

Abstract
Background
Cardiac hypertrophy increases the risk of developing heart failure and cardiovascular death. The neutrophil inflammatory protein, lipocalin‐2 (LCN2/NGAL), is elevated in certain forms of cardiac hypertrophy and acute heart failure. However, a specific role for LCN2 in predisposition and etiology of hypertrophy and the relevant genetic determinants are unclear. Here, we defined the role of LCN2 in concentric cardiac hypertrophy in terms of pathophysiology, inflammatory expression networks, and genomic determinants.
Methods and Results
We used 3 experimental models: a polygenic model of cardiac hypertrophy and heart failure, a model of intrauterine growth restriction and Lcn2‐knockout mouse; cultured cardiomyocytes; and 2 human cohorts: 114 type 2 diabetes mellitus patients and 2064 healthy subjects of the YFS (Young Finns Study). In hypertrophic heart rats, cardiac and circulating Lcn2 was significantly overexpressed before, during, and after development of cardiac hypertrophy and heart failure. Lcn2 expression was increased in hypertrophic hearts in a model of intrauterine growth restriction, whereas Lcn2‐knockout mice had smaller hearts. In cultured cardiomyocytes, Lcn2 activated molecular hypertrophic pathways and increased cell size, but reduced proliferation and cell numbers. Increased LCN2 was associated with cardiac hypertrophy and diastolic dysfunction in diabetes mellitus. In the YFS,LCN2 expression was associated with body mass index and cardiac mass and with levels of inflammatory markers. The single‐nucleotide polymorphism, rs13297295, located near LCN2 defined a significant cis‐eQTL for LCN2 expression.
Conclusions
Direct effects of LCN2 on cardiomyocyte size and number and the consistent associations in experimental and human analyses reveal a central role for LCN2 in the ontogeny of cardiac hypertrophy and heart failure.
Keywords: concentric hypertrophy, C‐reactive protein, gene coexpression networks, GlycA, hypertrophy, lipocalin‐2, NGAL, systems biology
Subject Categories: Hypertrophy, Animal Models of Human Disease, Basic Science Research, Inflammation, Translational Studies
Clinical Perspective
What is New?
Using several animal models, in vitro and human studies, we identified LCN2 as a central gene in the developmental origins of cardiac hypertrophy leading to heart failure.
Increased LCN2 expression has defined effects of cardiomyocyte proliferation and hypertrophy that might explain cardiac hypertrophy and is likely to reflect chronic activation of inflammatory pathways.
What are the Clinical Implications?
The experimental effects of LCN2 on cultured cardiomyocytes and in hearts of neonatal animals need to be corroborated in clinical studies of relationships between LCN2, heart size, and, if possible, cardiomyocyte numbers.
LCN2 could be targeted as a therapeutic target and also developed as an early marker for cardiac hypertrophy and heart failure.
Introduction
Cardiac hypertrophy is, after age, the single most important risk factor for cardiovascular death,1 often as a result of heart failure. Hypertrophic remodeling of the heart is usually in response to increased workload, and the response to such stress has been shown to involve inflammatory pathways.2, 3, 4 Indeed, chronic inflammatory processes have been implicated not only in response to stress, but also more generally as primary etiological factors in cardiovascular disease (CVD).5 Cardiovascular remodeling depends on refashioning the interstitium, and inflammation stimulates molecules, such as matrix metalloproteinase‐9 (MMP9), that degrade the interstitial matrix.6 MMP9 levels have been associated with cardiovascular disease prognosis,7 and MMP9 is stimulated by the protein lipocalin‐2 (LCN2), also known as neutrophil gelatinase‐associated lipocalin (NGAL).8, 9 LCN2 levels have been used to reflect tissue damage, particularly of the kidney, but more recently also for CVD manifestations,10 including hypertensive cardiac hypertrophy,11 coronary artery disease12 and acute heart failure.13 LCN2 has also been associated with long‐term mortality following acute heart failure, independent of renal function.14 However, it is unclear whether LCN2 is simply a marker of an inflammatory process or capable of direct effects on the heart that might contribute to cardiac hypertrophy and failure.
In this study, we investigated the association of LCN2 with concentric cardiac hypertrophy in genetic and environmental experimental models and in relation to the normal variation of human heart size and cardiac hypertrophy in diabetes mellitus. We examined transcriptional associations with LCN2 and identified genetic polymorphisms influencing LCN2 expression. We determined the direct cellular effects of LCN2 in cultured cardiomyocytes. Our findings reveal increased LCN2 levels as a consistent association with cardiac hypertrophy in a variety of models and human cohorts, and our in vitro studies support a direct role for LCN2 in the origins of cardiomyocyte hypertrophy and reduced cardiomyocyte proliferation.
Methods
Detailed methods are available in the Data S1.
Genetic Model of Cardiac Hypertrophy and Heart Failure
The hypertrophic heart rat (HHR) is a normotensive inbred polygenic model of adult cardiac hypertrophy, heart failure, and premature death generated by us (Prof Stephen Harrap and Prof Lea Delbridge, University of Melbourne, Melbourne, Parkville, Australia).15 HHRs have a reduced endowment of cardiomyocytes from very early life, a situation predisposing to hypertrophy and failure in later life.15, 16 Aged‐matched male animals were sampled during the following periods: neonatal (postnatal day 2, n=11 HHR, n=10 Normal Heart Rat [NHR]), adolescent (4 weeks old, n=4 HHR and n=4 NHR for cardiomyocyte isolation), young adult (13 weeks old, n=7 HHR, n=7 NHR; 35 weeks old, n=8 NHR, n=11 HHR), and old adult (50 weeks old, n=11 HHR, n=10 NHR).
Animals were euthanized by decapitation (neonatal) or with an overdose of pentobarbitone (Lethobarb; adult animals). The heart was immediately removed, and ventricles were dissected from the atria. Cardiac weight index (mg/g) was calculated from the total heart weight (mg) relative to total body weight (g) of the animal. The studies involving animals were approved by the Animal Ethics Committee of Deakin University and the University of Melbourne and ratified at Federation University Australia. They were performed according to the “Code of Practice for the Care and Use of Animals for Scientific Purposes” from the National Health & Medical Research Council of Australia.
Rat Microarray Experiments
RNA was extracted from the left ventricle of 2‐day‐old HHRs and NHRs (n=8/group, no pooling), and Affymetrix GeneChip Rat Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA) was used to assess genes differentially expressed with the assistance of the Ramaciotti Centre for Gene Function Analysis. The data set obtained has been deposited in the National Center for Biotechnology Information Gene Expression Omnibus database according to Minimum Information About a Microarray Experiment guidelines with series accession number GSE38607. Differentially expressed genes were identified using a 2‐sample t test in the Partek Genomics Suite (version 6.6; Partek Inc, Chesterfield, MO), with Bonferroni‐adjusted P<0.05 and fold difference higher than 2.
Lcn2 mRNA and Protein Levels in Models of Cardiac Hypertrophy and Heart Failure
Primers and conditions used for all real‐time quantitative PCR (qPCR) are shown in Table S1. Amplification reactions used the SensiFast SYBR Low‐ROX Kit qPCR reagent system (Bioline Reagents Ltd, London, UK) in a Viia7 qPCR instrument (Life Technologies, Life Technologies, Carlsbad, CA). Immunohistochemistry was performed using an anti‐LCN2 Rabbit Polyclonal antibody (1:200 dilution, TA322583; OriGene Technologies, Rockville, MD), followed by the EnVision+System‐HRP. Western blots were performed as previously described17 using anti‐LCN2 Rabbit Polyclonal antibody or β‐actin (Cell Signaling Tecnology, Danvers, MA). Lcn2 plasma and left ventricle (LV) protein levels were measured by ELISA in duplicates in neonatal and adult HHR and NHR using the Lipocalin‐2 Rat ELISA Kit (Abcam, Cambridge, UK) according to the supplier. Sanger sequencing was used to sequence 10 000 base pairs (bp) before and 2000 bp after the Lcn2 gene in the HHR and NHR (Table S1).
Lcn2‐Knockout
Whole body and heart size of adult (12‐ to 13‐week‐old) Lcn2‐KO (n=6) and age‐matched wild‐type mice C57BL/6 (n=4), generously donated by Prof Alan Aderem (Institute for Systems Biology, University of Washington, Seattle, WA), were measured upon death, and cardiac weight index was calculated as described above.
Intrauterine Growth Restriction Rat Model
An environmental model of cardiac hypertrophy was developed using Wistar Kyoto rats by intrauterine growth restriction, induced by uteroplacental insufficiency on day 18 of pregnancy (term being 22 days), was also investigated.18, 19 Six‐month‐old operated female and male rats (n=9) were compared to Wistar Kyoto female and male sham rats (n=16).
In Vitro Experiments
The pExpress vector containing the cDNA for the rat Lcn2 (2 ng/mL, MRN1768‐98079404; Thermo Fisher Scientific, Waltham, MA) or empty vector (pExpress) were transfected into rat embryonic ventricular myocardial cells (H9c2) using Lipofectamine 2000 (Life Technologies). We counted the number of cells by hemocytometry with the use of the Countess Automated Cell Counter (Life Technologies). Wheat germ agglutinin and Hoechst staining was used to measure cell size,20 and phospho‐histone H3 staining was used to determine cell proliferation.20 Apoptosis was investigated by flow cytometry using an Annexin‐V: FITC Apoptosis Detection Kit I. All in vitro experiments were independently repeated 3 times, each time in triplicates.
