

ABSTRACT
Understanding the mechanisms of controlled expansion and differentiation of basal progenitor cell populations during organogenesis is essential for developing targeted regenerative therapies. Since the cytokeratin 5-positive (K5+) basal epithelial cell population in the salivary gland is regulated by retinoic acid signaling, we interrogated how isoform-specific retinoic acid receptor (RAR) signaling impacts the K5+ cell population during salivary gland organogenesis to identify RAR isoform-specific mechanisms that could be exploited in future regenerative therapies. In this study, we utilized RAR isoform-specific inhibitors and agonists with murine submandibular salivary gland organ explants. We determined that RARα and RARγ have opposing effects on K5+ cell cycle progression and cell distribution. RARα negatively regulates K5+ cells in both whole organ explants and in isolated epithelial rudiments. In contrast, RARγ is necessary but not sufficient to positively maintain K5+ cells, as agonism of RARγ alone failed to significantly expand the population. Although retinoids are known to stimulate differentiation, K5 levels were not inversely correlated with differentiated ductal cytokeratins. Instead, RARα agonism and RARγ inhibition, corresponding with reduced K5, resulted in premature lumenization, as marked by prominin-1. With lineage tracing, we demonstrated that K5+ cells have the capacity to become prominin-1+ cells. We conclude that RARα and RARγ reciprocally control K5+ progenitor cells endogenously in the developing submandibular salivary epithelium, in a cell cycle-dependent manner, controlling lumenization independently of keratinizing differentiation. Based on these data, isoform-specific targeting RARα may be more effective than pan-RAR inhibitors for regenerative therapies that seek to expand the K5+ progenitor cell pool. Summary statement: RARα and RARγ reciprocally control K5+ progenitor cell proliferation and distribution in the developing submandibular salivary epithelium in a cell cycle-dependent manner while regulating lumenization independently of keratinizing differentiation.
KEYWORDS: atR, Abranching morphogenesis, cytokeratin 5, RARα, RARγ, salivary gland
INTRODUCTION
Retinoic acid, or all-trans retinoic acid (atRA), is a morphogen derived from Vitamin A that is important for the organogenesis of many systems, including the hematopoietic system, the brain, skin, lung, kidney, and the submandibular salivary gland (SMG).1-12 AtRA is generated following a two-step oxidation of vitamin A by retinol dehydrogenase (RDH) into the intermediate all-trans-retinal (atRAL); with the second oxidation by retinaldehyde dehydrogenase (RALDH) resulting in generation of atRA. AtRA is the primary physiological ligand for RAR, of which there are three main isoforms, alpha (RARα), beta (RARβ) and gamma (RARγ). Individual isoforms have both overlapping and distinct cellular effects, and differ in their ligand binding pockets and AF-2 domains such that selective pharmaceuticals can be used to target individual isoforms.13 RARs function as transcription factors, heterodimerizing with retinoid X receptor (RXR), of which there are also three isoforms (α, β, γ). RAR/RXR heterodimers bind DNA at defined retinoic acid response elements (RAREs) and affect transcription from target genes.14 Previous studies have indicated that Vitamin A and RAR signaling are essential for development of many organs during embryogenesis.3,8,15,16 Characterization of RAR signaling during organogenesis is complicated by the requirement for retinoic acid signaling during embryonic development as lack of atRA causes widespread developmental defects and embryonic lethality. AtRA supplementation can subvert embryonic lethality in mice defective in atRA synthesis, including retinaldehyde dehydrogenase 2 (RALDH2) knock-out mice and retinol dehydrogenase 10 (RDH10) mutant mice,17,18 and withdrawal of atRA supplementation in RALDH2−/− mice prevents lung development.19
The importance of the RARα and RARγ isoforms in developing salivary submandibular gland (SMG) was revealed with RARα and RARγ double knockout mice that showed SMG developmental defects, including shortening of the main duct.2,3 Lineage analysis using a RA response-element (RARE)-driven Cre also indicated that the SMG responded to atRA during branching morphogenesis,20 implying RAR functionality during early SMG organogenesis. To circumvent the need for atRA supplementation, a recent study interfered with atRA production using a RDH10 hypomorph, revealing decreased retinoic acid signaling in the developing SMG that was accompanied by decreased growth and branching morphogenesis.8 A recent ex vivo study using a pan-RAR pharmacological antagonist to inhibit all RAR isoform activity in early SMG embryonic organ explants showed that decreased overall RAR signaling resulted in decreased branching morphogenesis and increased expression of the KRT5 gene.9 Together, these studies indicate that retinoic acid has a critical role in SMG organogenesis and implicate RAR signaling in regulation of K5+ progenitor cells, however, RAR isoform-specific control of progenitor cell function during SMG development has not yet been identified.
Mouse SMG branching morphogenesis begins with protrusion of the oral epithelium into the surrounding mesenchyme at embryonic day 11 (E11), followed by formation of initial bud-on-stalk structure at E12-E12.5. Development and differentiation proceed resulting in an arborized ductal system that terminates in secretory acini that produce saliva in the adult21. Elaboration of the gland structure depends upon the controlled proliferation and differentiation of progenitor cells. One SMG epithelial progenitor cell type is characterized by expression of cytokeratin 5 (K5), the K5+ population. Initially present in the majority of the cells in the placode, the K5+ population becomes ductally restricted, forming a basal layer in the large and intercalated ducts as development proceeds and in the adult22,23. K5 is a marker of basal epithelial progenitor cells in several organs, including the mammary24 and the salivary25 glands. Cells of the K5 lineage were previously shown by lineage tracing to have the capacity to contribute to all epithelial cell types early in development of the submandibular gland, where their expansion was promoted by EGFR-dependent signaling from the parasymapathetic innervation25; however, since K5+ cells persist in the absence of parasympathetic innervation, there are other uncharacterized regulators of this cell type.
In this study, we investigated the effects of RAR isoform-specific signaling on the distribution of K5+ cells in the mouse SMG during organogenesis. To directly examine RAR isoform-mediated signaling effects on the K5+ cells without impacts from global embryonic effects, we used ex vivo E12.5 submandibular salivary gland (SMG) organ explants that were treated with isoform-specific RAR inhibitors and agonists. We here report that RARα negatively regulates the abundance and the proliferation of K5+ cells. In contrast, RARγ is necessary, but not sufficient, to positively maintain the accumulation and the proliferation of K5+ cells. The regulation of K5+ cells by both isoforms is independent of innervation. Additionally, RARα promotes the expression of prominin-1 (PROM-1) protein, a previously described SMG ductal lumenization marker26-28, without promoting differentiated ductal cytokeratins. Conversely, inhibition of RARγ with a selective inhibitor also promoted PROM-1 without promoting increased ductal cytokeratins, consistent with a RARα- and RARγ-induced decoupling of ductal differentiation from lumenization. We demonstrate that RARα and RARγ have opposing effects on K5+ progenitor cell expansion and proliferation, and inversely promote lumenization during early submandibular salivary gland branching morphogenesis.
