

 Unstrained C–C activation via putative radical cation formation promotes a directed radical fluorination event using Selectfluor, catalytic 9-fluorenone, and light.
Unstrained C–C activation via putative radical cation formation promotes a directed radical fluorination event using Selectfluor, catalytic 9-fluorenone, and light.
Abstract
Expanding the repertoire of controlled radical fluorination techniques, we present a photosensitized unstrained C–C bond activation/directed monofluorination method using Selectfluor and 9-fluorenone. The reaction is amenable to the opening of multiple 1-acetal-2-aryl substituted rings to yield ω-fluoro carboxylic acids, esters, alcohols, and ketones with relative ease. Initial mechanistic insight suggests radical ion intermediates.
The foundation of organic chemistry lies on C–C bond formation; it is the spirit of total synthesis, the valued ability to create intricate molecules from simple, cheap starting materials. Alternatively, selective C–C bond cleavage (C–C bond activation, in modern parlance) is more seldom discussed, as it is ostensibly a difficult undertaking and its importance is less immediately intuitive to students of organic chemistry. Yet, the field of C–C bond activation1 is beginning to receive a considerable amount of attention in the contemporary world as chemists are finding unique opportunities to construct complex molecules via C–C fragmentation that are not easily accessible by other means. For instance, our laboratory,2 among others,3 has recently become interested in using cyclopropane ring-opening chemistry to, in turn, achieve site-selective fluorination. In any event, it would seem that the application of this chemistry to larger ring systems (5, 6, etc.) that are more readily accessible, but experience relatively little angle strain, is more ambitious. However, the ability to use substituted cyclopentane and cyclohexane rings (or perhaps larger rings) as synthons en route to more complex molecules would prove handy and fundamentally interesting to synthetic chemists, especially with respect to fluorine chemistry. Given the mechanistic insight provided by our laboratory and others on the fluorination of alkyl radicals vis-à-vis C–H activation4 (or other approaches5), we postulated that site-selective fluorination via unstrained C–C fragmentation might be achieved through the photochemical generation of radical cations with the appropriate substituents.
In fact, there is literature precedent (albeit with limited examples) for selective formation of 1,5- and 1,6-radical cations from 5- and 6-membered rings, respectively, employing a putative one-electron photoxidant and proper placement of either acetal or methoxy moieties on the rings.6 The concept is simple – if the substrate undergoes a one-electron oxidation, the die is cast and the C–C bond productively elongates to form a stable radical (e.g., benzylic or tertiary) and resonance stabilized cation. Both the Albini7 and Perrott8 laboratories have shown photosensitized opening of rings followed by hydrogenation. Considering these precedents, we imagined that fluorination of a radical cation should be possible instead, thus allowing highly selective sp3 C–F formation (Scheme 1).
Scheme 1. Concept for selective C–C fragmentation/C–F formation.

Our initial screen for reaction conditions began with 6-phenyl-1,4-dioxaspiro[4.5]decane. We examined a variety of putative photoxidants in MeCN (300 nm light provided by a Rayonet reactor) in the presence of Selectfluor. Upon assessing the viability of such conventional photoxidants as 1,4-dicyanobenzene,8 1,2,4,5-tetracyanobenzene,9 xanthone,10 anthraquinone,11 acetophenone,12 and 9-fluorenone,13 we quickly found the most success with 2.2 equiv. of Selectfluor and 0.2 equiv. of 9-fluorenone in producing the desired ring-opened, fluorinated product by 19F NMR (Table 1). It is important to point out that similar conditions used by our laboratory in the C–C cleavage/fluorination of cyclopropane compounds resulted in a significantly lower yield when applied to this unstrained substrate (Table 1, Entry 2). Remarkably, the 19F NMR yield was comparable when a 14 Watt compact fluorescent light bulb was used as the light source instead, denoting the use of visible light as a more accessible alternative. Also note that none of the desired fluorinated product formed in the absence of light or 9-fluorenone, and heating the substrate with Selectfluor in MeCN to 100 °C only resulted in minor α-fluoro ether products (Scheme 2).14 Upon aqueous workup, we found that the resultant ethylene glycol ester was susceptible to varying degrees of oxidation in the presence of Selectfluor and thus proved difficult to isolate. Consequently, we altered the workup procedure to conduct a mild saponification with 5.0 equiv. aq. LiOH,15 in order to form the more easily isolable (and more synthetically useful) carboxylic acid 1 in 60% yield (Scheme 3).16
Table 1. Screening for photoxidants.
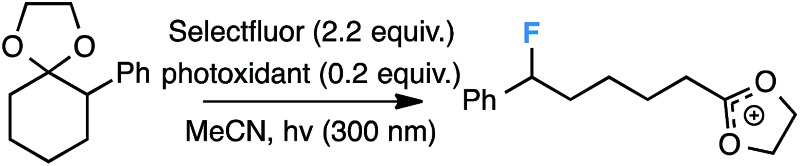
| ||
| Entry | Photoxidant | 19F NMR Yield (%) |
| 1 | — | 0 |
| 2 | 1,2,4,5-Tetracyanobenzene | 30 |
| 3 | 1,4-Dicyanobenzene | 31 |
| 4 | Acetophenone | 28 |
| 5 | Anthraquinone | 23 |
| 6 | Xanthone | 33 |
| 7 | 9–Fluorenone | 60 |
Scheme 2. Control reaction.

Scheme 3. Standard reaction conditions.

