

Abstract
Vallisneria spinulosa is a freshwater aquatic plant of ecological and economic importance. However, there is limited cytogenetic and genomics information on Vallisneria. In this study, we measured the nuclear DNA content of Vallisneria spinulosa by flow cytometry, performed a de novo assembly, and annotated repetitive sequences by using a combination of next-generation sequencing (NGS) and bioinformatics tools. The genome size of Vallisneria spinulosa is approximately 3,595 Mbp, in which nearly 60% of the genome consists of repetitive sequences. The majority of the repetitive sequences are LTR-retrotransposons comprising 43% of the genome. Although the amount of sequencing data used in this study was not sufficient for a whole-genome assembly, it could generate an overview of representative elements in the genome. These results will lay a new foundation for further studies on various species that belong to the Vallisneria genus.
Keywords: Genome size, C value, Repetitive sequences, Vallisneria spinulosa, RepeatExplorer
Introduction
Vallisneria, commonly called eelgrass, is a genus of freshwater aquatic plant. This genus consists of over 12 species worldwide and is widely distributed in tropical and subtropical regions of Asia, Africa, Europe, and North America (Les et al., 2008). Vallisneria spinulosa is of interest because of its importance in biodiversity and is of major human concern. The species has great impact on fisheries, wildlife, water resources, etc. (Baron et al., 2002). It usually occurs sympatrically in the middle to lower reaches of the Yangtze River in China (Wang et al., 2010) and is thought to be endemic to China (Xie, Deng & Wang, 2007). This species can provide food for waterfowl, nursery habitats for fish, and a substrate for invertebrates and may have a strong influence on water quality (Wang et al., 2010). Because of its ecological and economic importance, the interest of study V. spinulosa has raised greatly. Population genetic analysis revealed that V. spinulosa maintained high levels of genetic variation within populations and low subdivision among populations in ten lakes separated by approximately 900 km in the middle-lower reaches of the Yangtze River (Chen, Xu & Huang, 2007). Microsatellite primers were developed for studies of population genetic structure in the Vallisneria genus (Wang et al. , 2011). In other experiments, adaptive mechanisms in V. spinulosa operated via growth strategies and physiological responses in evading or adapting to Pb stress in a heterogeneously contaminated patch so that they could be chosen as suitable species in ecological restorations of heterogeneously contaminated habitats (Yan et al., 2006). However, the genetic and genomic knowledge of this genome is still limited. As an example, genome size of Vallisneria spinulosa is still unknown, whereas only the number of chromosomes was reported (Liang, 1991).
Recent advances in next-generation sequencing (NGS) technology and improvements in assembly strategies have resulted in the construction of complete genome sequences for more than 100 model and non-model plant species (Michael & VanBuren, 2015). Although the importance of aquatic plants has been noted because of their ability to yield a large amount of biomass without competing for agricultural land, relatively few aquatic plants have been subjected to genome sequencing projects. Aquatic plants which produce considerable amounts of biomass without competing with the agricultural land have drawn increasing attention. However, at present, relatively few aquatic plants except duckweeds (Wang et al., 2014; Van Hoeck et al., 2015) have been incorporated in sequencing projects exploring genome dynamics and the species’ roles in evolution and speciation. Undoubtedly, the eventual Vallisneria spinulosa genome sequence will serve as a reference genome for other species in the genus of Vallisneria. However, a complete genome sequencing project is still challenging with limited sequencing data. Apart from coding sequences that generally make up just a small fraction of the genome, repetitive sequences can account for more than 90% of a genome. De novo assembly and annotation of these repetitive sequences can be achieved effectively at reasonable costs by combining low-pass NGS (reviewed by Kelly & Leitch, 2011) and a series of software programs and pipelines (Novák, Neumann & Macas, 2010; Zytnicki, Akhunov & Quesneville, 2014).
Here we report the genome size, and compositions and fractions of various repetitive sequences in Vallisneria spinulosa genome. Our results lay a foundation for further researches on Vallisneria spinulosa. It will be also useful for genome studies of other species of the Vallisneria genus.
Material and Methods
Material
Vallisneria spinulosa was collected from E119°09.186′, N33°09.855′, Baima Lake, Jiangsu Province, China. Seedlings were grown in the green house until enough leaves were collected.
