

Abstract
Background
Pulmonary arterial hypertension (PAH) is a lethal disease characterized by obstructive pulmonary vascular remodeling and right ventricular (RV) dysfunction. Although RV function predicts outcomes in PAH, mechanisms of RV dysfunction are poorly understood, and RV‐targeted therapies are lacking. We hypothesized that in PAH, abnormal microtubular structure in RV cardiomyocytes impairs RV function by reducing junctophilin‐2 (JPH2) expression, resulting in t‐tubule derangements. Conversely, we assessed whether colchicine, a microtubule‐depolymerizing agent, could increase JPH2 expression and enhance RV function in monocrotaline‐induced PAH.
Methods and Results
Immunoblots, confocal microscopy, echocardiography, cardiac catheterization, and treadmill testing were used to examine colchicine's (0.5 mg/kg 3 times/week) effects on pulmonary hemodynamics, RV function, and functional capacity. Rats were treated with saline (n=28) or colchicine (n=24) for 3 weeks, beginning 1 week after monocrotaline (60 mg/kg, subcutaneous). In the monocrotaline RV, but not the left ventricle, microtubule density is increased, and JPH2 expression is reduced, with loss of t‐tubule localization and t‐tubule disarray. Colchicine reduces microtubule density, increases JPH2 expression, and improves t‐tubule morphology in RV cardiomyocytes. Colchicine therapy diminishes RV hypertrophy, improves RV function, and enhances RV–pulmonary artery coupling. Colchicine reduces small pulmonary arteriolar thickness and improves pulmonary hemodynamics. Finally, colchicine increases exercise capacity.
Conclusions
Monocrotaline‐induced PAH causes RV‐specific derangement of microtubules marked by reduction in JPH2 and t‐tubule disarray. Colchicine reduces microtubule density, increases JPH2 expression, and improves both t‐tubule architecture and RV function. Colchicine also reduces adverse pulmonary vascular remodeling. These results provide biological plausibility for a clinical trial to repurpose colchicine as a RV‐directed therapy for PAH.
Keywords: pulmonary hypertension, right ventricular failure, right ventricular pressure overload, T‐tubules
Subject Categories: Basic Science Research, Mechanisms, Myocardial Biology, Translational Studies
Clinical Perspective
What Is New?
Microtubule remodeling leads to RV–specific misregulation of junctophilin‐2, resulting in t‐tubule disruptions and RV dysfunction in monocrotaline rats.
Colchicine‐induced microtubule depolymerization increases junctophilin‐2, improves t‐tubule morphology, and enhances RV function and exercise capacity in monocrotaline rats.
What Are the Clinical Implications?
These data suggest that colchicine could be repurposed as a RV–directed therapy for PAH.
Introduction
Pulmonary arterial hypertension (PAH) is a lethal cardiovascular disorder with a 1‐year mortality rate of 15% to 20% and a median survival of 5 to 7 years after diagnosis.1, 2, 3 In PAH, obstructive remodeling of the pulmonary vasculature and reduced PA compliance increase pulmonary arterial pressures and right ventricular (RV) workload.4, 5 As PAH progresses, RV function declines, and many patients succumb from RV failure.6, 7 Although RV dysfunction is repeatedly identified as the major risk factor for mortality in PAH,8, 9, 10 the 4 classes of approved pulmonary hypertension medications primarily target the pulmonary vasculature, with little evidence of direct benefit to the RV. Although there are data that phosphodiesterase‐5 inhibitors improve RV function in animal models of PAH,11 an effect on the RV in humans has not been proven. Thus, an ideal therapeutic agent for PAH would not only reduce pulmonary vascular resistance but also augment RV function.
There is pathological remodeling of the microtubule cytoskeleton in the RV in pulmonary artery–banded felines12 and rats with monocrotaline (MCT)‐induced PAH.13 This suggested the possibility that targeting microtubules with colchicine, a microtubule‐depolymerizing agent,14 might improve RV function. Microtubule remodeling causes improper trafficking and subsequent misregulation of junctophilin‐2 (JPH2) in mouse models of left ventricular (LV) dysfunction15, 16; however, the role of microtubules in RV dysfunction is unknown. JPH2 is a protein that is essential for life17 because of its critical roles in maintaining cardiomyocyte t‐tubule structure and regulating calcium handling by gating the ryanodine receptor.18 Moreover, transgenic19 and virally mediated overexpression20 of JPH2 augments LV function in mice with LV dysfunction caused by aortic banding. Although there is strong evidence that JPH2 plays a significant role in LV dysfunction, little is known about the role of microtubule structure and JPH2 expression in RV dysfunction associated with PAH. Colchicine treatment reduces microtubule density and normalizes JPH2 subcellular localization, which improves t‐tubule architecture in LV cardiomyocytes in mouse models of LV dysfunction.15, 16 However, the effects of colchicine on the RV are unknown.
Here, we investigated the hypothesis that in PAH abnormal microtubulular structure in RV cardiomyocytes impairs RV function by reducing JPH2 expression and adversely altering t‐tubule structure. Conversely, we assessed whether colchicine could improve RV function in PAH by reducing the microtubule density and increasing JPH2 expression in RV cardiomyocytes. We created PAH using the well‐validated MCT rat model, which we selected because it results in RV failure. We injected rats with either phosphate‐buffered saline (PBS) or colchicine 1 week after MCT injection, the time point when pulmonary vascular disease is first detectable.21, 22 After 3 weeks of treatment, we examined the effects of colchicine therapy on microtubule density, JPH2 protein levels and localization patterns, t‐tubule morphology, RV size and function, pulmonary vascular disease severity, RV–pulmonary artery (PA) coupling, exercise capacity, and survival. In MCT‐PAH we show a RV‐restricted reduction in JPH2 expression leading to chamber‐specific t‐tubule derangements. Colchicine therapy in vivo increases JPH2 expression and attenuates t‐tubule remodeling. The beneficial effects of RV cardiomyocyte ultrastructure are associated with improvements in RV function. RV‐PA coupling is enhanced in rats treated with colchicine, indicating a direct beneficial effect on the RV. However, an additional advantage of colchicine is that it reduces the severity of pulmonary vascular disease. Finally, colchicine treatment improves exercise capacity. The observed benefits of colchicine on JPH2 expression, t‐tubule structure, RV‐PA coupling, and functional capacity suggest that a trial to repurpose colchicine to treat PAH may be reasonable.
Materials and Methods
Animal Models
Male Sprague‐Dawley rats were purchased from Charles Rivers Laboratories (Charles River, QC, Canada). Rats received subcutaneous injections of MCT (60 mg/kg; Sigma‐Aldrich, St. Louis, MO) (n=52) or PBS (n=16). Experiments were conducted in accordance with published guidelines of the Canadian Council of Animal Care and approved by Queen's University Animal Care Committee.
Chronic Colchicine Treatment
One week after induction of PAH with MCT, a time point that corresponds to the onset of detectable pulmonary vascular disease but early in the course of evolution of PAH,21, 22 rats were treated with intraperitoneal injections of 0.5 mg/kg filter‐sterilized colchicine (Sigma‐Aldrich, St. Louis, MO) dissolved in PBS (n=24) or an equivalent volume of PBS (n=28) on Monday, Wednesday, and Friday for 3 weeks and were then analyzed. A sample size of 25 to 30 was targeted for 3 reasons: there were 3 groups in the analysis, to reduce effects of the heterogeneity with colchicine treatment, and to account for possible deaths during the experimental time frame. The dosing scheme was implemented as similar doses were shown to depolymerize the microtubule cytoskeleton in cardiomyocytes in rats23, 24 but less than doses that are associated with significant cardiotoxicity in rats.25 Four weeks after MCT injection was the end point of the study because a previous study had shown that time frame was associated with severe pulmonary hypertension.21
Antibodies
Polyclonal antibodies for voltage‐gated calcium channel (Sigma‐Aldrich, St. Louis, MO) and JPH2 (ThermoScientific, Waltham, MA) and monoclonal antibodies for α‐tubulin (Sigma‐Aldrich) and β‐tubulin (Sigma‐Aldrich) were used. Secondary antibodies, Alexa Fluor‐488 or Alexa Fluor‐568 conjugated antirabbit or antimouse antibodies, were purchased from Molecular Probes (Eugene, OR). Infrared dye–conjugated antimouse and antirabbit antibodies were purchased from LICOR Biosciences (Lincoln, NE).