We then surveyed a variety of substrates to probe the stereoelectronic dependence on each substituent (Table 2). Interestingly, there was no evidence of C–C fragmentation/fluorination of 1,4-dioxaspiro[4.5]decane or 6-methyl-1,4-dioxaspiro[4.5]decane, suggesting that the aryl group may be essential to stabilize a putative radical cation enough for productive cleavage. In support of this claim, the extent of C–C bond elongation calculated for each radical cation at B3PW91/6-311++G** (MeCN) follows the trend in radical stability (2° benzylic (2.94 Å) ≫ 2° (1.94 Å) > 1° (1.64 Å)).
Table 2. Screening for substituents.
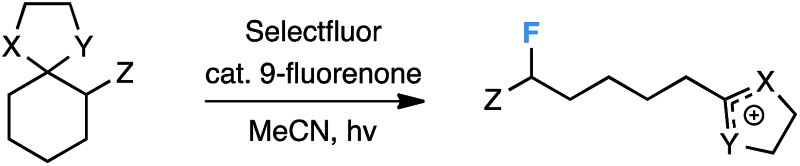
| |||
| X | Y | Z | 19F NMR Yield (%) |
| O | O | H | 0 |
| O | O | Me | 0 |
| O | O | Ph | 60 |
| O | O | OMe | 0 |
| O | O | NPhth | 0 |
| O | O | COOEt | 0 |
| O | NH | Ph | 0 |
| O | NAc | Ph | 50 |
On the other hand, we found that traditional non-aryl resonance stabilizers in the α-position (i.e. –OMe, –NPhth, and –COOEt) proved ineffective for C–C bond fragmentation/fluorination. The corresponding N,O-acetal also failed to produce any of the desired product under reaction conditions unless the nitrogen atom was substituted with an electron-withdrawing group (e.g. an acetyl group). Still, the acylated N,O-acetal performed less well than the O,O-acetal. The ideal substituents for C–C fragmentation/fluorination are therefore an aryl moiety and the easily accessible O,O-acetal.17
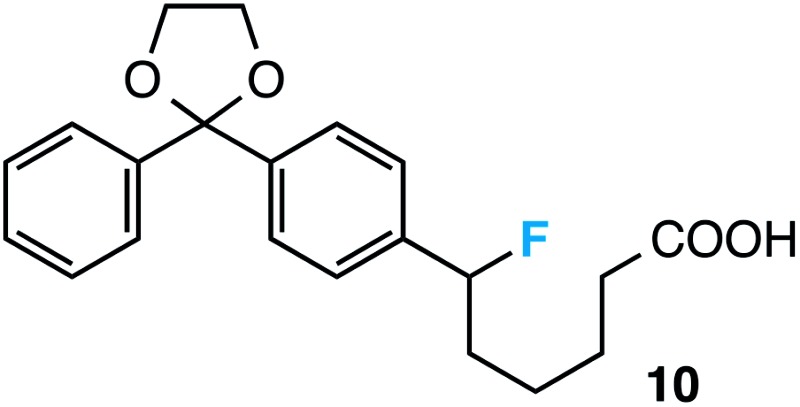
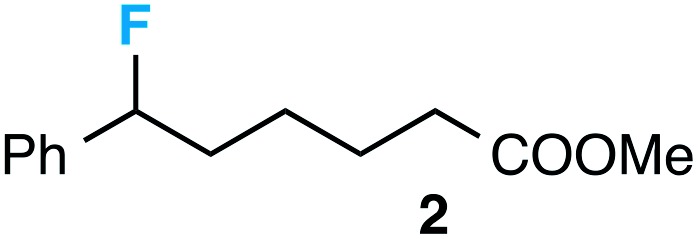
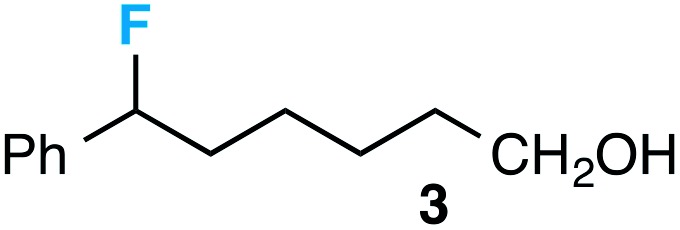
Preceding our evaluation of substrate scope, the success of the aq. LiOH quench for the isolation of the ω-fluoro carboxylic acid from 6-phenyl-1,4-dioxaspiro[4.5]decane also prompted a brief investigation of other quenching reagents as means to isolate molecules with diverse functional groups directly from the same reaction. To our satisfaction, quenching with either 5.0 equiv. aq. LiOMe or 6.0 equiv. LiAlH4 (with the crude reaction mixture redissolved in anhydrous THF) affords the ω-fluoro ester 2 or ω-fluoro alcohol 3 and in comparable yields (Table 3).
Table 3. Substrate scope a .