MATERIALS AND METHODS
Ex vivo gland culture
Embryonic day 12.5 glands were dissected from timed pregnant CD-1 mice (Charles River Laboratories, Wilmington, MA) with E0 defined as day of plug discovery following procedures approved by the University at Albany, SUNY IACUC committee. E12.5 glands were chosen to examine RAR isoform function during organogenesis, after both placode and organ anlage formation. Whole organ explants were placed on top of porous polycarbonate filters (Nuclepore, Whatman WHA110405) in MatTek dishes on serum-free 1:1 DMEM/Ham's F12 medium (F12) (Invitrogen 21041–025) containing penicillin (100 μg/ml), and streptomycin (100 μg/ml), ascorbic acid (150 µg/mL) and transferrin (50 µg/mL) for 24 to 96 hours. For isolated epithelial rudiment experiments, epithelial rudiments were isolated via manual dissection following a mild 15 minute enzymatic digestion in dispase 0.4% v/v. Rudiments were then embedded in a Matrigel/DMEM (50% v/v) gel on top of a Nuclepore filter and cultured as described above with the addition of FGF-7 (200 ng/mL) (Peprotech) and EGF (20 ng/mL) (R&D Systems) to the culture media and cultured for 48 hours. Pharmacological antagonists were solubilized in vehicle (DMSO) in stock concentrations of 10 mM and were aliquoted, protected from light, and frozen at −20°C until use, including RARα inhibitor BMS195614 (Tocris), RARβ inhibitor LE135 (Tocris), RARγ inhibitor MM11253 (Tocris), RARα agonist AM80 (Tocris), and RARγ agonist BMS961 (Tocris). RARγ agonist CD1530 (Tocris) was made in an 8 mM stock in DMSO with 11.2 mM NaOH, aliquoted, protected from light, and frozen at −20°C until future use. After performing dose response experiments, working concentrations used in experiments were 1 µM RARα inhibitor BMS195614, 1 µM RARβ inhibitor LE135, 1 µM RARγ inhibitor MM11253, 10 nM RARα agonist AM80, 10 nM RARγ agonist BMS961, and 10 nM RARγ agonist CD1530 with 14 nM NaOH. All treated samples were compared to their corresponding vehicle. Dose response experiments were completed for all agonists and antagonists, and the lowest effective dose was used for further experiments.
Immunocytochemistry
Glands were fixed in either ice-cold methanol at −20°C or in 4% paraformaldehyde (w/v) (Electron Microscopy Sciences, Hatfield, PA) for 18 minutes, followed by two washes in 0.5% tween in phosphate buffered saline (PBS-T). We permeabilized paraformaldehyde-fixed tissues with 0.1% triton in PBS or 1% NP-40 in PBS, followed by washing in PBS-T. Blocking was then performed using 3% bovine serum albumin (BSA) (Sigma) in PBS. Primary antibodies diluted in 3% BSA in PBS (Table S1) were applied and incubated at 4C overnight. After washing in PBS-T, secondary antibodies were applied for 1–2 hours at room temperature (1:200 dilution, Jackson ImmunoResearch Laboratories, West Grove, PA). Samples were washed twice in PBS-T, and DAPI (Life Technologies) was applied for nuclear staining. Samples were washed an additional two times in PBS-T prior to mounting on slides. Images were obtained using matched laser settings on a Zeiss 710 confocal microscope.
DNA labeling
EdU labeling was performed using whole glands cultured for 48 hours, with EdU incorporated into the media during the final two hours of culture. Staining for EdU was completed using Click-it following manufacturer's recommendations, with the exception of an increased permeabilization step of 30 minutes (Thermo C10337). Samples were quantified from a minimum of 3 separate biological repeats.
Image quantification and statistical analysis
Measurements of markers within ductal epithelial area were performed using the open source software, FIJI29, to quantify positive pixel area of the marker of interest in confocal images relative to DAPI. Epithelial area was manually circled using the freeform lasso tool based on the presence of the characteristic layer of outer columnar cells surrounding the developing epithelial duct on DAPI-stained images, with the main duct identified as extending to the first ductal branch point. Regions of interest selected on the DAPI channel were saved and transferred to marker-containing channels. The total marker-positive area within that region was measured per channel after matched thresholding. Values were exported into Excel and graphed. Area of marker positivity was normalized to total ductal area per gland. Groups were averaged and graphed showing SEM. Sample size represents the number of explants quantified. A small number of explants were excluded from quantification due to physical damage to ducts during immunostaining based on pre-established criteria. Samples were quantified from a minimum of 3 separate biological repeats. Graphs were created using Microsoft Excel. Statistical analysis was performed using a two-tailed Student's t-test with VassarStats freeware30.
Western blot
Glands were lysed for total protein analysis in cold RIPA buffer (50 mM HEPES, 150 mM NaCl, 10% Glycerol, 1.5 mM MgCl, 1.5 mM EGTA, 1% Triton-100, 1% Na-deoxycholate, 0.1% SDS), with Roche protease and phosphatase inhibitor cocktails, (Thermo Sigma) and lysate was vortexed every ten minutes for 30 minutes followed by mild sonication. High-speed centrifugation (13,300 × g) was used to clear cell debris from the lysate, and the supernatant was used for further analysis. Lysate was processed into gel samples with 4x Laemmli Buffer (BioRad) with 5% β-mercaptoethanol (Sigma). Samples were run on precast gradient gels (BioRad cat#456–1086) with Tris/Glycine/SDS buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3) (BioRad) and were transferred to PVDF membrane (GE Healthcare) in cold Tris/Glycine buffer (25 mM Tris, 192 mM glycine, pH 8.3) (BioRad). Blocking was performed rocking for 1 hour at room temperature using 5% milk in 0.5% tween in Tris buffered saline pH 7.6 (TBS-T). Primary antibody (Table S1) was applied in 4 mL of 0.1% milk in TBS-T at 1:1000 and incubated in 50 mL conicals at 4C overnight, rolling. After four 15 minute washes in TBS-T, HRP secondary antibody (GE Healthcare) was applied for 1–2 hours in 3% milk in TBS-T at room temperature, rolling. Blots were washed for an additional four washes in TBS-T and ECL (Pierce) was applied. Loading control GAPDH was subsequently applied at 1:10,000 in 4 mL of 3% milk in TBS-T for 1 hour at room temperature and processed as described above. Blots were imaged with X-ray film, scanned, and quantified using Quantity One software (BioRad). Sample size represents total number of Western blots, with each representing approximately 5–6 explants per group per blot. Groups were averaged and graphed using Microsoft Excel showing SEM. A two-tailed Student's t-test was performed using with VassarStats freeware30.
RNA extraction and qPCR
RNA was extracted from the glands using the Ambion RNAqueous®- 4PCR kit, per the manufacturer's protocol. The extracted RNA was then treated with DNase I to eliminate any genomic contamination. PCR was performed on the DNase-treated RNA samples using Interleukin-2 genomic primers to confirm lack of genomic DNA contamination. The PCR reaction mixture was set up as shown in Table S2 and run in a GeneAmp PCR System 2400 thermocycler using the program described in Table S3. All primers used are listed in Table S4. The PCR samples were electrophoresed in a 1% agarose gel in a 1X TAE buffer (Tris-acetate-EDTA), pH of 8.3 and stained with 0.2 µg/mL of ethidium bromide. RNA was made into cDNA according to the New England Biolabs (NEB) First Strand cDNA Synthesis protocol using RNAse inhibitor (NEB), M-MuLV reverse transcriptase (NEB) and 10x stock M-MuLV buffer (NEB), 10 mM stock dNTP mix (NEB), and 100 µM stock oligo dT (IDT). Quantitative PCR was performed on the cDNA samples in a 384 well plate from Applied Biosystems using the Biorad CFX384 thermocycler. The reaction mixture was set up per the manufacturer's protocol using iQ SYBR Green Supermix (BioRad). The CFX384 thermocycler was run for 40 cycles, denaturing at 95°C and annealing/extending at 65°C. All curves were normalized to the reference mRNA, GAPDH. Change in fluorescence was determined using the ΔΔCq method31. Sample size represents biological replicates comprised of 5–6 explants per group, with each biological replicate run in technical triplicate then averaged. Values were graphed with SD in Microsoft Excel. Statistical analysis was performed using a two-tailed Student's t-test with VassarStats freeware30.
