

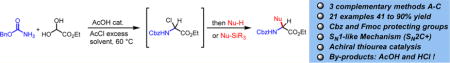
Abstract
An acetyl chloride-mediated cascade transformation involving a primary carbamate, ethyl glyoxylate, and various types of nucleophiles is reported for the synthesis of orthogonally protected α-amino esters. These reactions proceeded rapidly to afford the pivotal α-chloroglycine intermediate in excellent yields, which can be directly functionalized in situ with various types of nucleophiles. A mild and unique AcOH(cat.)/AcCl system was found to promote an autocatalytic-like condensation and facilitate the multicomponent assembly of non-proteinogenic α-amino esters. To better understand this one-pot transformation and the orchestration of the components’ condensations, the investigation of a broader scope of nucleophiles and some kinetic studies are presented. Our findings suggest that the halogenation step toward the formation of α-chloroglycine is the rate-determining step likely proceeding through the formation of N-carbamoyl iminium. Also, the initial kinetic profiling for the nucleophilic substitution supports an SN1-like (SN2C+) mechanism in which nucleophiles add to the iminium–chloride tight ionic pair. These results lead ultimately to the design of a new protocol in which an achiral hydrogen bond donor thiourea catalyst was utilized to enhance the reaction scope and enable silylated nucleophiles to be efficiently exploited to synthesize novel non-proteinogenic α-amino esters.
Graphical abstract
INTRODUCTION
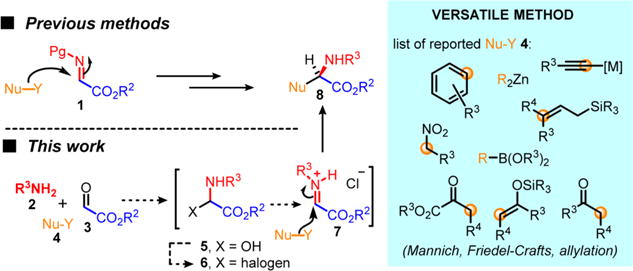
Non-proteinogenic α-amino acids (Xaas) represent an important class of organic compounds. They are prevalent foundational building blocks in many biologically active small molecules, nonribosomal peptides (NRPs) and pharmaceuticals (Figure 1).1 For example, NRPs containing α-aryl glycine units prove to typically have important antibacterial properties (e.g., amoxicillin, ampicillin, vancomycin, teicoplanin, ramoplanin) or anti-HIV properties (e.g., neuroprotectins A and B, feglymycin).2 Due to their extraordinary diversity (structural and functional), non-proteinogenic Xaas are also widely used as chiral building blocks in the synthesis of chiral auxiliaries, organocatalysts, and ligand backbones for metal catalysis, thus imparting them with a crucial role in modern organic synthesis. As a result, scalable and asymmetric synthesis of non-proteinogenic α-amino acids has attracted significant attention in the past decades.3 For example, asymmetric alkylations of a glycine-derived Schiff base4 or azlactone5 and NH-insertion6 of α-diazo esters are well-documented and useful methods for the catalytic asymmetric synthesis of α-amino esters. Imporantly, numerous asymmetric functionalizing methods of α-iminoglycinates 1 have also been reported (Strecker,7 Mannich,8 Petasis,9 hydrogenation,10 or aza-Friedel–Crafts11 reactions), demonstrating the prodigious versatility of this substrate for the synthesis of non-proteinogenic Xaas (Scheme 1). However, the instability of α-iminoglycinates 1, which require cumbersome preparation and some typical protecting groups manipulations, is an inherent limitation, leading overall to multipot operations to produce marketable α-amino esters 8.12 To overcome some of these issues, α-haloglycine esters13 5 and α-amido sulfones14 have been exploited as surrogates of α-iminoglycinates 1 (Scheme 1) in multipot operations. However, only sparse examples of cascade reactivity or multicomponent reactions (MCRs) to synthesize chiral α-amino esters in a single step have been reported.15 The most practical asymmetric MCRs for the synthesis of non-proteinogenic Xaas are certainly the Strecker-3CR (requiring one hydrolysis extra step) and the Petasis-3CR, which can accommodate several types of functionalizations. However, the vast majority of MCRs reported to date, using iminoglycinate 1 as the pivotal intermediate, are achiral and present a narrow scope of nucleophiles.16 Thus, we were drawn to establish a scalable and more versatile one-pot synthesis of α-amino esters, which could also be amenable to enantioselective catalysis17 to prepare Xaas bearing marketable carbamate protecting groups (e.g., N-protecting groups such as benzyloxycarbamate (Cbz) or fluorenylmethyloxycarbamate (Fmoc)).18 We now wish to expand the scope of this work and fully exploit our recently reported autocatalytic-like synthesis of α-chloroglycinate 6a to take advantage of the high reactivity of glycinate iminium 7 (Scheme 2). While α-halogenoglycine synthons have been extensively exploited in the 1980s, no kinetic studies have been previously reported and the mode of reactivity was not well understood.13,19 We hope to demonstrate that this novel method is truly amenable to the practical synthesis of numerous classes of α-amino esters and offers a great opportunity for some chiral variant to be developed.
Figure 1.