| Substrate | Product | Yield (%) | Substrate | Product | Yield (%) |
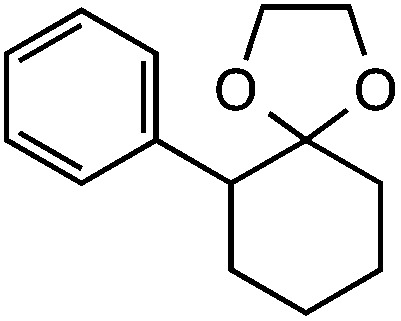
|
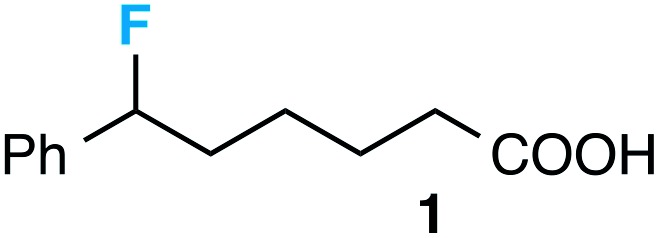
|
60 (52) 54 gram scale |
|
|
51 |
|
59 b | ||||
|
55 c | ||||
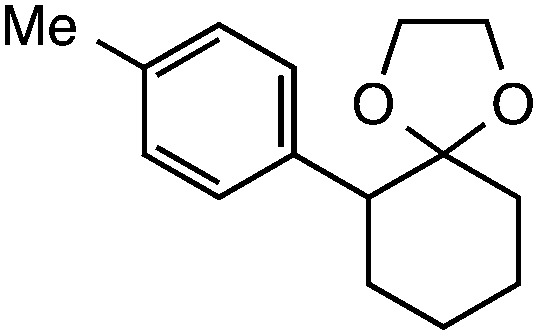
|
|
42 (40) |
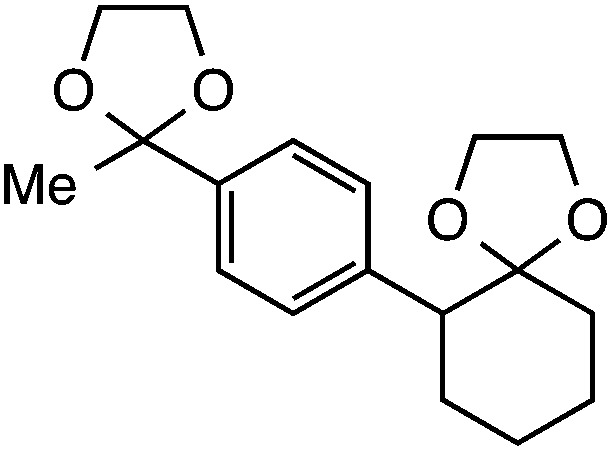
|
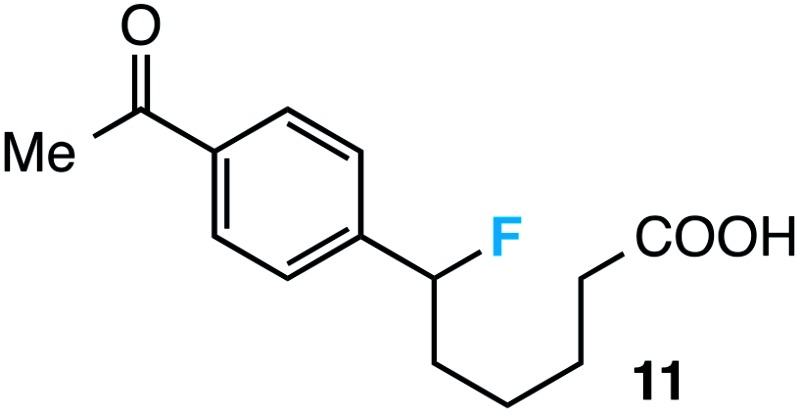
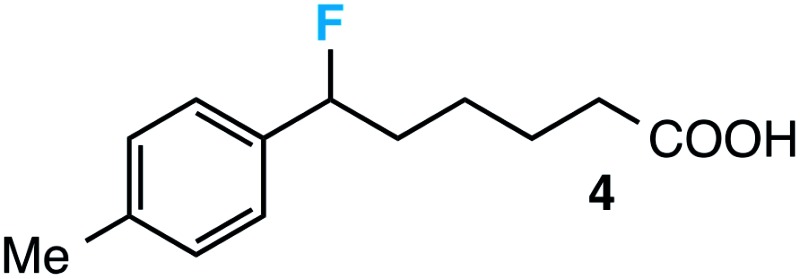
|
28 d |
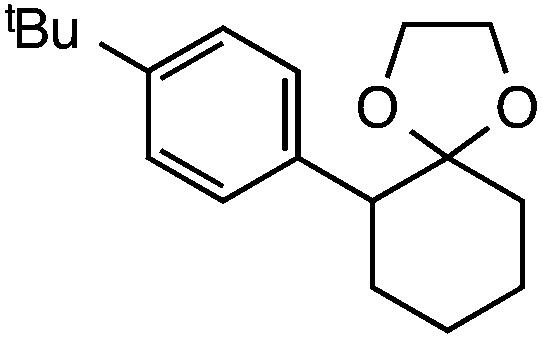
|
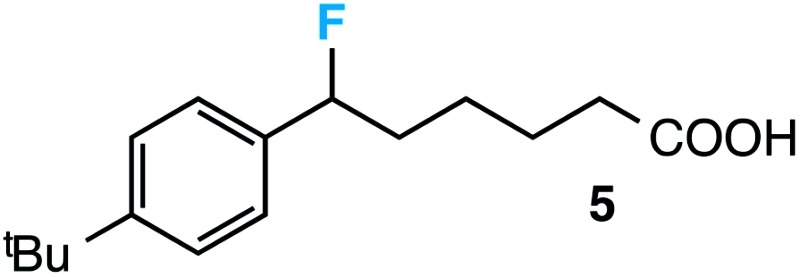
|
64 (58) |
|
|
40 |
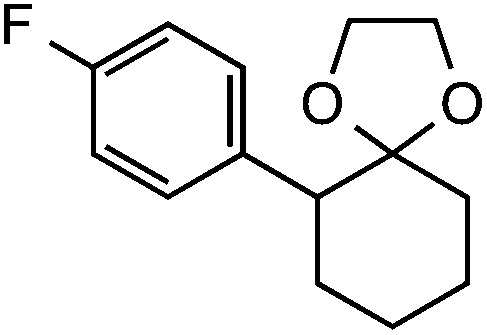
|
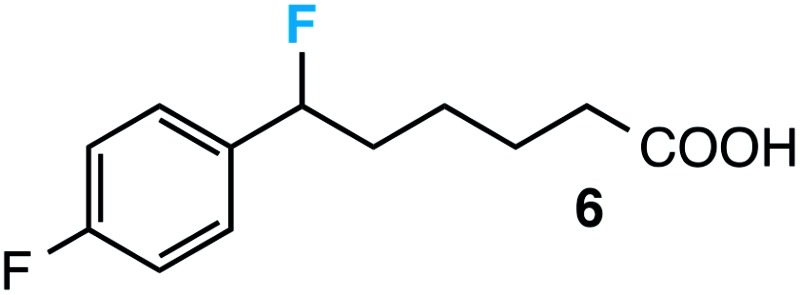
|
70 (68) |
|
|
58 e (60) |
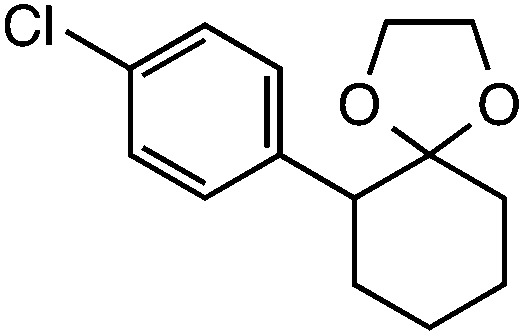
|
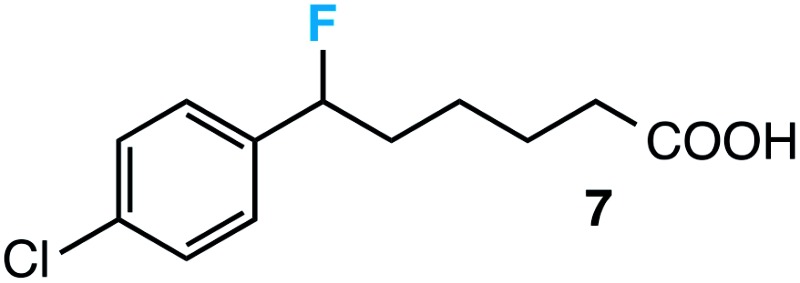
|
54 (49) |
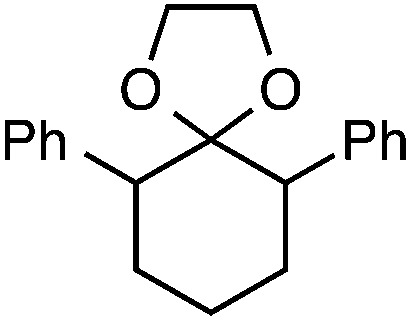
|