Lineage tracking using K5-CreERT2;ROSA26-TdTomato mice
K5-CreERT2 mice were obtained as a kind gift from Jianwen Que. Heterozygous Krt5tm1.1(cre/ERT2)Blh mice (K5-CreERT2) were crossed with B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J mice (ROSA26-TdTomato) (Jackson Labs) overnight with day of plug identified as E0. Embryos were harvested at E12.5 and genotyped for Cre and TdTomato using primers detailed in Table S4. K5CreERT2/+; ROSA26-TdTomato SMG organ explants were placed on top of Nuclepore filters (Whatman WHA110405) in MatTek dishes on serum-free 1:1 DMEM/Ham's F12 medium (F12) (Invitrogen 21041–025) containing penicillin (100 μg/ml), and streptomycin (100 μg/ml), ascorbic acid (150 µg/mL), and transferrin (50 µg/mL) and cultured for 72 hours. For K5-Cre induction, a pulse of 1 μM 4-OH tamoxifen (Sigma) solubilized in DMSO was incorporated into the media for the first four hours of culture. Culture media was rinsed and replaced for the remaining culture time.
RESULTS
RARα negatively regulates K5+ cells in developing salivary glands
To determine which isoforms of RAR are responsible for control of the K5+ population in the developing salivary gland, we manipulated RAR-mediated signaling in embryonic day 12.5 (E12.5) submandibular salivary gland organ explants cultured ex vivo. We manipulated the RAR isoforms independently with selective pharmacological inhibitors and agonists to assess their effects on K5+ cells after initial formation of the bud-on-stalk gland structure. Using immunocytochemistry (ICC) and confocal imaging, we first examined localization of RARα (Fig. S1). After verifying presence of RARα in the main duct, and after performing a dose response experiment (Fig. S2), we used the RARα-specific inhibitor BMS195614 in E12.5 gland cultures. Using ICC and confocal imaging, we observed an expansion of K5+ progenitors in the main duct of RARα-inhibited glands, corresponding to an increased K5+ epithelial area following quantification (Fig. 1a, b). Conversely, as first demonstrated in a dose response experiment (Fig. S2), activation of RARα using the selective agonist AM80 in E12.5 gland cultures decreased the expansion of K5+ cells (Fig. 1a, b). Western blotting also indicated a significant increase in K5 protein levels in RARα-inhibited explants as compared to control, while RARα agonism led to a significant decrease in K5 levels (Fig. 1c, d).
FIGURE 1.
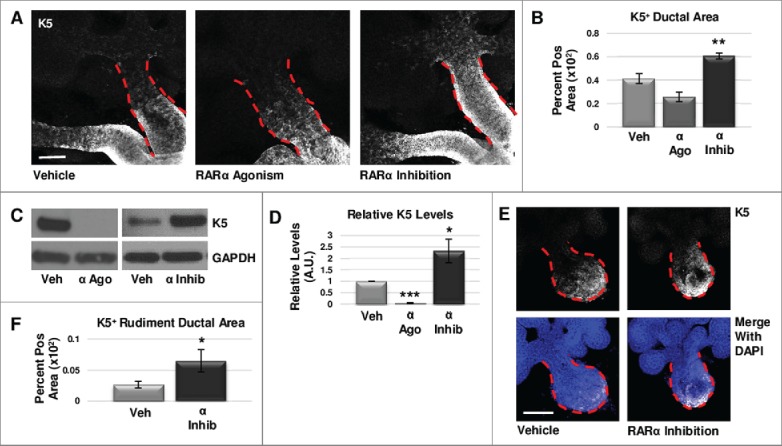
RARα negatively regulates K5+ salivary epithelial cells. (A) Whole explants cultured ex vivo for 72 hours with RARα (α ago) agonist show decreased expansion of K5+ cells in the main duct, while K5 extends beyond the primary duct relative to vehicle control (Veh) with RARα inhibition (α inhib) by ICC in single confocal sections. Ducts are outlined with red dotted line. Scale bar, 100 μm. (B) Quantification of K5+ area in the main duct normalized to the total main duct area indicates significantly increased K5+ with RARα inhibition. Veh n = 19, α ago n = 6, α inhib n = 15 explants. Statistical analysis completed using Student's two-tailed t-test. **p = 0.003, α ago p = 0.056. (C, D) E12.5 explants were cultured for 48 hours. Western blot and quantification of Western blot indicates significantly decreased levels of K5 with RARα agonism and significantly increased K5 levels with RARα inhibition as normalized to GAPDH levels. Mean represents three or more experiments with n ≥ 5 glands per condition. Statistical analysis completed using Student's two-tailed t-test. *p = 0.04, ***p<0.0001. (E, F) E12.5 epithelial rudiments were cultured for 48 hours. Quantification of the K5+ ductal area relative to total ductal area with RARα inhibition shows significantly increased K5+ area. Scale bar 100 μm. Veh n = 12, α inhib n = 11 explants. Statistical analysis completed using Student's two-tailed t-test. *p = 0.045.
Since maintenance of K5+ cells during SMG development is dependent on muscarinic signaling from developing parasympathetic innervation25, retinoic acid signaling is known to directly regulate innervation32, and RA-dependent signaling was detected in embryonic submandibular salivary gland epithelium, parasympathetic ganglion neurons, and mesenchyme9, we questioned whether retinoic acid signaling acts directly on epithelial cells during early salivary gland development. Although Western analysis did not reveal a change in βIII tubulin levels with RARα inhibition (Fig. S3), to confirm that RARα function is intrinsic to the epithelium and independent of innervation, we cultured isolated epithelial rudiments, devoid of nerves and mesenchyme, in the presence and absence of the RARα isoform-selective inhibitor BMS195614, where we observed an increase in the K5+ epithelial area similar to results observed with whole glands (Fig. 1 e, f). Together, these data indicate that RARα negatively regulates the expansion of K5+ progenitor cells, independently of innervation.
RARγ positively regulates K5+ cells in developing salivary glands
We next examined the effect of RARγ on the K5+ cell population. Immunostaining for RARγ showed localization in the main duct in areas normally positive for K5+ cells (Fig. S1). Using the RARγ-selective inhibitor MM11253, after performing a dose response experiment (Fig. S2), with ICC and confocal imaging in E12.5 whole explant cultures we observed greatly decreased K5+ epithelial area (Fig. 2a, b) and K5 levels by Western blotting (Fig. 2c, e). Conversely, activation of RARγ with the selective agonist CD1530, in accordance with the dose response experiment performed (Fig. S2), showed no significant change in K5+ cell area in the main duct of the gland using ICC (Fig. 2a, b) and Western blotting (Fig. 2c, d). Treatment of E12.5 explants in a separate experiment with another RARγ specific agonist, BMS961, produced similar results (data not shown). Isolated epithelial rudiments cultured with the RARγ-selective inhibitor MM11253 demonstrated reduced K5 expansion by ICC (Fig. 2e, f). Since no significant change was observed in βIII tubulin levels by Western blot with RARγ inhibition (Fig. S3), taken together, these data show that positive regulation of the K5 cells by RARγ is independent of innervation. Interestingly, manipulation of RARβ with the RARβ−specific antagonist, LE135, did not affect the levels of K5 (data not shown). We conclude that in contrast to RARα that negatively regulates the K5+ population, RARγ positively regulates K5+ cells, also independently of innervation.
FIGURE 2.