RNA‐Sequencing and Molecular Pathways
RNA was extracted from Lcn2‐KO mice and cells transfected with Lcn2 plasmid for 48 hours (and respective controls). RNA from 3 samples of each group was sent to RNA‐sequencing at the Australian Genome Research Facility using the Illumina HiSeq platform (v3 chemistry 100 bp paired‐end sequencing). Each sample was considered an individual sample and no pooling was performed. Analysis of differential expression was performed in the R statistical programming environment (version 3.1.0) using Rsubread (version 1.14.2) and edgeR (version 3.6.8) Bioconductor packages (Table S2).21 P values were adjusted for multiple testing using the Benjamini‐Hochberg correction with a false discovery rate <0.05. Gene ontology enrichment analysis was performed on filtered lists of differentially expressed genes to ask which pathways were enriched in genes differentially expressed.
Human Echocardiography Measurements
Briefly, 114 individuals with echocardiographic measures were selected from a prospective cohort of type 2 diabetic subjects22 whose basic characteristics are shown in Table S3. In addition, subjects with echocardiographic measures from the Young Finns Study (YFS) analyzed, shown in Table S4. The YFS is a longitudinal population‐based study of 3596 individuals recruited during childhood in 1980.23 Genome‐wide genotype data, transcriptome‐wide microarray profiling, C‐reactive protein (CRP), glycoprotein acetylation (GlycA), and echocardiographic measurements were available for different subsets of 2064 individuals aged 34 to 48 years, participating in the 2011 follow‐up study.24, 25, 26, 27 The cohort studies complied with the Declaration of Helsinki and were approved by the human ethics committee at each institution. All subjects gave informed consent.
In both cohorts, echocardiographic examinations were performed using transthoracic echocardiography by an Acuson Sequoia 512 (Acuson, Mountain View, CA) with a 3.5‐MHz scanning frequency phased‐array transducer. From the ultrasound images, LV structure, systolic, and diastolic function were measured following the guidelines of the American Society of Echocardiography, as previously described.28, 29 Cardiac hypertrophy was defined as LV mass indexed to the body surface of >95 g/m2 in women and >115 g/m2 in men.30 E/E′‐ratio was calculated using the average values of lateral and septal e′ velocity.29
Human Plasma LCN2 Measurement
Human plasma was used to measure LCN2 levels in duplicates in 121 subjects with type 2 diabetes mellitus using the Quantikine ELISA Human Lipocalin‐2 Immunoassay (R&D Systems, Minneapolis, MN), according to the supplier.
LCN2 mRNA Levels in Human Heart
We used data in the repository Gene Expression Omnibus series GSE1145 to investigate the levels of LCN2 in human idiopathic dilated hearts (n=11 control hearts and n=15 idiopathic dilated hearts). We performed a whole‐genome analysis using the Gene Expression Omnibus tools, including false discovery rate <0.05, to determine whether LCN2 was overexpressed in human idiopathic dilated hearts.
GlycA Measurement
GlycA reflects the integrated concentrations and glycosylation states of several of the most abundant inflammatory acute‐phase glycoproteins31, 32 measured with a proton nuclear magnetic resonance metabolomics platform.33
CRP Measurement
High‐sensitivity CRP was quantified from serum samples using an automated analyzer with a latex turbidimetric immunoassay kit.
Coexpression Networks and Quantitative Trait Loci
Transcriptome‐wide microarray profiling was performed on whole blood for 1650 individuals in the YFS as previously described.24 Briefly, stabilized total RNA was obtained from whole blood for individuals in the YFS. RNA was hybridized to Illumina HT‐12 (version 4; Illumina, San Diego, CA) BeadChip arrays, and raw probe data were exported with the Illumina BeadStudio software. Both positive and negative control probes were used to quantile normalize using the limma R package.34 Probe intensities were reported on a log2 scale.
Identification and characterization of the gene coexpression network analyzed in this study is described in Ritchie et al.32 Here, we defined the neutrophil module's coexpression as the Spearman's correlation coefficient between its 27 genes.32 The average expression was used for genes with multiple microarray probes. Edges in the coexpression network were defined as the magnitude of the correlation exponentiated to the power of 4. A vector summarizing module expression was calculated for association testing as the first eigenvector of a principal components analysis on module expression. This summary expression profile captured 57% of the total variation in module gene expression. Association analyses are described in the Statistical Analyses section below.
Genome‐wide genotyping was carried out on whole‐blood samples for 2442 individuals participating in the 2001 follow‐up study of the YFS as previously described.25 Sample and genotype quality control was performed for these 2442 individuals (Data S1). A combined total of 6 721 082 directly genotyped and imputed single‐nucleotide polymorphisms (SNPs) passed quality control.
Module quantitative trait loci (QTLs) were identified for 1386 individuals with matched genotype and gene expression data in the YFS through a genome‐wide scan for SNPs associated with the summary expression profile using PLINK 1.90 beta (version 3.32). Individual associations were tested using a linear model of minor allele dosage on neutrophil module summary expression. An SNP was considered a module QTL where P<5×10−8 (genome‐wide significance). Models were adjusted for age, sex, and the first 2 principal components of the genotype data. The module QTL, rs13297295, on chromosome 9 was further tested for an association with LCN2 expression levels using the same model. Rs13297295 was also tested for association with GlycA and CRP in the 1712 individuals with matched genotype and GlycA or CRP data.
Statistical Analyses
R software (version 3.13; R Foundation for Statistical Computing, Vienna, Austria) was used for the analyses of the YFS data. The NetRep package (version 0.54) was used for network analyses.35 Measurements of GlycA, routine lipids, CRP, body mass index (BMI), heart function (measured as early filling [E] to early diastolic mitral annular velocity [E′]—E/E′ ratio, and E to late [A] diastolic filling—E/A ratio) were normalized using a natural logarithm transformation, and all continuous measurements were standardized to SD units in both cohorts. Module associations with the inflammatory biomarkers were assessed by linear regression of: neutrophil module expression on GlycA and CRP; linear regression of LCN2 expression on GlycA and CRP. To assess whether LCN2 was a mediator of the relationship between these biomarkers and the neutrophil module, we used linear regression of: GlycA and CRP on LCN2 expression and neutrophil module expression; CRP on LCN2 expression and neutrophil module expression; and GlycA on LCN2 expression and neutrophil module expression. All terms in the models were additive, and all models were adjusted for age and sex. Matched gene expression, GlycA, and CRP data were available for 1650 individuals. Associations between LCN2 expression and echocardiographic measurements in the YFS (Table S5) were tested by linear regression of each echocardiographic measurement on LCN2 expression, adjusting for age and sex. Each association was considered significant where P<0.05. Matched gene expression and echocardiographic data were available for between 1482 and 1573 individuals depending on the LV phenotype.
Inter‐ and intraassay coefficients of variability were calculated for ELISAs, and only less than 15% variability was accepted (hence 7 human samples from the type 2 diabetes mellitus cohort were excluded from further analyses). Human plasma LCN2 levels were not normally distributed; therefore, LCN2 was log transformed for the association analyses presented in Table S3. An independent t test was used to assess differences in continuous variables between those with and without cardiac hypertrophy or chi‐square analyses for dichotomous variables. A general linear model analysis was performed to test for associations between presence of cardiac hypertrophy and plasma LCN2 levels after adjusting for variables from the univariable analysis with a P value of <1.0 (age, sex, BMI, estimated glomerular filtration rate, and systolic blood pressure). We used untransformed LCN2 levels to perform Spearman Rho correlations between human plasma LCN2 and LV left ventricle mass and function in the type 2 diabetes mellitus cohort (Table S6). Significance was set at P<0.05.
Results from the animal groups were tested for normal distribution using the Skewness and Kurtosis tests. Independent sample t tests (with Welch's correction in the case of different variance) and ANOVA were used to compare the data between the animal groups. A 2‐way ANOVA was used to compare between Lcn2 expression in the different cell types in HHRs and NHRs.
Results
Lcn2 Is Associated With Cardiac Size in Experimental Genetic and Environmental Models
HHR and NHR
A transcriptome analysis of neonatal P2 LV tissue identified 21 genes with significant differential expression between HHRs and NHRs (Table S7 and Figure S1) involving pathways for cardiovascular system development and function, and cell growth and proliferation (Tables S8 and S9), with Lcn2 showing the greatest differential expression (q=7×10−11; Table S7). Elevated cardiac Lcn2 expression was validated by qPCR at postnatal day 2 (Figure 1A) and was found to persist with established hypertrophy at 13 weeks of age and with the emergence of heart failure at 35 or 50 weeks of age (Figure 1B) and further confirmed by 25‐kDa Lcn2 monomer protein analyses (Figure 1C and 1D). Compared with its control strain, the NHR, we found significantly higher circulating Lcn2 in adult HHRs with established hypertrophy at 35 weeks of age (Figure 1E), but also soon after birth (Figure 1F) before hypertrophy is evident but cardiomyocyte numbers are already reduced. RNA and immunohistochemical studies (Figure 1G and 1H) showed Lcn2 expression in cardiomyocytes and noncardiomyocyte (fibroendothelial) cells. Correlation between cardiac Lcn2 mRNA and plasma Lcn2 in the HHRs and NHRs was r=0.996 (P<0.001).