Cardiomyocyte Isolation
Isolation of control RV cardiomyocytes was performed as described previously.26 Briefly, adult rat hearts were enzymatically digested via retrograde perfusion with collagenase. After digestion, the RV was isolated and then cut into small pieces and triturated to single cardiomyocytes. Cardiomyocytes were plated on laminin‐coated coverslips, and after 1 hour, cells were bathed with M‐199 media (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10 mmol/L HEPES, 26.2 mmol/L sodium bicarbonate, 0.02% bovine serum albumin, and 50 U/mL penicillin‐streptomycin, insulin (5 μg/mL), transferrin (5 μg/mL), and selenite (5 ng/mL).
Paclitaxel Treatment in Isolated Cardiomyocytes
Isolated control cardiomyocytes were cultured as described above. To evaluate the effects of microtubule stabilization, which is a pathologic feature of RV dysfunction, we administered the microtubule‐stabilizing agent paclitaxel (Cytoskeleton, Denver, CO)27 (20 μmol/L) dissolved in dimethyl sulfoxide (DMSO) (Sigma‐Aldrich) or an equivalent volume of DMSO for a total of 24 hours.
Western Blot Analysis and Quantification
Immunoblots were performed on RV and LV specimens, as described.16 Protein concentration was determined using a bicinchoninic acid protein assay kit (Pierce, Waltham, MA). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) was performed with 25 μg of protein extracts and then transferred to nitrocellulose membrane. Membranes were washed/blocked in 5% milk in PBS for 1 hour and incubated with primary antibodies for 1 hour at room temperature. Subsequently, membranes were again washed and blocked twice for 10 minutes in 5% milk in PBS and then incubated with infrared secondary antibodies for 30 minutes at room temperature. Finally, membranes were washed twice for 5 minutes in Tris‐buffered saline containing 0.1% Tween (Sigma‐Aldrich). Quantification of Western blots was performed on an Odyssey Infrared Imaging system (Lincoln, NE). Posttransfer SDS‐PAGE gels were stained with Coomassie brilliant blue and imaged at the 700‐nm wavelength on the Odyssey Imaging system as the loading control, with the band corresponding to the myosin heavy chain used as the reference.16
T‐Tubule Analysis
Primary cardiomyocytes and 10‐μm RV and LV cryosections were fixed with 4% paraformaldehyde for 10 minutes at 37°C, washed twice for 5 minutes with PBS, incubated with Alexa Fluor‐488 conjugated wheat germ agglutinin (Sigma‐Aldrich) for 10 minutes at room temperature, and then washed in PBS twice for 5 minutes and mounted in Anti‐Fade Reagent (Molecular Probes). Images were collected using a Bio‐Rad (Hercules, CA) MRC 1000 scan head mounted on an upright Nikon Optishot (Tokyo, Japan) microscope at the University of Minnesota Imaging Center. Z‐stacks were collected and converted into a Z‐projection using ImageJ (National Institutes of Health, Bethesda, MD). T‐tubule structure and regularity were quantified using the TTPower plugin on ImageJ, which uses fast Fourier transformation to quantitate the regularity of t‐tubules as previously described.16, 28
Immunofluorescence Analysis
Primary cardiomyocytes and 10‐μm RV or LV cryosections were fixed in 4% paraformaldehyde for 10 minutes at 37°C, permeabilized with 1% Triton X‐100 (Sigma‐Aldrich) in PBS, blocked in 5% bovine serum albumin (BSA) in PBS for 10 minutes 3 times, and incubated with primary antibodies overnight at 4°C. Slides were then washed and blocked with 5% BSA in PBS for 10 minutes 3 times and then incubated with Alexa Fluor‐488 or Alexa Fluor‐568 conjugated secondary antibodies for 30 minutes at 37°C. Then, slides were washed with PBS and mounted in Anti‐Fade Reagent (Molecular Probes). Confocal micrographs were collected as described above. JPH2 localization was quantified using the TTPower plugin on ImageJ as described.16
Image Processing
Confocal images were collected and processed using identical settings to optimize image quality in Adobe Photoshop Version CS6 (San Jose, CA).
Echocardiography
Echocardiography was performed using a Vevo2100 ultrasound system with a 37.5‐MHz transducer (Visual Sonics Inc, Toronto, ON, Canada). Rats were lightly anesthetized with isoflurane (1.6% to 2.0%), and chest hair was removed using a depilatory cream (Nair®; Carter‐Horner, Mississauga, ON, Canada). M‐mode and 2‐D modalities were applied to measure RV free wall (RVFW) thickness during end diastole and end systole and tricuspid annular plane systolic excursion (TAPSE). PA diameter was measured at the level of pulmonary outflow tract during midsystole. Pulsed‐wave Doppler was used to measure PA acceleration time and PA flow velocity time integral. RV ejection time was measured as the interval from the onset to the end of ejection in milliseconds, as previously described.21 Stroke volume, cardiac output, and cardiac index were calculated as previously described.21 Representative images are depicted in Figure S1.
Right Ventricular‐Pulmonary Artery Coupling
RV‐PA coupling was defined 2 ways: TAPSE divided by invasively measured mean pulmonary artery pressure (mPAP) and percentage change in RVFW thickness/mPAP as previously described.29, 30, 31, 32, 33
Treadmill Walk Distance
Exercise capacity was tested by measuring maximal distance run on a motorized treadmill (Simplex II Instrument; Columbus Instruments, Columbus, OH). The initial treadmill speed was 10 m/min and increased 5 m/min every 5 minutes for 30 minutes or until the rat fatigued.
Right Heart Catheterization
Rats were anesthetized with intraperitoneal injection of a cocktail containing ketamine (75 mg/kg) and xylazine (10 mg/kg). During catheterization, rats were intubated and ventilated. A fluid‐filled polyethylene catheter was used to measure RV pressure, systolic pulmonary artery pressure and mPAP in anesthetized, closed‐chest rats. High‐fidelity pressures were recorded and analyzed using a PowerLab 8/35 data acquisition module and LabChart 8.1 software (ADInstruments, Colorado Spring, CO). Cardiac output was measured by the thermodilution method.21 Total pulmonary resistance was calculated as mPAP/cardiac output.
Survival Analysis
Rats were defined as dead for purposes of survival calculation if they were found dead by investigators or were too ill for continued study and thus required euthanasia per criteria established by the Queen's University Veterinary staff (including more than 10% loss in body weight and failure to feed).
Lung Histology
Lungs were perfused with 10% formalin at a pressure of 25 cm H2O for 1 hour. After fixation in formalin, lungs were frozen in melting isopentane, cryosectioned, and stained with hematoxylin and eosin. Percentage medial thickness of small pulmonary arterioles (20 to 200 μm in diameter) was calculated as 100 times (outer diameter−inner diameter)/outer diameter,34 using ImageJ software (NIH).
Cardiomyocyte Fiber Diameter Analysis
RV cardiomyocyte diameter was quantified from transverse sections of the RVFW. The extracellular matrix was labeled with Alexa Fluor‐488 conjugated wheat germ agglutinin, and confocal micrographs were collected as described above. The greatest diameter of the cardiomyocytes was calculated using ImageJ software (NIH).
Statistics
All data are represented as mean±standard error of the mean. Unpaired t test was used for comparing means of 2 groups, and analysis of variance with Tukey post hoc test was used to compare means among 3 groups. The relationship between mPAP and RV function was analyzed using linear correlational analysis. A Kaplan‐Meier survival curve was constructed to determine survival differences using a log‐rank test to statistically assess differences in survival. All statistical analysis was performed using GraphPad Prism (V7.01; LaJolla, CA). Statistical significance was defined by P<0.05.
Results
Consistent with a previous report,13 the microtubule cytoskeleton was pathologically remodeled in the RV of MCT rats. Relative to controls, RV expression of α‐ and β‐tubulin, individual microtubule subunits, was elevated over 2.5‐fold in MCT RV extracts (Figure S2A and S2B). Moreover, immunofluorescence analysis of RV cryosections revealed an increase in the microtubule density in the RVs of MCT rats (Figure S2C).
Next, we assessed the effects of colchicine on JPH2 and t‐tubule morphology in the RV of MCT‐PAH rats. Colchicine reduced microtubule density in the RV (Figure 1A) and significantly increased JPH2 expression in MCT RV extracts, measured in arbitrary units (AU) (PBS 100±11 AU, MCT 44±8 AU, and MCT‐colchicine 81±9 AU) (Figure 1B and 1C). Levels of another important t‐tubule protein, voltage‐gated calcium channels, were unaltered in RV extracts from MCT‐PAH and did not change with colchicine therapy (Figure 1B and 1C). Analysis of the subcellular localization of JPH2 revealed loss of the regular striated staining pattern in MCT RVs, which colchicine significantly improved (PBS 70.6±1.6 AU, MCT 25.2±3.0 AU, MCT‐colchicine 36.6±2.7 AU) (Figure 1D and 1E). Finally, t‐tubule staining showed near complete loss of t‐tubule architecture in the RVs of MCT rats, which colchicine significantly mitigated (PBS 65.7±2.9 AU, MCT 30.6±2.7 AU, MCT‐colchicine 50.1±3.5 AU) (Figure 1F and 1G).