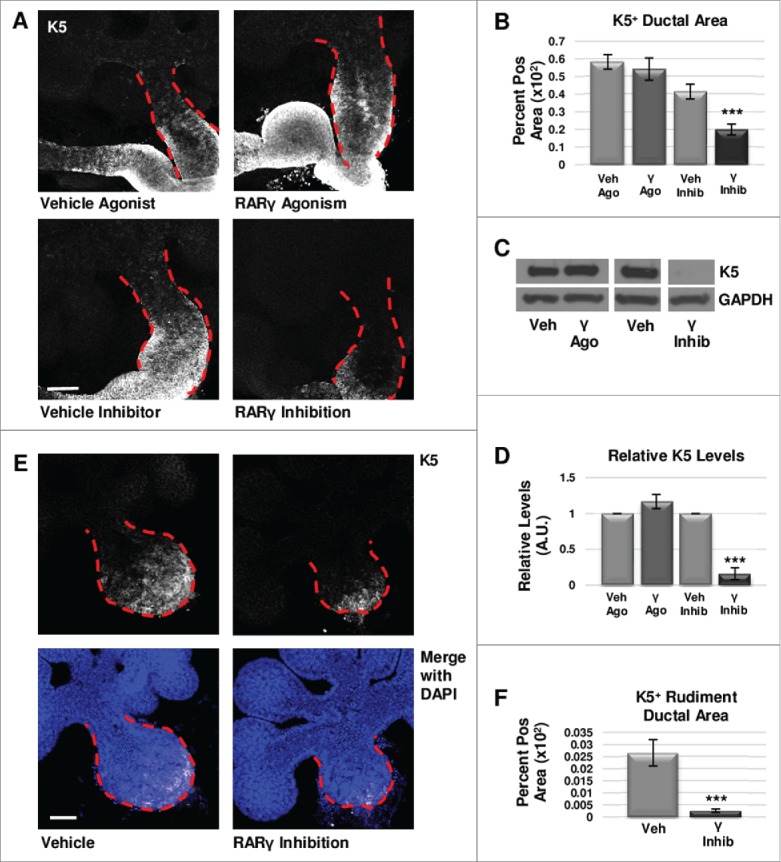
RARγ positively regulates K5+ salivary epithelial cells. (A) ICC for K5 in whole glands cultured ex vivo for 72 hours shows slightly increased expansion of K5+ cells with RARγ agonist (γ ago) in the main duct, and decreased expansion of K5 in the main duct of RARγ-inhibited (γ inhib) glands. Ducts are outlined with red dotted line. Scale bar, 100 μm. (B) Quantification of K5+ area in the main duct of RARγ-inhibited whole glands shows significantly decreased K5+ area, indicating that signaling through RARγ may be necessary but not sufficient to maintain K5+ cells. Ago veh n = 12, γ ago n = 9, veh inhib n = 19, γ inhib n = 14 explants. Statistical analysis completed using Student's two-tailed t-test. ***p = 0.0006. (C, D) E12.5 glands were cultured ex vivo for 48 hours. Western blot and quantification indicates a slight increase in K5 levels with RARγ agonist while K5 levels are almost absent with RARγ inhibitor as normalized to GAPDH levels. n ≥ 3 experiments with n ≥ 5 glands per condition. Statistical analysis completed using Student's two-tailed t-test. ***p < 0.0001. (E) E12.5 epithelial rudiments were cultured for 48 hours. ICC for K5 shows decreased K5+ ductal area with RARγ inhibition as compared to vehicle control. (F) Quantification of ICC for K5 in the ductal area of isolated epithelial rudiments shows significantly decreased K5 with RARγ inhibition indicating RARγ actions are endogenous to the epithelium. K5+ area normalized to total main duct area. Scale bar 100 μm. Veh n = 12 γ inhib n = 11 explants. Statistical analysis completed using Student's two-tailed t-test. *** p = 0.0004.
RARα negatively regulates cell cycle progression
To determine if regulation of cell proliferation contributed to changes in RARα-mediated levels of K5 in developing glands, we examined markers of cell cycle progression. Since Ki67 is present during all phases of the cell cycle except G033, we first analyzed Ki67 expression in ex vivo organ explants after 24 hours of treatment with the RARα-specific inhibitor BMS195614, where we observed that Ki67 mRNA expression was significantly increased as compared to control explants (Fig. 3a). To quantify cells in S-phase, we incorporated EdU, a thymidine analog, into the media during the last two hours of culture followed by quantitative ICC in explants cultured for 48 hours34, which revealed a significant increase in EdU staining in the main ducts of RARα-inhibited glands where K5+ cells are concentrated (Fig. 3b, c). To quantify cells in M-phase, we used quantitative ICC to examine phosphorylation of histone H3 (pHH3), which is phosphorylated on Ser10 specifically during mitosis35, where we observed a significant increase in pHH3 with RARα inhibition as compared to control (Fig. 3d, e). Analysis of cleaved caspase 3 (CC3), an executioner caspase that is activated by cleavage by both extrinsic and intrinsic apoptosis pathways36, with quantitative ICC indicated that programmed cell death does not play a major role in changing K5 levels. The CC3-positive area is unchanged with decreasing levels of K5 observed with RARα agonism; however, the CC3-positive area is decreased significantly with the increasing K5 levels observed with RARα inhibition (Fig. S4). These data are consistent with RARα negatively regulating cell cycle progression.
FIGURE 3.
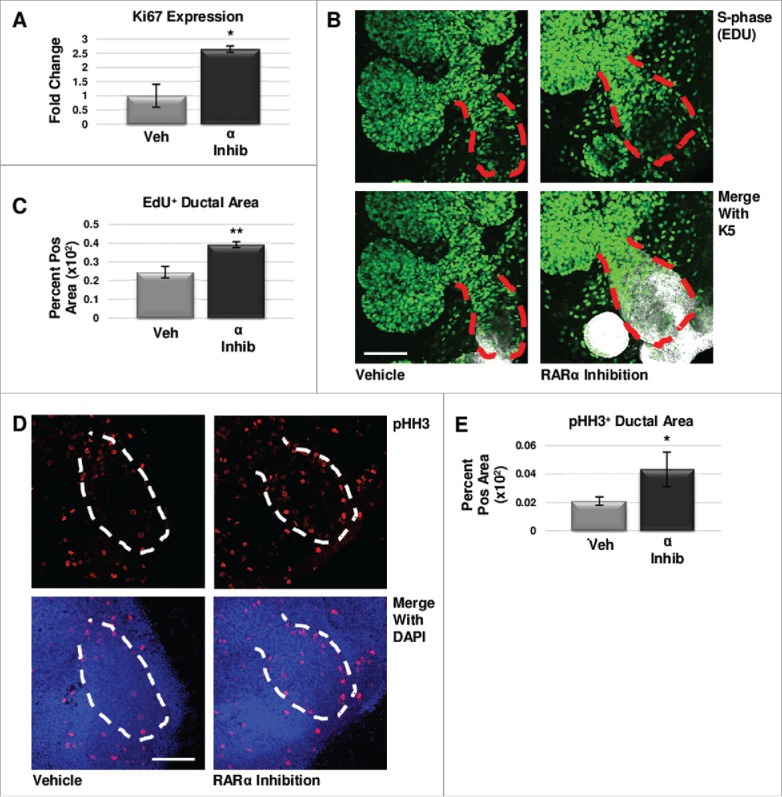
RARα maintains K5+ cells through negative cell cycle regulation. (A) E12.5 glands were cultured for 24 hours with or without RARα inhibitor. Analysis of non-phase specific cell cycle marker Ki67 using qPCR indicates RARα inhibition increases Ki67 expression, suggesting an overall increase in cells traversing the cell cycle. Mean represents two experiments run in triplicate with n ≥ 5 glands per condition. Statistical analysis performed using Student's two-tailed t-test. p = 0.03. (B) E12.5 explants were cultured for 48 hours with or without RARα inhibitor. EdU was incorporated into the media during the last two hours of culture to mark cells in S-phase showing increased staining in the main duct (outlined in red) with RARα inhibition. Scale bar,100 μm. (C) Quantification of ICC indicates significantly increased EdU staining in the main duct of RARα-inhibited glands as compared to vehicle control, indicating increased numbers of cells in S-phase. EdU+ area normalized to total main duct area. Veh n = 23, α ago n = 13, α inhib n = 9 explants. Statistical analysis completed using Student's two-tailed t-test. **p = 0.005. (D) ICC for M-phase marker, phospho-histone H3 (pHH3), shows an increase in staining in the main duct (outlined in white) of RARα-inhibited glands as compared to control. Scale bar, 100 μm. (E) Quantitative ICC for pHH3 shows an increasing trend in the main duct of RARα-inhibited glands as compared to control. pHH3+ area normalized to total main duct area. Veh n = 22, α ago n = 13, α inhib n = 9 explants. Statistical analysis completed using Student's two-tailed t-test. *p = 0.02.