Figure 1.

Overexpression of lipocalin‐2 (Lcn2) in a polygenic model of cardiac hypertrophy. A, Relative expression levels of Lcn2 mRNA measured by real‐time PCR in the heart of 2‐day‐old hypertrophic heart rat (HHR; n=10) compared to normal heart rat (NHR; n=8; P<0.0001), (B) 13‐week‐old (P=0.016; n=9/strain), 35‐week‐old (P<0.001; n=8 NHR and n=11 HHR), and 50‐week old (P=0.0015; n=8 NHR and n=11 HHR) HHR compared to NHR. Heart Lcn2 (25‐kDa monomer) protein is significantly higher in the HHR compared with NHR, measured by both (C) western blot (P=0.039; n=3/strain) and (D) ELISA (P=0.029; n=4/strain). E, Rat plasma Lcn2 in 35‐week‐old (P=0.0009; n=6/strain) and (F) 2‐day‐old (P<0.0001; n=5/strain) HHR compared to NHR. G, Lcn2 mRNA in cardiomyocytes (P=0.013; n=4/strain) and noncardiomyocytes (P=0.03) in the NHR and HHR. The interaction explained 5.135% of total variation (P=0.138), the cell type 6.91% of variation (P=0.0899), and the strain explained the majority of variation (63.58%; P=0.0001). H, Lcn2 staining in NHR and HHR hearts (×400 magnification; scale bar=200μm). *P<0.05; **P<0.01; ***P<0.001. Data shown as mean and error bars represent SEM.
Sequencing the HHR and NHR Lcn2 genes for comparison with the published sequence for spontaneously hypertensive rats and Fisher 344 (original progenitors of HHR and NHR), we found 3 unique SNPs in the HHR, all inherited from the spontaneously hypertensive rats, with 1 being intronic and 2 being upstream of the coding sequence (Figure 2A through 2D). In the HHR heart, both Lcn2 mRNA and pre‐mRNA levels were increased (Figure 2E), suggesting a transcriptional dysregulation of Lcn2 in the HHR. Transcription Factor Affinity Prediction (sTRAP)36, 37 analysis suggested that one of these SNPs (rs196968512) created a binding site for the enhancer, RAR‐related orphan receptor A (Figure 2B and 2F).38
Figure 2.
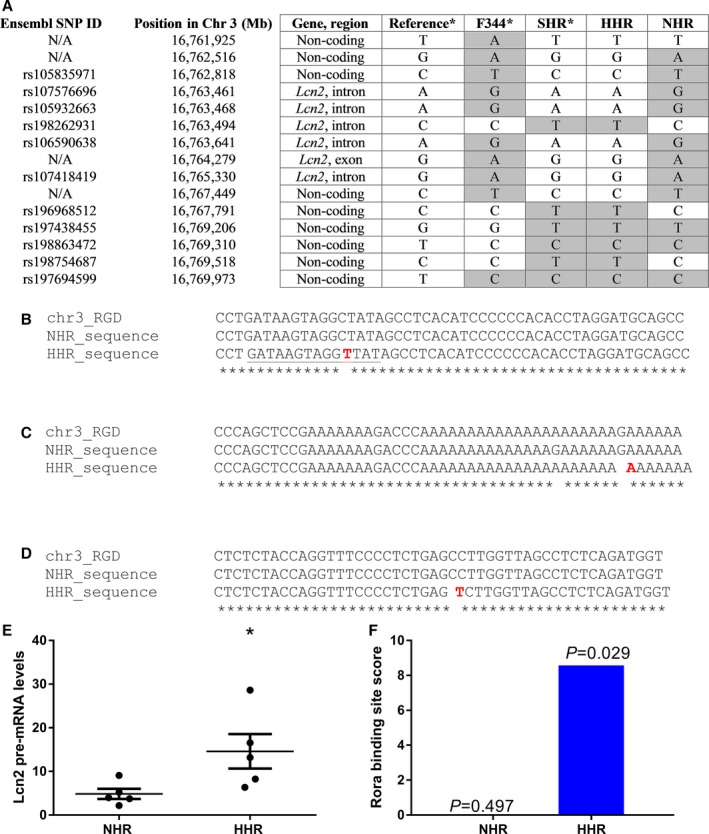
Variants in the lipocalin‐2 (Lcn2) gene, showing regions with variants in the HHR, according to the Rat Genome Database (RGD; version 5). A, Genotype analysis of the region of 10 000 bp around the Lcn2 gene, showing the origin of the variants observed in the HHR. Highlighted in gray are variants that differ from the reference genome, showing that the HHR carries 3 unique variants which were inherited from the SHR. B, Single‐nucleotide polymorphism (SNP) on chr3: 16 767 791 (rs196968512 C/T) 1401 bp upstream of Lcn2 gene. C, SNP on chr3: 16 767 398 (G/A) in a highly conserved region 1001 bp upstream the Lcn2 gene. D, Nonfunctional intronic SNP originally from SHR on position chr3: 16 763 494 (rs198262931 C/T). E, Lcn2 pre‐mRNA is also upregulated in the HHR compared to the NHR (n=5/strain), suggesting that it is dysregulated at the transcriptional level. F, The SNP, rs196968512, creates a new binding site for the transcription factor, Rora (region underlined in B), which acts as an enhancer for expression of Lcn2 and is exclusive of the HHR. The figure shows the binding site score and the P values for the binding of Rora to that region. chr3 indicates rat chromosome 3; F344, Fisher 344 rat; HHR, hypertrophic heart rat; N/A, nonannotated SNP; NHR, normal heart rat; RGD, reference sequence from the Rat Genome Database v5; SHR, spontaneously hypertensive rat.
Lcn2 knockout mice
Hearts from adult mice with double knockout of the Lcn2 gene (Lcn2‐KO)39 were significantly smaller than age‐matched wild‐type mice (cardiac weight index 5.4 versus 5.9 mg/g; P=0.03; Figure 3A). RNA‐sequencing of murine heart tissue from Lcn2‐KO versus wild‐type identified 16 significantly differentially expressed genes (Table S10) that are relevant to pathways related to hypertrophic cardiomyopathies (Table S11; Figure 3B and 3C).
Figure 3.
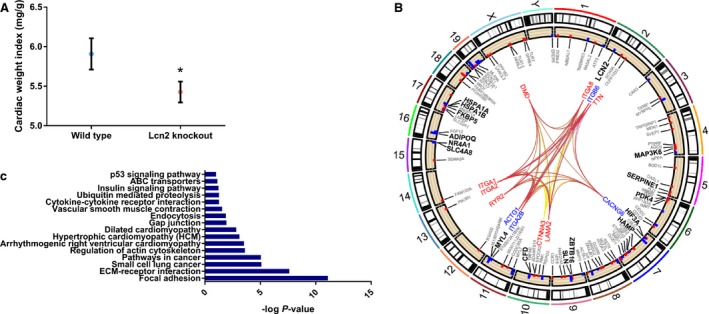
Heart size and associated pathways in lipocalin‐2 (Lcn2)‐knockout (KO) mice. A, Adult Lcn2‐knockout mice have smaller hearts (*P=0.033; n=4 wild‐type and n=6 Lcn2 KO). Data shown as mean and error bars represent standard error of mean. B, Genes and pathways differentially regulated in the heart of Lcn2‐knockout. Hypertrophic cardiomyopathy (KEGG mmu05410, P=0.0007) is shown in red, dilated cardiomyopathy in blue (Kyoto Encyclopedia of Genes and Genomes [KEGG] mmu05412; P=0.008) and arrhythmogenic right ventricular cardiomyopathy in yellow (KEGG mmu05412; P=0.0003). The genes of dilated cardiomyopathy and hypertrophic cardiomyopathy pathways were the same, and therefore lines are overlapped. Each edge point indicates the chromosomal location for genes identified in specific pathways from the differentially expressed genes. Bar plots are the differentially expressed genes in each pathway. Red depicts genes upregulated, and blue those downregulated, represented as log2 fold change. C, Gene ontology analysis, showing −log P value. ABC indicates ATP‐binding cassette; ECM extracellular matrix.
Intrauterine growth restricted rats
Last, in an environmental model of intrauterine growth restriction,18, 19 we demonstrated that subsequent adult cardiac hypertrophy was associated with significantly higher levels of cardiac Lcn2 mRNA (P=0.0214; Figure 4).
Figure 4.

Environmental model of cardiac hypertrophy overexpresses lipocalin‐2 (Lcn2) mRNA (*P=0.0214; n=16 sham and 9 restricted mice). Data shown as mean and error bars represent SEM.