C-value measurement by flow cytometry
For nuclear DNA content determinations, flow cytometric analysis was performed as described in Li & Arumugnathan (2000). The seeds of Pisum sativum (Cultivar Ctirad) were kindly provided by Prof. Ing. Doležel. Olomouc, Czech Republic. The C-value of Pisum sativum was used as a standard. Briefly, young Vallisneria spinulosa and Pisum sativum leaf tissues (approximately 30 mg of each sample) were hand-scraped on ice with a sharp razor blade in 1.5 ml of Tris-MgCl2 buffer (Pfosser et al., 1995). The nuclear suspension was filtered through a 30-µm mesh size nylon cloth into a labeled tube. Following filtration, the supernatant was centrifuged at 3,000 rpm at 4 °C for 1.5 min, and nuclei were resuspended in 450 µl of Tris-MgCl2 buffer. In this step, 50 µl of RNase A (50 µg/ml) was added to prevent the staining of double-stranded RNA. After resuspension, the suspension was stained with 5 µl of propidium iodide (PI) and was incubated in the dark at 37 °C for 15 min.
Nuclei were analyzed using FACSVerse™ flow cytometer (BD Biosciences, San Jose, CA, USA) with an excitation wavelength of 488 nm. Four independent samples were measured three times each. The nuclear DNA contents of each sample were calculated using the flowing formula:
NGS
Herbarium vouchers of Vallisneria spinulosa were prepared and deposited in the cytogenetic lab of Huai’an Research Centre, Institute of Hydrobiology, Chinese Academy of Sciences, China. Genomic DNA was extracted using a DNeasy plant mini kit from Qiagen, Valencia, CA, USA. Sequencing library was prepared using NEBNext® Ultra™DNALibrary Prep Kit Illumina (New England, Biolabs, Ipswich, MA, USA). Paired-end sequencing (2X150 bp, 350–400 bp insert size) of total genomic DNA was performed by Novogene (Tianjin, China) on the Illumina HiSeq 2500 platform on a single lane. Clean sequencing data were supplied in FASTQ format without adapters. The raw data has been deposited in NCBI SRA database (accession number: SRR6038670).
Data analysis
The RepeatExplorer pipeline (Novak et al., 2013) (http://repeatexplorer.org/) was used to cluster next-generation sequencing reads into groups of similar reads and to assemble contigs from these reads. As shown in Fig. 1, a subset of Illumina paired-end reads from Vallisneria spinulosa were preprocessed, randomly selected and clustered into repeat families using RepeatExplorer (Novák, Neumann & Macas, 2010; Novak et al., 2013) with default setting. The minimum overlap length for clustering is 55 bp, and the minimal overlap for assembly is 40 bp. Repeat clusters with genome proportions of no less than 0.01% were detail annotated. Repeat clusters with known protein domains can be classified by RepeatExplorer pipeline directly. Other clusters were subjected to analysis with similarity searches against GenBank databases (Nt and Nr) using Blastn and Blastx (Altschul et al., 1990) with E-value at 1e−5 manually. The consensus DNA sequences of chromovirus were classified using reverse transcriptase (RT) domain (Macas, Neumann & Navrátilová, 2007). Protein sequences of RT cores were downloaded from Gypsy Database (GyDB) (Llorens et al., 2011) and used as custom database for BLAST. The RT cores of Vallisneria spinulosa were achieved by BLASTx using consensus sequences of clusters as query (E-value at 1e−5). Alignment of RTs was carried out with CLustalX (Thompson et al., 1997) and the phylogenetic trees were calculated in Geneious (version 5.5.6) using neighbor-joining method.
Figure 1. Workflow for repeat analysis in this study.

Raw data from next-generation sequencing were uploaded to the Galaxy-based RepeatExplorer platform. The FASTQ QC: READ QC tool was then used to verify the quality of the reads before removing unnecessary sequences (i.e., adapter sequences) from the ends of each read using the FASTQ Trimmer tool. The QC analysis was then repeated, and the FASTQ to FASTA converter tool was used to convert each read into FASTA format. Using these DNA sequence reads as input, sequences undergo clustering, during which an “all-to-all” sequence comparison is performed, and similar sequences are grouped together into clusters.
Results and Discussion
C-value measurement in Vallisneria spinulosa
Vallisneria spinulosa Yan (Hydrocharitaceae) is a submerged macrophyte, which is also an endemic and dominant species in the Yangtze River Basin in China. There has been no DNA content of the members of genus Vallisneria recorded until the present study (last accessed: 2017.08.30, http://data.kew.org/cvalues/). In this study, the nuclear DNA content of Vallisneria spinulosa was measured by flow cytometry. Fluorescence histograms representing genome size and the internal standards used are shown in Fig. 2. The haploid genome size value (1C) in Vallisneria spinulosa is 3.68 pg, which equals 3,595 Mbp (1 pg = 978 Mbp (Dolezel et al., 2003)). The genome size is in the range of intermediate genome sizes (3.51–13.99 pg) (Soltis et al., 2003). Compared to the other aquatic plant species with known genome size (Wang, Kerstetter & Michael, 2011), the genome size of Vallisneria spinulosa is at least twice larger than the duckweeds (Wang, Kerstetter & Michael, 2011). However, more genome size data is needed to compare the genome evolution and intraspecific variation in the Vallisneria genus.