RARγ positively regulates cell cycle progression
To determine if regulation of cell proliferation contributed to the changes in RARγ-mediated levels of K5 in developing glands, we examined markers of cell cycle progression. Using quantitative ICC, we examined S-phase with an EdU pulse and M-phase using pHH3 antibody. In RARγ-inhibited explants grown for 48 hours with EdU incorporated into the media for the final two hours, we observed a significant decrease in EdU staining in the main duct relative to vehicle control-treated glands (Fig 4a, c). Additionally, we observed significantly reduced pHH3 levels in the main duct (Fig. 4b, d). Like manipulation of RARα, changes in CC3 staining with manipulation of RARγ signaling was not correlated with changes in K5 expansion, showing no significant change in RARγ agonism, while RARγ inhibition significantly decreased CC3 staining in the main duct (Fig. S4). In contrast to RARα, these results suggest that RARγ positively regulates cell cycle progression, and RARγ inhibition leads to cell cycle arrest in early developing salivary glands, consistent with loss of K5 expression.
FIGURE 4.
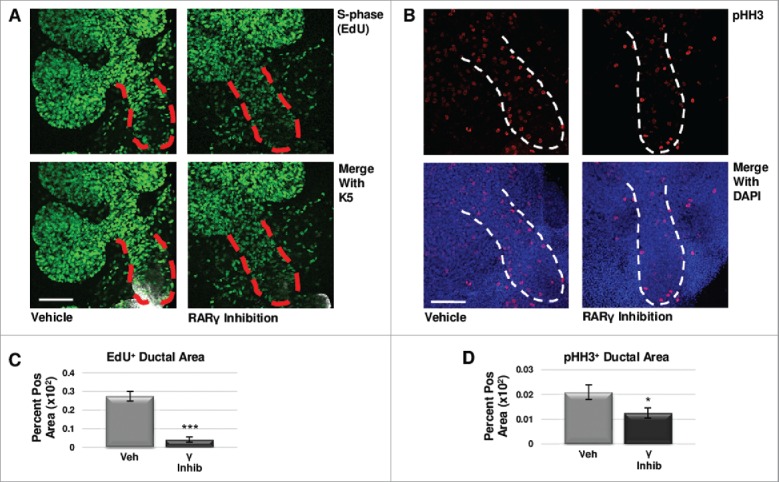
RARγ Maintains K5+ Cells Through Cell Cycle Regulation. (A) E12.5 explants were cultured for 48 hours with or without RARγ inhibitor with the incorporation of EdU for the last two hours. Scale bar, 100 μm. (B) ICC for M-phase marker pHH3 shows a decrease in positive staining in the main duct of RARγ-inhibited glands as compared to control. Scale bar, 100 μm. (C) Quantitative immunocytochemistry indicates significantly decreased EdU staining in the main duct of RARγ-inhibited glands as compared to vehicle control, indicating decreased numbers of cells in S-phase. EdU+ area normalized to total main duct area. Veh inhib n = 23, γ inhib n = 9 explants. Statistical analysis completed using Student's two-tailed t-test. ***p ≤ 0.001. (D) Quantitative immunocytochemistry for pHH3 shows a significant decrease in pHH3 positively stained area in the main duct of RARγ-inhibited glands as compared to control, indicating significantly fewer cells in M-phase. pHH3+ area normalized to total main duct area. Veh inhib n = 22, γ inhib n = 15 explants. Statistical analysis completed using Student's two-tailed t-test. *p = 0.04. Taken together, these results suggest G1 mediated cell cycle arrest.
RAR isoforms reciprocally regulate lumenization of the primary duct
Since retinoic acid is known to stimulate differentiation6,37-39 and since K5+ cells acquire expression of K19 as the ductal epithelia mature and differentiate along the ductal lineage25 we next examined if manipulation of RAR isoforms influenced ductal differentiation using quantitative ICC for K19. In conditions previously shown to decrease K5 levels, such as RARα agonism and RARγ inhibition, decreased K5 levels correspond with decreased K19 levels; however, K19 expression was not absent (Fig. 5a-c). Increased K5 levels seen in RARα inhibition and RARγ agonism, however, corresponded with no significant change in K19 (Fig. 5a-c). Taken together, these results indicate that decreased levels of K5 correspond to decreased K19, regardless of isoform-specificity, consistent with a loss of K5+ progenitor cells leading to a loss of downstream K19+ ductal differentiation. However, our data indicate that neither RARα nor RARγ specifically regulate duct-specific cytokeratin expression during ductal differentiation.
FIGURE 5.