Lcn2 Overexpression Induces Hypertrophy in Cardiomyocytes and reduces proliferation
We next sought to determine whether increased Lcn2 transcription in cardiomyocytes results in a hypertrophic phenotype. We transfected rat embryonic ventricular myocardial cells with a plasmid containing the Lcn2 mRNA sequence and performed imaging and RNA‐sequencing analyses. Significant increase in the expression of Lcn2 in transfected cells (log fold change=4.44; q=0.0005; Figure S2) resulted in a significant increase in the size of transfected cells (Figure 5A and Figure S3A). In these hypertrophic cells, we found significantly increased expression of 2 genes previously linked with hypertrophic cardiomyopathy—thrombospondin 2 (log fold change=−0.36; q=0.0005) and dynamin 1 (log fold change=−0.48; q=0.044).40, 41 In addition to hypertrophy, overexpression of Lcn2 resulted in a significant decrease in cell numbers (Figure 5B) with a concomitant reduction in cells positive for phosphorylated histone H3, reflecting reduced cell mitosis (Figure 5C and Figure S3B). No change in apoptosis was observed (Figure S4). Pathway enrichment analysis of the 529 genes with nominally significant evidence of differential expression (unadjusted P<0.05) between transfected and control myocytes suggested dysregulation of genes related to cell cycle (Kyoto Encyclopedia of Genes and Genomes rno04110; P=0.006). There was suggestive evidence for genes related to hypertrophic cardiomyopathy (Kyoto Encyclopedia of Genes and Genomes rno05410; P=0.07) and dilated cardiomyopathy (Kyoto Encyclopedia of Genes and Genomes rno05414; P=0.09; Figure 5D and 5E; Table S12).
Figure 5.
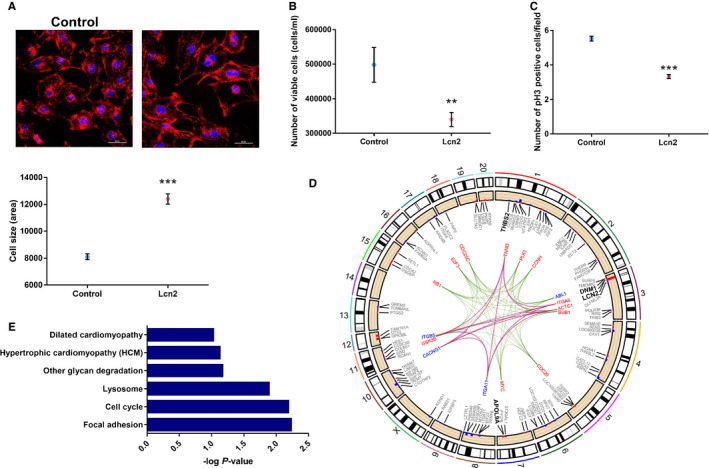
Role of lipocalin‐2 (Lcn2) in cardiac cells. A, Representation of wheat‐germ agglutinin (red) and DAPI (blue) staining, used to estimate cell size (×400 magnification; scale bar=60 μm). Overexpression of Lcn2 increased the size of the cells when compared with cells transfected with the empty plasmid (P<0.0001). B, Overexpression of Lcn2 reduced the number of cells measured by hemocytometer (P=0.0052). C, Overexpression of Lcn2 resulted in cell‐cycle arrest, observed by reduced phosphorylation of histone H3 (pH3; ×200 magnification; scale bar=100 μm; P<0.0001). D, Genes and pathways differentially regulated with overexpression of Lcn2. Cell cycle (Kyoto Encyclopedia of Genes and Genomes [KEGG] rno04110; P=0.006) is shown in green, hypertrophic cardiomyopathy (KEGG rno05410, P=0.07) in red, and dilated cardiomyopathy (KEGG rno05414; P=0.09) in blue. Genes of dilated cardiomyopathy and hypertrophic cardiomyopathy pathways were the same, and therefore lines overlapped. Each edge point indicates the chromosomal location for genes identified in specific pathways from the differentially expressed genes. Bar plots are the differentially expressed genes in each pathway. Red depicts genes upregulated, and blue those downregulated, represented as log2 fold change. E, Gene ontology analysis, showing −log P value. All experiments were run in 3 independent experiments, with at least 3 replicates each (total, 9 replicates). For experiments involving confocal microscopy, 10 different fields were analyzed per replicate. Positive controls were added to all experiments. **P<0.01; ***P<0.001. Data shown as mean and error bars represent SEM.
Human LCN2 Is Associated With LV Hypertrophy in Diabetes Mellitus
Obesity and type 2 diabetes mellitus have been associated with elevated levels of plasma LCN2.42, 43 Independently from those findings, cardiac hypertrophy has been associated with diastolic dysfunction and is recognized as a diabetic complication.44 However, whether cardiac hypertrophy and diastolic dysfunction in type 2 diabetic patients is associated with high LCN2 levels is not known. Echocardiographic assessment of 114 patients with type 2 diabetes mellitus and normal renal function revealed significantly higher levels of mean plasma LCN2 in the 30 diabetic subjects with LV hypertrophy than those 84 without 44.0 ng/mL [95% CI, 38.3−50.6] versus 36.0 ng/mL [33.1−39.2] P=0.017) that remained after adjustment for age, sex, BMI, estimated glomerular filtration rate, and systolic blood pressure (P=0.034; Figure 6A). There was a positive correlation between LCN2 levels and LV mass (n=114; Spearman's r=0.22; P=0.018; Figure 6B and Table S6). In diabetic subjects with cardiac hypertrophy, there was evidence of diastolic dysfunction (Table S3) with a significantly increased E/E′ ratio (mean±SD 15.2±4.5 versus 11.4±3.5; P<0.0001), but there was no association between these measurements and LCN2 (Table S6).
Figure 6.
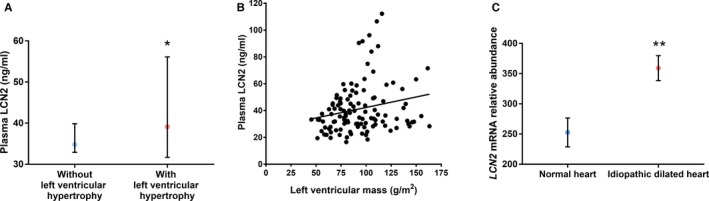
Lipocalin‐2 (LCN2) is associated with human cardiac hypertrophy. A, Plasma levels of LCN2 were higher in patients with echocardiographically determined left ventricular hypertrophy (n=30) compared with those without (n=84; P=0.017, showing the median and 95% CI). B, There was a positive correlation between LCN2 levels and left ventricular mass (n=114; Spearman's r=0.22; P=0.018). C, LCN2 was overexpressed in human idiopathic dilated hearts compared with normal hearts (P=0.008 after adjustment for false discovery rate). Data shown as mean and error bars represent SEM. *P<0.05; **P<0.01.
LCN2 mRNA Is the Human Heart
From subjects with idiopathic dilated cardiomyopathy, cardiac RNA expression data in a public repository (GSE1145) revealed overexpression of cardiac LCN2 after adjustment for multiple comparisons (Figure 6C; false discovery rate, q=0.008).
LCN2 Expression, Cardiac Size and Function, and BMI in the YFS
In the 1590 YFS individuals (mean age, 42 years) with matched echocardiographic and whole‐blood gene expression data, linear regression analysis adjusted for age and sex showed that LCN2 expression was associated with various structural and functional LV phenotypes (Table S5). Higher LCN2 expression was associated with increased heart rate (P=6×10−6), LV end‐diastolic volume (P=0.02), and cardiac output (P=3×10−6). LV mass (P=5×10−5) and thickness of the interventricular septum (P=8×10−4) were also positively correlated with LCN2 expression. Although the negative correlation between LCN2 expression and E/A ratio (P=5×10−4) suggested diastolic impairment, this might have been confounded by the increased heart rate,45 given that other measures of diastolic function (E/E′ ratio, mitral E‐wave declaration time, and isovolumic relaxation time) did not show significant correlations with LCN2 expression (Table S5). There was also no significant correlation between LCN2 expression and indices of systolic function, including ejection fraction and systolic wall velocities (Table S5). In addition, LCN2 expression correlated significantly with BMI (r=0.33; 95% CI, 0.28–0.37; P=8×10−42). In terms of LV phenotypes, BMI was associated with increased LV size, heart rate, end‐diastolic volume, and cardiac output (data not shown). There was also more consistent evidence of reduced diastolic function with increasing BMI, although systolic function was normal. Regression models that included LCN2 expression and BMI revealed that the correlation between BMI and LCN2 expression could account for the associations observed for LCN2 expression alone (data not shown). This has been reported previously and interpreted as part of the low‐grade inflammatory activation that accompanies obesity and predisposes to insulin resistance and type 2 diabetes mellitus.42
LCN2 Is Central to a Neutrophil Gene Coexpression Network and Is Under Genetic Control
Previous analysis of whole‐blood gene expression data identified a reproducible tightly coexpressed gene module associated with elevated levels of inflammatory markers in 2 independent healthy population studies.32 This module was significantly enriched for genes involved in the innate immune response, in particular neutrophil function.32 Among these, the expression of LCN2 showed high centrality to the neutrophil module (Figure 7), with a scaled connectivity (Data S1) of 0.65 and a correlation of 0.89 with the module's summary expression profile. In a healthy population of 1650 individuals from the YFS cohort (Table S4), we found that the module summary expression was independently associated with both GlycA (β=0.16; 95% CI, 0.11– 0.21; P=2×10−9) and CRP (β=0.15; 95% CI, 0.093–0.20; P=5×10−8) when both were included in the same model, suggesting the module is related to inflammatory processes reflected by both biomarkers. Although LCN2 expression itself was independently associated with both GlycA (β=0.18; 95% CI, 0.12–0.23; P=7×10−11) and CRP (β=0.19; 95% CI, 0.14–0.24; P=3×10−12), GlycA and CRP were no longer significant when LCN2 was included in the model. This suggests that LCN2 on its own is a better predictor of the module summary profile. To determine the potential genetic determinants of the neutrophil module function, we performed a QTL scan on the neutrophil module's summary expression (Data S1) in 1650 healthy individuals from the YFS cohort. We found that rs13297295, the top module QTL, was located 750 kb downstream from LCN2 to which it was a cis‐eQTL, with each “C” allele at rs13297295 increasing expression of LCN2 by 0.39 SD (P=2×10−9; adjusted for age, sex, and 2 genetic principal components). There was no detectable association between rs13297295 and CRP or GlycA. Taken together, these results suggest that increased LCN2 expression is central to the inflammatory gene module.