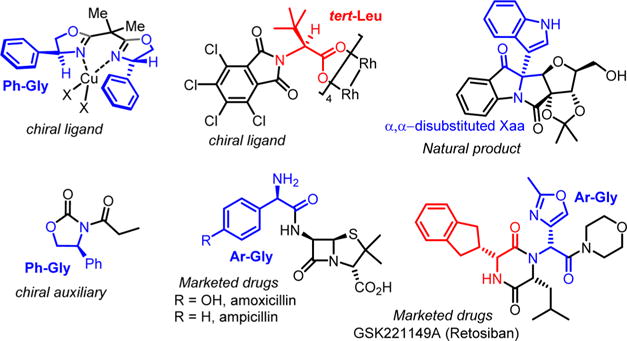
Chemicals derived from non-proteinogenic Xaa structural motifs.
Scheme 1. Multicomponent Assembly of Non-proteinogenic α-Amino Esters 7.
Scheme 2. One-Pot Synthesis of α-Chloroglycine Ester 6aa.
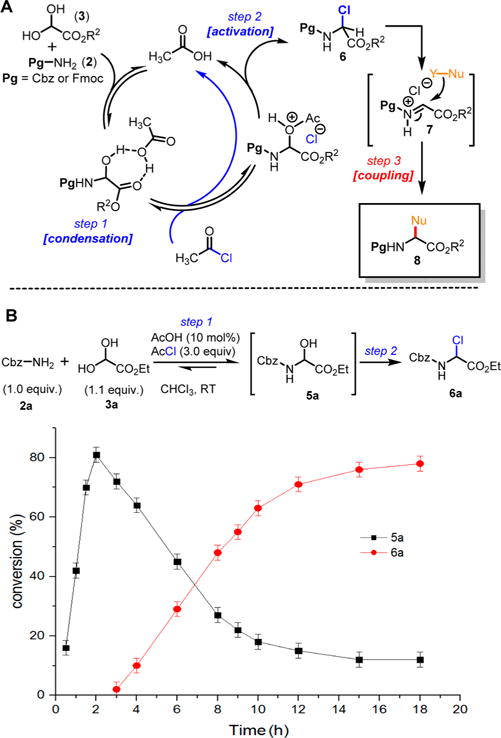
aReaction mixture aliquots were taken during the course of the reaction, and the solvent was evaporated under vacuum. Mesitylene was added as an internal standard, and conversions were calculated from 1H NMR using a quantitative protocol with the appropriate acquisition parameters (see the Experimental Section).
RESULTS AND DISCUSSION
Summary of Previous Results
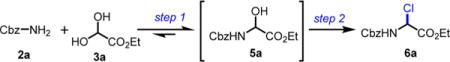
Recently, our group established a suitable system for a practical racemic route to α-aryl α-amino esters bearing carbamate protecting groups.18 We have previously shown that primary carbamates 2, ethyl glyoxylate (hydrate) 3, and arene nucleophiles 4 have been condensed to achieve a stepwise three-component synthesis of numerous α-aryl α-amino esters 8 in a single-pot transformation (Scheme 2A). The reaction advances through a pathway involving three main steps: (1) the condensation between 2 and 3, followed by (2) the activation of the α-hydroxyglycine 5 to α-chloroglycine 6, and the final (3) the nucleophile addition. As proposed in Scheme 2A, a system of AcOH(catalyzed)/AcCl was previously found to assist both steps 1 and 2, while enhancing the overall reaction rate, leading us to propose a new mechanism for this transformation. We have previously suggested that the condensation between 2 and 3 catalyzed by acetic acid (10 mol %) afforded modest conversion to α-hydroxyglycine 5a (72% conversion) due to the reversibility of the initial condensation step.20 On the other end, the catalyzed reaction with acid and excess acetyl chloride proceeded more rapidly to a full conversion in 5a and further afforded the desired α-chloroglycine 6a in quantitative yields (Scheme 2B). To rationalize these results, we propose that the catalytic amount of acid facilitates the condensation between benzyl carbamate 2a and ethyl glyoxylate 3a to rapidly form the first intermediate α-hydroxyglycine 5a (less than 2 h), which further irreversibly evolves by the reaction with acetyl chloride (catalyzed by AcOH and HCl) to deliver α-chloroglycine ester 6a in an autocatalytic-like manner.21 The preliminary kinetic experiments support that the halogenation is the rate-determining step (persistent hemiaminal 5a >80% formation in 2 h) and that acetyl chloride not only promotes the condensation step between 2a and 3a but also traps the water formed during the reaction, producing more acetic acid byproduct, which autocatalyzes and accelerates the overall cascade (slope characteristic of a rate acceleration in the α-chloroglycine 6a formation; Scheme 2B).
Optimization of Reaction Conditions
From an extensive screening of hemiaminal 5a activation (previous publication18) to chloro-, bromo-, or acetoxy-aminals, acetyl chloride was found to be the most efficient reagent in the prospect of a tandem process. During the investigation, we faced the expected problem of full conversion to the desired α-chloroglycine 6a and proposed that the modest yields observed for the condensation between carbamate 2a and ethyl glyoxylate 3a, 62% and 82% yield in chloroform and ethyl acetate, respectively, are due to the reversibility of the condensation (Table 1, entries 1 and 3).20 Upon activation with acetyl chloride, the reactions proceeded to full conversion and further afforded the desired α-chloroglycine 6a with a conversion of 4% and 19%, respectively (Table 1, entries 2 and 4).22 Under strict anhydrous conditions at 60 °C, α-chloroglycine 6a was obtained in a quantitative manner in ethyl acetate and chloroform (Table 1, entries 5 and 6).23 Several other solvents were tested (Table 1, entries 7–9), and α-chloroglycine 6a was also synthesized quantitatively in CH3CN in 24 h. After optimizations, two sets of conditions in CHCl3 and CH3CN were established for the quantitative preparation of α-chloroglycine ester 6a in a single step, which represents an advancement from the previous literature (2 steps).24
Table 1.
