

Abstract
Background
Acute kidney injury (AKI) following primary percutaneous coronary intervention (pPCI) is frequently interpreted as contrast‐induced AKI but may result from other insults. We aimed to determine the causal association of contrast material exposure and the incidence of AKI following pPCI using a control group of propensity score–matched patients with ST‐segment–elevation myocardial infarction who were not exposed to contrast material.
Methods and Results
We studied 2025 patients with ST‐segment–elevation myocardial infarction who underwent pPCI and 1025 patients receiving fibrinolysis or no reperfusion who were not exposed to contrast material during the first 72 hours of hospital stay (control group). AKI was defined as creatinine of ≥0.5 mg/dL or >25% rise within 72 hours. AKI rates were similar in the pPCI and control groups (10.3% versus 12.1%, respectively; P=0.38). Propensity score matching resulted in 931 matched pairs with PCI and no PCI, with balanced baseline covariates (standardized difference <0.1). Among propensity score–matched patients, AKI rates were not significantly different with and without PCI (8.6% versus 10.9%, P=0.12). In the pPCI cohort, independent predictors of AKI included age ≥70 years, insulin‐treated diabetes mellitus, diuretic therapy, anterior infarction, baseline estimated glomerular filtration rate, and variables related to the presence of pump failure (higher Killip class, intra‐aortic balloon pump use) and reduced left ventricular ejection fraction but not contrast material dose. A risk score based on the PCI cohort had similar discriminatory capacity for AKI in the control group (C statistic 0.81±0.02 and 0.78±0.02, respectively; P=0.26).
Conclusions
The development of AKI in patients with ST‐segment–elevation myocardial infarction undergoing pPCI is mainly related to older age, baseline estimated glomerular filtration rate, heart failure, and hemodynamic instability. Risk for AKI is similar among ST‐segment–elevation myocardial infarction patients with and without contrast material exposure.
Keywords: contrast media, contrast‐induced nephropathy, myocardial infarction, primary percutaneous coronary intervention
Subject Categories: Nephrology and Kidney, Percutaneous Coronary Intervention, Coronary Artery Disease, Myocardial Infarction
Clinical Perspective
What Is New?
In patients with ST‐segment–elevation myocardial infarction treated with primary percutaneous coronary intervention (PCI), acute kidney injury (AKI) is frequently interpreted as being induced by contrast media but may result from multiple other contributing factors. Previous studies linking CM use to AKI in the setting of primary PCI inherently lacked a control group of patients in whom CM was not administered.
The current study of patients with ST‐segment–elevation myocardial infarction undergoing primary PCI (patients exposed to contrast media) and propensity score–matched patients treated with thrombolysis or without reperfusion (patients not exposed to contrast media), demonstrates similar incidence of AKI.
The impact of AKI on clinical outcomes was similar among patients with and without contrast exposure.
What Are the Clinical Implications?
It is unlikely that contrast media exposure is a primary pathogenic factor responsible for renal dysfunction following primary PCI.
The worse clinical outcomes after AKI are independent of contrast material exposure.
Attempts to reduce AKI rates in patients with ST‐segment–elevation myocardial infarction likely require targeting mechanisms that are unrelated to contrast media.
Introduction
Patients undergoing primary percutaneous coronary intervention (pPCI) are considered to be at higher risk of contrast medium (CM)–induced acute kidney injury (CI‐AKI) compared with those undergoing elective interventions.1 Several studies have shown that CI‐AKI following pPCI is associated with increased mortality.2, 3 In these studies, AKI is frequently interpreted as CI‐AKI.1, 3 However, CI‐AKI is defined as an acute deterioration of renal function after administration of radiocontrast media in the absence of other alternative explanations for renal impairment.4, 5 Although this may be a reasonable assumption in patients undergoing elective PCI, deterioration of renal function after pPCI may be the result of multiple other contributing factors including hemodynamic alterations, neurohormonal activation, and initiation of nephrotoxic drugs.
Recent studies evaluating the propensity of contrast exposure to induce AKI in noncardiovascular settings demonstrated that intravenous contrast is not an independent risk factor for AKI, dialysis, or mortality.6, 7, 8 Importantly, because studies linking CM use to AKI in the setting of pPCI lack a control group in which CM was not administered, it is impossible to distinguish CM‐dependent AKI from multiple additional risk factors other than exposure to contrast media.9 Consequently, studies that include ST‐segment–elevation myocardial infarction (STEMI) patients who were not exposed to CM are desirable.
The aim of the present study was to identify the specific clinical correlates that produced AKI in patients with acute ST‐elevation myocardial infarction undergoing pPCI, and the relative contribution of contrast media exposure to the development of AKI. We also sought to compare the incidence of AKI between STEMI patients with and without CM exposure.
Methods
We used a prospective database consisting of all STEMI patients admitted to the intensive care unit at the Rambam Medical Center, Haifa, Israel, from January 2000 and September 2015. The investigational review committee on human research approved the study protocol, and the need for written informed consent was specifically waived.
The decision to use pPCI was based on the practice at the time the patient was admitted. In the beginning of the period, patients eligible for reperfusion (generally those admitted within 12 hours from symptom onset) were treated mainly with thrombolytic therapy, although patients perceived to be at high risk (eg, those with hemodynamic instability, large anterior infarction) were submitted to pPCI. Over the years, the proportion of primary PCI as the preferred treatment modality in STEMI increased, and from mid‐2007, all patients underwent pPCI.
Primary PCI Cohort
The cohort included consecutive STEMI patients who underwent pPCI. All patients received nonionic, low‐osmolar, iodinated contrast agents.
Thrombolysis/No Reperfusion Cohort
This cohort included patients who were admitted during the same period, who were treated with thrombolysis or who received no reperfusion therapy at all, and who were not exposed to CM during the first 72 hours of hospital stay. This cohort served to study the risk of developing AKI in the absence of contrast exposure in the setting of STEMI.
Assessment of Renal Function
Venous blood samples were obtained at admission and at 24, 48, and 72 hours thereafter. After the first 72 hours from admission, creatinine measurements were checked only when clinically indicated. Estimated glomerular filtration rate (eGFR) was calculated based on the abbreviated MDRD (Modification of Diet in Renal Disease) study equation.
Definitions
AKI was defined as an increase in serum creatinine concentration ≥0.5 mg/dL compared with admission value or a >25% relative rise during the first 72 hours after the procedure.3
Several predictive indices for AKI after radiocontrast administration were calculated for each patient:
Maximum contrast dose using an empirical formula: maximum contrast dose (mL)=(5×body weight [kg])/serum creatinine (mg/dL).10 The contrast ratio is determined by dividing the administered contrast amount by the calculated maximum contrast dose.
Total volume of contrast given divided by creatinine clearance (shown as CrCl), as estimated by the Cockcroft‐Gault method: V/CrCl.