Figure 7.

Neutrophil module: An inflammatory biomarker associated coexpression network is under genetic control of a cis‐eQTL of lipocalin‐2 (LCN2). A, coexpression heatmap (Spearman's correlation) and scaled network connectivity (Methods) of genes composing the neutrophil module in the YFS (n=1650). B, Box plots of age‐ and sex‐adjusted LCN2 expression in individuals with differing dosages of the rs13297295 minor allele (“C”). C, Locus zoom plot of the 1‐MB region around LCN2 showing association on the y‐axis (−log10 P value) between each single‐nucleotide polymorphism (points) and LCN2 expression, recombination rate in the EUR population in that region (blue line underneath the points), and r 2 between each SNP and rs13297295 (point color).
Discussion
Our experimental and human studies reveal increased levels of LCN2 as a consistent correlate of cardiac hypertrophy (summarized in Figure 8). This relationship existed in the HHR polygenic model of spontaneous cardiac hypertrophy leading to heart failure, in Lcn2 gene knockout mice, but also in an environmental model of cardiac hypertrophy following intrauterine growth retardation. In human studies, LCN2 was associated with cardiac hypertrophy in healthy subjects of the YFS and in patients with type 2 diabetes mellitus. Importantly, our in vitro studies of Lcn2 overexpression showed that it can activate hypertrophic pathways and cause an increase in cardiomyocyte size but a decrease in their proliferation. Irrespective of the primary cause of increased LCN2, these direct cellular effects provide a common fundamental pathophysiology for the contribution of LCN2 to cardiac hypertrophy.
Figure 8.
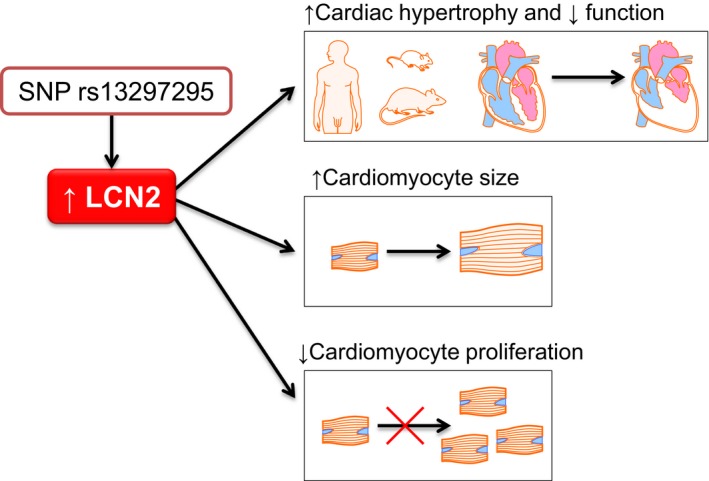
Diagram showing summary of findings. LCN2 indicates lipocalin‐2; SNP, single‐nucleotide polymorphism.
This is the first time that a specific cardiomyocyte hypertrophic effect of LCN2 has been demonstrated. Previous studies have focused on the effects of LCN2 on the interstitial matrix through induction of the proteinase, MMP9. This is relevant to the degradation of intercellular matrix as part of the remodeling of the heart during the development of hypertrophy. However, we could find no significant variation in MMP9 expression in association with changes in Lcn2 in HHR and Lcn2 knockout mice (data not shown). It would also be beneficial to submit Lcn2 knockout mice to stressors such as transverse aortic constriction to further understand the role of Lcn2 in heart disease, but this was outside the scope of this study.
Interestingly, we observed that Lcn2 expression reduced in vitro cardiomyocyte proliferation and cell numbers. It might seem counterintuitive that a limitation of cardiomyocyte numbers would contribute to cardiac hypertrophy. However, in very early life, when cardiomyocyte replication establishes the endowment of cardiac contractile cells, the actions of Lcn2 to reduce cell numbers could have long‐lasting effects.46 Fewer cells means greater individual workload resulting in hypertrophy. We have shown previously that the HHR has a reduced complement of cardiomyocytes in early postnatal life, an age at which we discovered cardiac Lcn2 to be highly expressed. Although we have no measurements of cardiomyocyte numbers in the early postnatal period following intrauterine growth restriction, other studies have shown that birth weight is associated with reduced numbers of cardiomyocytes,47 and very early protein restriction has been associated with increased cardiomyocyte apoptosis.48 Therefore, increased Lcn2 very early in life (whether genetic or environmental in origin) could predispose to hypertrophy through effects on cell number. The propensity for hypertrophy would be magnified by any persistent increase in Lcn2 levels into adulthood, as we saw in the HHR and in adult animals that had experienced intrauterine growth retardation.
In our human analyses, we found that LCN2 expression correlated significantly with cardiac size in healthy subjects in the YFS. Cardiac size also correlated with BMI in these subjects. Increased BMI is known to augment LCN2, probably as part of the induction of a chronic mild inflammatory state.49 Given the direct effects of LCN2 on cardiomyocyte hypertrophy, it is not unreasonable to suggest that at least part of the influence of BMI on heart size might be mediated through increases in LCN2. Diabetes mellitus is also characterized as a state of chronic inflammation and cardiac hypertrophy is a common finding patients with type 2 diabetes mellitus.50 We found that diabetic patients with cardiac hypertrophy had significantly higher plasma concentrations of LCN2, even after adjustment of BMI, renal function, and blood pressure. In the absence of other measures of inflammatory markers, we cannot be certain of the explanation of the elevated LCN2 in those diabetics with cardiac hypertrophy.
The factors that might increase LCN2 deserve consideration. LCN2 exhibits complex and tissue‐specific regulation and pathophysiology relevant to a broad portfolio of biological functions and disease involvements, including bacterial infection, inflammation, cancer, renal damage, obesity, and, more recently, CVD.51
In the YFS, the associations between LCN2 and the biomarkers, CRP and GlycA, suggest that underlying activation of inflammatory pathways might be responsible. LCN2 is released from neutrophils in response to inflammatory cytokines, including tumor necrosis factor‐α, interleukin‐1β and interleukin‐6.51 We analyzed the transcriptional subnetworks associated with the biomarkers of low‐grade inflammation in otherwise healthy individuals of the YFS. LCN2 expression appeared central to a coexpression module representing proteins secreted or expressed by neutrophils as part of the innate immune response.32 The expression of this neutrophil module was significantly associated with levels of both CRP and GlycA. Expression QTL analyses identified a common human genetic variant that increased LCN2 levels (rs13297295 “C” allele) and was also positively associated with module expression. Despite limited power to detect hypertrophic phenotype associations, further studies of this LCN2 eQTL in human cardiomyocytes is warranted.
In relation to increased Lcn2 expression in the HHR, evidence of underlying activation of inflammatory pathways is less clear. We could find no significant differential cardiac expression in neonatal HHR of the cytokines, tumor necrosis factor‐α, interleukin‐1β, or interleukin‐6, or the acute‐phase reactants that comprise the majority of GlycA (data not shown). Nor was there any significant association of Lcn2 with MMP9 expression. Given the polygenic nature of the HHR, other genetic factors are presumed to drive the higher Lcn2 expression. In a simple candidate gene approach, we identified 3 differences in DNA sequence in and around the Lcn2 gene between HHR and NHR, 1 of which (rs196968512) created a binding site for the transcriptional enhancer, RAR‐related orphan receptor A. The significance of this SNP and other polymorphisms requires further investigation in cross‐breeding linkage analyses.