Figure 1.
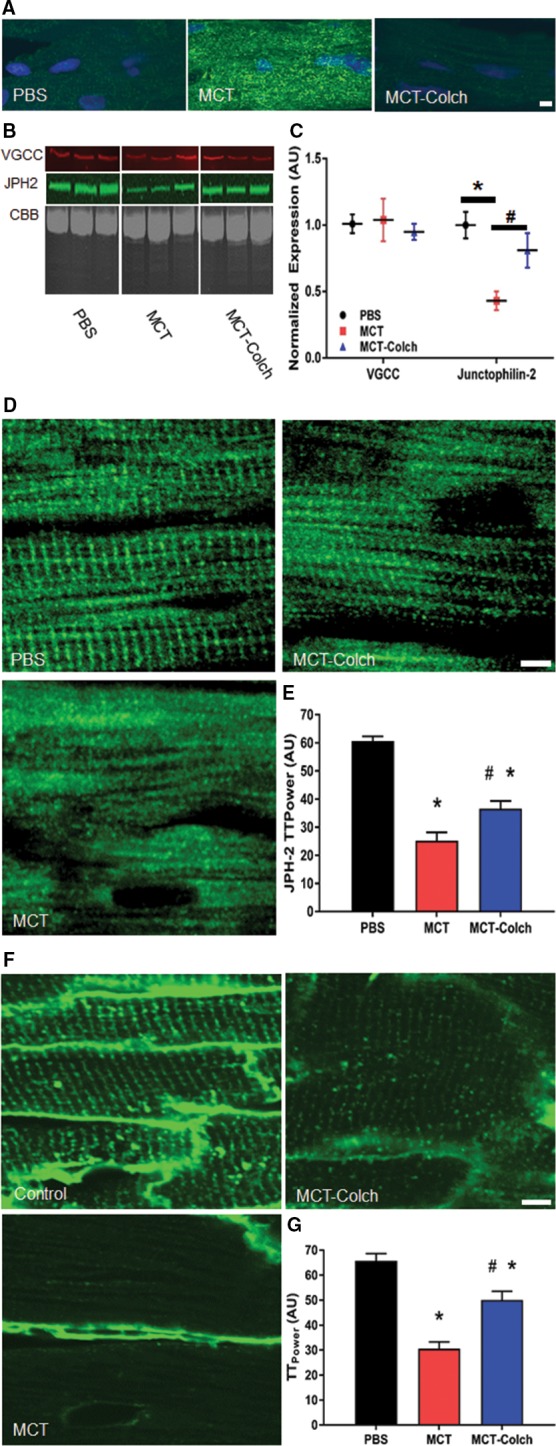
Colchicine increases JPH2 protein levels, restores JPH2 to t‐tubules, and mitigates pathological t‐tubule remodeling in the RV in MCT rats. A, Confocal micrographs of RV sections stained with a β‐tubulin antibody (green) to delineate microtubules. Microtubules are depolymerized in the MCT‐colchicine rats, and MCT rats have increased microtubule density as compared with PBS controls. B, Representative Western blots from 3 PBS, MCT, and MCT‐colchicine RV extracts and (C) quantification of VGCC (PBS 101±7 AU, n=5; MCT 104±16 AU, n=5; and MCT‐colchicine 95±6 AU, n=5) and JPH2 (PBS 100±11 AU, n=5, MCT 44±8 AU, n=10; and MCT‐colchicine 81±9 AU, n=10). Colchicine treatment significantly increases JPH2 expression. D, Confocal micrographs of RV sections stained for JPH2 (green). The striated pattern of JPH2 is partially restored with colchicine treatment. (PBS 70.6±1.6 AU, n=54 cells from 3 animals, MCT 25.2±3.0 AU, n=50 cells from 3 animals, MCT‐colchicine 36.6±2.7 AU, n=46 cells from 3 animals) (E). Confocal micrographs of RV sections stained with WGA (green) to label t‐tubules (F). Colchicine treatment improves t‐tubule architecture (PBS 65.7±2.9 AU, n=49 cells from 3 animals, MCT 30.6±2.7 AU, n=50 cells from 3 animals, MCT‐colchicine 50.1±3.5 AU, n=45 cells from 3 animals) (G). *Significantly different from PBS; #significantly different from MCT rats as determined by 1‐way ANOVA with Tukey post hoc analysis. Scale bar: 5 μm in all images. AU indicates arbitrary units; CBB, Coomassie brilliant blue; Colch, colchicine; JPH2, junctophilin‐2; MCT, monocrotaline; PBS, phosphate‐buffered saline; RV, right ventricle; VGCC, voltage‐gated calcium channel; WGA, wheat germ agglutinin.
To further test the relationship between microtubular remodeling and JPH2 and t‐tubule morphology in RV cardiomyocytes, we examined the effects of paclitaxel on JPH2 localization patterns and t‐tubule morphology in control isolated RV cardiomyocytes. After exposure to paclitaxel for 24 hours, the regular striated pattern of JPH2 was decreased (DMSO 75.4±1.9 AU, paclitaxel 68.1±1.9 AU, P=0.01). This was accompanied by t‐tubule remodeling characterized by the emergence of discontinuous t‐tubules (DMSO 55.2±1.6 AU, paclitaxel 44.9±3.2 AU, P=0.006) (Figure 2A through 2D). Thus, the adverse effects of microtubule stabilization on JPH2 and t‐tubules were observed whether microtubules were chemically disordered by paclitaxel in normal myocytes or as part of the disease process in cardiomyocytes from MCT RVs.
Figure 2.
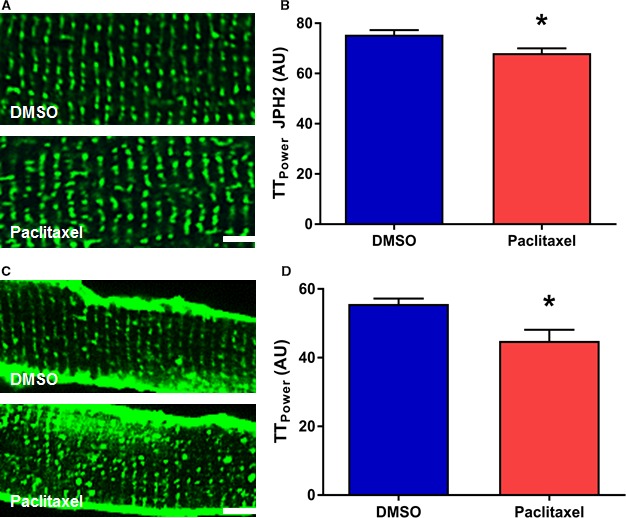
Paclitaxel induces JPH2 mislocalization and t‐tubule disruptions in cultured control rat RV cardiomyocytes. A, Representative confocal micrographs of isolated RV cardiomyocytes stained with JPH2 antibody (green). B, Quantification of JPH2 localization patterns (DMSO 75.4±1.9, paclitaxel 68.1±1.9, P=0.01). Paclitaxel treatment partially disrupts the striated JPH2 staining pattern. C, Confocal micrographs of isolated cardiomyocytes stained with WGA (green) to delineate t‐tubules. D, Quantification of t‐tubule architecture (DMSO 55.2±1.6, paclitaxel 44.9±3.2, P=0.006). Paclitaxel treatment induces t‐tubule remodeling. Scale bar: 5 μm. *P<0.05 as determined by t test. DMSO indicates dimethyl sulfoxide; JPH2, junctophilin‐2; RV, right ventricle; WGA, wheat germ agglutinin.
Next, we analyzed the chamber specificity of microtubule and JPH2 misregulation and subsequent t‐tubule morphology. In LV extracts, α‐tubulin was nonsignificantly reduced in the MCT LV but was significantly reduced in the MCT‐colchicine LV (PBS 1.0±0.08, MCT 0.88±0.06 AU, and MCT‐colchicine 0.59±0.07 AU) (Figure S3A and S3B). In contrast to the RV, JPH2 protein levels were significantly elevated in MCT LV and were even higher in MCT‐colchicine LV (PBS 1.0±0.07, MCT 1.6±0.14, MCT‐colchicine 2.1±0.08) (Figure S3A and S3B). There were no significant differences in JPH2 localization patterns (PBS 47.1±2 AU, MCT 51.7±2.8 AU, MCT‐colchicine 51.5±3.3 AU) and t‐tubule architecture (PBS 58.8±1.2 AU, MCT 61.2±1.7 AU, MCT‐colchicine 60.2±1.3 AU) (Figure S3C through S3F) among control, MCT, and MCT‐colchicine LVs. Thus, the misregulation of JPH2 and changes in t‐tubule architecture were specific to the RV in MCT rats.