The total volume of contrast given divided by eGFR: V/eGFR.11
Statistical Analysis
The baseline characteristics of groups were compared using the unpaired t test for continuous variables and the χ2 statistic for noncontinuous variables. The area under the receiver operating characteristic (ROC) curve was used as a measure that reflects the discriminatory power of each predictive index for the diagnosis of AKI. Areas under the ROC curves were compared using the DeLong method.
The amount of contrast agent was modeled using an indicator variable (quartiles of the distribution). Logistic regression was performed with AKI as the dependent variable to model the association between contrast volume and other potential risk factors and the development of AKI. Stepwise variable selection with a significance level of 0.05 (Wald test) for variable retention was used to develop parsimonious predictor models. All variables in Table 1 were considered for the model. Model discrimination was assessed by the area under the ROC curve.
Table 1.
Baseline Clinical Characteristics of the pPCI Cohort
| Characteristics | No AKI (n=1816) | AKI (n=209) | P Value |
|---|---|---|---|
| Age, y | 59±12 | 66±13 | <0.0001 |
| Female sex | 324 (18) | 51 (24) | 0.02 |
| Weight, kg | 81±16 | 79±17 | 0.16 |
| Hypertension | 806 (44) | 122 (61) | <0.0001 |
| Diabetes mellitus | |||
| Noninsulin treated | 380 (21) | 46 (22) | 0.72 |
| Insulin treated | 66 (4) | 26 (12) | <0.0001 |
| Killip Class II–III | 185 (10) | 53 (25) | <0.0001 |
| Killip Class IV or IABP use | 82 (6) | 57 (27) | <0.0001 |
| Previous infarction | 384 (19) | 50 (24) | 0.10 |
| Anterior infarction | 858 (47) | 134 (64) | <0.0001 |
| Left ventricular ejection fraction <45% | 556 (31) | 124 (59) | <0.0001 |
| Baseline renal function | |||
| Baseline creatinine, mg/dL | 0.99±0.32 | 1.32±0.87 | <0.0001 |
| Baseline eGFR, mL/min per 1.73 m2 | 82±22 | 65±30 | <0.0001 |
| eGFR <60 mL/min per 1.73 m2 | 850 (47) | 143 (68) | <0.0001 |
| Baseline hemoglobin | 14.0±1.6 | 13.5±2.0 | 0.0001 |
| Medical therapy | |||
| Antiplatelet agents | 1811 (100) | 209 (100) | 0.45 |
| Beta blockers | 764 (89) | 84 (81) | 0.01 |
| ACE inhibitors/ARBs | 766 (89) | 85 (82) | 0.02 |
| Diuretics | 240 (28) | 49 (47) | <0.0001 |
Data are number (%) or mean±SD. ACE indicates angiotensin‐converting enzyme; AKI, acute kidney injury; ARB, angiotensin‐receptor blocker; eGFR, estimated glomerular filtration rate; IABP, intra‐aortic balloon pump; pPCI, primary percutaneous coronary intervention.
An additive risk score for the prediction of AKI was developed on the pPCI cohort from the multiple logistic regression model by using a regression coefficient–based scoring method.12 For simplicity of use, scores were approximated by rounding coefficients to the nearest half integer. The risk score was used to predict AKI in the cohort of patients who were not exposed to CM.
To investigate the effect of contrast exposure on the change in creatinine levels in the first 72 hours as a continuous variable, we adopted a linear mixed‐model approach with random slopes and intercepts. The model included terms for pPCI versus no pPCI, baseline creatinine, time, and an interaction term between pPCI and time. This model also included other covariates that potentially affected the change in creatinine as fixed effects.
Propensity score estimates representing the probability of pPCI were generated in both the CM (pPCI) and non‐CM groups by using a logistic regression model derived from 16 clinical variables (Table 1). Following propensity score generation, patients were matched by using 1:1 nearest neighbor (greedy type) matching and a caliper width of 0.1 standard deviation of the propensity score logit. Matching was performed without replacement, and nonmatched results were discarded.
We assessed the success of the matches by examining standardized differences (measured in percentage points) in the observed confounders between the matched pPCI and non‐pPCI groups.13 Small (<10%) standardized differences support the assumption of balance between treatment groups based on observed confounders. The incidence of AKI was compared between CM and non‐CM groups following propensity score matching by using the Fisher exact test.
Event‐free survival curves were estimated by the Kaplan–Meier method and compared with the log‐rank test. Univariate and multivariate Cox proportional hazards models were used to model the relationship between contrast volume and 1‐year composite major adverse cardiac events (consisting of death, readmission for heart failure, and reinfarction). The model adjusted for all variables in Table 1. Variables found to show marginal association with mortality in univariate analysis (P<0.20) were used in the multivariate model.
Differences were considered statistically significant at the 2‐sided P<0.05 level. Statistical analyses were performed using the Stata statistical software (version 13.1).
Results
Between January 2000 and December 2015, a total of 2200 patients presenting with acute STEMI underwent pPCI. Patients were excluded because of missing creatinine measurements (no repeated creatinine measurement; n=54) or contrast volume data (n=121). The remaining 2025 patients composed the pPCI cohort. The thrombolysis/no reperfusion cohort included 1025 STEMI patients admitted during the same period, with 403 treated with thrombolytic agents and 622 who did not receive any reperfusion therapy.
Characteristics of the pPCI cohort according to AKI status are shown in Table 1. Patients who developed AKI during their hospital course were older and had a higher prevalence of hypertension and lower eGFR; they presented with higher Killip class and were more likely to have anterior infarction, present with cardiogenic shock, and to receive treatment with an intra‐aortic balloon pump. Patients with AKI were less likely to receive β‐blockers or angiotensin‐converting enzyme inhibitors and more likely to receive diuretics.
Relationship of Contrast Volume and AKI
The median amount of contrast administered was 150 mL (interquartile range 100–200 mL; range 35–550 mL). Of the total 3050 patients, 22 (0.7%), 265 (8.7%), and 771 (25.3%) were missing creatinine values at the 24, 48, and 72 hours, respectively. Using the cutoff of ≥0.5 mg/dL or a 25% increase in serum creatinine, 209 of the 2025 pPCI patients (10.3%) developed AKI. The median contrast dose was 160 mL in patients with AKI (interquartile range 120–210 mL) and 150 mL (interquartile range 120–200 mL) in patients without AKI (P=0.12).
ROC analysis demonstrated that contrast volume alone was not a predictor of AKI (Figure 1). The calculation of V/eGFR displayed the best discriminatory power for AKI but with low accuracy overall (Figure 1).
Figure 1.