The downstream effects of Lcn2 on cardiac expression pathways were reasonably consistent with changes in the pathways typically associated with hypertrophy observed in the HHR, the Lcn2 knockout mice, and in cultured cardiomyocytes. Of particular interest, in vitro Lcn2 overexpression downregulated the genes for dynamin 1 and thrombospondin 2 in cardiomyocytes. Mice with a mutation in the gene for dynamin 1 develop dilated cardiomyopathy through mitochondria defect and thus energy deficiency in the heart.40 thrombospondin 2‐knockout mice also display dilated cardiomyopathy with progressive cardiomyocyte stress and death.41
Our findings add to the evidence supporting a role for LCN2 in CVD, likely through inflammatory processes. Previous studies have shown LCN2 to be associated with atherosclerosis and plaque instability, endothelial dysfunction, oxidative stress,52 interstitial fibrosis,53 myocarditis, cardiac remodeling, and heart failure.54, 55 Baseline circulating LCN2 levels have been independently associated with subsequent development of CVD in 1 population study56 and in a 4‐year follow‐up after cerebrovascular ischemia study.57
Perspectives
The actions of LCN2 that we have observed on cardiomyocyte division and growth have implications for the development of cardiac hypertrophy and failure. The timing of the actions of LCN2 is also important. In the perinatal period, LCN2 could reduce the endowment of cardiomyocytes. In adulthood, LCN2 would tend to augment cardiomyocyte hypertrophy in response to any form of myocardial injury. The stimuli to LCN2 expression are both genetic—as in the HHR polygenic model in rats and possibly in the case of rs13297295 in man—and also environmental, as in the case of early‐life deprivation or other inflammatory activators, such as obesity and type 2 diabetes mellitus. Unfortunately, there are no human data regarding LCN2 in the neonatal period, and it is yet to be determined whether adult human cardiac hypertrophy might have its origin in reduced numbers of cardiomyocytes, but this emerges as a fascinating and important possibility.
Sources of Funding
This work was supported by grants from the National Health & Medical Research Council of Australia (project grant APP1034371, APP509252), the National Heart Foundation (project grant G10M5155, GM6368), and the Federation University Australia “Self‐sustaining Regions Research and Innovation Initiative,” an Australian Government Collaborative Research Network (CRN). Marques is supported by NHMRC (APP1052659) and National Heart Foundation (PF12M6785) co‐shared Early Career Fellowships, and a National Heart Foundation Future Leader and Baker Fellowships. Prestes is supported by a Robert HT Smith Fellowship from the Federation University Australia. Patel is supported by a Melbourne University Career Development Fellowship. Inouye was supported by a Career Development Fellowship from the National Health & Medical Research Council of Australia and National Heart Foundation (APP1061435). Byars and Inouye were supported by an NHMRC Project Grant (APP1062227). Wlodek was supported by a National Health & Medical Research Council of Australia Project Grant (APP400003). Salomaa was supported by the Finnish Foundation for Cardiovascular Research. Kettunen was supported by the Academy of Finland (grant 283045). Ala‐Korpela receives funds from the University of Bristol and UK Medical Research Council (MC_UU_12013/1). Würtz was supported by the Novo Nordisk Foundation. The Young Finns Study was financially supported by the Academy of Finland: grants 286284, 134309 (Eye), 126925, 121584, 124282, 129378 (Salve), 117787 (Gendi), and 41071 (Skidi); the Social Insurance Institution of Finland; Kuopio, Tampere, and Turku University Hospital Medical Funds (grant X51001); Juho Vainio Foundation; Paavo Nurmi Foundation; Finnish Foundation of Cardiovascular Research; Finnish Cultural Foundation; Tampere Tuberculosis Foundation; Emil Aaltonen Foundation; Yrjö Jahnsson Foundation; and Signe and Ane Gyllenberg Foundation. The quantitative serum NMR metabolomics platform and its development was supported by the Academy of Finland, TEKES—the Finnish Funding Agency for Technology and Innovation, Sigrid Juselius Foundation, Novo Nordisk Foundation, Finnish Diabetes Research Foundation, Paavo Nurmi Foundation, and strategic and infrastructural research funding from the University of Oulu, Finland, British Heart Foundation, Wellcome Trust, and Medical Research Council, UK.
Disclosures
Würtz, Kangas, Soininen, and Ala‐Korpela are shareholders of Brainstake Ltd. (www.brainshake.fi), a company offering NMR‐based metabolic profiling. Würtz, Kangas, Soininen, and Kettunen report employment for Brainstake Ltd.
Supporting information
Data S1. Supplemental methods.
Table S1. Primers and Conditions Used to Validate Microarray Gene Expression Results
Table S2. RNA‐Sequencing Alignment Characteristics of Individual Sample
Table S3. Characteristics of the Participants in the Type 2 Diabetes Mellitus Cohort
Table S4. Characteristics of Individuals From the YFS Cohort
Table S5. Linear Regression of LV Phenotypes on LCN2 Expression in the YFS. Models Were Adjusted for Age and Sex. Effect Size Indicates SD Change in Left Ventricular (LV) Phenotype Per SD Increase in LCN2 Expression
Table S6. Correlation of Plasma LCN2 With Echocardiogram Parameters in the Type 2 Diabetes Mellitus Cohort
Table S7. Transcriptome‐Wide Gene Expression Array Results Showing Genes Differentially Expressed in the Heart of Neonatal HHR (n=8) and NHR (n=8; Only Showing Genes With P Value Adjusted by Bonferroni <0.05 and Fold Difference >2)
Table S8. Gene Ontology Analysis With All Genes Differentially Expressed With Bonferroni‐Adjusted P<0.05
Table S9. Gene Set Enrichment Analysis, Based on Gene Ontology, of the Gene List for Differentially Expressed Genes Between HHR and NHR (P<0.05)
Table S10. Genes Differentially Expressed Between Lcn2‐Knockout Compared to Wild‐Type Mice Identified by RNA‐Sequencing (n=3/Group). Here Shown Only Genes With False Discovery Rate (FDR) q<0.05
Table S11. Gene Set Enrichment Analysis With Differentially Expressed Genes Between Lcn2‐Knockout Compared to Wild‐Type Mice
Table S12. Gene Set Enrichment Analysis With the Differentially Expressed Genes Between Cells Transfected With Lcn2 or Empty Plasmid
Figure S1. Hierarchical clustering showing that neonatal hypertrophic heart rat (HHR; blue) and normal heart rat (NHR; orange) form distinct groups based on gene expression (genes with Bonferroni adjustment <0.05). Numbers on the right are probe‐set numbers. Red depicts genes upregulated and green those downregulated.
Figure S2. Overexpression of Lcn2 in vitro. Lcn2 was overexpressed in H9c2 cells with the use of a plasmid. A, Optimization of the concentration of Lcn2 plasmid in vitro. B, There were higher Lcn2 mRNA in cells transfected with LCN2 plasmid. C, Overexpression of in cells Lcn2 visualized by immunofluorescence. D, There was no difference in apoptotic cells measured by flow cytometry in cells transfected with Lcn2 compared to those transfected with empty plasmid. ***P<0.001. Error bars represent SEM.
Figure S3. Representative of confocal microscopy images after transfection with Lcn2 plasmid, which provided results for Figure 4 in the main paper. A, Wheat‐germ agglutinin (WGA; red) and DAPI (blue) staining, used to estimate cell size (×400 magnification; scale bar=60 μm). B, Phospho‐histone H3 (pH3; green) and DAPI (blue), used to estimate cell proliferation and arrest (×200 magnification; scale bar=100μm).
Figure S4. There was no difference in apoptotic cells measured by flow cytometry in cells transfected with Lcn2 compared to those transfected with empty plasmid, demonstrated by Annexin V bound to apoptotic cells determined by flow cytometry. Data shown as number of cells as a percentage of the total number of cells. Error bars represent SEM.
Acknowledgments
We thank the Ramaciotti Centre for Gene Function Analysis for the help with arrays and the Australian Genome Research Facility for the help with Sanger and next‐generation sequencing. The authors thank Irina Lisinen for the expert technical assistance in the statistical analyses.