Figure 2. Fluorescence histograms of the genome size assessments in Vallisneria spinulosa by flow cytometry using propidium iodide.
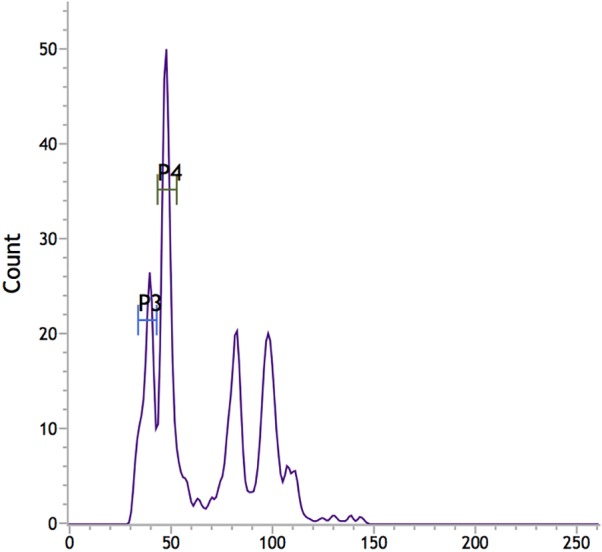
P3, Vallisneria spinulosa; P4, Pisum sativum.
Graph-based sequence clustering and genome repeat composition analysis of Vallisneria spinulosa genome
The genome sizes of wetland plants are usually large (Hidalgo et al., 2015), making it difficult to analyze the genome using traditional molecular methods. Thus, we employed the latest next generation sequencing technology and a series of bioinformatics tools to reveal the genome structure of Vallisneria spinulosa. The recent advent of NGS methods has, for the first time, analyzed in details for any genome a possibility with reasonable costs. Although Vallisneria spinulosa is a non-model plant of great interest, there is no whole-genome sequencing project for Vallisneria proposed.
Table 1 shows the major cytogenetic and genomic parameters of Vallisneria spinulosa. Our Illumina sequencing returned 35,196,639 150-bp paired-end reads (70,393,278 reads in total) with more than 10 Gbp of raw DNA sequences. These sequence results constitute an essential genome resource for further study of this species in the future.
Table 1. Cytogenetics, genomic and sequencing features of Vallisneria spinulosa.
| Species | Vallisneria spinulosa |
|---|---|
| Chromosome number (2n) | 20 |
| 1C value (pg) | 3.68 |
| Genome size (Mbp) | 3,595 |
| Sequenced read number | 70,393,278 |
| Genome coverage (%) of reads analyzed | 18 |
| Repetitive DNA cluster numbers (≥0.01% genome proportion) | 184 |
One of the aims of this study was to annotate the repetitive sequences of Vallisneria spinulosa to investigate the structure of its genome. RepeatExplorer (Novák, Neumann & Macas, 2010), a graph-based clustering approach to identify repetitive sequences, was employed to analyze the repetitive sequences in Vallisneria spinulosa genome.
As shown in Fig. 1, more than 70 million paired end reads from next-generation sequencing were uploaded to the Galaxy-based RepeatExplorer platform (Novak et al., 2013). The FastQC tool was then used to verify the quality of the reads (i.e., adapter sequences, etc.). Illumina sequencing can cause bias in the beginning and end of reads (Hansen, Brenner & Dudoit, 2010). Therefore, we trimmed the first 9 bp of each read based on the FastQC results. The FASTQ to FASTA converter tool was then used to convert each read into FASTA format. RepeatExplorer can predict the repeat composition from a typical 2–5% of genome coverage sequencing data (Novak et al., 2013). Therefore, 2,100,000 DNA sequence reads were selected randomly as input, which was equal to 8.2% of the predicted genome (141 bp × 2,100,000/3,595 Mbp × 100% of the genome). All paired sequences underwent clustering, during which an “all-to-all” sequence comparison was performed, and similar sequences were grouped together into clusters. The genome proportion of each cluster was calculated as the percentage of reads. In our study, the RepeatExplorer results in 1,241,356 reads in 77,554 clusters and nearly 60% of the genome were determined to be repetitive sequences (Fig. 3), in which there were 184 separated clusters with genome proportions of no less than 0.01% each. The top 184 clusters in total represented approximately 48% of the genome (Fig. 3 and Table 2). The graphical figures and three most abundant consensus sequences of each cluster could be found in Fig. S1 and Data S1 separately. Based on the analyzed data, the frequency of singletons should represent the low copy fraction of the genome, which resulted in approximately 31% of genome in Vallisneria spinulosa.