Then, we examined the impact of colchicine on RV morphology in MCT rats. Colchicine significantly reduced RV cardiomyocyte diameter (PBS 15.1±0.2 μm, MCT 23.9±0.3 μm, MCT‐colchicine 21.0±0.2 μm) (Figure 3A and 3B). Colchicine also significantly blunted RV hypertrophy as quantified by diastolic RVFW thickness (PBS 0.6±0.03 mm, MCT‐PBS 1.5±0.10 mm, MCT‐colchicine 1.0±0.07 mm) (Figure 3C) and the Fulton index (PBS 0.21±0.01, MCT‐PBS 0.67±0.02, MCT‐colchicine 0.48±0.03) (Figure 3D).
Figure 3.
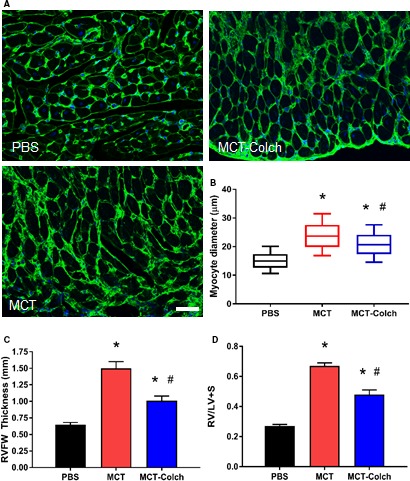
Colchicine reduces right ventricular hypertrophy in MCT rats. A, WGA stained RVFW sections. B, Quantification of cardiomyocyte diameter using a box‐and‐whisker plot. Whiskers extend from 10th to 90th percentile. (PBS 15.1±0.2 μm, n=617 cells from 3 animals, MCT 23.9±0.3 μm, n=474 cells from 3 animals, MCT‐colchicine 21.0±0.2 μm, n=584 cells from 3 animals.) Colchicine significantly reduces RV cardiomyocyte diameter. C, Quantification of diastolic RVFW thickness on echocardiography. (PBS 0.6±0.03 mm, n=16, MCT‐PBS 1.5±0.10 mm, n=21, MCT‐colchicine 1.0±0.07 mm, n=22.) D, Fulton index in isolated hearts. (PBS 0.21±0.01, n=12, MCT 0.67±0.02, n=15, MCT‐colchicine 0.48±0.03, n=16. Colchicine blunts RV hypertrophy. *Significantly different from PBS; #significantly different from MCT rats as determined by 1‐way ANOVA with Tukey post hoc analysis. Scale bar: 50 μm. Colch indicates colchicine; MCT, monocrotaline; PBS, phosphate‐buffered saline; RVFW, right ventricle free wall; RV/LV+S, RV free wall mass/LV and septal mass; WGA, wheat germ agglutinin.
Colchicine treatment augmented RV function in vivo as measured by echocardiography and cardiac catheterization. All indices of RV function were significantly improved in MCT‐PAH rats treated with colchicine: TAPSE (PBS 2.9±0.01 mm, MCT 1.9±0.08 mm, MCT‐colchicine 2.4±0.10 mm) (Figure 4A), percentage change in RVFW thickness (PBS 94±9%, MCT 24±3%, MCT‐colchicine 54±6%) (Figure 4B), and cardiac output (PBS 133±7 mL/min, MCT 67±3 mL/min, MCT‐colchicine 93±6 mL/min) (Figure 4C). Consistent with echocardiography data, colchicine significantly enhanced invasively measured hemodynamics: thermodilution cardiac output (PBS 130±8 mL/min, MCT 42±5 mL/min, MCT‐colchicine 67±8 mL/min) (Figure 4D) and cardiac index (PBS 0.27±0.02 mL/[min·g], MCT 0.11±0.10 mL/[min·g], MCT‐colchicine 0.19±0.02 mL/[min·g]) (Figure 4E).
Figure 4.
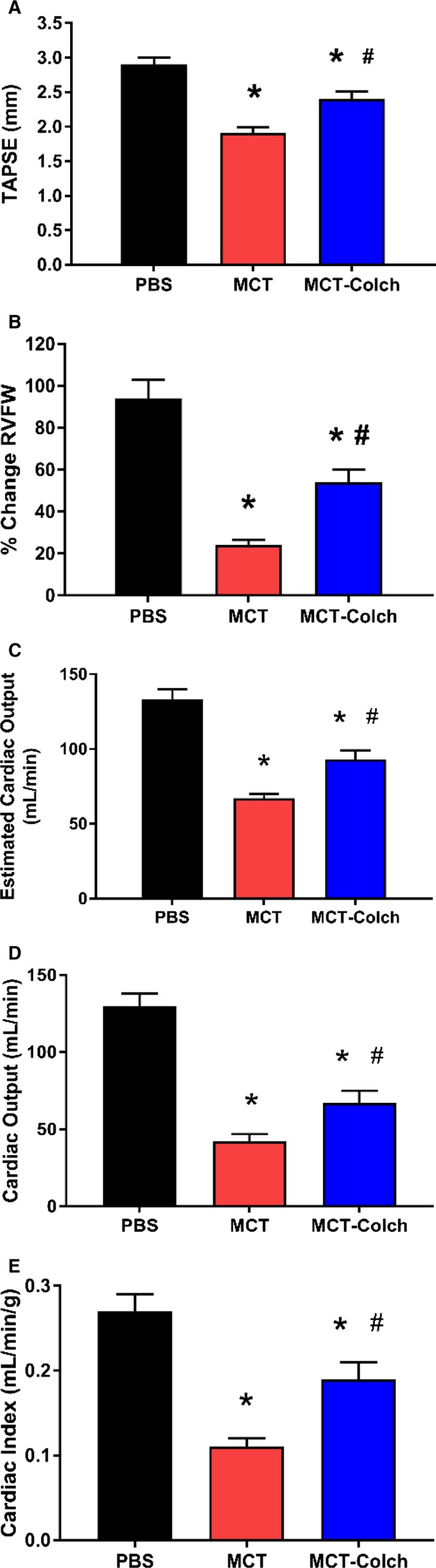
Colchicine therapy improves in vivo RV function in MCT rats. On echocardiography, TAPSE (PBS 2.9±0.01 mm, n=16, MCT 1.9±0.08 mm, n=21, MCT‐colchicine 2.4±0.10 mm, n=22) (A), percentage change in RVFW thickness (PBS 94±9%, n=16, MCT 24±3%, n=21, MCT‐colchicine 54±6%, n=22) (B), and estimated cardiac output (CO) (PBS 133±7 mL/min, n=16, MCT 67±3 mL/min, n=21, MCT‐colchicine 93±6 mL/min, n=22) (C) are all improved with colchicine treatment in MCT rats. Right heart catheterization‐derived thermodilution CO (PBS 130±8 mL/min, n=5, MCT 42±5 mL/min, n=10, MCT‐colchicine 67±8 mL/min, n=13) (D) and cardiac index (PBS 0.27±0.02 mL/[min·g], n=5, MCT 0.11±0.10 mL/[min·g], n=10, MCT‐colchicine 0.19±0.02 mL/[min·g], n=13) (E) are increased with colchicine treatment in MCT rats. *Significantly different from PBS, #significantly different from MCT rats as determined by 1‐way ANOVA with Tukey post hoc analysis. Colch indicates colchicine; MCT, monocrotaline; PBS, phosphate‐buffered saline; RVFW, right ventricle free wall; TAPSE, tricuspid annular plane systolic excursion.
We also examined the relationships between RV JPH2 expression and RV size and function in vivo. There was an inverse association between JPH2 levels and diastolic RVFW thickness (r=−0.58, P=0.002) (Figure 5A). RV function, as determined by percentage change in RVFW thickness (r=0.67, P=0.0003) (Figure 5B) and TAPSE (r=0.54, P=0.005) (Figure 5C), was positively associated with JPH2 expression. Thus, reduced levels of JPH2 were associated with RV hypertrophy and RV dysfunction.
Figure 5.