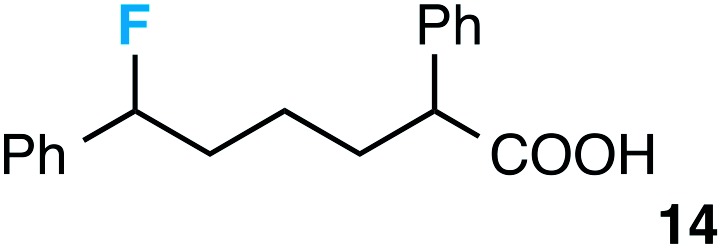
|
56 e |
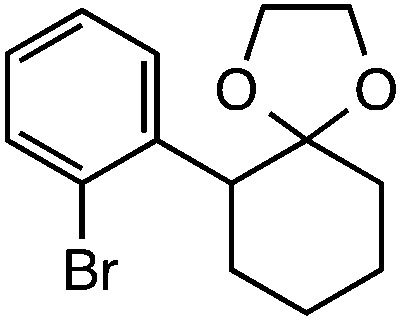
|
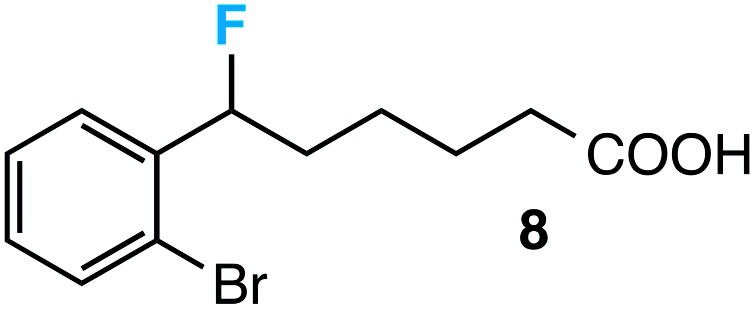
|
63 |
|
|
30 |
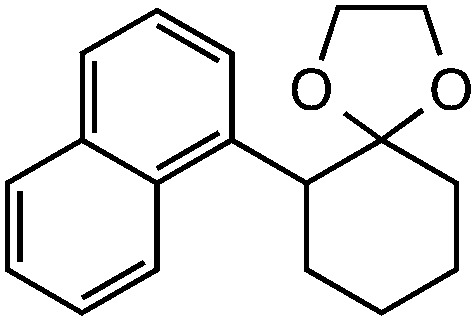
|
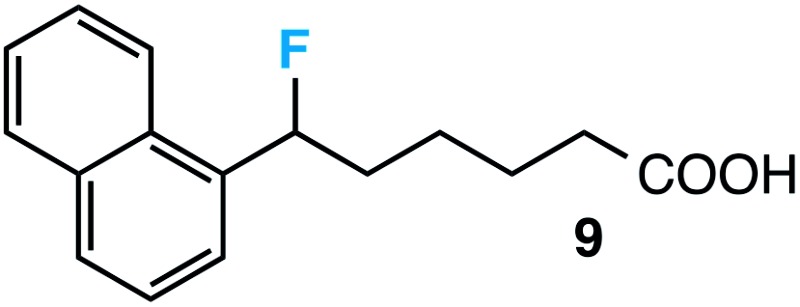
|
47 |
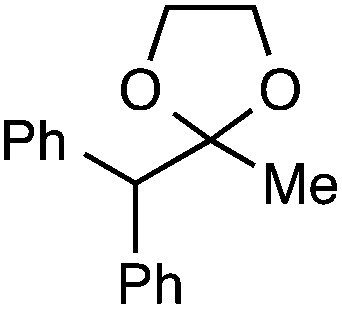
|
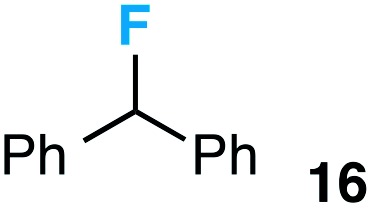
|
28 f |
aUnless otherwise stated, all reactions were conducted using Selectfluor (2.2 equiv.) and 9-fluorenone (0.2 equiv.) in anhydrous MeCN under UV-irradiation (300 nm, Rayonet reactor) and inert N2 atmosphere for 12 h, followed by dilution with H2O and 25 min of stirring with LiOH (5.0 equiv.) to yield carboxylic acids. Isolated yields reported. Yields in parentheses are from reactions using a visible light source (14 Watt CFL).