Figure 3. Repeat composition of clusters generated in RepeatExplorer (similarity-based partitioning) of 2.1 M reads.

x-axis: cumulative proportion of clusters of the genome. y-axis: numbers of reads. The color of the bars shows the types of repetitive elements.
Table 2. Repeat composition of the Vallisneria spinulosa genome estimated from the Illumina sequencing data.
| Repeat type | Lineage | Genome proportion (%) |
|---|---|---|
| Retroelements | ||
| Ty-3/Gypsy | ||
| Chromovirus | 31.582 | |
| Tat/Ogre | 0 | |
| Athila | 0 | |
| Unclassified | 0.020 | |
| Ty-1/Copia | ||
| Maximus | 6.427 | |
| Angela | 0 | |
| Bianca | 0 | |
| Tork | 1.905 | |
| Ivana/Oryco | 0 | |
| AleII | 0.928 | |
| TAR | 0.185 | |
| AleI/Retrofit | 1.812 | |
| Unclassified | 0.161 | |
| LINE | 0.460 | |
| SINE | 0 | |
| Other | 0.023 | |
| DNA transposons | 0.514 | |
| Satellite repeats | 0.013 | |
| rDNA | 0.351 | |
| Unclassified | 0.977 | |
| Chloroplast | 2.704 | |
| Total | 48.062 |
Characterization of the LTR-retrotransposons of Vallisneria spinulosa
The top 184 clusters were further characterized and annotated by searching the sequence-similarity of the assembled contigs against GenBank using Blastn and Blastx (Altschul et al., 1990). No coding gene was found except ribosomal RNA gene and plastid genes among top 184 clusters. The genome proportions of each type of repetitive sequence in Vallisneria spinulosa are shown and detailed in Table 2. The majority of the repeats are LTR-retrotransposons, representing in 43% of the genome. Concerning the two main superfamilies of LTR-retrotransposons, Gypsy-related contigs are more represented than Copia ones in this species. Gypsy elements belong to three main lineages, while Copia ones belong to seven lineages (Wicker et al., 2007). The greatest majority of repeats divided by lineage is the Chromovirus, in which repeats comprise greater than 31.5% of the genome proportion. Furthermore, we compared Vallisneria spinulosa Chromoviruses with known plant clades (Llorens et al., 2011) (Fig. S2). All the Chromovirus in Vallisneria spinulosa are from Tekay (Del) clade. It has been reported that Tekay is the most abundant Chromovirus in Orobanche (Piednoël, Carrete-Vega & Renner, 2013) and banana (Hribová et al., 2010). But in Rumex acetosa, CRM clade is the more abundant than rest of Chromovirus (Steflova et al., 2013). The most abundant Ty-1/Copia element is Maximus, which accounts for 6.4% of the genome proportion. The genome consists of 0.46% LINEs, 0.514% DNA transposons, 0.013% Satellite repeats and 0.351% rDNA. Several comparative analysis of repeats from different species revealed that there are quantitative differences and sequence variations detected for classified repeat families (Novák et al., 2014; Kelly et al., 2015; Macas et al., 2015). But it is still possible that some differences in repeat abundance estimates can be also attributed to incompleteness of assembly and biased composition of sequences in the genome assembly (Novák et al., 2014). In addition, 2.7% chloroplast DNA was found. It has been reported that chloroplast DNA could be found integrated into the nuclear genome (Roark et al., 2010). However, the significantly high proportion of chloroplast DNA suggests that it might have come from the DNA extraction process. Since it was reported B chromosomes in plant are enriched with chloroplast and mitochondria DNA (Klemme et al., 2013). FISH using plastid DNA as a probe could be performed to test either possibility. Many elements in the repeat lineages were not found by our analysis, such as Tat/Ogra, Athila, Angela, Bianca and Ivana/Oryco, as we only annotated repeats with genome proportions greater than 0.01% of the genome. Therefore, these elements are likely to be present in the genome, but with minor proportions of each. Moreover, approximately 1% of the genome is unclassified to any of major lineages according to the RepeatExplorer analysis. More extensive sequencing efforts will be required to conclusively