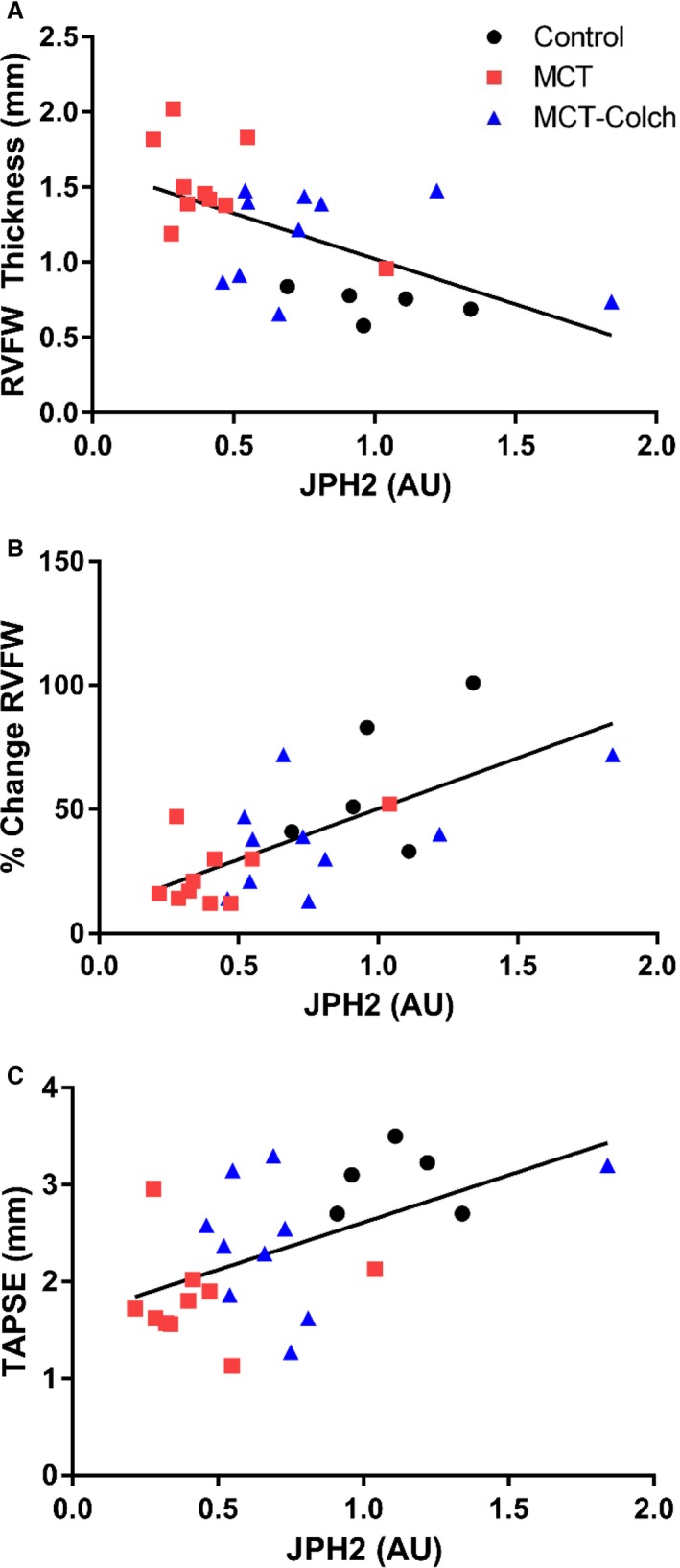
Reduced JPH2 expression is associated with right ventricular hypertrophy and RV dysfunction in vivo. JPH2 levels are inversely associated with diastolic RVFW thickness (r=−0.58, P=0.002) (A) and are positively correlated with RV function as determined by percentage change in RVFW thickness (r=0.67, P=0.0003) (B) and TAPSE (r=0.54, P=0.005) (C). AU indicates arbitrary units; JPH2, junctophilin‐2; RVFW, right ventricle free wall; TAPSE, tricuspid annular plane systolic excursion.
In addition to the beneficial effects on the RV, colchicine mitigated adverse pulmonary vascular remodeling. The percentage medial thickness of small intrapulmonary arterioles was significantly reduced with colchicine treatment (PBS 13.5±0.6%, MCT 43.4±1.5%, MCT‐colchicine 39.5±1.1%) (Figure 6A and 6B). Consistent with this finding, colchicine significantly prolonged PA acceleration time (PBS 33±1 milliseconds, MCT 17±1 milliseconds, MCT‐colchicine 22±1 milliseconds) (Figure 6C). Likewise, colchicine significantly reduced mPAP (PBS 12±1 mm Hg, MCT 41±3 mm Hg, MCT‐colchicine 29±3 mm Hg) (Figure 6D) and total pulmonary resistance (PBS 0.1±0.01 mm Hg/[mL·min], MCT 0.9±0.1 mm Hg/[mL·min], MCT‐colchicine 0.5±0.1 mm Hg/[mL·min]) (Figure 6E).
Figure 6.
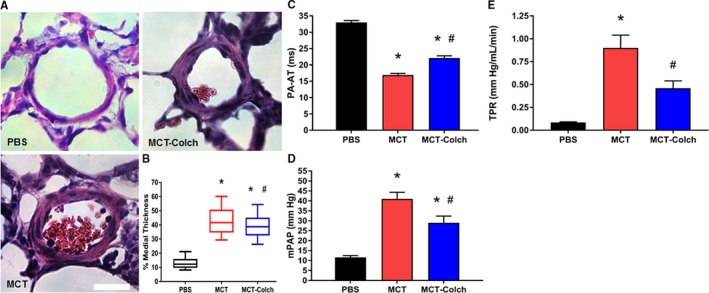
Colchicine reduces severity of pulmonary vascular disease in MCT rats. A, Representative hematoxylin and eosin–stained lung sections showing pulmonary arterioles. B, Box‐and‐whisker plots of percentage medial thickness (PBS 13.5±0.6% n=85 arterioles from 3 different animals, MCT 43.4±1.5% n=78 arterioles from 3 animals, MCT‐colchicine 39.5±1.1% n=81 arterioles from 3 different animals). Whiskers extend from 10th to 90th percentiles. Colchicine reduces pulmonary arteriolar medial thickness. C, Colchicine increases PA acceleration time (PBS 33±1 milliseconds, n=16, MCT 17±1 milliseconds, n=21, MCT‐colchicine 22±1 milliseconds n=22). Quantification of (D) mPAP (PBS 12±1 mm Hg, n=5, MCT 41±3 mm Hg, n=13, MCT‐colchicine 29±3 mm Hg n=14) and (E) TPR (PBS 0.1±0.01 mm Hg/[mL·min], n=5, MCT 0.9±0.1 mm Hg/[mL·min], n=9, MCT‐colchicine 0.5±0.1 mm Hg/[mL·min], n=11) from right heart catheterization. Colchicine improves pulmonary hemodynamics. *Significantly different from PBS, #significantly different from MCT rats as determined by 1‐way ANOVA with Tukey post hoc analysis. Scale bar 25 μm. Colch indicates colchicine; MCT, monocrotaline; mPAP, mean pulmonary arterial pressure; PBS, phosphate‐buffered saline; TPR, total pulmonary resistance.
To determine whether the observed improvements in RV function were simply due to regression of pulmonary vascular disease, we analyzed RV‐PA coupling. Consistent with a direct effect on the RV, colchicine improved RV‐PA coupling as quantified by TAPSE/mPAP (PBS 0.24±0.03 mm/mm Hg, MCT 0.05±0.01 mm/mm Hg, MCT‐colchicine 0.11±0.02 mm/mm Hg) (Figure 7A) or percentage change in RVFW thickness/mPAP (PBS 8.9±1.4%/mm Hg, MCT 0.6±0.1%/mm Hg, MCT‐colchicine 2.7±0.5%/mm Hg) (Figure 7B). To further probe RV‐PA coupling, the relationship between RV function (TAPSE and percentage change in RVFW thickness) and mPAP was examined. There were inverse relationships among TAPSE (r=−0.73, P=0.0003), percentage change in RVFW thickness (r=−0.77, P<0.0001), and mPAP when PBS and MCT rats were combined (Figure 7C and 7D); however, in the MCT‐colchicine rats the relationships between RV function and mPAP were lost (TAPSE r=0.17, P=0.56, percentage change in RVFW thickness r=−0.35, P=0.22) (Figure 7E and 7F).
Figure 7.