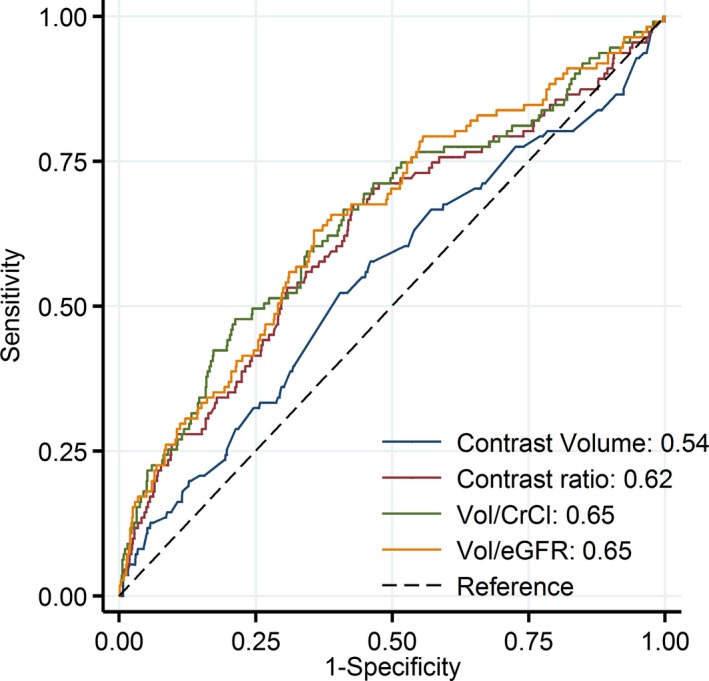
Comparison of areas under the receiver operator characteristic curves that relate various contrast media indices to the development of acute kidney injury. CrCl indicates creatinine clearance; eGFR, estimated glomerular filtration rate; Vol, volume.
The association of clinical variables and quartiles of V/eGFR with the risk of developing AKI was assessed using a logistic regression model. Significant univariable predictors of AKI included age, insulin‐treated diabetes mellitus, lower hemoglobin levels, anterior infarction, Killip class and intra‐aortic balloon pump (IABP) use, diuretic therapy, eGFR, and V/eGFR (Table 2, model 1).
Table 2.
Univariable and Multivariate Logistic Regression Model for AKI
| Variable | Unadjusted | Multivariate Results | ||
|---|---|---|---|---|
| Odds Ratio (95% CI) | P Value | Odds Ratio (95% CI) | P Value | |
| Model 1: primary PCI patients | ||||
| Age ≥70 y | 3.33 (2.47–4.88) | <0.0001 | 1.89 (1.33–2.71) | <0.0001 |
| Hypertension | 1.94 (1.45–2.60) | <0.0001 | ··· | ··· |
| Diabetes mellitus | ||||
| No diabetes mellitus | 1.0 (Referent) | ··· | 1.0 (Referent) | ··· |
| Noninsulin treated | 1.37 (0.98–1.91) | 0.07 | 0.90 (0.61–1.32) | 0.59 |
| Insulin treated | 3.34 (1.93–5.77) | <0.0001 | 2.03 (1.06–3.89) | 0.03 |
| Anterior infarction | 2.00 (1.48–2.69) | <0.0001 | 1.51 (1.06–2.13) | 0.02 |
| eGFR, mL/min per 1.73 m2 | ||||
| ≥60 | 1.0 (Referent) | ··· | 1.0 (Referent) | ··· |
| 30–59 | 3.41 (2.23–5.20) | <0.0001 | 1.71 (1.17–2.50) | 0.005 |
| <30 | 15.8 (6.19–40.30) | <0.0001 | 6.27 (3.15–12.49) | <0.0001 |
| Hemoglobin (per 1‐g/L increase) | 0.81 (0.74–0.88) | <0.0001 | ··· | ··· |
| Killip class I | 1.0 (Referent) | ··· | 1.0 (Referent) | |
| Killip class II–III | 4.47 (3.10–6.47) | <0.0001 | 2.19 (1.45–3.31) | <0.0001 |
| Killip class IV or IABP use | 10.85 (7.31–16.09) | <0.0001 | 6.59 (4.30–10.08) | <0.0001 |
| LVEF <45% | 3.31 (2.27–4.43) | <0.0001 | 1.68 (1.18–2.39) | 0.004 |
| Diuretic therapy | 4.02 (2.96–5.40) | <0.0001 | 1.81 (1.27–2.57) | 0.001 |
| V/eGFR quartile | ||||
| First (<2.0) | 1.0 (Referent) | ··· | ··· | ··· |
| Second (2.0–2.8) | 0.940 (0.54–1.51) | 0.79 | ··· | ··· |
| Third (2.9–3.8) | 1.53 (0.97–2.43) | 0.07 | ··· | ··· |
| Fourth (>3.8) | 3.40 (1.24–5.16) | <0.0001 | ··· | ··· |
| Model 2: all patients | ||||
| Age ≥70 y | 3.57 (2.83–4.51) | <0.0001 | 1.96 (1.48–2.60) | <0.0001 |
| Hypertension | 2.00 (1.58–2.52) | <0.0001 | 1.38 (1.05–1.80) | 0.02 |
| Diabetes mellitus | ||||
| No diabetes mellitus | 1.0 (Referent) | ··· | 1.0 (Referent) | ··· |
| Noninsulin treated | 1.25 (0.96–1.64) | 0.10 | 0.83 (0.62–1.13) | 0.24 |
| Insulin treated | 3.60 (2.36–5.47) | <0.0001 | 2.08 (1.38–3.37) | 0.003 |
| Anterior infarction | 1.91 (1.52–2.41) | <0.0001 | 1.53 (1.21–2.00) | 0.002 |
| eGFR, mL/min per 1.73 m2 | ||||
| ≥60 | 1.0 (Referent) | ··· | 1.0 (Referent) | ··· |
| 30–59 | 3.65 (2.84–4.68) | <0.0001 | 1.71 (1.27–2.30) | <0.0001 |
| <30 | 7.97 (5.03–12.63) | <0.0001 | 3.01 (1.75–5.17) | <0.0001 |
| Hemoglobin (per 1‐g/L increase) | 0.85 (0.80–0.91) | <0.0001 | 0.98 (0.91–1.05) | 0.56 |
| Killip class I | 1.0 (Referent) | ··· | 1.0 (Referent) | ··· |
| Killip class II–III | 4.73 (3.32–5.77) | <0.0001 | 1.99 (1.45–2.73) | <0.0001 |
| Killip class IV or IABP use | 12.18 (8.63–17.19) | <0.0001 | 7.43 (5.10–10.84) | <0.0001 |
| LVEF <45% | 2.88 (2.28–3.63) | <0.0001 | 1.44 (1.09–1.90) | 0.011 |
| Diuretic therapy | 3.59 (2.85–4.54) | <0.0001 | 1.75 (1.32–2.32) | <0.0001 |
| Primary PCI vs no PCI | 0.84 (0.66–1.06) | 0.14 | 0.79 (0.60–1.05) | 0.10 |
ACE indicates angiotensin‐converting enzyme; AKI, acute kidney injury; ARB, angiotensin receptor blocker; CI, confidence interval; eGFR, estimated glomerular filtration rate; IABP, intra‐aortic balloon pump; LVEF, left ventricular ejection fraction; PCI, percutaneous coronary intervention.
