouse) would influence intravasal oncotic pressure, we applied the same amount of DNase as a control; however, DNase did not reduce edema formation, indicating that a potential increase in the intravasal oncotic pressure following administration of this low amount of RNase‐1 may not be responsible for the observed effects. DNase was also not effective in reducing VP in vitro and in a rat model of focal cerebral ischemia.12 Furthermore, the enzymatic activity of RNase‐1 is responsible for the effects on VP, as a recombinant enzymatically inactive mutant of RNase‐1 was shown to be ineffective in preventing the eRNA‐induced increase in VP in vitro and edema formation in vivo.15 These data indicate specific adverse effects of eRNA, possibly in direct and indirect fashions, that can be abolished by RNase‐1 treatment. Moreover, and in accordance with the data on edema formation after administration of eRNA in the present study, systemic administration of eRNA was shown previously to induce myocardial, lung, and liver tissue edema formation, providing additional in vivo evidence for the (VEGF‐dependent) endothelial permeability–increasing activity of eRNA.28
Besides extracellular edema formation caused by increased VP, myocardial water content increases acutely in response to ischemia also caused by cell swelling and the development of an intracellular edema.1 Moreover, a biphasic/bimodal mode of edema formation after ischemia or IRI has been described in large animal models.27, 31 Nevertheless, given a lack of longitudinal time course data in the current study (the time course of edema formation may differ in small versus large animal models) and a lack of appropriate methods available to distinguish between intracellular and extracellular edema formation, the exact time‐ and compartment‐dependent modes of action of RNase treatment will have to be assessed in future studies.
In summary, we provided evidence that the release of eRNA after MI induces myocardial edema formation, contributing to reduced tissue perfusion and exacerbated ischemic myocardial tissue damage, especially after prolonged periods of vessel occlusion. Reduction or degradation of eRNA by systematic administration of RNase‐1 reduces VP and edema formation, preserves myocardial function, and increases overall survival in mice. Because no effective strategy is currently available to prevent myocardial edema formation, RNase‐1, as an endogenous nontoxic and highly stable as well as potentially well‐tolerated nuclease, holds promise for the effective management of edema formation and ischemic injury following MI.
Sources of Funding
This work was supported by grants SFB‐547‐A10, by the Cluster of Excellence‐147 Cardio‐Pulmonary Systems (Giessen, Germany), by the International Research Training Group‐1566 “PROMISE” and SFB/TRR79 (subproject Z3) from the German Research Foundation (DFG, Bonn, Germany) to Sedding, Preissner, Cabrera‐Fuentes, Langheinrich, and Kampschulte, by the “Anschubfinanzierung des Fachbereichs Medizin” (Giessen, Germany) to Stieger, and by the DFG‐Cluster of Excellence REBIRTH‐2 (from Regenerative Biology to Reconstructive Therapy) grant to Sedding, Daniel, Wollert, and Bauersachs. Preissner was supported by a grant from the LOEWE network “Medicinal RNomics” (Wiesbaden, Germany). Cabrera‐Fuentes is funded by a Startup Grant of the “Excellence Cluster Cardio‐Pulmonary System” (ECCPS) from the German Research Foundation (DFG, Bonn, Germany) and “Peter and Traudl Engelhorn‐Stiftung” (Weilheim, Germany). Part of the work from Cabrera‐Fuentes was supported by the Russian Government Program for competitive growth of Kazan Federal University.
Disclosures
None.