Optimizations To Synthesize the Pivotal α-Chloroglycin Ester 6a
| ||||
|---|---|---|---|---|
|
| ||||
| entry | solvent, tempa | activator | time (h) | conversion (%)b |
| 1 | CHCl3, rt | 12 | 5a (62)c | |
| 2 | CHCl3, rt | AcCl | 18 | 5a (76); 6a (4) |
| 3 | CH3CO2Et, rt | 12 | 5a (82)c | |
| 4 | CH3CO2Et, rt | AcCl | 18 | 5a (59); 6a (19) |
| 5 | CH3CO2Et, 60 °C | AcCl | 9 | 6a (100) |
| 6 | CHCl3, 60 °C | AcCl | 12 | 6a (100) |
| 7 | THF, 60 °C | AcCl | 12 | 5a (61); 6a (25)d |
| 8 | toluene, 60 °C | AcCl | 12 | 5a (44); 6a (56) |
| 9 | CH3CN, 60 °C | AcCl | 24 | 6a (100) |
Reactions were performed with 2a (1.0 mmol), 3a (1.3 mmol), acetyl chloride (2.5 mmol), and acetic acid (10 mol %).
Conversions were reported due to the innate instability of chloroaminal 6a, using a quantitative 1H NMR technique21 with mesitylene as an internal standard.
Isolated yields of pure hemiaminal 5a were 60% and 81% for entries 1 and 3, respectively.
Decomposition and possible polymerization were observed.
Scope and Limitation of the Methods A and B
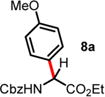
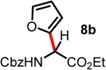
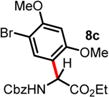
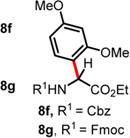
We then turned our attention to the aza-Friedel–Crafts alkylation of the in situ generated α-chloroglycine ester 6a with a series of arene nucleophiles (Table 2). Experiments were conducted with a reduced amount of ethyl glyoxylate 3a (1.05 equiv) to avoid the formation of direct aza-Friedel–Crafts byproducts.25 According to the empirical nucleophilicity scale established by Mayr,26 the reactivity of carbocations can be predicted using the correlation equation log k(20°C) = sN(E + N), in which the strength of the N-carbamoyliminium electrophile 9 would be characterized by the parameter E and the various π-nucleophile arenes tested would be characterized by two parameters N and sN. For each substrate tested in the arylation (Table 2), both solvent conditions were compared, CHCl3 (method A) and CH3CN (method B), and the reactivity parameters were used to rationalize the scope of the iminium arylations depending on solvent polarity. The experiments summarized in Table 2 revealed that the most reactive arenes (electron rich; N > 3.0) reacted in CHCl3 at low temperatures (method A), while less reactive arenes (electron poor; N < 1.5) reacted more cleanly in CH3CN and at higher temperatures (method B). Several phenolic derivatives were selected to compare their innate reactivity (electronic and steric factors), while heteroaromatic substrates also showed a broad scope of reactivity as demonstrated by the range of temperature utilized (entries 1–5). For example, anisole (N = −1.2, CH2Cl2),27 furan (N = 1.4, CH2Cl2),28 anthracene (N = 2.0, CH2Cl2),29 and bromoresorcinol and a chalcone substrate showed a very modest reactivity in CHCl3 as anticipated from the corresponding nucleophilic factor (N); therefore, these substrates were subjected to more efficient reaction conditions in CH3CN (method B). We then observed a complete switch of reactivity for the most electronically rich arenes, for which the mildest reaction conditions in CHCl3 (method A) afford the best results in terms of yield and regioselectivity (entries 6–12). Pyrrole (N = 4.6, CH3CN), N-methylpyrrole (N = 5.8, CH3CN),30 indole (N = 5.6, CH2Cl2), and N-methylindole (N = 5.7, CH2Cl2)31 are extremely powerful π-nucleophiles, which facilitate the synthesis of the corresponding indolyl (8k,l) and pyrrolyl (8i,m) α-amino esters in high yields (62–79% yields) as single regioisomers. Finally, several MCRs were examined using dimethoxybenzene as a nucleophile (N = 2.5, CH2Cl2)32 (entry 6) in CH3CN and CHCl3, and it was found that a particular carbamate protecting group also affects the reaction rate. While the Cbz-protected amino ester 8f was obtained in 9 h (82% yield), the corresponding Fmoc-protected product 8g was produced more slowly over 36 h in 61% yield. Another limitation for both methods A and B is the overreaction of electronically rich arenes (indole and pyrrole), which tend to competently polymerize while electron-deficient heteroarenes (oxazole and thiazole) are unreactive even at elevated temperatures.