After adjustments for eGFR alone, V/eGFR was no longer a significant predictor of AKI (P=0.47). In the final multivariable model, independent predictors of AKI included age ≥70 years, insulin‐treated diabetes mellitus, diuretic therapy, anterior infarction, baseline eGFR, and variables related to the presence of pump failure (higher Killip class and use of IABP) and reduced left ventricular ejection fraction (Table 2, model 1).
Several sensitivity analyses were conducted for the logistic regression models with varying CM cutoffs and definitions of AKI. When contrast exposure was modeled in the whole study population, the adjusted odds ratio for AKI in patients undergoing primary PCI versus no PCI was 0.79 (95% confidence interval [CI], 0.60–1.05, P=0.10; Table 2, model 2).
We analyzed the effect of CM as a dichotomous variable above and below median value. Compared with patients with contrast volume below median value, the adjusted odds ratio for AKI was 1.15 (95% CI 0.83–1.59, P=0.39) in patients with contrast volume above median. When AKI was defined using a higher increments of serum creatinine (≥1.0 mg/dL or a ≥50% increase), the adjusted odds ratio for AKI in the upper quartile compared with the lower quartile of contrast volume was 1.18 (95% CI, 0.71–1.96; P=0.52).
We also tested the interaction between the effect of CM exposure and risk factors for AKI. There was no significant interaction between the effect of CM and eGFR <60 mL/min per 1.73 m2 (P=0.39), diabetes mellitus (P=0.18), use of diuretics (P=0.07), age (P=0.12), Killip class (P=0.20), or reduced left ventricular ejection fraction (P=0.14).
Predictors of AKI in STEMI Patients Without pPCI
Table 3 shows the clinical characteristics of STEMI patients who underwent pPCI compared with those who did not undergo pPCI. Patients who were not treated with pPCI had more risk factors for AKI, including older age, worse baseline renal function, and higher rates of Killip class II or III. However, they were less likely to have cardiogenic shock, IABP use, or anterior infarction. Baseline renal function was slightly better in the pPCI cohort, and the use of β‐blockers, angiotensin‐converting enzyme inhibitors, or angiotensin II receptor blockers was more frequent.
Table 3.
Baseline Clinical Characteristics in Patients With and Without pPCI
| Characteristics | Before Propensity Score Matching | After Propensity Score Matching | ||||
|---|---|---|---|---|---|---|
| pPCI (n=2025) | Thrombolysis/No Reperfusion (n=1025) | P Value | pPCI (n=931) | Thrombolysis/No Reperfusion (n=931) | P Value | |
| Age, y | 60±13 | 63±13 | <0.0001 | 62±12 | 62±13 | 0.33 |
| Female sex | 375 (19) | 263 (26) | <0.0001 | 222 (24) | 222 (24) | 1.0 |
| Hypertension | 993 (46) | 481 (47) | 0.66 | 434 (47) | 427 (46) | 0.75 |
| Diabetes mellitus | ||||||
| Noninsulin treated | 426 (21) | 275 (27) | <0.0001 | 229 (25) | 239 (26) | 0.59 |
| Insulin treated | 92 (5) | 30 (3) | 0.03 | 25 (3) | 28 (3) | 0.68 |
| Killip class II–III | 238 (12) | 195 (19) | <0.0001 | 144 (15) | 151 (16) | 0.66 |
| Killip class IV or IABP use | 139 (7) | 28 (3) | <0.0001 | 23 (2) | 27 (3) | 0.57 |
| Previous infarction | 398 (20) | 232 (23) | 0.06 | 209 (22) | 209 (22) | 1.00 |
| Anterior infarction | 992 (49) | 328 (32) | <0.0001 | 312 (34) | 306 (33) | 0.77 |
| LVEF <45% | 680 (34) | 351 (34%) | 0.71 | 305 (33) | 328 (35) | 0.26 |
| Baseline renal function | ||||||
| Baseline creatinine, mg/dL | 1.03±0.42 | 1.06±0.37 | 0.03 | 1.04±0.35 | 1.04±0.46 | 0.67 |
| Baseline eGFR, mL/min per 1.73 m2 | 80±23 | 74±24 | <0.0001 | 77±22 | 77±23 | 0.78 |
| eGFR categories, mL/min per 1.73 m2 | ||||||
| >60 | 1633 (80) | 738 (62) | <0.0001 | 719 (77) | 698 (75) | 0.25 |
| 45–60 | 226 (11) | 154 (15) | 0.002 | 123 (13) | 135 (15) | 0.42 |
| 30–44 | 118 (6) | 95 (9) | <0.0001 | 59 (6) | 72 (8) | 0.24 |
| <30 | 48 (2) | 38 (4) | 0.04 | 30 (3) | 26 (3) | 0.59 |
| Baseline hemoglobin | 14.0±1.2 | 13.3±2.0 | <0.0001 | 13.9±1.7 | 14.0±1.7 | 0.20 |
| Medical therapy in hospital | ||||||
| Antiplatelet agents | 2020 (100) | 986 (96) | <0.0001 | 928 (100) | 925 (99) | 0.32 |
| Beta blockers | 1811 (89) | 841 (82) | <0.0001 | 801 (86) | 786 (84) | 0.33 |
| ACE inhibitors/ARBs | 1825 (90) | 828 (81) | <0.0001 | 791 (85) | 778 (84) | 0.41 |
| Diuretics | 637 (31) | 234 (23) | <0.0001 | 246 (26) | 258 (28) | 0.53 |
Data are number (%) or mean±SD. ACE indicates angiotensin‐converting enzyme; ARB, angiotensin receptor blocker; eGFR, estimated glomerular filtration rate; IABP, intra‐aortic balloon pump; LVEF, left ventricular ejection fraction; pPCI, primary percutaneous coronary intervention.
AKI occurred in 124 (12.1%) patients receiving either thrombolytic therapy or no reperfusion (P=0.14 compared with the pPCI cohort). The multivariable logistic model in Table 2 was used to generate a risk score for AKI based on the multivariate‐adjusted coefficients as follows: age ≥70 years, 7 points; anterior infarction, 4 points, eGFR 30 to 60 mL/min per 1.73 m2, 5 points; eGFR <30 mL/min per 1.73 m2, 18 points; insulin therapy, 7 points; Killip class II or III, 8 points; Killip class IV or IABP use, 19 points; left ventricular ejection fraction <45%, 5 points; diuretic therapy, 6 points. Application of the risk score that was derived from the pPCI patients to the thrombolysis/no reperfusion population revealed a similar discriminatory capacity for AKI. The area under the ROC curve was 0.81±0.02 in the derivation pPCI group and 0.78±0.02 in thrombolysis/no reperfusion patients (P=0.26; Figure 2).