(J Am Heart Assoc. 2017;6:e004541 DOI: 10.1161/JAHA.116.004541.)28637776
References
- 1. Garcia‐Dorado D, Andres‐Villarreal M, Ruiz‐Meana M, Inserte J, Barba I. Myocardial edema: a translational view. J Mol Cell Cardiol. 2012;52:931–939. [DOI] [PubMed] [Google Scholar]
- 2. Dongaonkar RM, Stewart RH, Geissler HJ, Laine GA. Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc Res. 2010;87:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mehlhorn U, Geissler HJ, Laine GA, Allen SJ. Myocardial fluid balance. Eur J Cardiothorac Surg. 2001;20:1220–1230. [DOI] [PubMed] [Google Scholar]
- 4. Garcia‐Dorado D, Oliveras J. Myocardial oedema: a preventable cause of reperfusion injury? Cardiovasc Res. 1993;27:1555–1563. [DOI] [PubMed] [Google Scholar]
- 5. Seal JB, Gewertz BL. Vascular dysfunction in ischemia‐reperfusion injury. Ann Vasc Surg. 2005;19:572–584. [DOI] [PubMed] [Google Scholar]
- 6. Cabrera‐Fuentes HA, Aragones J, Bernhagen J, Boening A, Boisvert WA, Botker HE, Bulluck H, Cook S, Di Lisa F, Engel FB, Engelmann B, Ferrazzi F, Ferdinandy P, Fong A, Fleming I, Gnaiger E, Hernandez‐Resendiz S, Kalkhoran SB, Kim MH, Lecour S, Liehn EA, Marber MS, Mayr M, Miura T, Ong SB, Peter K, Sedding D, Singh MK, Suleiman MS, Schnittler HJ, Schulz R, Shim W, Tello D, Vogel CW, Walker M, Li QO, Yellon DM, Hausenloy DJ, Preissner KT. From basic mechanisms to clinical applications in heart protection, new players in cardiovascular diseases and cardiac theranostics: meeting report from the third international symposium on “new frontiers in cardiovascular research”. Basic Res Cardiol. 2016;111:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Preissner KT. Extracellular RNA. A new player in blood coagulation and vascular permeability. Hamostaseologie. 2007;27:373–377. [PubMed] [Google Scholar]
- 8. Deindl E, Fischer S, Preissner KT. New directions in inflammation and immunity: the multi‐functional role of the extracellular RNA/RNase system. Indian J Biochem Biophys. 2009;46:461–466. [PubMed] [Google Scholar]
- 9. Fischer S, Cabrera‐Fuentes HA, Noll T, Preissner KT. Impact of extracellular RNA on endothelial barrier function. Cell Tissue Res. 2014;355:635–645. [DOI] [PubMed] [Google Scholar]
- 10. Cabrera‐Fuentes HA, Lopez ML, McCurdy S, Fischer S, Meiler S, Baumer Y, Galuska SP, Preissner KT, Boisvert WA. Regulation of monocyte/macrophage polarisation by extracellular RNA. Thromb Haemost. 2015;113:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, von Bruehl ML, Sedding D, Massberg S, Gunther A, Engelmann B, Preissner KT. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA. 2007;104:6388–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fischer S, Gerriets T, Wessels C, Walberer M, Kostin S, Stolz E, Zheleva K, Hocke A, Hippenstiel S, Preissner KT. Extracellular RNA mediates endothelial‐cell permeability via vascular endothelial growth factor. Blood. 2007;110:2457–2465. [DOI] [PubMed] [Google Scholar]
- 13. Muller J, Gorressen S, Grandoch M, Feldmann K, Kretschmer I, Lehr S, Ding Z, Schmitt JP, Schrader J, Garbers C, Heusch G, Kelm M, Scheller J, Fischer JW. Interleukin‐6‐dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res Cardiol. 2014;109:440. [DOI] [PubMed] [Google Scholar]
- 14. Fischer S, Gesierich S, Griemert B, Schanzer A, Acker T, Augustin HG, Olsson AK, Preissner KT. Extracellular RNA liberates tumor necrosis factor‐alpha to promote tumor cell trafficking and progression. Cancer Res. 2013;73:5080–5089. [DOI] [PubMed] [Google Scholar]
- 15. Kleinert E, Langenmayer MC, Reichart B, Kindermann J, Griemert B, Blutke A, Troidl K, Mayr T, Grantzow T, Noyan F, Abicht JM, Fischer S, Preissner KT, Wanke R, Deindl E, Guethoff S. Ribonuclease (RNase) prolongs survival of grafts in experimental heart transplantation. J Am Heart Assoc. 2016;5:e003429 DOI: 10.1161/JAHA.116.003429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walberer M, Tschernatsch M, Fischer S, Ritschel N, Volk K, Friedrich C, Bachmann G, Mueller C, Kaps M, Nedelmann M, Blaes F, Preissner KT, Gerriets T. RNase therapy assessed by magnetic resonance imaging reduces cerebral edema and infarction size in acute stroke. Curr Neurovasc Res. 2009;6:12–19. [DOI] [PubMed] [Google Scholar]