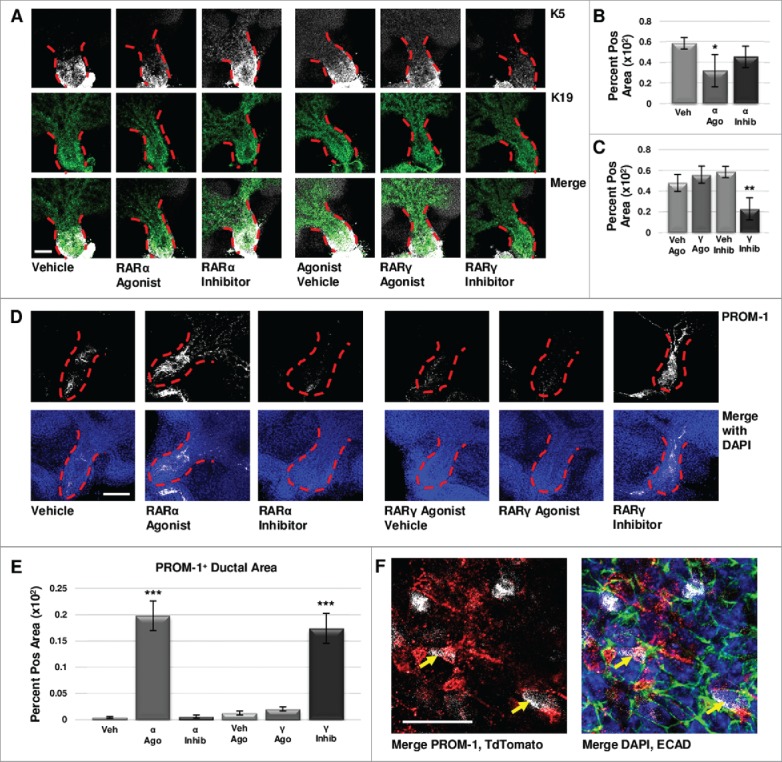
Retinoid Signaling Promotes Lumenizing Cell Differentiation. (A) E12.5 explants were cultured for 72 hours and immunostained for basal marker K5 and ductal marker K19. Scale bar, 100 μm. (B) Quantification of K19-positive area in the main duct with RARα agonist shows significantly decreased K19, correlating with decreased K5, while no significant change in K19 is observed with RARα inhibition. Veh n = 11, α ago n = 6, α inhib n = 5 explants. Statistical analysis completed using Student's two-tailed t-test. *p = 0.02. (C) Quantification of K19-positive area in the main duct with RARγ agonist treatment indicates no significant change in K19, similar to RARα inhibition. With RARγ inhibition, K19 levels are significantly decreased corresponding to decreased K5. Ago veh n = 12, γ ago n = 9, veh inhib n = 11, γ inhib n = 5 explants. Statistical analysis completed using Student's two-tailed t-test. **p = 0.005. This data indicates that loss of K5 does not correspond with a gain of K19. (D) E12.5 explants cultured for 72 hours show increased expression of PROM-1 with RARα agonism and RARγ inhibition under conditions that show significantly decreased K5. Increased PROM-1 is not observed with RARα inhibition or RARγ agonism where K5 was increased. Scale bar, 100 μm. (E) Quantification of PROM-1 positive area in the main duct of the gland in cultures at both 72 hours and 96 hours (data not shown) indicates a significant increase in PROM-1 positive area with RARα agonist or RARγ inhibitor as compared to control. Veh n = 11, α ago n = 17, α inhib n = 10, ago veh n = 10, γ ago n = 10, veh inhib n = 15, γ inhib n = 15 explants. Statistical analysis completed using Student's two-tailed t-test. ***p < 0.0001. This data indicates RAR isoform-mediated decreases in K5 result in increased PROM-1 expression. (F) Lineage tracking using E12.5 K5- CreERT2;ROSA26-TdTomato mice cultured ex vivo for 68 hours following a four hour tamoxifen induction (1 μM) shows TdTomato reporter expression (white) in a handful of PROM-1 (red) positive cells in the main duct (yellow arrows), with ECAD (green) and DAPI (blue), indicating that these cells were derived from the K5 lineage. Scale bar, 50 μm.
As both epithelial maturation and lumenization occur during the development of the salivary duct, we next examined the effect of isoform-specific RAR signaling on the presence of PROM-1, an apical lumenizing surface marker of the ductal epithelium in the salivary gland and other stratified epithelia26-28. Using quantitative ICC of E12.5 explants cultured under conditions resulting in decreased K5, (i.e. RARα agonism and RARγ inhibition), significantly increased PROM-1 levels were observed within the main duct (Fig. 5d, e). PROM-1 was relatively absent in conditions corresponding to increased K5, including RARα inhibition and RARγ agonism (Fig. 5d, e). Lineage tracking using tamoxifen inducible K5CreERT2/+; ROSA26-TdTomato mice indicated that in cultured E12.5 explants, PROM-1 and TdTomato co-localize in a subset of cells after a short pulse of tamoxifen at the start of culture to induce Cre expression in K5+ cells (Fig. 5 f), demonstrating that K5+ cells have the capacity to become PROM-1 expressing cells. Taken together, these results indicate that signaling through RARα restricts K5+ cell expansion and promotes lumenization, while RARγ signaling promotes K5+ cell expansion and negatively regulates lumenization.
DISCUSSION
Many FDA-approved pharmaceuticals target nuclear receptors, including estrogen receptor (ER), progesterone receptor (PR), glucocorticoid receptor (GCR), peroxisome proliferator-activated receptor (PPAR), RAR, and others, although the exact pharmaceutical mechanisms of action or isoform specificity in some cases is unknown due to unresolved receptor activity or use of pan-receptor ligands. Although in vivo RAR isoforms bind identical ligands, biochemical characterization revealed differences in isoform ligand binding domain sequences, allowing for design of synthetic isoform-specific retinoid agonists and inhibitors40. In regenerative therapies, atRA initially demonstrated the capacity to regenerate alveoli in rat41 and mouse42. After demonstrating efficacy in a human case study where atRA was used to initiate regeneration after damage from emphysema43, clinical trials using atRA were completed44,45. Retinoids are FDA approved for topical application in the form of tretinoin, and synthetic Tazarotene (AGN 190299), and are currently in Phase I and II clinical trials used alone and in combination for several conditions including breast carcinoma (Phase Ib NCT02876640) and multiple myeloma (Phase I and II NCT02751255). However, use of atRA as a pan-RAR agonist has not been entirely successful. Initial trial results using atRA to induce lung regeneration indicated that atRA was unsuccessful in improving patient lung conditions or function44. Recent studies have begun targeting RARs in an isoform-specific manner using isoform-specific pharmacological agonists and inhibitors for improved selectivity and function. Regenerative studies in skeletal muscle in the mouse have recently demonstrated that selective receptor isoform agonism of RARγ improved muscle regeneration46, as well as moving to RARγ isoform-selective Phase I and Phase II trials in lung regeneration47, clinical trials achieving positive results yet limited by small patient cohort48. Targeting of specific RAR isoforms has not yet been tested as a strategy for salivary gland regeneration; however, our results indicate that the behavior of K5+ cells is under complex control by RARs during development.
In this study, we examined isoform-specific RAR effects on SMG organogenesis in organ explants to investigate RAR isoform-specific control of K5+ progenitor cells during development with the intent to inform future salivary gland regenerative therapies. We report that during organogenesis, RARα and RARγ have opposing effects on cell cycle progression and expansion of K5+ cells, in that RARα negatively regulates cell cycle progression and K5+ expansion, while RARγ positively regulates cell cycle progression and K5+ cell expansion. Inhibition of pan-RAR signaling in embryonic SMG organ explants was recently shown to inhibit cell proliferation and reduce FGF-dependent signaling during branching morphogenesis while transcriptionally upregulating K5, independent of innervation9. Here, we have isolated the impacts of individual RAR isoforms on cell cycle and K5+ cell distribution, uncovering that RARα and RAR have antagonizing effects on cell cycle progression and K5+ progenitor cell expansion. Our data cannot discriminate whether the expansion of K5+ cells during early SMG development results from expansion of existing K5+ progenitors, progressive specification of K5+ progenitors, or both, at this stage of development. Nevertheless, we have shown that accumulation of K5+ cells is dependent on RAR isoform-specific signaling, and our results imply that the previously reported expansion of K5+ cells with pan-RAR inhibitor9 is due primarily to manipulation of RARα. Thus for future therapeutic restoration of salivary gland function, inhibition of endogenous RARα function may be effective to expand K5+ progenitor cells, whereas RARγ may not be an effective clinical target for progenitor cell expansion, suggesting that RARα inhibitors will be superior to pan-RAR modulation for restoration of impaired salivary function.
Controlled regulation of the cell cycle is important for proper differentiation during development37,49,50, while in other contexts differentiation impacts cell cycle progression. Retinoic acid is well known to induce differentiation downstream of cell cycle arrest through tissue-specific mechanisms38,51. Interestingly, we did not detect an associated change in differentiation of K5+ cells into K19+ ductal cells with RAR-mediated manipulation of the cell cycle, indicating that RAR-induced manipulation of cell proliferation does not necessarily lead to keratin-marked differentiation, consistent with recently published data that RAR signaling does not affect keratin-marked differentiation in salivary glands9. These data are also consistent with atRA manipulation of keratinocytes, where atRA administration decreased both basal and suprabasal cytokeratins52. Interestingly, we did observe premature ductal lumenization with RARα agonism or RARγ inhibition. During SMG development, signaling through RARγ may normally restrict ductal lumenization to counterbalance lumenizing signals provided by RARα, providing fine control over timing of ductal lumenization during normal development. The lack of correlation between the lumenizing marker, PROM-1, with the ductal marker, K19, with RAR manipulation indicates that keratin-marked ductal fate and cell lumenization are uncoupled events during ductal development. Importantly, K19 levels did not decrease with retinoid-expanded K5 levels, and lineage tracking revealed K5+ cells can acquire the PROM-1 lumenization marker with normal development, indicating that the expanded K5+ progenitor cells can differentiate into PROM-1+ duct cells. Together, these data suggest that isoform-specific RARα inhibition holds promise for clinical application to expand K5+ endogenous progenitor cells to enhance salivary gland regeneration and restore function in patients suffering from salivary hypofunction.