Figure 2.
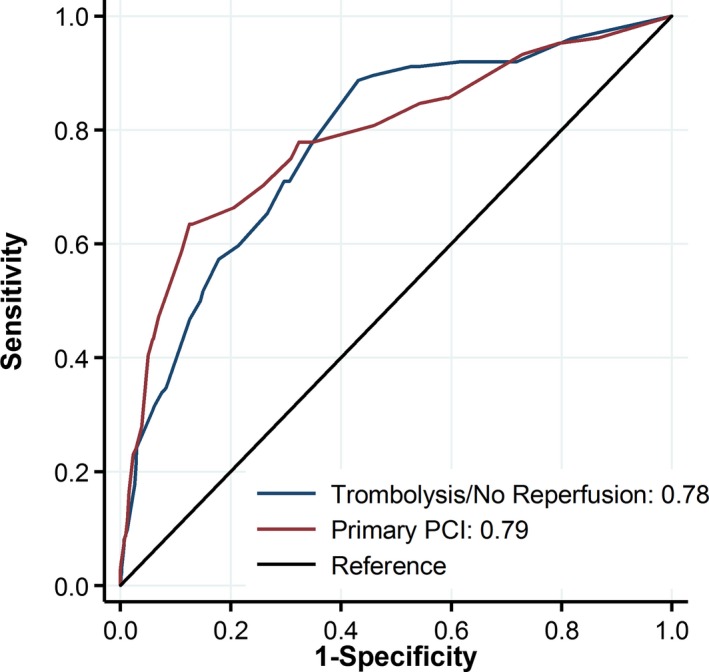
ROC curves of the simplified scoring model for acute kidney injury in the primary percutaneous coronary intervention (PCI) and thrombolysis/no reperfusion groups.
Propensity Score Matching
One‐to‐one matching of the propensity score yielded a total of 931 patient pairs. Patients were well matched with respect to the 16 variables included in the propensity model (Figure 3 and Table 3, right panel), with a mean difference in paired propensity scores of 0.8% (95% CI, −2.1% to 0.5%). Following propensity score matching, the incidence of AKI in both the CM group (80 AKI events, 8.6%) and the non–CM group (101 AKI events, 10.9%) was similar (odds ratio: 0.77; 95% CI, 0.56–1.06; P=0.12).
Figure 3.
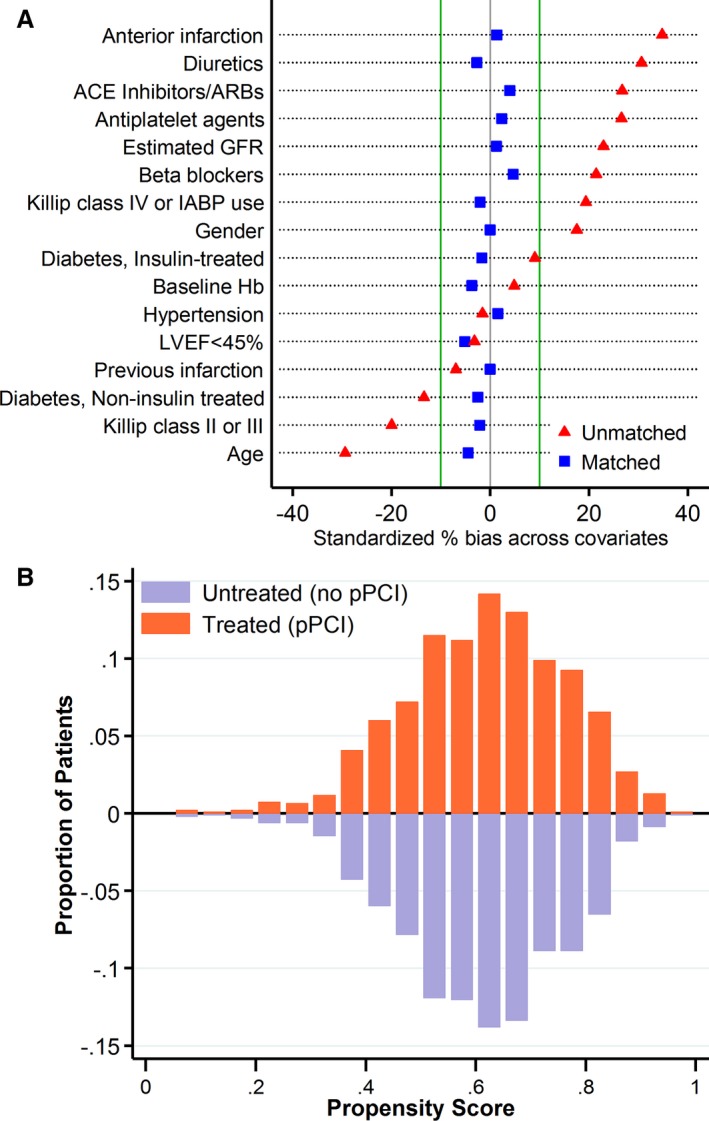
A, Covariable balance before (red triangles) and after (blue squares) matching. The standardized difference after propensity matching (blue squares) are all well within 10%. B, Mirrored histogram of distribution of propensity scores for primary percutaneous coronary intervention (pPCI; bars above the zero line) vs no‐pPCI (bars below the zero line). ACE indicates angiotensin‐converting enzyme; ARB, angiotensin receptor blocker; GFR, glomerular filtration rate; Hb, hemoglobin; IABP, intra‐aortic balloon pump; LVEF, left ventricular ejection fraction.
Assessment of Creatinine Changes as a Continuous Variable
We also modeled the change in renal function over the first 72 hours of hospital stay as a continuous variable in patients with and without CM exposure, using linear mixed models. Serum creatinine trajectory was linear over the first 72 hours of hospitalization, with a modest increase in both the pPCI and non‐PCI groups of 0.036 mg/dL per day (95% CI, 0.023–0.050) and 0.037 mg/dL per day (95% CI, 0.027–0.047), respectively. There was no significant interaction between PCI and time with regard to creatinine change (P=0.97), indicating that the rate of change in serum creatinine was similar with and without PCI (Figure 4). Similar results were obtained after propensity score matching (P=0.77).
Figure 4.