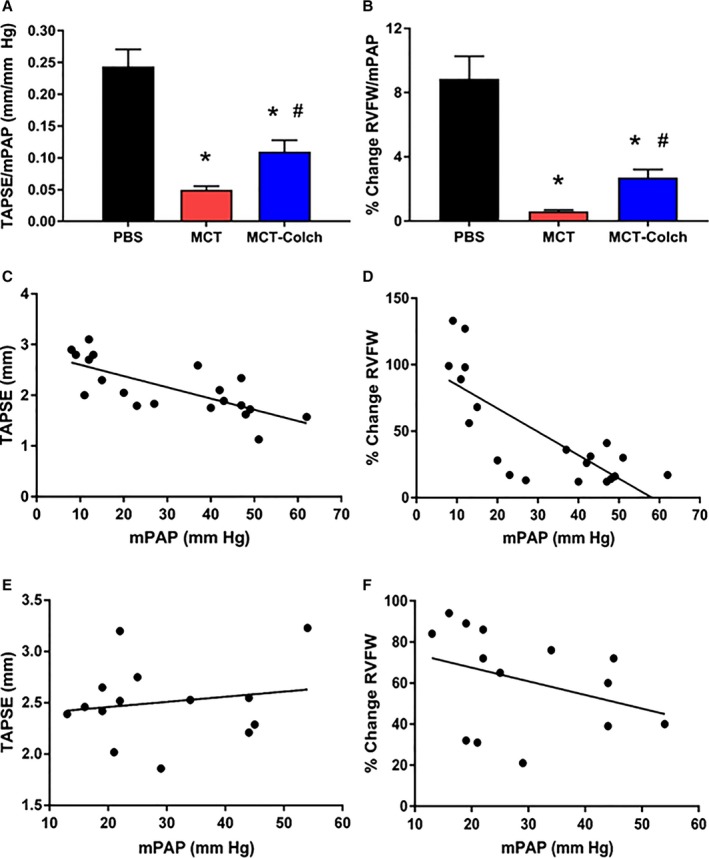
RV‐PA coupling is enhanced with colchicine treatment. Colchicine improves RV‐PA coupling as quantified by TAPSE/mPAP (PBS 0.24±0.03 mm/mm Hg, n=7, MCT 0.05±0.01 mm/mm Hg, n=13, MCT‐colchicine 0.11±0.02 mm/mm Hg, n=14) (A) or percentage change in RVFW thickness/mPAP (PBS 8.9±1.4%/mm Hg, n=7, MCT 0.6±0.1%/mm Hg, n=13, MCT‐colchicine 2.7±0.5%/mm Hg, n=14) (B). There are significant inverse relationships between (C) TAPSE (r=−0.73, P=0.0003) and mPAP and (D) percentage change in RVFW thickness and mPAP in control and MCT rats (r=−0.77, P<0.0001) but not in MCT‐colchicine rats: TAPSE and mPAP (r=0.17 P=0.56) (E) and percentage change in RVFW thickness and mPAP (r=−0.35 P=0.22) (F). *Significantly different from PBS, #significantly different from MCT rats as determined by 1‐way ANOVA with Tukey post hoc analysis. Colch indicates colchicine; MCT, monocrotaline; mPAP, mean pulmonary arterial pressure; PA, pulmonary artery; PBS, phosphate‐buffered saline; RVFW, right ventricle free wall; TAPSE, tricuspid annular plane systolic excursion.
Finally, we probed whole‐animal physiology to assess tolerability and therapeutic efficacy of colchicine. Colchicine was well tolerated as there was no difference in end‐of‐study mass between MCT and MCT‐colchicine rats (PBS 464±13 g, MCT 350±10 g, and MCT‐colchicine 344±8 g) (Figure 8A). Colchicine significantly improved treadmill walk distance when compared with MCT (PBS 192±15 m, MCT 9±2 m, MCT‐colchicine 44±11 m) (Figure 8B). Finally, there was a trend for reduced mortality in the colchicine‐treated group (mortality rates at study completion: PBS 0%, MCT 29%, and MCT‐colchicine 12%, P=0.13) (Figure S4).
Figure 8.
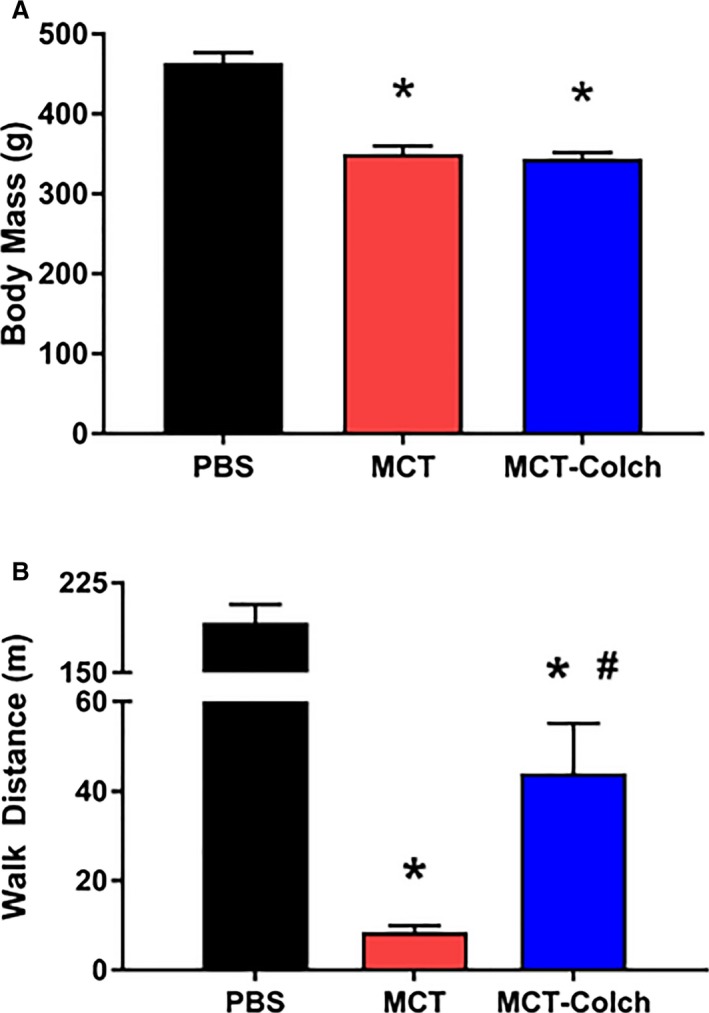
Colchicine is well tolerated and increases exercise capacity in MCT rats. A, No significant difference in mass at end of study with colchicine treatment (PBS 464±13 g, MCT 350±10 g, and MCT‐colchicine 344±8 g); (B) treadmill walk distance is improved in colchicine‐treated MCT rats (PBS 192±15 m, n=7, MCT 9±2 m, n=20, MCT‐colchicine 44±11 m, n=14). Colch indicates colchicine; MCT, monocrotaline; PBS, phosphate‐buffered saline.
Discussion
In this study we show that pathologic and chamber‐specific remodeling of the microtubule cytoskeleton is associated with reduced JPH2 expression and t‐tubule disarray in RV cardiomyocytes in MCT‐PAH. These molecular and cellular changes are accompanied by RV dysfunction. We further demonstrate that colchicine‐induced microtubule depolymerization increases JPH2 expression and improves t‐tubule architecture in the RV. Moreover, in vivo colchicine therapy improves RV function and augments cardiac output while decreasing RV hypertrophy. Colchicine also has beneficial effects on the pulmonary vasculature with a decrease in percentage medial thickness of small pulmonary arterioles, a reduction in mPAP and total pulmonary resistance, and prolongation of the PA acceleration time. However, colchicine's beneficial effects on RV function are not solely due to reduced afterload, as is evident from its ability to enhance RV‐PA coupling. Finally, colchicine increases exercise capacity. Collectively, these data suggest that colchicine has the potential to improve RV function in PAH by increasing JPH2 and improving t‐tubule morphology.
Our findings provide further evidence that pathological microtubule cytoskeletal remodeling causes JPH2 misregulation and t‐tubule disruptions in cardiomyocytes. In vivo microtubule depolymerization with colchicine restores JPH2 localization to the t‐tubules and corrects t‐tubule morphology in an aortic‐banding heart failure model.15 Our findings are consistent with prior studies showing that nocodazole‐mediated microtubule depolymerization rescues JPH2 mislocalization and t‐tubule remodeling in cultured cardiomyocytes.15 Conversely, microtubule stabilization (achieved using paclitaxel) recapitulates the microtubule phenotype observed in pressure‐overloaded and failing cardiomyocytes leading to JPH2 mislocalization and t‐tubule irregularities.15 Consistent with this, we demonstrate that paclitaxel induces JPH2 mislocalization and t‐tubule disarray in control RV cardiomyocytes (Figure 2). Thus, there is a clear relationship between microtubule structure and JPH2 expression in both RV and LV cardiomyocytes.