Supplementary Material
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
ACKNOWLEDGMENTS
This manuscript is dedicated to Anthony Sauro. The authors acknowledge Hae Ryong Kwon for assistance in epithelial rudiment dissections and the Rangan lab, especially Pooja Floora, for helpful discussions.
FUNDING
This work was supported by The University at Albany, SUNY, NIH RO1 DE022467 (to M.L.), and NIH C06 RR015464.
REFERENCES
- [1].Niederreither K, Vermot J, Le Roux I, Schuhbaur B, Chambon P, Dollé P. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Dev Camb Engl. 2003;130(11):2525-2534. [DOI] [PubMed] [Google Scholar]
- [2].Lohnes D, Kastner P, Dierich A, Mark M, LeMeur M, Chambon P. Function of retinoic acid receptor gamma in the mouse. Cell. 1993; 73(4):643-658. [DOI] [PubMed] [Google Scholar]
- [3].Lohnes D, Mark M, Mendelsohn C, Dollé P, Dierich A, Gorry P, Gansmuller A, Chambon P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Dev Camb Engl. 1994; 120(10):2723-2748. [DOI] [PubMed] [Google Scholar]
- [4].Schuger L, Varani J, Mitra R, Gilbride K. Retinoic acid stimulates mouse lung development by a mechanism involving epithelial-mesenchymal interaction and regulation of epidermal growth factor receptors. Dev Biol. 1993; 159(2):462-473. doi: 10.1006/dbio.1993.1256. [DOI] [PubMed] [Google Scholar]
- [5].Gijbels MJ, van der Ham F, van Bennekum AM, Hendriks HF, Roholl PJ. Alterations in cytokeratin expression precede histological changes in epithelia of vitamin A-deficient rats. Cell Tissue Res. 1992; 268(1):197-203. [DOI] [PubMed] [Google Scholar]
- [6].Kautsky MB, Fleckman P, Dale BA. Retinoic acid regulates oral epithelial differentiation by two mechanisms. J Invest Dermatol. 1995; 104(2):224-230. [DOI] [PubMed] [Google Scholar]
- [7].Malpel S, Mendelsohn C, Cardoso WV. Regulation of retinoic acid signaling during lung morphogenesis. Dev Camb Engl. 2000; 127(14):3057-3067. [DOI] [PubMed] [Google Scholar]
- [8].Wright DM, Buenger DE, Abashev TM, Lindeman RP, Ding J, Sandell LL. Retinoic acid regulates embryonic development of mammalian submandibular salivary glands. Dev Biol. 2015; 407(1):57-67. doi: 10.1016/j.ydbio.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Abashev TM, Metzler MA, Wright DM, Sandell LL. Retinoic acid signaling regulates Krt5 and Krt14 independently of stem cell markers in submandibular salivary gland epithelium. Dev Dyn Off Publ Am Assoc Anat. 2017; 246(2):135-147. doi: 10.1002/dvdy.24476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wongtrakool C, Malpel S, Gorenstein J, Sedita J, Ramirez MI, Underhill TM, Cardoso WV. Down-regulation of Retinoic Acid Receptor α Signaling Is Required for Sacculation and Type I Cell Formation in the Developing Lung. J Biol Chem. 2003; 278(47):46911-46918. doi: 10.1074/jbc.M307977200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kastner P, Lawrence HJ, Waltzinger C, Ghyselinck NB, Chambon P, Chan S. Positive and negative regulation of granulopoiesis by endogenous RARalpha. Blood. 2001; 97(5):1314-1320. [DOI] [PubMed] [Google Scholar]
- [12].Rosselot C, Spraggon L, Chia I, Batourina E, Riccio P, Lu B, Niederreither K, Dolle P, Duester G, Chambon P, et al.. Non-cell-autonomous retinoid signaling is crucial for renal development. Dev Camb Engl. 2010; 137(2):283-292. doi: 10.1242/dev.040287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bourguet W, Germain P, Gronemeyer H. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci. 2000; 21(10):381-388. [DOI] [PubMed] [Google Scholar]
- [14].Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995; 83(6):841-850. [DOI] [PubMed] [Google Scholar]
- [15].See AW-M, Kaiser ME, White JC, Clagett-Dame M. A nutritional model of late embryonic vitamin A deficiency produces defects in organogenesis at a high penetrance and reveals new roles for the vitamin in skeletal development. Dev Biol. 2008; 316(2):171-190. doi: 10.1016/j.ydbio.2007.10.018. [DOI] [PubMed] [Google Scholar]
- [16].Chytil F. Retinoids in lung development. FASEB J Off Publ Fed Am Soc Exp Biol. 1996; 10(9):986-992. [DOI] [PubMed] [Google Scholar]
- [17].Niederreither K, Subbarayan V, Dollé P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet. 1999; 21(4):444-448. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- [18].Sandell LL, Lynn ML, Inman KE, McDowell W, Trainor PA. RDH10 Oxidation of Vitamin A Is a Critical Control Step in Synthesis of Retinoic Acid during Mouse Embryogenesis. PLoS ONE. 2012; 7(2):e30698. doi: 10.1371/journal.pone.0030698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang Z, Dollé P, Cardoso WV, Niederreither K. Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev Biol. 2006; 297(2):433-445. doi: 10.1016/j.ydbio.2006.05.019. [DOI] [PubMed] [Google Scholar]
- [20].Dollé P, Fraulob V, Gallego-Llamas J, Vermot J, Niederreither K. Fate of retinoic acid-activated embryonic cell lineages. Dev Dyn Off Publ Am Assoc Anat. 2010; 239(12):3260-3274. doi: 10.1002/dvdy.22479. [DOI] [PubMed] [Google Scholar]
- [21].Knosp WM, Knox SM, Hoffman MP. Salivary gland organogenesis. Wiley Interdiscip Rev Dev Biol. 2012; 1(1):69-82. doi: 10.1002/wdev.4. [DOI] [PubMed] [Google Scholar]
- [22].Nelson DA, Manhardt C, Kamath V, Sui Y, Santamaria-Pang A, Can A, Bello M, Corwin A, Dinn SR, Lazare M, et al.. Quantitative single cell analysis of cell population dynamics during submandibular salivary gland development and differentiation. Biol Open. 2013; 2(5):439-447. doi: 10.1242/bio.20134309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gervais EM, Desantis KA, Pagendarm N, Nelson DA, Enger T, Skarstein K, Liaaen Jensen J, Larsen M. Changes in the Submandibular Salivary Gland Epithelial Cell Subpopulations During Progression of Sjögren's Syndrome-Like Disease in the NOD/ShiLtJ Mouse Model. Anat Rec Hoboken NJ 2007. 2015; 298(9):1622-1634. doi: 10.1002/ar.23190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rios AC, Fu NY, Lindeman GJ, Visvader JE. In situ identification of bipotent stem cells in the mammary gland. Nature. 2014; 506(7488):322-327. doi: 10.1038/nature12948. [DOI] [PubMed] [Google Scholar]