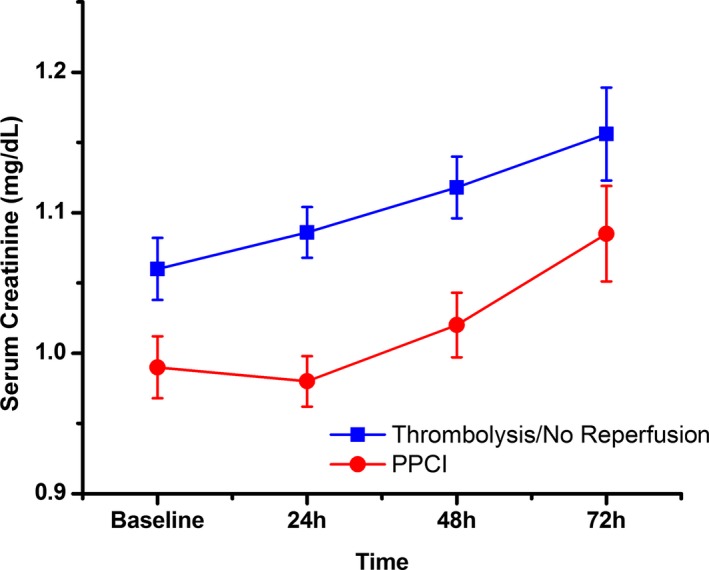
Fitted mean multivariate‐adjusted trajectories of serum creatinine change over time in the primary percutaneous coronary intervention (pPCI) and thrombolysis/no reperfusion groups, based on the results from linear mixed models. The slops of the lines were similar (P=0.97 for interaction).
CM Exposure and Clinical Outcomes
Patients were followed for 1 year after study entry. During follow‐up, the primary composite end point occurred in 589 patients (19.3%). Patients treated with pPCI and those who did not develop AKI had better clinical outcomes (Figure 5). In the group of patients who were treated with pPCI, the rates of the primary composite end point were 12.5% among those without AKI and 42.1% among those with AKI. Among patients not treated with pPCI, the respective rates of events were 22.1% versus 62.1%. These differences are presented graphically in Figure 5 in terms of the cumulative incidence of the primary end point in the 4 groups. Within the pPCI group, there was no significant variation in the 1‐year event rate across quartiles of contrast volume (log rank, P=0.81).
Figure 5.
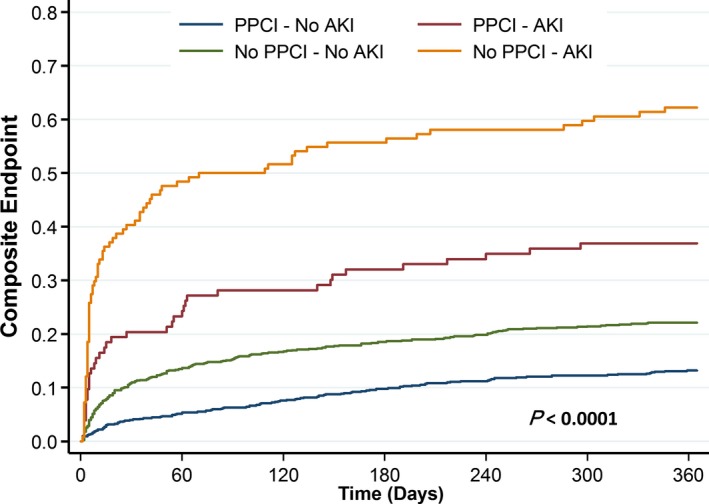
Cumulative incidence of mortality, readmission for heart failure, and recurrent myocardial infarction, according primary percutaneous coronary intervention (pPCI) treatment and occurrence of acute kidney injury (AKI).
In a multivariable Cox model, the adjusted hazard ratio [HR] for the primary end point was significantly increased with AKI among participants with pPCI (adjusted HR: 2.09; 95% CI, 1.59–2.75; P<0.0001) and without pPCI (HR: 2.01; 95% CI, 1.45–2.79; P<0.0001). There was no interaction between pPCI and AKI with regard to the primary end point; the interaction HR (ie, the ratio of the HRs for risk of AKI in the pPCI and no‐pPCI groups) was 1.03 (95% CI, 0.68–1.58; P=0.86 for interaction), indicating that the hazard associated with AKI was similar in patients with and without contrast exposure.
Discussion
The current analysis of STEMI patients undergoing pPCI and those treated with thrombolysis or without reperfusion provides no evidence that CM exposure is associated with increased rates of AKI. In an analysis of AKI events that accrued among propensity score–matched patients who were or were not exposed to CM, the rates of AKI were not significantly different. The slope of creatinine rise over the first 72 hours of hospitalization was similar in patients with and without CM exposure. AKI after pPCI was strongly associated with baseline eGFR, age, heart failure and hemodynamic instability, and reduced left ventricular function but not with contrast volume. Risk factors for AKI were similar among STEMI patients regardless of CM exposure. Finally, the use of a higher contrast dose was not associated with increased mortality rates, and AKI incurred similar risk for adverse events in patients with and without PCI.
In the radiology literature, <1% of all publications concerning CI‐AKI had control groups.9 These controlled studies demonstrated similar rates of AKI, dialysis, and death between patients in the groups that received CM and the control groups of patients who did not receive CM,7, 8 underscoring the crucial need for a control group of participants who do not receive CM.
To our knowledge, our study represents the first to include a control group of patients who were not exposed to CM in the setting of STEMI. We found that AKI occurred as frequently with and without CM exposure. A potential explanation for these findings is that there may be no appreciable increased risk of AKI associated with CM administration due to shared, and potentially more powerful, CM‐independent risk factors for AKI in STEMI patients. Age, baseline renal dysfunction, and clinical evidence of heart failure or hemodynamic instability were major determinants of AKI risk in all STEMI patients. These results suggest CM‐independent renal insults can explain most post‐pPCI AKI events14; therefore, AKI in the setting of pPCI likely represents 1 facet of the spectrum of the type 1 cardiorenal syndrome.15
Animal models generally require that additional insults to the kidney be combined with contrast exposure to reliably induce AKI.16 Similarly, in clinical studies including elective procedures performed on stable patients, the development of CI‐AKI largely depends on coexisting risk factors such as intravascular volume depletion, baseline renal dysfunction, heart failure, and diabetes mellitus.9
The term CI‐AKI implies impairment of renal function occurring within 3 to 5 days following the administration of CM in the absence of an alternative etiology16, 17; however, it is unlikely that CM is the sole factor responsible for renal dysfunction in the patient population undergoing pPCI. Conditions resulting in low effective circulating volume, hypotension, hemodynamic instability or cardiogenic shock, acute heart failure, hemorrhage, and initiation of medical therapies are additional contributing factors that may lead directly to renal dysfunction or potentiate the effects of CM.18 Consequently, in the setting of pPCI, most AKI events likely have alternative and multiple causes, and the use of the term CI‐AKI in this context should be avoided in most cases.