bQuenched with LiOMe (5.0 equiv.).
cUpon reaction completion, reaction mixture was concentrated, redissolved in anhydrous THF, and stirred with LiAIH4 (6.0 equiv.) for an additional 1 h.
dAcetal removed during workup/isolation.
eIsolated as a 1 : 1 mixture of diastereomers.
f 19F NMR yield.
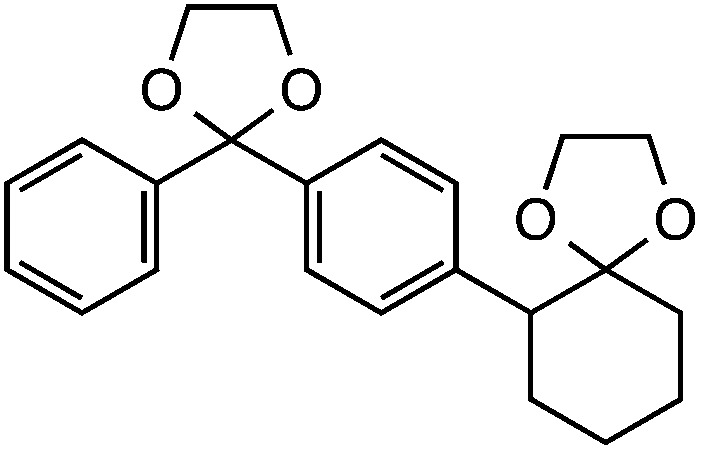
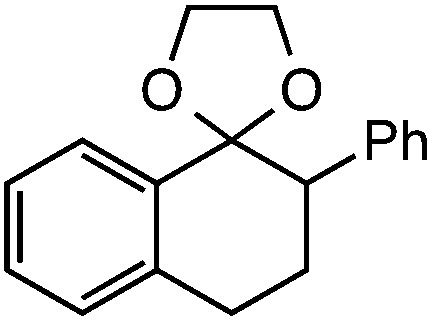
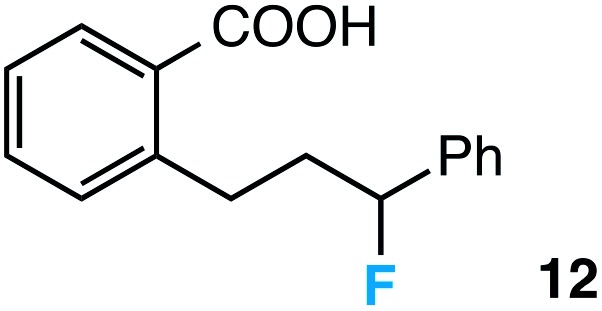
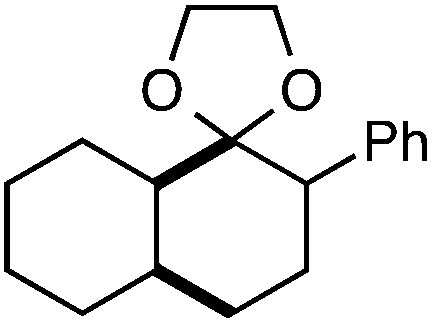
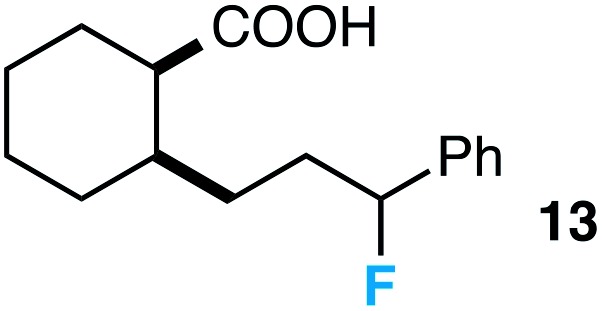
Further assessment of the scope of the reaction revealed several interesting features. For one, conditions are easily amenable to gram scale synthesis with no major sacrifice in yield, as highlighted in Table 3, demonstrating the practicality of this method. In terms of electronic effects, the aryl substituent may be adorned with mildly electron donating (4–5), mildly electron withdrawing (6–8), or electron neutral groups (the extremes typically perform less favorably, e.g. –OMe and –CF3); the system is also tolerant of polyaromatic substituents such as naphthalene (9). Regarding the chemoselectivity of the reaction, only the desired C–C bond fragments in the presence of other aliphatic and aryl-substituted acetal functional groups (10–11). Even more exceptionally, if other secondary benzylic positions are present on the substrate, barely any direct sp3 C–H benzylic fluorination is observed, as C–C fragmentation dominates.18 This is exemplified in the β-phenyl-substituted α-tetralone derivative 12, which, along with the cis-decalin derivative 13, additionally exemplifies the utility of this method in forming complex, substituted rings (e.g. benzene and cyclohexane) from commercially available polycyclic substrates. Lastly, to our knowledge, none of the products we present have been synthesized using direct sp3 C–H benzylic fluorination methods to date.