Table 2.
Scope for the Synthesis of Racemic α-Aryl α-Amino Esters 8a–la
| Entry | N factorb | Method A — Step 3 — Method B
|
Product | |
|---|---|---|---|---|
| (Time, Temp, yield) | (Time, Temp, yield) | |||
| 1 | −1.18 | 10 d, RT, <10% | 24 h, 60 °C, 48%e (6:1 rr)c,d |
|
| 2 | 1.36 | 14 d, RT, <10% | 17 h, RT, 59%c,e |
|
| 3 | ND | 2 d, 60 °C, 34% (2:1 rr)d |
16 h, RT, 72%e |
|
| 4 | 2.00 | 1.5 d, 60 °C, 74% | 18 h, 60 °C, 82%e |
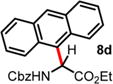
|
| 5 | ND | 48 h, 40 °C, 10% | 96 h, 0 °C, 41%e |
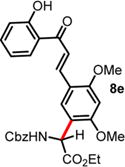
|
| 6 | 2.48 | 9 h, 60 °C, 82%e (5:1 rr)d 36 h, 60 °C, 61% (10:1 rr)d |
2 h, RT, 74% (5:1 rr)d — |
|
| 7 | ND | 1.5 d, 60 °C, 71%e (>20:1 rr)d |
4 h, 60 °C, 74% (2:1 rr)d |
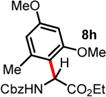
|
| 8 | 4.63 | 0.5 h, −60 °C, 67%e | 10 min, −40 °C, 45% |
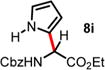
|
| 9f | 5.50 | 24 h, 40 °C, 46%e | 12 h, RT, 66% |
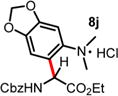
|
| 10 | 5.55 | 2 h, 0 °C, 64%e | 15 min, −40 °C, 42% |
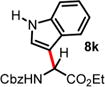
|
| 11 | 5.75 | 3 h, −45 °C, 79%e | 30 min, −40 °C, 56% |
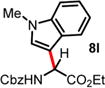
|
| 12 | 5.85 | 1.5 h, 0 °C, 62%e | 15 h, −40 °C, 58% |
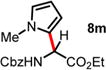
|
Method A: reaction performed in CHCl3, steps 1/2 for 12 h. Method B: reaction performed in CH3CN, steps 1/2 for 24 h. Both methods are followed by the addition of arenes (step 3) for the specified time and temperature. Isolated yields are reported.
Factors of nucleophilicity have been previously reported in CH3CN or CH2Cl2.
An excess of arenes (3.0 equiv) was used, and the isolated yields for 8a and 8b are 60% and 71%, respectively.
Ratio of regioisomers (para/ortho) are reported (rr) based on 1H NMR integration values.
Synthesis of these compounds was previously reported. See ref 18.
The employed nucleophile is aniline hydrochloric salt.
Mechanism Investigation
Altogether, the results shown above demonstrate that the empirical Mayr’s scale of nucleophilicity seems extremely useful to predict the nucleophile reactivity with the N-carbamoyl iminium electrophile 9 (Table 2). Therefore, by using the approximate equation of reactivity that dictates the reaction advancement in which electrophiles and nucleophiles have to fulfill the conditions N + E ≥ −5, we were able to estimate the electrophilic parameter of iminium 9. To this aim, several arene nucleophiles previously reported and characterized by Mayr were tested in the reaction with N-Cbz α-chloroglycine ester 6a (precursor of the proposed N-carbamoyl iminium electrophile 9) at room temperature in CH2Cl2 for 2 days.21,33 Reactions using anisole (N = −1.2, CH2Cl2) and thiofuran (N = −1.0, CH2Cl2) as nucleophiles only faintly proceeded, while the reaction with a furan nucleophile (N = 1.4, CH2Cl2) delivered the corresponding α-amino esters under the same conditions (11% isolated yield).21 Thus, it is reasonable to consider that an electrophilic value E can be estimated for the N-carbamoyl iminium electrophile 9, in a tight ionic pair as E ≈ −5 − 1.4 = −6.4 (Figure 2). In comparison, the most reactive iminium characterized by Mayr to date (N,N-dimethyl-substituted iminium 10; E = −9.3) is likely in the range of 4 orders of magnitude less reactive than the present N-carbamoyl iminium 9. Even though iminiums such as 9 have been previously proposed as the highly reactive intermediate for the formation of C–C bonds,34 no concrete spectroscopic or kinetic evidence has been reported to support this hypothesis. Therefore, we undertook a preliminary