- 17. Simsekyilmaz S, Cabrera‐Fuentes HA, Meiler S, Kostin S, Baumer Y, Liehn EA, Weber C, Boisvert WA, Preissner KT, Zernecke A. Role of extracellular RNA in atherosclerotic plaque formation in mice. Circulation. 2014;129:598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laine GA, Allen SJ. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ Res. 1991;68:1713–1721. [DOI] [PubMed] [Google Scholar]
- 19. Rassaf T, Totzeck M, Hendgen‐Cotta UB, Shiva S, Heusch G, Kelm M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res. 2014;114:1601–1610. [DOI] [PubMed] [Google Scholar]
- 20. Cabrera‐Fuentes HA, Ruiz‐Meana M, Simsekyilmaz S, Kostin S, Inserte J, Saffarzadeh M, Galuska SP, Vijayan V, Barba I, Barreto G, Fischer S, Lochnit G, Ilinskaya ON, Baumgart‐Vogt E, Boning A, Lecour S, Hausenloy DJ, Liehn EA, Garcia‐Dorado D, Schluter KD, Preissner KT. RNase1 prevents the damaging interplay between extracellular RNA and tumour necrosis factor‐alpha in cardiac ischaemia/reperfusion injury. Thromb Haemost. 2014;112:1110–1119. [DOI] [PubMed] [Google Scholar]
- 21. Friedrich MG. Myocardial edema–a new clinical entity? Nat Rev Cardiol. 2010;7:292–296. [DOI] [PubMed] [Google Scholar]
- 22. Gansler J, Preissner KT, Fischer S. Influence of proinflammatory stimuli on the expression of vascular ribonuclease 1 in endothelial cells. FASEB J. 2014;28:752–760. [DOI] [PubMed] [Google Scholar]
- 23. Chen C, Feng Y, Zou L, Wang L, Chen HH, Cai JY, Xu JM, Sosnovik DE, Chao W. Role of extracellular RNA and TLR3‐Trif signaling in myocardial ischemia‐reperfusion injury. J Am Heart Assoc. 2014;3:e000683 DOI: 10.1161/JAHA.113.000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Higgins CB, Herfkens R, Lipton MJ, Sievers R, Sheldon P, Kaufman L, Crooks LE. Nuclear magnetic resonance imaging of acute myocardial infarction in dogs: alterations in magnetic relaxation times. Am J Cardiol. 1983;52:184–188. [DOI] [PubMed] [Google Scholar]
- 25. Garcia‐Dorado D, Oliveras J, Gili J, Sanz E, Perez‐Villa F, Barrabes J, Carreras MJ, Solares J, Soler‐Soler J. Analysis of myocardial oedema by magnetic resonance imaging early after coronary artery occlusion with or without reperfusion. Cardiovasc Res. 1993;27:1462–1469. [DOI] [PubMed] [Google Scholar]
- 26. Tilak GS, Hsu LY, Hoyt RF Jr, Arai AE, Aletras AH. In vivo T2‐weighted magnetic resonance imaging can accurately determine the ischemic area at risk for 2‐day‐old nonreperfused myocardial infarction. Invest Radiol. 2008;43:7–15. [DOI] [PubMed] [Google Scholar]
- 27. Fernandez‐Jimenez R, Garcia‐Prieto J, Sanchez‐Gonzalez J, Aguero J, Lopez‐Martin GJ, Galan‐Arriola C, Molina‐Iracheta A, Doohan R, Fuster V, Ibanez B. Pathophysiology underlying the bimodal edema phenomenon after myocardial ischemia/reperfusion. J Am Coll Cardiol. 2015;66:816–828. [DOI] [PubMed] [Google Scholar]
- 28. Fischer S, Nishio M, Peters SC, Tschernatsch M, Walberer M, Weidemann S, Heidenreich R, Couraud PO, Weksler BB, Romero IA, Gerriets T, Preissner KT. Signaling mechanism of extracellular RNA in endothelial cells. FASEB J. 2009;23:2100–2109. [DOI] [PubMed] [Google Scholar]
- 29. Inserte J, Cardona M, Poncelas‐Nozal M, Hernando V, Vilardosa U, Aluja D, Parra VM, Sanchis D, Garcia‐Dorado D. Studies on the role of apoptosis after transient myocardial ischemia: genetic deletion of the executioner caspases‐3 and ‐7 does not limit infarct size and ventricular remodeling. Basic Res Cardiol. 2016;111:18. [DOI] [PubMed] [Google Scholar]
- 30. Navar PD, Navar LG. Relationship between colloid osmotic pressure and plasma protein concentration in the dog. Am J Physiol. 1977;233:H295–H298. [DOI] [PubMed] [Google Scholar]
- 31. Fernandez‐Jimenez R, Sanchez‐Gonzalez J, Aguero J, Garcia‐Prieto J, Lopez‐Martin GJ, Garcia‐Ruiz JM, Molina‐Iracheta A, Rossello X, Fernandez‐Friera L, Pizarro G, Garcia‐Alvarez A, Dall'Armellina E, Macaya C, Choudhury RP, Fuster V, Ibanez B. Myocardial edema after ischemia/reperfusion is not stable and follows a bimodal pattern: imaging and histological tissue characterization. J Am Coll Cardiol. 2015;65:315–323. [DOI] [PubMed] [Google Scholar]