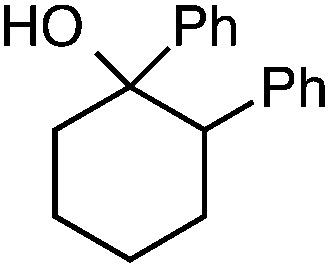
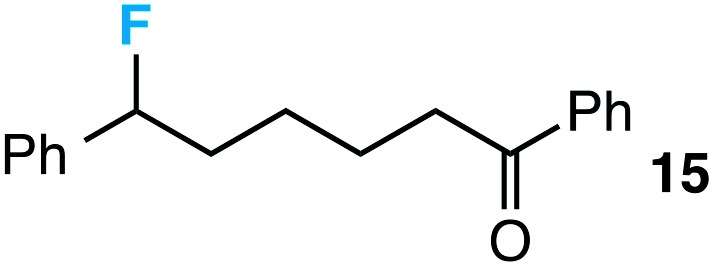
We also provide an example where C–C fragmentation/fluorination can be accomplished from an aryl substituted tertiary alcohol, in lieu of the acetal (15). Although the yield is slightly lower in this instance, it exhibits the ability of this method to access ω-fluoro-ω-aryl ketones in addition to ω-fluoro-ω-aryl carboxylic acids, esters, and alcohols. On another note, the acetal functional group can act as an unconventional “leaving group” concomitant with fluorine installation (16), if the desired reaction does not call for the opening of a ring.
Although our initial efforts focused primarily on cleavage of ever-pervasive 6-membered rings, we subsequently examined the application to both smaller and larger rings (Table 4). Both 5- and 7-membered rings (also common in natural products) underwent ring-opening/fluorination with very similar efficacy to the 6-membered rings vide supra (17 and 18). Remarkably, the reaction also proved amenable to 8- and 12-membered rings (19 and 20). Conceivably, the accessibility of a variety of linear ω-fluoro-ω-aryl carbonyl derivatives as a function of initial ring size using this method may prove particularly useful in the synthesis of fatty acid derivatives (possibly of pharmaceutical or cosmetic interest).19 In our experience, C–C cleavage/fluorination of the larger rings may also offer a distinct advantage over existing sp3 C–H benzylic fluorination methods, as these substrates are not prone to competitive sp3 C–H fluorination along the chain under our specified conditions.
Table 4. Application to 5-, 7-, 8-, and 12-membered rings a .
| Entry | Substrate | Product | Yield (%) |
| 1 |
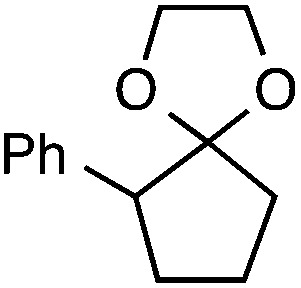
|
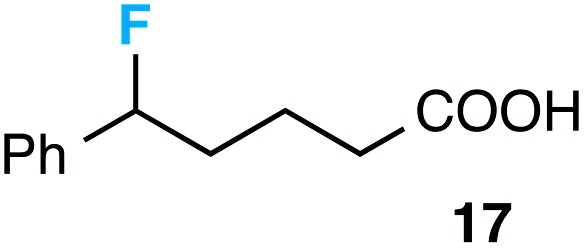
|
58 (62) |
| 2 |
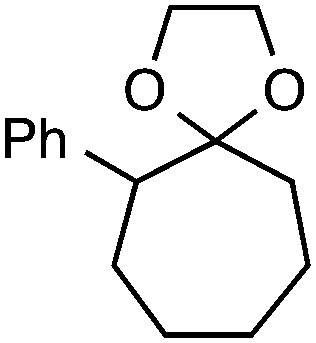
|
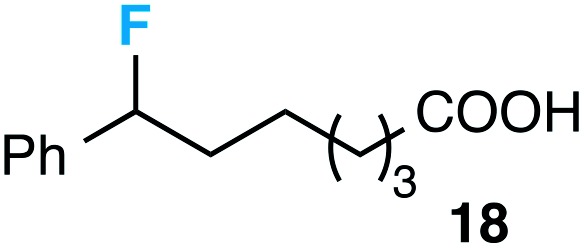
|
57 (53) |
| 3 |
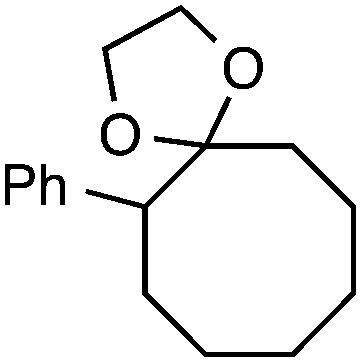
|
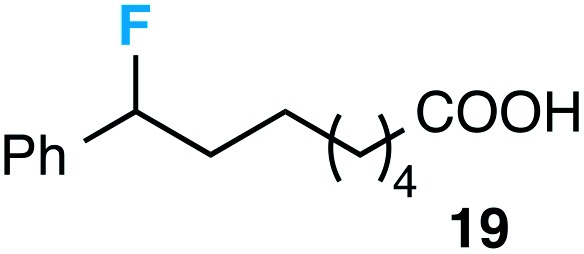
|
46 |
| 4 |
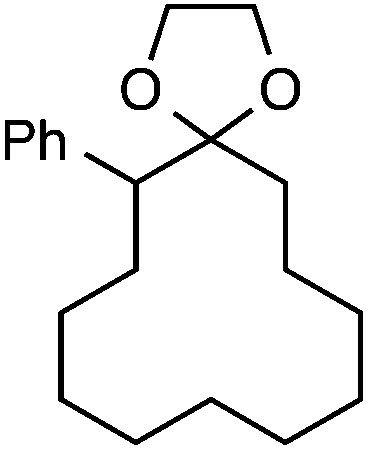
|
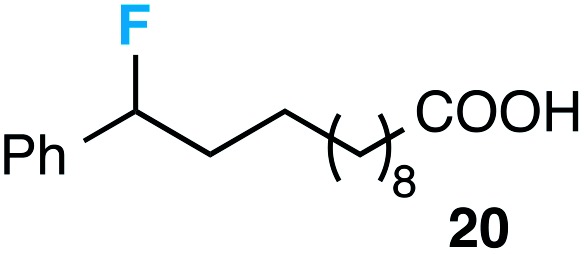
|
30 |
aIsolated yields reported. Yields in parentheses are from reactions using a visible light source (14 Watt CFL).