Colchicine‐induced microtubule depolymerization increases JPH2 and corrects t‐tubule morphology in vivo in a mouse model of Duchenne muscular dystrophy16 and aortic banding.15 Our study extends these findings to the RV and demonstrates that proper regulation of JPH2 and subsequent t‐tubule structure requires a normal microtubule cytoskeleton in both ventricles. However, although the molecular pathway may be similar in LV and RV dysfunction, the changes in the microtubules‐JPH2–t‐tubule pathway in MCT‐PAH are chamber specific in that the LV is spared (Figure S3). The current findings of chamber‐specific increases in microtubule density, JPH2 dysregulation, and t‐tubule remodeling (Figure 1 and Figure S2) indicate that JPH2 downregulation is relevant to RV dysfunction in experimental PAH, analogous to its role in the LV disease in various cardiomyopathies.
The ability of colchicine to improve RV‐PA coupling suggests that colchicine has a direct effect on the RV. However, colchicine also reduces pulmonary vascular remodeling (Figure 6), likely due to colchicine's antimitotic effects,14, 35 but that does not fully explain the enhanced RV‐PA coupling in MCT‐colchicine rats. The ability of colchicine to dissociate the very strong relationship between RV afterload and RV function demonstrates that colchicine improves RV‐PA coupling (Figure 7). We propose that increases in JPH2 and improvements in t‐tubule morphology mediate the augmented RV function, which is further supported by the significant relationships between JPH2 and RV function (Figure 5). These results agree with several studies showing that reduced levels of JPH2 are associated with left ventricular dysfunction in animal models36, 37, 38, 39, 40 and human disease states15, 41 and that increased expression of JPH2, achieved via transgenic overexpression,19 antagonism of miR‐24,42 viral‐mediated overexpression,20 or inhibition of calpain proteases,43 improves cardiac function in animal models of LV dysfunction. Thus, increased JPH2 likely mediates the improved RV‐PA coupling in MCT‐colchicine rats.
Although several studies show t‐tubule disruption to be associated with LV dysfunction, there is less information about the role of t‐tubule disarray in RV dysfunction in PAH. Pathological t‐tubule remodeling in the RV of MCT rats is associated with reduced RV fractional shortening.44 Furthermore, sildenafil treatment early in the development of PAH normalizes t‐tubule structure and increases RV fractional shortening; however, if sildenafil treatment is initiated later in the disease, t‐tubule morphology and RV function do not improve to the same extent.44 Here, we show that improvements in t‐tubule structure due to colchicine treatment are associated with augmented RV function. Thus, there appears to be a direct relationship between t‐tubule architecture and RV function in PAH.
Finally, our finding that colchicine improves RV function in PAH is easily translatable into human patients with PAH or other World Health Organization etiologies of pulmonary hypertension. Because colchicine is approved by the Food and Drug Administration, a clinical trial using colchicine in PAH could be conducted. Dosing would need to be altered when planning a human trial using colchicine to treat PAH because the dose used in our study could cause significant toxicity in humans. However, a 6‐month course of colchicine (0.5 mg twice a day) is well tolerated in stable heart failure patients and causes small but significant improvements in LV remodeling.45 However, in this study the overall functional capacity and rates of hospitalization and death did not change with colchicine treatment.45 Although colchicine does not improve outcomes in patients with left heart failure, there are reasons to believe that it may be efficacious in PAH. First, the best evidence for colchicine's benefits derive from preclinical heart failure models induced by pressure overload.15 This contrasts with the etiology of left heart failure patients, which is usually due to intrinsic LV dysfunction rather than LV pressure overload. In contrast, PAH is by definition a pressure overload state.46 A second reason to believe that colchicine might be beneficial in the RV failure associated with PAH is the finding that colchicine reduces adverse pulmonary vascular remodeling (Figure 6) in addition to its direct beneficial effect on RV function through increased JPH2 expression. Thus, colchicine could target both afterload and direct cardiac disease mechanisms in PAH, which may not be the case in many patients with LV failure.
Limitations
Because colchicine was administered in the early stages in PAH in the MCT model, it is unclear whether colchicine prevented or regressed RV t‐tubule remodeling as a basis for the observed improvement in RV function.
Conclusion
Colchicine improves RV function and regresses pulmonary vascular disease in the MCT‐PAH model. However, enhanced RV‐PA coupling in rats treated with colchicine provides evidence of a RV‐specific effect. Therefore, colchicine has promise as an RV‐targeted therapy for PAH, and thus, a clinical trial repurposing this therapy could be considered.
Sources of Funding
Prins is funded by NIH F32 HL129554. Tian and Wu are funded by Canadian Vascular Network Scholar Awards. Thenappan is funded by American Heart Association Scientist Development Grant 15SDG25560048. Metzger is funded by NIH RO1s HL122323 and HL132874. Archer is supported by Canada Foundation for Innovation (229252 and 33012), NIH RO1s HL113003 and HL071115, a Tier 1 Canada Research Chair in Mitochondrial Dynamics and Translational Medicine (950‐229252), and a grant from the William J. Henderson Foundation.
Disclosures
Thenappan received a modest honorarium from Gilead. No other author has any disclosures.
Supporting information
Figure S1. Representative images from echocardiograms from PBS, MCT, and MCT‐Colch rats. Colch indicates colchicine; MCT, monocrotaline; PBS, phosphate‐buffered saline.
Figure S2. RV microtubule misregulation in MCT rats. MCT indicates monocrotaline; RV, right ventricle.
Figure S3. The LV is spared from JPH2‐mediated t‐tubule disruptions in MCT rats. JPH2 indicates junctophilin‐2; LV, left ventricle; MCT, monocrotaline.
Figure S4. Colchicine treatment leads to a nonsignificant reduction in mortality. Mortality rates at study completion: phosphate‐buffered saline 0%, monocrotaline 29%, and monocrotaline‐colchicine 12%, P=0.13.
(J Am Heart Assoc. 2017;6:e006195 DOI: 10.1161/JAHA.117.006195.)28566298
References
- 1. Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg‐Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35:1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Benza RL, Miller DP, Gomberg‐Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long‐Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122:164–172. [DOI] [PubMed] [Google Scholar]
- 3. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud‐Gaubert M, Haloun A, Laurent M, Hachulla E, Simonneau G. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–1030. [DOI] [PubMed] [Google Scholar]
- 4. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. [DOI] [PubMed] [Google Scholar]
- 5. Thenappan T, Prins KW, Pritzker MR, Scandurra J, Volmers K, Weir EK. The critical role of pulmonary arterial compliance in pulmonary hypertension. Ann Am Thorac Soc. 2016;13:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ryan JJ, Huston J, Kutty S, Hatton ND, Bowman L, Tian L, Herr JE, Johri AM, Archer SL. Right ventricular adaptation and failure in pulmonary arterial hypertension. Can J Cardiol. 2015;31:391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forfia PR, Fisher MR, Mathai SC, Housten‐Harris T, Hemnes AR, Borlaug BA, Chamera E, Corretti MC, Champion HC, Abraham TP, Girgis RE, Hassoun PM. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174:1034–1041. [DOI] [PubMed] [Google Scholar]
- 9. van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG, Spreeuwenberg MD, Postmus PE, Vonk‐Noordegraaf A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28:1250–1257. [DOI] [PubMed] [Google Scholar]
- 10. van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, Boonstra A, Marques KM, Westerhof N, Vonk‐Noordegraaf A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. 2011;58:2511–2519. [DOI] [PubMed] [Google Scholar]
- 11. Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JR, Michelakis ED. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116:238–248. [DOI] [PubMed] [Google Scholar]
- 12. Tsutsui H, Ishihara K, Cooper G. Cytoskeletal role in the contractile dysfunction of hypertrophied myocardium. Science. 1993;260:682–687. [DOI] [PubMed] [Google Scholar]