kinetic profiling of the aza-Friedel–Crafts reactions to assess the different mechanistic possibilities (Scheme 3). The observation of characteristic spectral features of α-chloroglycine ester 6a, N-methylindole, and product 8l by in situ 1H NMR spectroscopy enabled a kinetic analysis of the reaction progress under synthetically relevant conditions (CDCl3–benzene-d6 solvent mixture at room temperature). The kinetic order of each of the reagents was determined via “different-excess” experiments, in which the initial concentration of N-methylindole was varied, while the concentration of 6a was kept constant. Four distinct experiments were performed and plotted with the N-methylindole concentration varying from 0.75 to 0.3 M to measure the formation of product 8l and determine the kinetic order in α-chloroglycine ester 6a (overlay shown Scheme 3A). As shown in Scheme 3B, the plots of reciprocal concentration in [6a]t versus time exhibit slightly offset straight lines with different slopes, which fit well with straight line regressions. This concludes that the kinetic order in α-chloroglycine 6a for the reaction must be one, while the reaction’s kinetics follow a second-order rate law.21 When a larger excess of N-methylindole nucleophile was used (3 equiv; green stars), while maintaining the concentration of α-chloroglycine [6a] at 0.1 M, the reaction appears to reach a pseudo-first-order condition as shown by the exponential plot of reciprocal concentration in [6a]t versus time (Scheme 3B). More surprisingly, the kinetic plots of reciprocal concentration in [Nu]t versus time also fit some straight line regressions that correlates to the same kinetic order one in N-methylindole (see Supporting Information, Figures SI-3a, SI-7a, SI-10a, SI-14a). Taken together, these results support that the reaction displays first-order dependence on both concentrations of the α-chloroglycine ester electrophile, [6a], and the N-methylindole nucleophile [Nu]. The results are consistent with an SN1-like mechanism (or SN2C+ with a rate = k[6a] × [Nu]), which can be rationalized by suggesting that α-chloroglycine [6a] might undergo heterolysis to the corresponding iminium 9, in a tight ionic pair with the chloride anion, which likely reduces the iminium reactivity, hence the increase of the energy barrier for the nucleophilic attack to occur in the second step. In this scenario, the SN1-like mechanism supports the fact that both steps of heterolysis and the C–C bond formation are rate determining.35 Furthermore, the reaction with 2 equiv of the N-methylindole nucleophile was reproduced with the addition of an external source of the chloride anion to test for a possible ion return to the iminium 9 (tetrabutyl ammonium chloride: 20 mol %). This experiment shown in Figure 3 demonstrates that ion return might be evoked in this reaction, while the overall rate acceleration of the reaction is likely due to a salt effect on the solvent ionic strength.36 The plot of a general second-order expression ln{[Nu][6a]0/[6a][Nu]0} with time for the reaction without an additive fits with the straight line, suggesting that the reaction follows a second-order kinetic (see eq 1).
Figure 2.
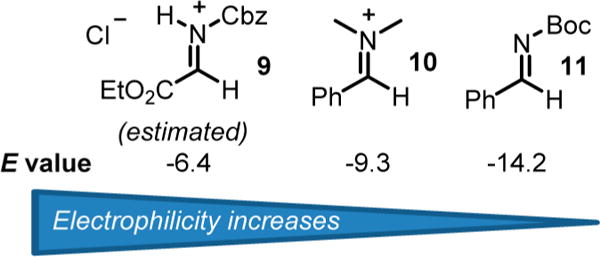
Comparison of iminium and imine electrophilic reactivities E.
Scheme 3. Kinetic Profiling To Assess the Friedel–Crafts Reaction Mechanism.
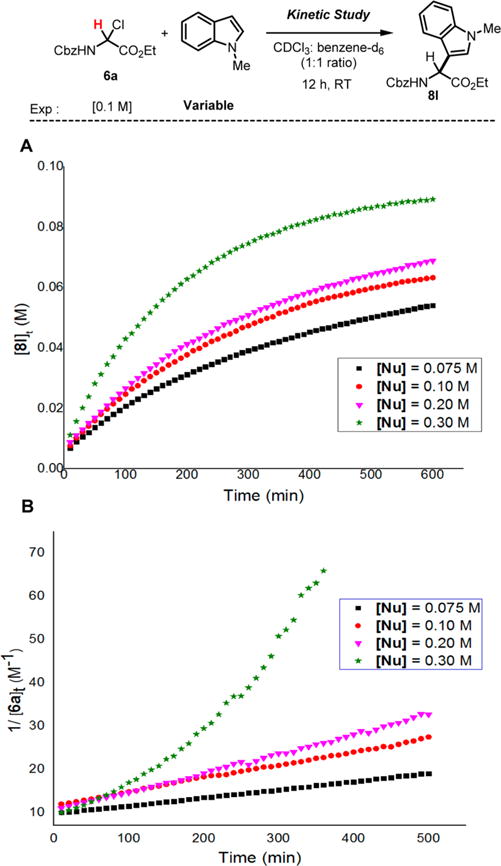
Figure 3.
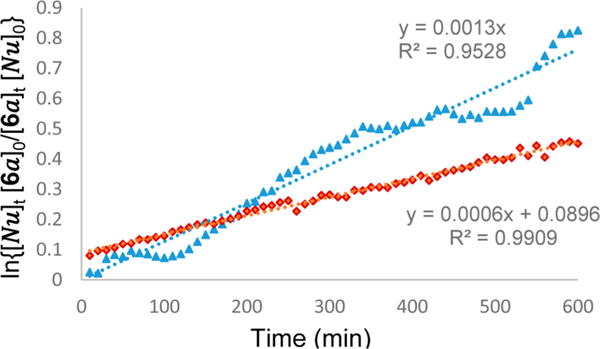
Salt effect on the Friedel–Crafts reaction of α-chloroglycine and N-methylindole to support the reaction mechanism.