annotate these repeats. Additionally, we compared repeat composition of Vallisneria spinulosa with other well-studied monocot plant genomes (Table 3). It has been reported that in small-size (<1 Gb) plant genomes, there is a linear dependency between genome size and LTR retrotransposon content (Table 3) (Wang et al., 2014). The genome size of Vallisneria spinulosa is almost five times as big as that of Sorghum bicolor. But the proportion of retroelements in Vallisneria spinulosa is less than that of Sorghum bicolor. Vallisneria spinulosa appear to contain the highest proportion of Gypsy elements compared to other plant genomes (Table 3). But the Gypsy/copia ratio is not as high as Sorghum bicolor (Table 3). The proportion of other transposons in Vallisneria spinulosa is much lower than that of the rest analyzed plant genomes. It is possible that most of the transposons are not present as high copy number in the genome of Vallisneria spinulosa. They might be in the repeat catalogue that contains the repetitive sequences with the grequency of less than 0.01% of the genome. Differences in TE content, especially the LTR retrotransposons make up the difference in genome size variation in angiosperms. For example, repeat content explains 94.5% of the genome size difference between Spirodela polyrhiza (2n = 40; 158 Mbp) and Lemna minor genome (2n = 40; 481 Mbp). But our clustering data only explained approximately 60% of the genome in Vallisneria spinulosa (Fig. 3). There are considerable amounts of sequences are neither repetitive sequence detected by RepeatExplorer nor genes. Because genomic repeat abundances contain phylogenetic signatures (Dodsworth et al., 2015), the dynamics of LTR retrotransposons and their contributions to genome evolution could be used to analyze the speciation of Vallisneria in the future once more sequence information is available for this genus. Since Vallisneria spinulosa is a member of the order Alismatales, which are basal monocots, genome sequence of Vallisneria spinulosa can also contribute to the understanding of the genome evolution of monocots as well.
Table 3. Repeat composition of Vallisneria spinulosa compared with other monocot plant genomes.
| Genome size | Vallisneria spinulosa 3,595 Mbp | Sorghum bicolor 730 Mbp | Lemna minor 481 Mbp | Oryza sativa 430 Mbp | Brachypodium distachyon 355 Mbp | Spirodela polyrhiza 158 Mbp | |
|---|---|---|---|---|---|---|---|
| Retroelements (%) | 43.50 | 54.50 | 31.20 | 32.10 | 23.30 | 13.06 | |
| LTR | 43.02 | 54.47 | 29.57 | 30.85 | 21.39 | 13.06 | |
| Gypsy | 31.60 | 19.00 | 10.59 | 9.06 | 13.46 | 6.06 | |
| Copia | 11.42 | 5.18 | 18.79 | 3.32 | 5.13 | 1.72 | |
| Gypsy/copia ratio | 2.77 | 3.67 | 0.56 | 2.73 | 2.62 | 3.50 | |
| non-LTR | 0.46 | 0.06 | 1.62 | 1.24 | 1.94 | n.a. | |
| Transposon (%) | 0.51 | 7.50 | 5.08 | 10.10 | 4.80 | n.a. |
Notes.
Values are represented as percentage of genome.
Annotation of a typical Ty1/copia TAR repeat
The position of the reverse transcriptase (RT) gene in relation to the integrase (INT) gene of pol was used to classify the retrotransposon families into Ty1-copia (PROT-INT-RT) and Ty3-gypsy (PROT-RT-INT) (Wicker et al., 2007). Figure 4A shows the plotted graph of VpCL96, in which each dot represents a sequence read and each line represents similarity hits between dots. This shows a Ty1/copia TAR repeats with complete typical copia protein domains, including GAG, PROT, INT, RT and RH in order. The LTRs at both ends have high similarity as shown in a circle graph (Fig. 4A). It has been reported that 52 copia families from Triticeae, rice, and Arabidopsis could be classified into six ancient lineages (Bianca, TAR, Angela, Ale, Ivana, and Maximus). While many of the contigs from RepeatExplorer clusters are truncated without having the protein domains. CL96 is intact, with a dominant contig that is shown as a wider bar in Fig. 4B. Additionally, the length of the contig is greater than 5,000 bp (Fig. 4B).
Figure 4. Annotation of a TAR element VsCL96.