(J Am Heart Assoc. 2017;6:e005971 DOI: 10.1161/JAHA.117.005971.)28615213
References
- 1. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. [DOI] [PubMed] [Google Scholar]
- 2. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation. 2015;131:1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palmieri V, Tracy RP, Roman MJ, Liu JE, Best LG, Bella JN, Robbins DC, Howard BV, Devereux RB; Strong Heart S . Relation of left ventricular hypertrophy to inflammation and albuminuria in adults with type 2 diabetes: the Strong Heart Study. Diabetes Care. 2003;26:2764–2769. [DOI] [PubMed] [Google Scholar]
- 4. Salles GF, Fiszman R, Cardoso CR, Muxfeldt ES. Relation of left ventricular hypertrophy with systemic inflammation and endothelial damage in resistant hypertension. Hypertension. 2007;50:723–728. [DOI] [PubMed] [Google Scholar]
- 5. Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C‐reactive protein, albumin, or leukocyte count with coronary heart disease: meta‐analyses of prospective studies. JAMA. 1998;279:1477–1482. [DOI] [PubMed] [Google Scholar]
- 6. Halade GV, Jin YF, Lindsey ML. Matrix metalloproteinase (MMP)‐9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol Ther. 2013;139:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blankenberg S, Rupprecht HJ, Poirier O, Bickel C, Smieja M, Hafner G, Meyer J, Cambien F, Tiret L; AtheroGene I . Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation. 2003;107:1579–1585. [DOI] [PubMed] [Google Scholar]
- 8. Folkesson M, Kazi M, Zhu C, Silveira A, Hemdahl AL, Hamsten A, Hedin U, Swedenborg J, Eriksson P. Presence of NGAL/MMP‐9 complexes in human abdominal aortic aneurysms. Thromb Haemost. 2007;98:427–433. [PubMed] [Google Scholar]
- 9. Bu DX, Hemdahl AL, Gabrielsen A, Fuxe J, Zhu C, Eriksson P, Yan ZQ. Induction of neutrophil gelatinase‐associated lipocalin in vascular injury via activation of nuclear factor‐kappaB. Am J Pathol. 2006;169:2245–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bolignano D, Coppolino G, Lacquaniti A, Buemi M. From kidney to cardiovascular diseases: NGAL as a biomarker beyond the confines of nephrology. Eur J Clin Invest. 2010;40:273–276. [DOI] [PubMed] [Google Scholar]
- 11. Leoncini G, Mussap M, Viazzi F, Fravega M, Degrandi R, Bezante GP, Deferrari G, Pontremoli R. Combined use of urinary neutrophil gelatinase‐associated lipocalin (uNGAL) and albumin as markers of early cardiac damage in primary hypertension. Clin Chim Acta. 2011;412:1951–1956. [DOI] [PubMed] [Google Scholar]
- 12. Hemdahl AL, Gabrielsen A, Zhu C, Eriksson P, Hedin U, Kastrup J, Thoren P, Hansson GK. Expression of neutrophil gelatinase‐associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol. 2006;26:136–142. [DOI] [PubMed] [Google Scholar]
- 13. Maisel AS, Mueller C, Fitzgerald R, Brikhan R, Hiestand BC, Iqbal N, Clopton P, van Veldhuisen DJ. Prognostic utility of plasma neutrophil gelatinase‐associated lipocalin in patients with acute heart failure: the NGAL EvaLuation Along with B‐type NaTriuretic Peptide in acutely decompensated heart failure (GALLANT) trial. Eur J Heart Fail. 2011;13:846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minana G, Rumiz E, Palau P, Valero E, Bodi V, Nunez E, Sanchis J, Nunez J. Plasma neutrophil gelatinase‐associated lipocalin and long‐term mortality in patients with acute heart failure and normal renal function. Int J Cardiol. 2016;214:51–53. [DOI] [PubMed] [Google Scholar]
- 15. Harrap SB, Danes VR, Ellis JA, Griffiths CD, Jones EF, Delbridge LM. The hypertrophic heart rat: a new normotensive model of genetic cardiac and cardiomyocyte hypertrophy. Physiol Genomics. 2002;9:43–48. [DOI] [PubMed] [Google Scholar]
- 16. Porrello ER, Bell JR, Schertzer JD, Curl CL, McMullen JR, Mellor KM, Ritchie RH, Lynch GS, Harrap SB, Thomas WG, Delbridge LM. Heritable pathologic cardiac hypertrophy in adulthood is preceded by neonatal cardiac growth restriction. Am J Physiol Regul Integr Comp Physiol. 2009;296:R672–R680. [DOI] [PubMed] [Google Scholar]
- 17. Markus MA, Marques FZ, Morris BJ. Resveratrol, by modulating RNA processing factor levels, can influence the alternative splicing of pre‐mRNAs. PLoS One. 2011;6:e28926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA, Moritz KM. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J Am Soc Nephrol. 2007;18:1688–1696. [DOI] [PubMed] [Google Scholar]
- 19. Wlodek ME, Westcott K, Siebel AL, Owens JA, Moritz KM. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008;74:187–195. [DOI] [PubMed] [Google Scholar]
- 20. Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, Dorn GW II, van Rooij E, Olson EN. MiR‐15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res. 2011;109:670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Srivastava PM, Calafiore P, Macisaac RJ, Patel SK, Thomas MC, Jerums G, Burrell LM. Prevalence and predictors of cardiac hypertrophy and dysfunction in patients with Type 2 diabetes. Clin Sci (Lond). 2008;114:313–320. [DOI] [PubMed] [Google Scholar]
- 23. Raitakari OT, Juonala M, Ronnemaa T, Keltikangas‐Jarvinen L, Rasanen L, Pietikainen M, Hutri‐Kahonen N, Taittonen L, Jokinen E, Marniemi J, Jula A, Telama R, Kahonen M, Lehtimaki T, Akerblom HK, Viikari JS. Cohort profile: the cardiovascular risk in Young Finns Study. Int J Epidemiol. 2008;37:1220–1226. [DOI] [PubMed] [Google Scholar]
- 24. Raitoharju E, Seppala I, Oksala N, Lyytikainen LP, Raitakari O, Viikari J, Ala‐Korpela M, Soininen P, Kangas AJ, Waldenberger M, Klopp N, Illig T, Leiviska J, Loo BM, Hutri‐Kahonen N, Kahonen M, Laaksonen R, Lehtimaki T. Blood microRNA profile associates with the levels of serum lipids and metabolites associated with glucose metabolism and insulin resistance and pinpoints pathways underlying metabolic syndrome: the cardiovascular risk in Young Finns Study. Mol Cell Endocrinol. 2014;391:41–49. [DOI] [PubMed] [Google Scholar]
- 25. Smith EN, Chen W, Kahonen M, Kettunen J, Lehtimaki T, Peltonen L, Raitakari OT, Salem RM, Schork NJ, Shaw M, Srinivasan SR, Topol EJ, Viikari JS, Berenson GS, Murray SS. Longitudinal genome‐wide association of cardiovascular disease risk factors in the Bogalusa heart study. PLoS Genet. 2010;6:e1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wurtz P, Wang Q, Kangas AJ, Richmond RC, Skarp J, Tiainen M, Tynkkynen T, Soininen P, Havulinna AS, Kaakinen M, Viikari JS, Savolainen MJ, Kahonen M, Lehtimaki T, Mannisto S, Blankenberg S, Zeller T, Laitinen J, Pouta A, Mantyselka P, Vanhala M, Elliott P, Pietilainen KH, Ripatti S, Salomaa V, Raitakari OT, Jarvelin MR, Smith GD, Ala‐Korpela M. Metabolic signatures of adiposity in young adults: Mendelian randomization analysis and effects of weight change. PLoS Med. 2014;11:e1001765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruohonen S, Koskenvuo JW, Wendelin‐Saarenhovi M, Savontaus M, Kahonen M, Laitinen T, Lehtimaki T, Jokinen E, Viikari J, Juonala M, Taittonen L, Tossavainen P, Kallio M, Bax JJ, Raitakari O. Reference values for echocardiography in middle‐aged population: the cardiovascular risk in Young Finns Study. Echocardiography. 2016;33:193–206. [DOI] [PubMed] [Google Scholar]
- 28. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ; Chamber Quantification Writing G, American Society of Echocardiography's G, Standards C and European Association of E . Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–1463. [DOI] [PubMed] [Google Scholar]
- 29. Maragiannis D, Nagueh SF. Echocardiographic evaluation of left ventricular diastolic function: an update. Curr Cardiol Rep. 2015;17:3. [DOI] [PubMed] [Google Scholar]
- 30. Wai B, Patel SK, Ord M, MacIsaac RJ, Jerums G, Srivastava PM, Burrell LM. Prevalence, predictors and evolution of echocardiographically defined cardiac abnormalities in adults with type 1 diabetes: an observational cohort study. J Diabetes Complications. 2014;28:22–28. [DOI] [PubMed] [Google Scholar]
- 31. Otvos JD, Shalaurova I, Wolak‐Dinsmore J, Connelly MA, Mackey RH, Stein JH, Tracy RP. GlycA: a composite nuclear magnetic resonance biomarker of systemic inflammation. Clin Chem. 2015;61:714–723. [DOI] [PubMed] [Google Scholar]
- 32. Ritchie SC, Wurtz P, Nath AP, Abraham G, Havulinna AS, Fearnley LG, Sarin AP, Kangas AJ, Soininen P, Aalto K, Seppala I, Raitoharju E, Salmi M, Maksimow M, Mannisto S, Kahonen M, Juonala M, Ripatti S, Lehtimaki T, Jalkanen S, Perola M, Raitakari O, Salomaa V, Ala‐Korpela M, Kettunen J, Inouye M. The biomarker GlycA is associated with chronic inflammation and predicts long‐term risk of severe infection. Cell Syst. 2015;1:293–301. [DOI] [PubMed] [Google Scholar]