A general second-order integrated rate law can be expressed as follows:
| (1) |
where [6a]0 ≠ [Nu]0 and slope = kobs[xs]; where [xs] = ([Nu]0 − [6a]0)
However, the presence of a chloride source caused a significant deviation from the linear regression, which could be attributed to a change in solvent polarity over time (ionic strength leading to increased rate), while a more pronounced equilibrium between the iminium ionic pair 9 and chloroaminal 6a might have simultaneously occurred (SN2C+).
Complementary Method C: Use of an H-Bond Donor Catalyst
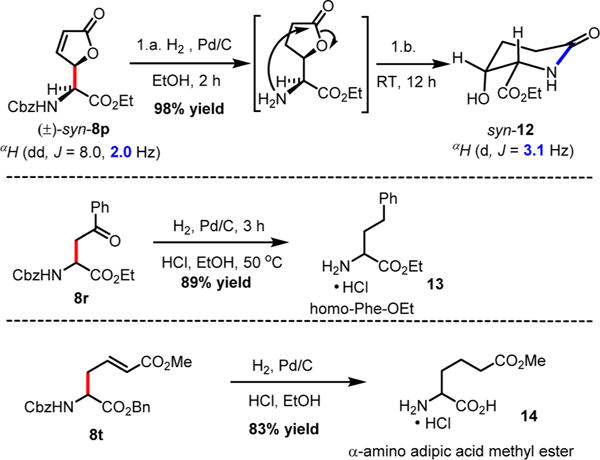
Both methods (A and B), employed for a stepwise multicomponent synthesis of α-aryl α-amino esters 8a–m, were then expanded upon to other types of nonaromatic π-nucleophiles (silylenols and silyl ketene acetals). We rapidly faced the issues of silylated-nucleophile stability due to the enhanced acidity of the reaction media resulting from the stoichiometric amounts of HCl and AcOH generated overtime. A practical solution to this problem arose from our abilities to generate the stable α-chloroaminal intermediate 9, which can be stripped to dryness to remove traces of acids, therefore allowing for successive functionalization to occur in the same pot under neutral reaction conditions catalyzed by a hydrogen bond-donor catalysts (Scheme 4). By doing so, we postulated that the addition of an achiral H-bond donor thiourea catalyst T could facilitate the chloroaminal X intermediate activation to generate in situ the reactive N-carbamoyl iminium (e.g., 9) in a tight ionic pair species I. The Schreiner’s thiourea catalyst T was selected for its abilities of anion-binding to promote this complementary approach (method C), which relies on the chloride leaving group activation by the catalyst to assist the functionalization stage and deliver the α-amino ester product P. This third method represents a merging of two successive catalysis in the same pot. To test this method, both arenes 1,3-dimethoxybenzene and indole nucleophiles have been tested and were successfully reacted to afford products 8n and 8o in 71% and 66% yields, respectively, under the thiourea catalysis approach (Scheme 5). As mentioned above, the reactivity of N-Fmoc-protected chloroglycine is much less than its N-Cbz-protected counterpart (Table 2, entries 6 and 10), which demonstrates the strength of the achiral thiourea catalyst T. This expansion of the methodology also allowed us to prepare α-amino esters orthogonally protected (N-Fmoc and benzyl ester) and incorporate different acid-labile side chain nucleophiles that could not be installed otherwise. Thus, seven novel products 8p–8v have been synthesized with yields ranging from 41% to 90% and diastereoselectivity up to >20:1 (Scheme 5). Interestingly, the γ-butyrolactone-derived product 8p was obtained in 70% yield as a single syn-diastereomer,37 while the other dimedone-derived product 8u, which could also be formed through a Diels–Alder-like transition state, was obtained as a 1:1 mixture of diastereomers in 74% yield. Furthermore, silylenol ethers and silyl ketene acetals could be added smoothly to synthesize the α-amino ester products 8q, 8r, and 8s in 90%, 41%, and 61% yields, respectively. Noticeably, in each case, the reactions catalyzed by the achiral Schreiner’s thiourea were much more efficient and faster as shown by the synthesis of 8t isolated in 55% yield as a single regioisomer (catalyzed 100% conversion versus uncatalyzed 56% conversion). Finally, selected compounds were derivatized to show the applicability of our method in producing non-proteinogenic α-amino esters with site-specific designed protecting groups (Scheme 6). γ-Butyrolactone-derived α-amino ester 8p was transformed to a rearranged product analogue to pipecolic ester 12 in one pot with 98% yield.37 Spectral analysis of the cyclic product 12 confirmed the relative syn-stereochemical assignment made for the γ-butyrolactone-derivative 8p. Homo-Phe-OEt 13 was prepared from α-amino ester 8r by a simple and efficient hydrogenation protocol. Also, the δ-monoprotected derivative of α-amino adipic acid 14 was prepared in 83% yield by hydrogenation of the synthetic α-amino ester 8t.
Scheme 4. One-Pot Synthesis of Non-proteinogenic α-Amino Esters P Using a Tandem Catalysis.