(A) Graphical 2D projection of the structure of VsCL96, a Ty1/copia TAR repeat with complete protein domains including GAG, PROT, INT, RT and RH in order. Each dot represents a sequence read and each line represents similarity hits between dots. The protein domains are highlighted in different colors. (B) Read depth of each contig within CL96. x-axial: read depth, y-axial: contig length.
Our database of repetitive sequences can be a useful resource for further investigation of localization and visualization of these sequences in the chromosomes of Vallisneria spinulosa chromosomes (Becher et al., 2014).
Conclusion
Although the amount of sequencing data of Vallisneria spinulosa used in this study was not sufficient for whole-genome assembly, it still enabled us to generate an overview of representative elements in the genome. We also measured the genome size of this aquatic plant. The genome size and the genomic data described here will become a valuable resource for further studies of Vallisneria spinulosa and other species of the genus.
Supplemental Information
Acknowledgments
We thank Dr. Yan Wang (Institute of Hydrobiology, Chinese Academy of Sciences) for her assistant in flow cytometry. We also thank two anonymous reviewers for the helpful comments.
Funding Statement
This work was supported by the project “Technical support for the maintenance and monitoring of the key aquatic communities in Yanlong Lake” (YCCG1507-81). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests. Ruijuan Feng is an employee of Jiangsu Tianshen Co., Ltd, China.
Author Contributions
RuiJuan Feng conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
Xin Wang performed the experiments, contributed reagents/materials/analysis tools, reviewed drafts of the paper.
Min Tao and Guanchao Du performed the experiments, analyzed the data, reviewed drafts of the paper.
Qishuo Wang conceived and designed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
DNA Deposition
The following information was supplied regarding the deposition of DNA sequences:
NCBI SRA: SRR6038670.
Data Availability
The following information was supplied regarding data availability:
Wang, Qishuo (2017): F_H33NLALXX_L3_1.clean.fq.gz. figshare. https://dx.doi.org/10.6084/m9.figshare.5387008.v1.
Wang, Qishuo (2017): F_H33NLALXX_L3_2.clean.fq.gz. figshare. https://dx.doi.org/10.6084/m9.figshare.5387041.v1.
References
- Altschul et al. (1990).Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baron et al. (2002).Baron JS, LeRoy Poff N, Angermeier PL, Dahm CN, Gleick PH, Hairston NG, Jackson RB, Johnston CA, Richter BD, Steinman AD. Meeting ecological and societal needs for freshwater. Ecological Applications. 2002;12:1247–1260. doi: 10.1890/1051-0761(2002)012[1247:MEASNF]2.0.CO;2. [DOI] [Google Scholar]
- Becher et al. (2014).Becher H, Ma L, Kelly LJ, Kovarik A, Leitch IJ, Leitch AR. Endogenous pararetrovirus sequences associated with 24 nt small RNAs at the centromeres of Fritillaria imperialis L. (Liliaceae), a species with a giant genome. The Plant Journal. 2014;80:823–833. doi: 10.1111/tpj.12673. [DOI] [PubMed] [Google Scholar]
- Chen, Xu & Huang (2007).Chen L, Xu L, Huang H. Genetic diversity and population structure in Vallisneria spinulosa (Hydrocharitaceae) Aquatic Botany. 2007;86:46–52. doi: 10.1016/j.aquabot.2006.09.001. [DOI] [Google Scholar]
- Dodsworth et al. (2015).Dodsworth S, Chase MW, Kelly LJ, Leitch IJ, Macas J, Novák P, Piednoël M, Weiss-Schneeweiss H, Leitch AR. Genomic repeat abundances contain phylogenetic signal. Systematic Biology. 2015;64:112–126. doi: 10.1093/sysbio/syu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolezel et al. (2003).Dolezel J, Bartos J, Voglmayr H, Greilhuber J. Nuclear DNA content and genome size of trout and human. Cytometry. Part A: The Journal of the International Society for Analytical Cytology. 2003;51:127–128. doi: 10.1002/cyto.a.10013. author reply 129. [DOI] [PubMed] [Google Scholar]