- [25].Knox SM, Lombaert IMA, Reed X, Vitale-Cross L, Gutkind JS, Hoffman MP. Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science. 2010; 329(5999):1645-1647. doi: 10.1126/science.1192046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jászai J, Janich P, Farkas LM, Fargeas CA, Huttner WB, Corbeil D. Differential expression of Prominin-1 (CD133) and Prominin-2 in major cephalic exocrine glands of adult mice. Histochem Cell Biol. 2007; 128(5):409-419. doi: 10.1007/s00418-007-0334-2. [DOI] [PubMed] [Google Scholar]
- [27].Karbanová J, Missol-Kolka E, Fonseca A-V, Lorra C, Janich P, Hollerová H, Jászai J, Ehrmann J, Kolár Z, Liebers C, et al.. The Stem Cell Marker CD133 (Prominin-1) Is Expressed in Various Human Glandular Epithelia. J Histochem Cytochem. 2008; 56(11):977-993. doi: 10.1369/jhc.2008.951897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nedvetsky PI, Emmerson E, Finley J, Ettinger A, Cruz-Pacheco N, Prochazka J, Haddox CL, Northrup E, Hodges C, Mostov KE, et al.. Parasympathetic innervation regulates tubulogenesis in the developing salivary gland. Dev Cell. 2014; 30(4):449-462. doi: 10.1016/j.devcel.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al.. Fiji – an Open Source platform for biological image analysis. Nat Methods. 2012; 9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lowry R. VassarStats.; 2008. http://vasssarstats.net. [Google Scholar]
- [31].Haimes J, Kelley M. Demonstration of a ddCq calculation method to compute relative gene expression from qPCR data. GE Healthc Tech Note. 2014. [Google Scholar]
- [32].Clagett-Dame M, McNeill EM, Muley PD. Role of all-trans retinoic acid in neurite outgrowth and axonal elongation. J Neurobiol. 2006; 66(7):739-756. doi: 10.1002/neu.20241. [DOI] [PubMed] [Google Scholar]
- [33].Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000; 182(3):311-322. doi:. [DOI] [PubMed] [Google Scholar]
- [34].Buck SB, Bradford J, Gee KR, Agnew BJ, Clarke ST, Salic A. Detection of S-phase cell cycle progression using 5-ethynyl-2’-deoxyuridine incorporation with click chemistry, an alternative to using 5-bromo-2’-deoxyuridine antibodies. BioTechniques. 2008; 44(7):927-929. doi: 10.2144/000112812. [DOI] [PubMed] [Google Scholar]
- [35].Tapia C, Kutzner H, Mentzel T, Savic S, Baumhoer D, Glatz K. Two mitosis-specific antibodies, MPM-2 and phospho-histone H3 (Ser28), allow rapid and precise determination of mitotic activity. Am J Surg Pathol. 2006; 30(1):83-89. [DOI] [PubMed] [Google Scholar]
- [36].Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997; 91(4):443-446. [DOI] [PubMed] [Google Scholar]
- [37].Janesick A, Wu SC, Blumberg B. Retinoic acid signaling and neuronal differentiation. Cell Mol Life Sci CMLS. 2015; 72(8):1559-1576. doi: 10.1007/s00018-014-1815-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wainwright LJ, Lasorella A, Iavarone A. Distinct mechanisms of cell cycle arrest control the decision between differentiation and senescence in human neuroblastoma cells. Proc Natl Acad Sci U S A. 2001; 98(16):9396-9400. doi: 10.1073/pnas.161288698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Boylan JF, Lufkin T, Achkar CC, Taneja R, Chambon P, Gudas LJ. Targeted disruption of retinoic acid receptor alpha (RAR alpha) and RAR gamma results in receptor-specific alterations in retinoic acid-mediated differentiation and retinoic acid metabolism. Mol Cell Biol. 1995; 15(2):843-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J Off Publ Fed Am Soc Exp Biol. 1996; 10(9):940-954. [PubMed] [Google Scholar]
- [41].Massaro GD, Massaro D. Retinoic acid treatment abrogates elastase-induced pulmonary emphysema in rats. Nat Med. 1997; 3(6):675-677. [DOI] [PubMed] [Google Scholar]
- [42].Hind M, Maden M. Retinoic acid induces alveolar regeneration in the adult mouse lung. Eur Respir J. 2004; 23(1):20-27. [DOI] [PubMed] [Google Scholar]
- [43].Frankenberger M, Heimbeck I, Möller W, Mamidi S, Kassner G, Pukelsheim K, Wjst M, Neiswirth M, Kroneberg P, Lomas D, et al.. Inhaled all-trans retinoic acid in an individual with severe emphysema. Eur Respir J. 2009; 34(6):1487-1489. doi: 10.1183/09031936.00105309. [DOI] [PubMed] [Google Scholar]
- [44].Mao JT, Goldin JG, Dermand J, Ibrahim G, Brown MS, Emerick A, McNitt-Gray MF, Gjertson DW, Estrada F, Tashkin DP, et al.. A pilot study of all-trans-retinoic acid for the treatment of human emphysema. Am J Respir Crit Care Med. 2002; 165(5):718-723. doi: 10.1164/ajrccm.165.5.2106123. [DOI] [PubMed] [Google Scholar]
- [45].Roth MD, Connett JE, D'Armiento JM, Foronjy RF, Friedman PJ, Goldin JG, Louis TA, Mao JT, Muindi JR, O'Connor GT, et al.. Feasibility of retinoids for the treatment of emphysema study. Chest. 2006; 130(5):1334-1345. doi: 10.1378/chest.130.5.1334. [DOI] [PubMed] [Google Scholar]
- [46].Di Rocco A, Uchibe K, Larmour C, Berger R, Liu M, Barton ER, Iwamoto M. Selective Retinoic Acid Receptor γ Agonists Promote Repair of Injured Skeletal Muscle in Mouse. Am J Pathol. 2015; 185(9):2495-2504. doi: 10.1016/j.ajpath.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hind M, Stinchcombe S. Palovarotene, a novel retinoic acid receptor gamma agonist for the treatment of emphysema. Curr Opin Investig Drugs Lond Engl. 2000. 2009; 10(11):1243-1250. [PubMed] [Google Scholar]
- [48].Oh DK, Kim Y-S, Oh Y-M. Lung regeneration therapy for chronic obstructive pulmonary disease. Tuberc Respir Dis. 2017; 80(1):1-10. doi: 10.4046/trd.2017.80.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ruijtenberg S, van den Heuvel S. Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle. 2016; 15(2):196-212. doi: 10.1080/15384101.2015.1120925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kim YH, Larsen HL, Rué P, Lemaire LA, Ferrer J, Grapin-Botton A. Cell Cycle–Dependent Differentiation Dynamics Balances Growth and Endocrine Differentiation in the Pancreas. PLoS Biol. 2015; 13(3). doi: 10.1371/journal.pbio.1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yu Z, Lin J, Xiao Y, Han J, Zhang X, Jia H, Tang Y, Li Y. Induction of cell-cycle arrest by all-trans retinoic acid in mouse embryonic palatal mesenchymal (MEPM) cells. Toxicol Sci Off J Soc Toxicol. 2005; 83(2):349-354. doi: 10.1093/toxsci/kfi030. [DOI] [PubMed] [Google Scholar]
- [52].Kopan R, Fuchs E. The use of retinoic acid to probe the relation between hyperproliferation-associated keratins and cell proliferation in normal and malignant epidermal cells. J Cell Biol. 1989; 109(1):295-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.