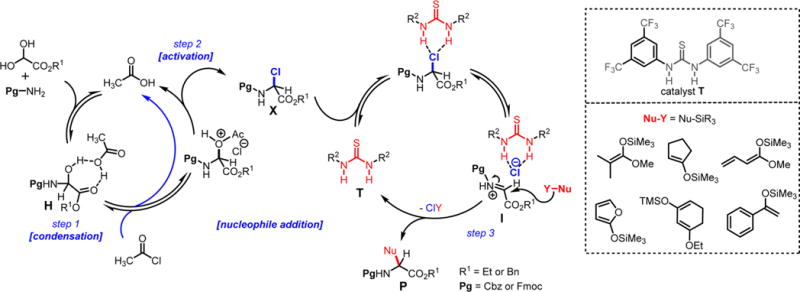
Scheme 5. Method C: One-Pot Synthesis of α-Amino Esters 8n–8u Catalyzed by Thioureaa,b,c.
a1H NMR yields of uncatalyzed versus catalyzed reactions were calculated from crude reaction mixtures using mesitylene as the internal standard. bIsolated yields from catalyzed reactions. cIsolated yield from the uncatalyzed reaction.
Scheme 6. Direct Derivatization of Non-Proteinogenic α-Amino Esters.
In summary, we reported a general strategy for the one-pot synthesis of non-proteinogenic α-amino esters bearing orthogonal N-carbamoyl protecting groups (Cbz or Fmoc) from commercially inexpensive starting materials. Our results demonstrate that an autocatalytic-like approach for the synthesis of the pivotal α-chloroglycine ester intermediate was efficient and practical. This general strategy entails two distinct methods: (i) a multicomponent-like process in which the three components are mixed in a temporally manner (methods A and B in CH3CN or CHCl3, respectively) and (ii) a one-pot synthesis catalyzed through anion-abstraction by a hydrogen bond donor thiourea catalyst (method C in CH2Cl2). These approaches are complementary and presumed to proceed via an activated iminium intermediate 9 that has been empirically characterized with an electrophilic value E = −6.4 (Mayr’s scale). Our findings through the initial kinetic profiling support an SN1-like mechanism (or SN2C+), which is consistent with the effect of the solvent on the reaction rates and the effect of the thiourea catalyst in facilitating the addition of weaker silylated nucleophiles. The discovery of the innate reactivity of α-chloroglycine esters via an SN1-like mechanism and the corresponding N-carbamoyl iminium–chloride tight ionic pair supports that the scope of this reaction will not be only limited to relatively strong nucleophiles. Therefore, the development of a reaction catalyzed by hydrogen bond donors appears attractive to enable a less reactive nucleophile to be incorporated (enantioselectively).17 This one-pot synthesis of α-amino esters proved to be efficient and versatile as highlighted by the synthetic scope of α-aryl and α-alkyl amino esters 8a–g prepared and the simplicity of functionalizations (e.g., 12–14) due to the protecting groups’ orthogonality. Ongoing studies are aimed at developing an asymmetric variant of this transformation and expanding the scope to additional classes of nucleophiles to prepare non-proteinogenic amino acids for nonribosomal peptide synthesis.
EXPERIMENTAL SECTION
Instrumentation and Methods
Infrared spectra were recorded on a Nicolet IS5 FT-IR spectrophotometer. 1H NMR spectra were recorded on a Varian Mercury400 (400 MHz) spectrometer and were reported in ppm using solvent as an internal standard (CDCl3 at 7.26 ppm and CD3CN at 1.94 ppm). Quantitative 1H NMR spectra were performed using standard parameters (pw = 90, at = 5, d1 = 15) and recorded on a Varian Mercury400. A careful treatment of the NMR signal was required to proceed to quantification. Data were reported as b = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, and m = multiplet. Coupling constants were reported in hertz (Hz, integration). 13C NMR spectra were recorded on a Varian Mercury400 (100 MHz) spectrometer. Chemical shifts were reported in ppm, with solvent resonance employed as the internal standard (CDCl3 at 77.0 ppm, CD3CN at 1.9 ppm, 118.7 ppm, and DMSO-d6 at 39.5 ppm). Melting points were determined using an MPA 160 digital melting point apparatus. The low-resolution ES-API mass spectra were performed on an FTICR mass spectrometer. Accurate mass (HRMS) was obtained from the University of Florida using an Agilent 6210 TOF instrument.
Reactions were performed in flame-dried glassware under a positive pressure of argon. Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise noted. Analytical TLC was performed on 0.25 mm glass backed 60 Å F-254 TLC plates. Flash chromatography was performed using 200–400 mesh silica gel (Silicycle, Inc.) or 60–325 mesh neutral alumina (Fisher, Inc.). Neutralized silica gel was prepared by adding 5 wt % triethylamine in a methylene chloride solution to a normal-phase silica gel and mixing for 30 min at room temperature. The plates were visualized by exposure to UV light (254 nm) and developed by a solution of phosphomolybdic acid in ethanol, vanillin/sulfuric acid in ethanol, ninhydrin in ethanol, potassium permanganate in water/potassium carbonate/sodium hydroxide, or cerium-ammonium-molybdate in water/sulfuric acid and heat.
Reagents and Solvents
All reagents used in the present paper were acquired from Alfa Aesar or Sigma-Aldrich. All bulk solvents were acquired from Fisher Scientific. Freshly distilled solvents were used in the reactions presented herein.