The reported incidence of AKI is 1% to 3% in patients undergoing elective PCI17, 19, 20, 21 but rises significantly to 10% to 16% in patients undergoing pPCI.3, 22, 23 The large increase in incident AKI is the result of heart failure, hemodynamic instability, and pharmacological interventions associated with STEMI rather than the use of greater contrast volume.
Unfortunately, the term CI‐AKI is used frequently to describe AKI that occurs following an interventional procedure even when other potential explanations for AKI are present.2, 3, 18 Because previous studies inherently lacked a control population of patients who did not receive CM, the relative contribution attributable to CM could not be extricated from CM‐independent causes. Furthermore, potent risk factors that directly cause AKI such as IABP use in the setting of cardiogenic shock, are often cited as risk factors for CI‐AKI.18
Few data are available with regard to risk factors for the development of AKI in patients undergoing pPCI. Our results are at odds with prior studies that reported a positive association between higher contrast volume and incident AKI in patients undergoing pPCI2, 3 although other risk factors for AKI were similar.18 In contrast, as in the present study, Shacham and colleagues could not find an independent association between V/eGFR and AKI.22 Furthermore, in randomized trials of complete versus lesion‐only revascularization in patients undergoing pPCI, contrast volume was higher in the multivessel‐PCI group with similar CI‐AKI rates.24, 25 Recently, Kooiman reported that a high contrast dose (contrast volume [in ml] ≥3× baseline eGFR) was only a marginal predictor of AKI after PCI, with an estimated attributable risk fraction to AKI of 10.6%.14
Although one cannot disagree with the need to avoid excess contrast volume in any patient undergoing pPCI, it is unlikely that CM is a primary pathogenic factor responsible for renal dysfunction in this patient population. Furthermore, the current results suggest that attempts to reduce AKI rates in STEMI patients likely require targeting mechanisms that are unrelated to CM.
AKI and Clinical Outcomes
The association between CI‐AKI and adverse clinical outcomes, including cardiovascular complications and death, has been amply demonstrated in previous studies of patients with acute myocardial infarction.26, 27, 28 These analyses, however, have not been sufficient to establish a causal relationship. More important, putative mechanisms for how CI‐AKI increases cardiovascular events have not been elucidated.
In the present study, there was no significant association between contrast volume and mortality. AKI was strongly predictive of adverse outcomes; however, the impact of AKI on clinical outcomes was similar among patients with and without contrast exposure. These results suggest that contrast volume did not directly contribute to the increase in adverse events. Of note, risk variables that predicted mortality were also strong predictors of AKI. This overlap of risk factors for the development of AKI and for mortality is very common in the setting of pPCI, as many patients are affected by cardiogenic shock, heart failure, and preexisting or acute renal failure.
Study Limitations
Some limitations of our study should be noted. Selection bias is an inherent concern in observational nonrandomized studies. Some patients were recruited during a period in which both pPCI and thrombolysis were acceptable reperfusion strategies. Consequently, treatment bias related to age, baseline renal function, infarct location, and hemodynamic status may have affected the decision to perform pPCI, resulting in heterogeneity caused by AKI‐predisposing comorbidities between contrast and noncontrast groups. The use of propensity score matching cannot fully correct for these imbalances; therefore, the results should be viewed as exploratory and hypothesis generating only.
Because all patients in the present study underwent emergency PCI, no protocol‐defined pre‐ or postprocedural hydration could be given. Total fluid volume and net fluid balance were not quantified in the study. Patients without heart failure may have been better hydrated, resulting in reduced incidence of AKI, although a recent study challenges the ability of hydration to prevent CI‐AKI.29
Conclusion
In this study, the development of AKI in STEMI patients after pPCI is mainly related to older age, baseline eGFR, heart failure, and hemodynamic instability. Higher contrast was not associated with higher AKI or mortality rates. Risk of AKI is similar among STEMI patients with and without CM exposure.
Disclosures
None.
(J Am Heart Assoc. 2017;6:e005715 DOI: 10.1161/JAHA.117.005715.)28647690
References
- 1. Marenzi G, Lauri G, Assanelli E, Campodonico J, De Metrio M, Marana I, Grazi M, Veglia F, Bartorelli AL. Contrast‐induced nephropathy in patients undergoing primary angioplasty for acute myocardial infarction. J Am Coll Cardiol. 2004;44:1780–1785. [DOI] [PubMed] [Google Scholar]
- 2. Marenzi G, Assanelli E, Campodonico J, Lauri G, Marana I, De Metrio M, Moltrasio M, Grazi M, Rubino M, Veglia F, Fabbiocchi F, Bartorelli AL. Contrast volume during primary percutaneous coronary intervention and subsequent contrast‐induced nephropathy and mortality. Ann Intern Med. 2009;150:170–177. [DOI] [PubMed] [Google Scholar]
- 3. Narula A, Mehran R, Weisz G, Dangas GD, Yu J, Genereux P, Nikolsky E, Brener SJ, Witzenbichler B, Guagliumi G, Clark AE, Fahy M, Xu K, Brodie BR, Stone GW. Contrast‐induced acute kidney injury after primary percutaneous coronary intervention: results from the HORIZONS‐AMI substudy. Eur Heart J. 2014;35:1533–1540. [DOI] [PubMed] [Google Scholar]
- 4. McCullough PA. Contrast‐induced acute kidney injury. J Am Coll Cardiol. 2008;51:1419–1428. [DOI] [PubMed] [Google Scholar]
- 5. Morcos SK, Thomsen HS, Webb JA. Contrast‐media‐induced nephrotoxicity: a consensus report. Contrast Media Safety Committee, European Society of Urogenital Radiology (ESUR). Eur Radiol. 1999;9:1602–1613. [DOI] [PubMed] [Google Scholar]
- 6. Prasad V, Gandhi D, Stokum C, Miller T, Jindal G. Incidence of contrast material‐induced nephropathy after neuroendovascular procedures. Radiology. 2014;273:853–858. [DOI] [PubMed] [Google Scholar]