Finally, as a preliminary mechanistic probe, we conducted a few competition experiments to obtain relative rate data, as the effect of structural modifications on reaction rate should provide information about reactive intermediates,20 viz. the postulated radical cation. In support of our hypothesis, the competition experiments (Scheme 4) resulted in a larger ratio of the product substituted with a mildly electron donating group (tBu) relative to the electron neutral product, as well as smaller relative ratios of the products substituted with electron withdrawing groups (Cl and CF3). In the most extreme case, hardly any CF3-substituted product was formed in the presence of the electron neutral species. This suggests a better ability of electron donating groups to stabilize an electron deficient intermediate.21 To further investigate the substituent effects on the postulated radical cation, we calculated a series of isodesmic equations.22 In each case, we consistently found a more stable radical cation with the more electron rich substitution at B3PW91/6-311++G** (Scheme 5).23
Scheme 4. Intermolecular competition experiments.

Scheme 5. Isodesmic analysis at B3PW91/6-311++G**(MeCN).

Conclusions
All in all, this photosensitized C–C bond cleavage reaction provides a mild, unique opportunity for the monofluorination of complex substrates, effortlessly opening classically stable rings in the presence of light. While initial mechanistic studies support the idea of a radical cation intermediate (based on substituent effects and DFT calculations), extensive studies are currently underway to provide a full Hammett analysis, KIE studies, computations, and spectroscopic data.
Acknowledgments
T. L. would like to thank the NSF (CHE 1152996) for support.
Footnotes
References
- (a) Marek I., Masarwa A., Delaye P.-O., Leibeling M. Angew. Chem., Int. Ed. 2015;54:414–429. doi: 10.1002/anie.201405067. [DOI] [PubMed] [Google Scholar]; (b) Drahl M. A., Manpadi M., Williams L. J. Angew. Chem., Int. Ed. 2013;52:11222–11251. doi: 10.1002/anie.201209833. [DOI] [PubMed] [Google Scholar]; (c) Schneider T. F., Kaschel J., Werz D. B. Angew. Chem., Int. Ed. 2014;53:5504–5523. doi: 10.1002/anie.201309886. [DOI] [PubMed] [Google Scholar]
- Bloom S., Bume D. D., Pitts C. R., Lectka T. Chem.–Eur. J. 2015;21:8060–8063. doi: 10.1002/chem.201501081. [DOI] [PubMed] [Google Scholar]
- (a) Zhao H., Fan X., Yu J., Zhu C. J. Am. Chem. Soc. 2015;137:3490–3493. doi: 10.1021/jacs.5b00939. [DOI] [PubMed] [Google Scholar]; (b) Ren S., Feng C., Loh T.-P. Org. Biomol. Chem. 2015;13:5105–5109. doi: 10.1039/c5ob00632e. [DOI] [PubMed] [Google Scholar]
- (a) Bloom S., Pitts C. R., Miller D., Haselton N., Holl M. G., Urheim E., Lectka T. Angew. Chem., Int. Ed. 2012;51:10580–10583. doi: 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]; (b) Bloom S., Pitts C. R., Woltornist R., Griswold A., Holl M. G., Lectka T. Org. Lett. 2013;15:1722–1724. doi: 10.1021/ol400424s. [DOI] [PubMed] [Google Scholar]; (c) Bloom S., Sharber S. A., Holl M. G., Knippel J. L., Lectka T. J. Org. Chem. 2013;78:11082–11086. doi: 10.1021/jo401796g. [DOI] [PubMed] [Google Scholar]; (d) Pitts C. R., Bloom S., Woltornist R., Auvenshine D. J., Ryzhkov L. R., Siegler M. A., Lectka T. J. Am. Chem. Soc. 2014;136:9780–9791. doi: 10.1021/ja505136j. [DOI] [PubMed] [Google Scholar]; (e) Pitts C. R., Ling B., Woltornist R., Liu R., Lectka T. J. Org. Chem. 2014;79:8895–8899. doi: 10.1021/jo501520e. [DOI] [PubMed] [Google Scholar]; (f) Liu W., Huang X., Cheng M., Nielson R. J., Goddard III W. A., Groves J. T. Science. 2012;337:1322. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]; (g) Liu W., Groves J. T. Angew. Chem., Int. Ed. 2013;52:6024–6027. doi: 10.1002/anie.201301097. [DOI] [PubMed] [Google Scholar]; (h) Amaoka Y., Nagamoto M., Inoue M. Org. Lett. 2013;15:2160–2163. doi: 10.1021/ol4006757. [DOI] [PubMed] [Google Scholar]; (i) Braun M.-G., Doyle A. J. Am. Chem. Soc. 2013;135:12990–12993. doi: 10.1021/ja407223g. [DOI] [PubMed] [Google Scholar]; (j) Xia J.-B., Ma Y., Chen C. Org. Chem. Front. 2014;1:468–472. doi: 10.1039/C4QO00057A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Rueda-Becerril M., Mahe O., Drouin M., Majewski M. B., West J. G., Wolf M. O., Sammis G. M., Paquin J.-F. J. Am. Chem. Soc. 2014;136:2637–2641. doi: 10.1021/ja412083f. [DOI] [PubMed] [Google Scholar]; (l) Halperin S. D., Fan H., Chang S., Martin R. E., Britton R. Angew. Chem., Int. Ed. 2014;53:4690–4693. doi: 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]; (m) Li Z., Song L., Li C. J. Am. Chem. Soc. 2013;135:4640–4643. doi: 10.1021/ja400124t. [DOI] [PubMed] [Google Scholar]