- Hansen, Brenner & Dudoit (2010).Hansen KD, Brenner SE, Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Research. 2010;38:e131–e131. doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo et al. (2015).Hidalgo O, Garcia S, Garnatje T, Mumbrú M, Patterson A, Vigo J, Vallès J. Genome size in aquatic and wetland plants: fitting with the large genome constraint hypothesis with a few relevant exceptions. Plant Systematics and Evolution. 2015;301:1927–1936. doi: 10.1007/s00606-015-1205-2. [DOI] [Google Scholar]
- Hribová et al. (2010).Hribová E, Neumann P, Matsumoto T, Roux N, Macas J, Dolezel J. Repetitive part of the banana (Musa acuminata) genome investigated by low-depth 454 sequencing. BMC Plant Biology. 2010;10:204. doi: 10.1186/1471-2229-10-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly & Leitch (2011).Kelly LJ, Leitch IJ. Exploring giant plant genomes with next-generation sequencing technology. Chromosome Research. 2011;19:939–953. doi: 10.1007/s10577-011-9246-z. [DOI] [PubMed] [Google Scholar]
- Kelly et al. (2015).Kelly LJ, Renny-Byfield S, Pellicer J, Macas J, Novák P, Neumann P, Lysak MA, Day PD, Berger M, Fay MF, Nichols RA, Leitch AR, Leitch IJ. Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. New Phytologist. 2015;208:596–607. doi: 10.1111/nph.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemme et al. (2013).Klemme S, Banaei-Moghaddam AM, Macas J, Wicker T, Novák P, Houben A. High-copy sequences reveal distinct evolution of the rye B chromosome. The New Phytologist. 2013;199:550–558. doi: 10.1111/nph.12289. [DOI] [PubMed] [Google Scholar]
- Les et al. (2008).Les DH, Jacobs SWL, Tippery NP, Chen L, Moody ML, Wilstermann-Hildebr M. Systematics of Vallisneria (Hydrocharitaceae) Systematic Botany. 2008;33:49–65. doi: 10.1600/036364408783887483. [DOI] [Google Scholar]
- Li & Arumugnathan (2000).Li L, Arumugnathan K. High recovery of large molecular weight DNA from sorted maize chromosomes. Plant Molecular Biology Reporter. 2000;18:41–45. doi: 10.1007/BF02825292. [DOI] [Google Scholar]
- Liang (1991).Liang W. Chromosome number and karyotype study of Vallisneria asiatica and V. spinulosa. Guihaia. 1991;11:153–156. [Google Scholar]
- Llorens et al. (2011).Llorens C, Futami R, Covelli L, Domínguez-Escribá L, Viu JM, Tamarit D, Aguilar-Rodríguez J, Vicente-Ripolles M, Fuster G, Bernet GP, Maumus F, Munoz-Pomer A, Sempere JM, Latorre A, Moya A. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Research. 2011;39:D70–D74. doi: 10.1093/nar/gkq1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas, Neumann & Navrátilová (2007).Macas J, Neumann P, Navrátilová A. Repetitive DNA in the pea (Pisum sativum L.) genome: comprehensive characterization using 454 sequencing and comparison to soybean and Medicago truncatula. BMC Genomics. 2007;8:427. doi: 10.1186/1471-2164-8-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas et al. (2015).Macas J, Novak P, Pellicer J, Cizkova J, Koblizkova A, Neumann P, Fukova I, Dolezel J, Kelly LJ, Leitch IJ. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe fabeae. PLOS ONE. 2015;10:e0143424. doi: 10.1371/journal.pone.0143424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael & VanBuren (2015).Michael TP, VanBuren R. Progress, challenges and the future of crop genomes. Current Opinion in Plant Biology. 2015;24:71–81. doi: 10.1016/j.pbi.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Novák et al. (2014).Novák P, Hřibová E, Neumann P, Koblížková A, Doležel J, Macas J. Genome-wide analysis of repeat diversity across the family musaceae. PLOS ONE. 2014;9:e98918. doi: 10.1371/journal.pone.0098918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák, Neumann & Macas (2010).Novák P, Neumann P, Macas J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics. 2010;11:378. doi: 10.1186/1471-2105-11-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak et al. (2013).Novak P, Neumann P, Pech J, Steinhaisl J, Macas J. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics. 2013;29:792–793. doi: 10.1093/bioinformatics/btt054. [DOI] [PubMed] [Google Scholar]
- Pfosser et al. (1995).Pfosser A, Amon A, Lelley T, Heberle-Bors E. Evaluation of sensitivity of flow cytometry in detecting aneuploidy in wheat using disomic and ditelosomic wheat-rye addition lines. Cytometry. 1995;21:387–393. doi: 10.1002/cyto.990210412. [DOI] [PubMed] [Google Scholar]