- 33. Bell JD, Brown JC, Nicholson JK, Sadler PJ. Assignment of resonances for ‘acute‐phase’ glycoproteins in high resolution proton NMR spectra of human blood plasma. FEBS Lett. 1987;215:311–315. [DOI] [PubMed] [Google Scholar]
- 34. Shi W, Oshlack A, Smyth GK. Optimizing the noise versus bias trade‐off for Illumina whole genome expression BeadChips. Nucleic Acids Res. 2010;38:e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ritchie SC, Watts S, Fearnley LG, Holt KE, Abraham G, Inouye M. A scalable permutation approach reveals replication and preservation of network modules. Cell Syst. 2016;3:71–82. [DOI] [PubMed] [Google Scholar]
- 36. Thomas‐Chollier M, Hufton A, Heinig M, O'Keeffe S, Masri NE, Roider HG, Manke T, Vingron M. Transcription factor binding predictions using TRAP for the analysis of ChIP‐seq data and regulatory SNPs. Nat Protoc. 2011;6:1860–1869. [DOI] [PubMed] [Google Scholar]
- 37. Manke T, Heinig M, Vingron M. Quantifying the effect of sequence variation on regulatory interactions. Hum Mutat. 2010;31:477–483. [DOI] [PubMed] [Google Scholar]
- 38. Giguere V, Tini M, Flock G, Ong E, Evans RM, Otulakowski G. Isoform‐specific amino‐terminal domains dictate DNA‐binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev. 1994;8:538–553. [DOI] [PubMed] [Google Scholar]
- 39. Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. [DOI] [PubMed] [Google Scholar]
- 40. Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, Lygate CA, Hough T, Townsend S, Williams D, Wells S, Norris D, Glyn‐Jones S, Land J, Barbaric I, Lalanne Z, Denny P, Szumska D, Bhattacharya S, Griffin JL, Hargreaves I, Fernandez‐Fuentes N, Cheeseman M, Watkins H, Dear TN. A mutation in the mitochondrial fission gene Dnm1 l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Swinnen M, Vanhoutte D, Van Almen GC, Hamdani N, Schellings MW, D'Hooge J, Van der Velden J, Weaver MS, Sage EH, Bornstein P, Verheyen FK, VandenDriessche T, Chuah MK, Westermann D, Paulus WJ, Van de Werf F, Schroen B, Carmeliet P, Pinto YM, Heymans S. Absence of thrombospondin‐2 causes age‐related dilated cardiomyopathy. Circulation. 2009;120:1585–1597. [DOI] [PubMed] [Google Scholar]
- 42. Yan QW, Yang Q, Mody N, Graham TE, Hsu CH, Xu Z, Houstis NE, Kahn BB, Rosen ED. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes. 2007;56:2533–2540. [DOI] [PubMed] [Google Scholar]
- 43. Taube A, Schlich R, Sell H, Eckardt K, Eckel J. Inflammation and metabolic dysfunction: links to cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2012;302:H2148–H2165. [DOI] [PubMed] [Google Scholar]
- 44. Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. [DOI] [PubMed] [Google Scholar]
- 45. Galderisi M, Benjamin EJ, Evans JC, D'Agostino RB, Fuller DL, Lehman B, Levy D. Impact of heart rate and PR interval on Doppler indexes of left ventricular diastolic filling in an elderly cohort (the Framingham Heart Study). Am J Cardiol. 1993;72:1183–1187. [DOI] [PubMed] [Google Scholar]
- 46. Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stacy V, De Matteo R, Brew N, Sozo F, Probyn ME, Harding R, Black MJ. The influence of naturally occurring differences in birthweight on ventricular cardiomyocyte number in sheep. Anat Rec (Hoboken). 2009;292:29–37. [DOI] [PubMed] [Google Scholar]
- 48. Cheema KK, Dent MR, Saini HK, Aroutiounova N, Tappia PS. Prenatal exposure to maternal undernutrition induces adult cardiac dysfunction. Br J Nutr. 2005;93:471–477. [DOI] [PubMed] [Google Scholar]
- 49. Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW, Chow WS, Wat NM, Xu JY, Hoo RL, Xu A. Lipocalin‐2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34–41. [DOI] [PubMed] [Google Scholar]
- 50. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. [DOI] [PubMed] [Google Scholar]
- 51. Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta. 2012;1826:129–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chan YK, Sung HK, Sweeney G. Iron metabolism and regulation by neutrophil gelatinase‐associated lipocalin in cardiomyopathy. Clin Sci (Lond). 2015;129:851–862. [DOI] [PubMed] [Google Scholar]
- 53. Tarjus A, Martinez‐Martinez E, Amador C, Latouche C, El Moghrabi S, Berger T, Mak TW, Fay R, Farman N, Rossignol P, Zannad F, Lopez‐Andres N, Jaisser F. Neutrophil gelatinase‐associated lipocalin, a novel mineralocorticoid biotarget, mediates vascular profibrotic effects of mineralocorticoids. Hypertension. 2015;66:158–166. [DOI] [PubMed] [Google Scholar]
- 54. Daniels LB, Barrett‐Connor E, Clopton P, Laughlin GA, Ix JH, Maisel AS. Plasma neutrophil gelatinase‐associated lipocalin is independently associated with cardiovascular disease and mortality in community‐dwelling older adults: the Rancho Bernardo Study. J Am Coll Cardiol. 2012;59:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alvelos M, Lourenco P, Dias C, Amorim M, Rema J, Leite AB, Guimaraes JT, Almeida P, Bettencourt P. Prognostic value of neutrophil gelatinase‐associated lipocalin in acute heart failure. Int J Cardiol. 2013;165:51–55. [DOI] [PubMed] [Google Scholar]
- 56. Wu G, Li H, Fang Q, Jiang S, Zhang L, Zhang J, Hou X, Lu J, Bao Y, Xu A, Jia W. Elevated circulating lipocalin‐2 levels independently predict incident cardiovascular events in men in a population‐based cohort. Arterioscler Thromb Vasc Biol. 2014;34:2457–2464. [DOI] [PubMed] [Google Scholar]
- 57. Falke P, Elneihoum AM, Ohlsson K. Leukocyte activation: relation to cardiovascular mortality after cerebrovascular ischemia. Cerebrovasc Dis. 2000;10:97–101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Table S1. Primers and Conditions Used to Validate Microarray Gene Expression Results
Table S2. RNA‐Sequencing Alignment Characteristics of Individual Sample
Table S3. Characteristics of the Participants in the Type 2 Diabetes Mellitus Cohort
Table S4. Characteristics of Individuals From the YFS Cohort
Table S5. Linear Regression of LV Phenotypes on LCN2 Expression in the YFS. Models Were Adjusted for Age and Sex. Effect Size Indicates SD Change in Left Ventricular (LV) Phenotype Per SD Increase in LCN2 Expression
Table S6. Correlation of Plasma LCN2 With Echocardiogram Parameters in the Type 2 Diabetes Mellitus Cohort
Table S7. Transcriptome‐Wide Gene Expression Array Results Showing Genes Differentially Expressed in the Heart of Neonatal HHR (n=8) and NHR (n=8; Only Showing Genes With P Value Adjusted by Bonferroni <0.05 and Fold Difference >2)
Table S8. Gene Ontology Analysis With All Genes Differentially Expressed With Bonferroni‐Adjusted P<0.05
Table S9. Gene Set Enrichment Analysis, Based on Gene Ontology, of the Gene List for Differentially Expressed Genes Between HHR and NHR (P<0.05)
Table S10. Genes Differentially Expressed Between Lcn2‐Knockout Compared to Wild‐Type Mice Identified by RNA‐Sequencing (n=3/Group). Here Shown Only Genes With False Discovery Rate (FDR) q<0.05
Table S11. Gene Set Enrichment Analysis With Differentially Expressed Genes Between Lcn2‐Knockout Compared to Wild‐Type Mice
Table S12. Gene Set Enrichment Analysis With the Differentially Expressed Genes Between Cells Transfected With Lcn2 or Empty Plasmid
Figure S1. Hierarchical clustering showing that neonatal hypertrophic heart rat (HHR; blue) and normal heart rat (NHR; orange) form distinct groups based on gene expression (genes with Bonferroni adjustment <0.05). Numbers on the right are probe‐set numbers. Red depicts genes upregulated and green those downregulated.
Figure S2. Overexpression of Lcn2 in vitro. Lcn2 was overexpressed in H9c2 cells with the use of a plasmid. A, Optimization of the concentration of Lcn2 plasmid in vitro. B, There were higher Lcn2 mRNA in cells transfected with LCN2 plasmid. C, Overexpression of in cells Lcn2 visualized by immunofluorescence. D, There was no difference in apoptotic cells measured by flow cytometry in cells transfected with Lcn2 compared to those transfected with empty plasmid. ***P<0.001. Error bars represent SEM.
Figure S3. Representative of confocal microscopy images after transfection with Lcn2 plasmid, which provided results for Figure 4 in the main paper. A, Wheat‐germ agglutinin (WGA; red) and DAPI (blue) staining, used to estimate cell size (×400 magnification; scale bar=60 μm). B, Phospho‐histone H3 (pH3; green) and DAPI (blue), used to estimate cell proliferation and arrest (×200 magnification; scale bar=100μm).
Figure S4. There was no difference in apoptotic cells measured by flow cytometry in cells transfected with Lcn2 compared to those transfected with empty plasmid, demonstrated by Annexin V bound to apoptotic cells determined by flow cytometry. Data shown as number of cells as a percentage of the total number of cells. Error bars represent SEM.