- (a) Rueda-Becerril M., Sazepin C. C., Leung J. C. T., Okbinoglu T., Kennepohl P., Paquin J.-F., Sammis G. M. J. Am. Chem. Soc. 2012;134:4026–4029. doi: 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]; (b) Yin F., Wang Z., Li Z., Li C. J. Am. Chem. Soc. 2012;134:10401–10404. doi: 10.1021/ja3048255. [DOI] [PubMed] [Google Scholar]; (c) Li Z., Wang Z., Zhu L., Tan X., Li C. J. Am. Chem. Soc. 2014;136:16439–16443. doi: 10.1021/ja509548z. [DOI] [PubMed] [Google Scholar]; (d) Phae-nok S., Soorukram D., Kuhakarn C., Reutrakul V., Pohmakotr M. Eur. J. Org. Chem. 2015;2015:2879–2888. doi: 10.1039/c5ob01574j. [DOI] [PubMed] [Google Scholar]; (e) Ventre S., Petronijevic F. P., MacMillan D. W. C. J. Am. Chem. Soc. 2015;137:5654–5657. doi: 10.1021/jacs.5b02244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Patel N. R., Flowers II R. A. J. Org. Chem. 2015;80:5834–5841. doi: 10.1021/acs.joc.5b00826. [DOI] [PubMed] [Google Scholar]
- For a review, see: Albini A., Mella M., Freccero M., Tetrahedron, 1994, 50 , 575 –607 . [Google Scholar]
- Mella M., Fasani E., Albini A. J. Org. Chem. 1992;57:3051–3057. [Google Scholar]
- Arnold D. R., Lamont L. J., Perrott A. L. Can. J. Chem. 1991;69:225–233. [Google Scholar]
- (a) Bloom S., Knippel J. L., Lectka T. Chem. Sci. 2014;5:1175–1178. [Google Scholar]; (b) Bloom S., McCann M., Lectka T. Org. Lett. 2014;16:6338–6341. doi: 10.1021/ol503094m. [DOI] [PubMed] [Google Scholar]
- Cantillo D., de Frutos O., Rincon J. A., Mateos C., Kappe O. C. J. Org. Chem. 2014;79:8486–8490. doi: 10.1021/jo5016757. [DOI] [PubMed] [Google Scholar]
- Kee C. W., Chin K. F., Wong M. W., Tan C.-H. Chem. Commun. 2014;50:8211–8214. doi: 10.1039/c4cc01848f. [DOI] [PubMed] [Google Scholar]
- Xia J.-B., Zhu C., Chen C. Chem. Commun. 2014;50:11701–11704. doi: 10.1039/c4cc05650g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J.-B., Zhu C., Chen C. J. Am. Chem. Soc. 2013;135:17494–17500. doi: 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers R. D., Okazoe T., Sanford G., Thomas E., Trmcic J. J. Fluorine Chem. 2010;131:933–936. [Google Scholar]
- Evans D. A. Aldrichimica Acta. 1982;15:23–32. [Google Scholar]
- In this instance, and with most other products in Table 3, the starting material was completely consumed by 1H NMR analysis of the crude reaction mixture. The remainder of the mass was accounted for in a small amount of unidentified ring-opened byproducts (some detected by 19F NMR) and/or was lost during workup/isolation
- Note that the penultimate 2-aryl ketone is straightforward to obtain either directly through a variety of Pd-catalyzed α-arylation reactions or through epoxide opening via arylmagnesium halides, followed by oxidation de choix. See ESI for details
- This is particularly exceptional given the plethora of direct sp3 C-H photosensitized benzylic fluorination reactions under similar conditions. See ref. 9a and 10–13 for some examples. On the other hand, minor direct benzylic fluorination at the primary position of compound 4 (post C-C bond cleavage/fluorination) was observed in the crude 19F NMR (∼10% yield)
- Alvarez A. M. R., Rodríguez M. L. G. Grasas Aceites. 2000;51:74–96. [Google Scholar]
- Anslyn E. V. and Dougherty D. A., Modern Physical Organic Chemistry, University Science Books, Sausalito, CA, 2006. [Google Scholar]
- Hammett L. P. J. Am. Chem. Soc. 1937;59:96–103. [Google Scholar]
- Wheeler S. E., Houk K. N., Schleyer P. v. R., Allen W. D., J. Am. Chem. Soc., 2009, 131 , 2547 –2560 , , and cited references therein . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew J. P., Chevary J. A., Vosko S. H., Jackson K. A., Pederson M. R., Singh D. J., Fiolhais C. Phys. Rev. B: Condens. Matter Mater. Phys. 1992;46:6671–6687. doi: 10.1103/physrevb.46.6671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.