- Piednoël, Carrete-Vega & Renner (2013).Piednoël M, Carrete-Vega G, Renner SS. Characterization of the LTR retrotransposon repertoire of a plant clade of six diploid and one tetraploid species. Plant Journal. 2013;75:699–709. doi: 10.1111/tpj.12233. [DOI] [PubMed] [Google Scholar]
- Roark et al. (2010).Roark LM, Hui AY, Donnelly L, Birchler JA, Newton KJ. Recent and frequent insertions of chloroplast DNA into maize nuclear chromosomes. Cytogenetic and Genome Research. 2010;129:17–23. doi: 10.1159/000312724. [DOI] [PubMed] [Google Scholar]
- Soltis et al. (2003).Soltis DE, Soltis PS, Bennett MD, Leitch IJ. Evolution of genome size in the angiosperms. American Journal of Botany. 2003;90:1596–1603. doi: 10.3732/ajb.90.11.1596. [DOI] [PubMed] [Google Scholar]
- Steflova et al. (2013).Steflova P, Tokan V, Vogel I, Lexa M, Macas J, Novak P, Hobza R, Vyskot B, Kejnovsky E. Contrasting patterns of transposable element and satellite distribution on sex chromosomes (XY1Y2) in the dioecious plant Rumex acetosa. Genome Biology and Evolution. 2013;5:769–782. doi: 10.1093/gbe/evt049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson et al. (1997).Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoeck et al. (2015).Van Hoeck A, Horemans N, Monsieurs P, Cao HX, Vandenhove H, Blust R. The first draft genome of the aquatic model plant Lemna minor opens the route for future stress physiology research and biotechnological applications. Biotechnology for Biofuels. 2015;8:188. doi: 10.1186/s13068-015-0381-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al. (2014).Wang W, Haberer G, Gundlach H, Gläßer C, Nussbaumer T, Luo MC, Lomsadze A, Borodovsky M, Kerstetter RA, Shanklin J, Byrant DW, Mockler TC, Appenroth KJ, Grimwood J, Jenkins J, Chow J, Choi C, Adam C, Cao X-H, Fuchs J, Schubert I, Rokhsar D, Schmutz J, Michael TP, Mayer KFX, Messing J. The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nature Communications. 2014;5:1–13. doi: 10.1038/ncomms4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Kerstetter & Michael (2011).Wang W, Kerstetter R, Michael TP. Evolution of genome size in duckweeds (Lemnaceae) Journal of Botany. 2011;2011:1–9. doi: 10.1155/2011/570319. [DOI] [Google Scholar]
- Wang et al. (2011).Wang B, Liao H, Zhao Y, Li W, Song Z. Microsatellite loci in Vallisneria natans (Hydrocharitaceae) and cross-reactivity with V. spinulosa and V. denseserrulata. American Journal of Botany. 2011;98 doi: 10.3732/ajb.1000441. e44–e47. [DOI] [PubMed] [Google Scholar]
- Wang et al. (2010).Wang B, Song Z, Liu G, Lu F, Li W. Comparison of the extent of genetic variation of Vallisneria natans and its sympatric congener V. spinulosa in lakes of the middle-lower reaches of the Yangtze River. Aquatic Botany. 2010;92:233–238. doi: 10.1016/j.aquabot.2009.12.006. [DOI] [Google Scholar]
- Wicker et al. (2007).Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH. A unified classification system for eukaryotic transposable elements. Nature Reviews. Genetics. 2007;8:973–982. doi: 10.1038/nrg2165-c4. [DOI] [PubMed] [Google Scholar]
- Xie, Deng & Wang (2007).Xie Y, Deng W, Wang J. Growth and root distribution of Vallisneria natans in heterogeneous sediment environments. Aquatic Botany. 2007;86:9–13. doi: 10.1016/j.aquabot.2006.08.002. [DOI] [Google Scholar]
- Yan et al. (2006).Yan X, Yu D, Wang H, Wang J. Response of submerged plant (Vallisneria spinulosa) clones to lead stress in the heterogenous soil. Chemosphere. 2006;63:1459–1465. doi: 10.1016/j.chemosphere.2005.09.030. [DOI] [PubMed] [Google Scholar]
- Zytnicki, Akhunov & Quesneville (2014).Zytnicki M, Akhunov E, Quesneville H. Tedna: a transposable element de novo assembler. Bioinformatics. 2014;30:2656–2658. doi: 10.1093/bioinformatics/btu365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The following information was supplied regarding data availability:
Wang, Qishuo (2017): F_H33NLALXX_L3_1.clean.fq.gz. figshare. https://dx.doi.org/10.6084/m9.figshare.5387008.v1.
Wang, Qishuo (2017): F_H33NLALXX_L3_2.clean.fq.gz. figshare. https://dx.doi.org/10.6084/m9.figshare.5387041.v1.