- 13. Stones R, Benoist D, Peckham M, White E. Microtubule proliferation in right ventricular myocytes of rats with monocrotaline‐induced pulmonary hypertension. J Mol Cell Cardiol. 2013;56:91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fitzgerald TJ. Molecular features of colchicine associated with antimitotic activity and inhibition of tubulin polymerization. Biochem Pharmacol. 1976;25:1383–1387. [DOI] [PubMed] [Google Scholar]
- 15. Zhang C, Chen B, Guo A, Zhu Y, Miller JD, Gao S, Yuan C, Kutschke W, Zimmerman K, Weiss RM, Wehrens XH, Hong J, Johnson FL, Santana LF, Anderson ME, Song LS. Microtubule‐mediated defects in junctophilin‐2 trafficking contribute to myocyte transverse‐tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation. 2014;129:1742–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prins KW, Asp ML, Zhang H, Wang W, Metzger JM. Microtubule‐mediated misregulation of junctophilin‐2 underlies T‐tubule disruptions and calcium mishandling in mdx mice. JACC Basic Transl Sci. 2016;1:122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. [DOI] [PubMed] [Google Scholar]
- 18. Quick AP, Landstrom AP, Wehrens XH. Junctophilin‐2 at the intersection of arrhythmia and pathologic cardiac remodeling. Heart Rhythm. 2016;13:753–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo A, Zhang X, Iyer VR, Chen B, Zhang C, Kutschke WJ, Weiss RM, Franzini‐Armstrong C, Song LS. Overexpression of junctophilin‐2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc Natl Acad Sci USA. 2014;111:12240–12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, Showell J, Barreto‐Torres G, Thuerauf DJ, Doroudgar S, Glembotski CC, Wehrens XH. Junctophilin‐2 gene therapy rescues heart failure by normalizing RyR2‐mediated Ca(2+) release. Int J Cardiol. 2016;225:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urboniene D, Haber I, Fang YH, Thenappan T, Archer SL. Validation of high‐resolution echocardiography and magnetic resonance imaging vs. high‐fidelity catheterization in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2010;299:L401–L412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, Thenappan T, Bache‐Wiig P, Piao L, Paul J, Chen CT, Archer SL. Lung 18F‐fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsutsui H, Ishibashi Y, Takahashi M, Namba T, Tagawa H, Imanaka‐Yoshida K, Takeshita A. Chronic colchicine administration attenuates cardiac hypertrophy in spontaneously hypertensive rats. J Mol Cell Cardiol. 1999;31:1203–1213. [DOI] [PubMed] [Google Scholar]
- 24. Scopacasa BS, Teixeira VP, Franchini KG. Colchicine attenuates left ventricular hypertrophy but preserves cardiac function of aortic‐constricted rats. J Appl Physiol (1985). 2003;94:1627–1633. [DOI] [PubMed] [Google Scholar]
- 25. Tochinai R, Suzuki K, Nagata Y, Ando M, Hata C, Komatsu K, Suzuki T, Uchida K, Kado S, Kaneko K, Kuwahara M. Cardiotoxic changes of colchicine intoxication in rats: electrocardiographic, histopathological and blood chemical analysis. J Toxicol Pathol. 2014;27:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang W, Barnabei MS, Asp ML, Heinis FI, Arden E, Davis J, Braunlin E, Li Q, Davis JP, Potter JD, Metzger JM. Noncanonical EF‐hand motif strategically delays Ca2+ buffering to enhance cardiac performance. Nat Med. 2013;19:305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parness J, Horwitz SB. Taxol binds to polymerized tubulin in vitro. J Cell Biol. 1981;91:479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pasqualin C, Gannier F, Malécot CO, Bredeloux P, Maupoil V. Automatic quantitative analysis of t‐tubule organization in cardiac myocytes using ImageJ. Am J Physiol Cell Physiol. 2015;308:C237–C245. [DOI] [PubMed] [Google Scholar]
- 29. Andersen MJ, Hwang SJ, Kane GC, Melenovsky V, Olson TP, Fetterly K, Borlaug BA. Enhanced pulmonary vasodilator reserve and abnormal right ventricular: pulmonary artery coupling in heart failure with preserved ejection fraction. Circ Heart Fail. 2015;8:542–550. [DOI] [PubMed] [Google Scholar]
- 30. Melenovsky V, Hwang SJ, Lin G, Redfield MM, Borlaug BA. Right heart dysfunction in heart failure with preserved ejection fraction. Eur Heart J. 2014;35:3452–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guazzi M, Bandera F, Pelissero G, Castelvecchio S, Menicanti L, Ghio S, Temporelli PL, Arena R. Tricuspid annular plane systolic excursion and pulmonary arterial systolic pressure relationship in heart failure: an index of right ventricular contractile function and prognosis. Am J Physiol Heart Circ Physiol. 2013;305:H1373–H1381. [DOI] [PubMed] [Google Scholar]
- 32. Guazzi M, Naeije R, Arena R, Corrà U, Ghio S, Forfia P, Rossi A, Cahalin LP, Bandera F, Temporelli P. Echocardiography of right ventriculoarterial coupling combined with cardiopulmonary exercise testing to predict outcome in heart failure. Chest. 2015;148:226–234. [DOI] [PubMed] [Google Scholar]
- 33. Prins KW, Weir EK, Archer SL, Markowitz J, Rose L, Pritzker M, Madlon‐Kay R, Thenappan T. Pulmonary pulse wave transit time is associated with right ventricular‐pulmonary artery coupling in pulmonary arterial hypertension. Pulm Circ. 2016;6:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin‐related protein 1‐mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110:1484–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bhattacharyya B, Panda D, Gupta S, Banerjee M. Anti‐mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev. 2008;28:155–183. [DOI] [PubMed] [Google Scholar]
- 36. Zhang R, Dai LZ, Xie WP, Yu ZX, Wu BX, Pan L, Yuan P, Jiang X, He J, Humbert M, Jing ZC. Survival of Chinese patients with pulmonary arterial hypertension in the modern treatment era. Chest. 2011;140:301–309. [DOI] [PubMed] [Google Scholar]
- 37. Landstrom AP, Kellen CA, Dixit SS, van Oort RJ, Garbino A, Weisleder N, Ma J, Wehrens XH, Ackerman MJ. Junctophilin‐2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium‐handling. Circ Heart Fail. 2011;4:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caldwell JL, Smith CE, Taylor RF, Kitmitto A, Eisner DA, Dibb KM, Trafford AW. Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN‐1). Circ Res. 2014;115:986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, Feng XH, Guo YB, Li SF, Lai ST, Zhou P, Li LL, Yang HQ, Luo GZ, Bai Y, Xi JJ, Gao W, Han QD, Zhang YY, Wang XJ, Meng X, Wang SQ. Mir‐24 regulates junctophilin‐2 expression in cardiomyocytes. Circ Res. 2012;111:837–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, De Almeida AC, Skapura DG, Rudy Y, Burns AR, Ackerman MJ, Wehrens XH. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, Ommen SR, Wehrens XH, Claycomb WC, Ko JK, Hwang M, Pan Z, Ma J, Ackerman MJ. Mutations in JPH2‐encoded junctophilin‐2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li RC, Tao J, Guo YB, Wu HD, Liu RF, Bai Y, Lv ZZ, Luo GZ, Li LL, Wang M, Yang HQ, Gao W, Han QD, Zhang YY, Wang XJ, Xu M, Wang SQ. In vivo suppression of microRNA‐24 prevents the transition toward decompensated hypertrophy in aortic‐constricted mice. Circ Res. 2013;112:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu CY, Chen B, Jiang YP, Jia Z, Martin DW, Liu S, Entcheva E, Song LS, Lin RZ. Calpain‐dependent cleavage of junctophilin‐2 and T‐tubule remodeling in a mouse model of reversible heart failure. J Am Heart Assoc. 2014;3:e000527 DOI: 10.1161/JAHA.113.000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xie YP, Chen B, Sanders P, Guo A, Li Y, Zimmerman K, Wang LC, Weiss RM, Grumbach IM, Anderson ME, Song LS. Sildenafil prevents and reverses transverse‐tubule remodeling and Ca(2+) handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension. 2012;59:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deftereos S, Giannopoulos G, Panagopoulou V, Bouras G, Raisakis K, Kossyvakis C, Karageorgiou S, Papadimitriou C, Vastaki M, Kaoukis A, Angelidis C, Pagoni S, Pyrgakis V, Alexopoulos D, Manolis AS, Stefanadis C, Cleman MW. Anti‐inflammatory treatment with colchicine in stable chronic heart failure: a prospective, randomized study. JACC Heart Fail. 2014;2:131–137. [DOI] [PubMed] [Google Scholar]
- 46. Rich JD, Rich S. Clinical diagnosis of pulmonary hypertension. Circulation. 2014;130:1820–1830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative images from echocardiograms from PBS, MCT, and MCT‐Colch rats. Colch indicates colchicine; MCT, monocrotaline; PBS, phosphate‐buffered saline.
Figure S2. RV microtubule misregulation in MCT rats. MCT indicates monocrotaline; RV, right ventricle.
Figure S3. The LV is spared from JPH2‐mediated t‐tubule disruptions in MCT rats. JPH2 indicates junctophilin‐2; LV, left ventricle; MCT, monocrotaline.
Figure S4. Colchicine treatment leads to a nonsignificant reduction in mortality. Mortality rates at study completion: phosphate‐buffered saline 0%, monocrotaline 29%, and monocrotaline‐colchicine 12%, P=0.13.