- 7. McDonald RJ, McDonald JS, Carter RE, Hartman RP, Katzberg RW, Kallmes DF, Williamson EE. Intravenous contrast material exposure is not an independent risk factor for dialysis or mortality. Radiology. 2014;273:714–725. [DOI] [PubMed] [Google Scholar]
- 8. McDonald JS, McDonald RJ, Carter RE, Katzberg RW, Kallmes DF, Williamson EE. Risk of intravenous contrast material‐mediated acute kidney injury: a propensity score‐matched study stratified by baseline‐estimated glomerular filtration rate. Radiology. 2014;271:65–73. [DOI] [PubMed] [Google Scholar]
- 9. Newhouse JH, RoyChoudhury A. Quantitating contrast medium‐induced nephropathy: controlling the controls. Radiology. 2013;267:4–8. [DOI] [PubMed] [Google Scholar]
- 10. Cigarroa RG, Lange RA, Williams RH, Hillis LD. Dosing of contrast material to prevent contrast nephropathy in patients with renal disease. Am J Med. 1989;86:649–652. [DOI] [PubMed] [Google Scholar]
- 11. Gurm HS, Dixon SR, Smith DE, Share D, Lalonde T, Greenbaum A, Moscucci M; Registry BMC . Renal function‐based contrast dosing to define safe limits of radiographic contrast media in patients undergoing percutaneous coronary interventions. J Am Coll Cardiol. 2011;58:907–914. [DOI] [PubMed] [Google Scholar]
- 12. Moons KG, Harrell FE, Steyerberg EW. Should scoring rules be based on odds ratios or regression coefficients? J Clin Epidemiol. 2002;55:1054–1055. [DOI] [PubMed] [Google Scholar]
- 13. Austin PC. Balance diagnostics for comparing the distribution of baseline covariates between treatment groups in propensity‐score matched samples. Stat Med. 2009;28:3083–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kooiman J, Seth M, Nallamothu BK, Heung M, Humes D, Gurm HS. Association between acute kidney injury and in‐hospital mortality in patients undergoing percutaneous coronary interventions. Circ Cardiovasc Interv. 2015;8:e002212. [DOI] [PubMed] [Google Scholar]
- 15. Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, Bellomo R, Berl T, Bobek I, Cruz DN, Daliento L, Davenport A, Haapio M, Hillege H, House AA, Katz N, Maisel A, Mankad S, Zanco P, Mebazaa A, Palazzuoli A, Ronco F, Shaw A, Sheinfeld G, Soni S, Vescovo G, Zamperetti N, Ponikowski P. Cardio‐renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010;31:703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Persson PB, Hansell P, Liss P. Pathophysiology of contrast medium‐induced nephropathy. Kidney Int. 2005;68:14–22. [DOI] [PubMed] [Google Scholar]
- 17. Mehran R, Nikolsky E. Contrast‐induced nephropathy: definition, epidemiology, and patients at risk. Kidney Int Suppl. 2006;S11–S15. [DOI] [PubMed] [Google Scholar]
- 18. Mehran R, Aymong ED, Nikolsky E, Lasic Z, Iakovou I, Fahy M, Mintz GS, Lansky AJ, Moses JW, Stone GW, Leon MB, Dangas G. A simple risk score for prediction of contrast‐induced nephropathy after percutaneous coronary intervention: development and initial validation. J Am Coll Cardiol. 2004;44:1393–1399. [DOI] [PubMed] [Google Scholar]
- 19. Rich MW, Crecelius CA. Incidence, risk factors, and clinical course of acute renal insufficiency after cardiac catheterization in patients 70 years of age or older. A prospective study. Arch Intern Med. 1990;150:1237–1242. [PubMed] [Google Scholar]
- 20. Parfrey PS, Griffiths SM, Barrett BJ, Paul MD, Genge M, Withers J, Farid N, McManamon PJ. Contrast material‐induced renal failure in patients with diabetes mellitus, renal insufficiency, or both. A prospective controlled study. N Engl J Med. 1989;320:143–149. [DOI] [PubMed] [Google Scholar]
- 21. Laskey WK, Jenkins C, Selzer F, Marroquin OC, Wilensky RL, Glaser R, Cohen HA, Holmes DR Jr. Volume‐to‐creatinine clearance ratio: a pharmacokinetically based risk factor for prediction of early creatinine increase after percutaneous coronary intervention. J Am Coll Cardiol. 2007;50:584–590. [DOI] [PubMed] [Google Scholar]
- 22. Shacham Y, Leshem‐Rubinow E, Gal‐Oz A, Arbel Y, Keren G, Roth A, Steinvil A. Acute cardio‐renal syndrome as a cause for renal deterioration among myocardial infarction patients treated with primary percutaneous intervention. Can J Cardiol. 2015;31:1240–1244. [DOI] [PubMed] [Google Scholar]
- 23. Sgura FA, Bertelli L, Monopoli D, Leuzzi C, Guerri E, Sparta I, Politi L, Aprile A, Amato A, Rossi R, Biondi‐Zoccai G, Sangiorgi GM, Modena MG. Mehran contrast‐induced nephropathy risk score predicts short‐ and long‐term clinical outcomes in patients with ST‐elevation‐myocardial infarction. Circ Cardiovasc Interv. 2010;3:491–498. [DOI] [PubMed] [Google Scholar]
- 24. Gershlick AH, Khan JN, Kelly DJ, Greenwood JP, Sasikaran T, Curzen N, Blackman DJ, Dalby M, Fairbrother KL, Banya W, Wang D, Flather M, Hetherington SL, Kelion AD, Talwar S, Gunning M, Hall R, Swanton H, McCann GP. Randomized trial of complete versus lesion‐only revascularization in patients undergoing primary percutaneous coronary intervention for STEMI and multivessel disease: the CvLPRIT trial. J Am Coll Cardiol. 2015;65:963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wald DS, Morris JK, Wald NJ, Chase AJ, Edwards RJ, Hughes LO, Berry C, Oldroyd KG; Investigators P . Randomized trial of preventive angioplasty in myocardial infarction. N Engl J Med. 2013;369:1115–1123. [DOI] [PubMed] [Google Scholar]
- 26. Parikh CR, Coca SG, Wang Y, Masoudi FA, Krumholz HM. Long‐term prognosis of acute kidney injury after acute myocardial infarction. Arch Intern Med. 2008;168:987–995. [DOI] [PubMed] [Google Scholar]
- 27. Goldberg A, Hammerman H, Petcherski S, Zdorovyak A, Yalonetsky S, Kapeliovich M, Agmon Y, Markiewicz W, Aronson D. Inhospital and 1‐year mortality of patients who develop worsening renal function following acute ST‐elevation myocardial infarction. Am Heart J. 2005;150:330–337. [DOI] [PubMed] [Google Scholar]
- 28. Goldberg A, Kogan E, Hammerman H, Markiewicz W, Aronson D. The impact of transient and persistent acute kidney injury on long‐term outcomes after acute myocardial infarction. Kidney Int. 2009;76:900–906. [DOI] [PubMed] [Google Scholar]
- 29. Nijssen EC, Rennenberg RJ, Nelemans PJ, Essers BA, Janssen MM, Vermeeren MA, Ommen VV, Wildberger JE. Prophylactic hydration to protect renal function from intravascular iodinated contrast material in patients at high risk of contrast‐induced nephropathy (AMACING): a prospective, randomised, phase 3, controlled, open‐label, non‐inferiority trial. Lancet. 2017;389:1312–1322. [DOI] [PubMed] [Google Scholar]