Chloroform was dried over CaCl2 overnight prior to distillation (bp 61 °C) and transferred under argon to a dark glass bottle with 3 Å molecular sieves for storage. Tetrahydrofuran was purified by refluxing with and distilling from sodium with benzophenone and transferred under argon to a dark glass bottle for storage. Dichloromethane was dried over CaCl2 overnight, prior to distillation (bp 40 °C), and transferred under argon to a dark glass bottle with 3 Å molecular sieves for storage. Toluene was dried over CaH2 and molecular sieves and transferred under argon to a dark glass bottle for storage. Full procedures can be found in Armarego, W. L. F.; Chai, C. L. L. Purification of Laboratory Chemicals, 6th ed.; Elsevier, 2009.
General Procedures A and B (Methods A and B): One-Pot Preparation of α-Amino Ester
In a flame-dried round-bottom flask under argon, a mixture of benzyl carbamate 2a (76 mg, 0.50 mmol, 1.0 equiv), ethyl glyoxylate 3a (108 μL, 0.05 mmol, 1.05 equiv), acetyl chloride (95 μL, 1.25 mmol, 2.5 equiv), and acetic acid (3 μL, 10 mol %, 0.1 equiv) in a CHCl3 (method A) or CH3CN (method B) solvent (5.0 mL, 0.1 M) was stirred for 24 or 12 h, respectively (steps 1/2), at 60 °C. After that point, the reaction mixture was cooled to rt. Then the arene nucleophile (1.5 equiv) was added to the reaction mixture (step 3) and stirred at the indicated temperature for the appropriate time (x h). After full consumption of chloroaminal intermediate 5 (observed by TLC, caution: same Rf value as the hydroxyaminal 3), the reaction mixture was quenched with a saturated solution of sodium bicarbonate (20.0 mL) and extracted with CH2Cl2 (3 × 10.0 mL). The combined organic layers were then dried over sodium sulfate, filtered, and concentrated under vacuum to afford the crude reaction mixture. The crude mixture was then purified by flash chromatography using an appropriate elution.
General Procedure C (Method C): One-Pot Synthesis of α-Amino Esters 8n–8u Catalyzed by the Schreiner’s Thiourea
In a flame-dried round-bottom flask under argon, a mixture of primary carbamate 2 (0.50 mmol, 1.0 equiv), glyoxylate 3 (0.05 mmol, 1.05 equiv), acetyl chloride (95 μL, 1.25 mmol, 2.5 equiv), and acetic acid (3 μL, 10 mol %, 0.1 equiv) was stirred in a CHCl3 solvent (5.0 mL, 0.1 M) for the appropriate time (× h, steps 1/2) at 60 °C. The reaction mixture was then cooled to rt, and the solvent was evaporated under vacuum. The crude reaction mixture was dissolved in the appropriate solvent (5.0 mL, 0.1 M), and the Schreiner’s thiourea catalyst (10 mol %) was added consecutively with a nucleophile (1.5 equiv) (step 3) in the reaction mixture. The mixture was stirred at the indicated temperature for the appropriate time (x h). After full consumption of chloroaminal intermediate 6 (observed by TLC, caution: same Rf value as the hydroxyaminal 5), the reaction mixture was quenched with a saturated solution of sodium bicarbonate (20.0 mL) and extracted with CH2Cl2 (3 × 10.0 mL). The combined organic layers were then dried over sodium sulfate, filtered, and concentrated under vacuum to afford the crude reaction mixture. The crude mixture was then purified by flash chromatography using an appropriate elution.
Compound Characterization
The synthesis and characterization of compounds 8a–8m (except 8g) was previously reported by our group.18
Benzyl Glyoxylate (3b)
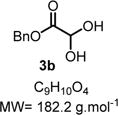 |
Dibenzyl 2,3-dihydroxysuccinate (330 mg, 1.0 mmol, 1.0 equiv) was dissolved in methanol (6.0 mL), and a solution of NaIO4 (321 mg, 1.5 mmol, 1.5 equiv) in H2O (3.0 mL) was added. The reaction was stirred for 6 h at 0 °C until the full consumption of the starting material. After that point, the reaction was partitioned between H2O and diethyl ether. The organic phase was dried (MgSO4), and the solvent was removed under reduced pressure to give a mixture of benzyl glyoxylate hydrate (3b) and aldehyde SI-1 (13:1) as a colorless liquid. The product was used without further purification in the synthesis for the next step. Rf = 0.35 (hexanes/ethyl acetate 70:30; UV active). IR (neat) νmax = 696, 737, 847, 908, 967, 1078, 1212, 1278, 1356, 1455, 1498, 1744, 3438 cm−1. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.53−7.32 (m, 5H), 5.25 (q, J = 12.1 Hz, 2H), 4.90 (d, J = 11.1 Hz, 1H), 3.78 (bs, 1H). (Note: The integration value does not match exactly due to the exchangeable –OH group.) 13C NMR (100 MHz, CDCl3): δ (ppm) 183.7, 170.5, 168.8, 168.8, 134.8, 134.7, 134.7, 128.6, 128.6, 128.5, 128.4, 128.39, 90.9, 88.7, 87.1, 67.9, 67.9, 67.6. HRMS (DART) m/z: [M + NH4]+ calcd for C9H14NO4, 200.0917; found, 200.0913 (−2.0 ppm).
Compound SI-1
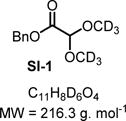 |
SI-1 was prepared by adding CD3OD to the crude reaction mixture of benzyl glyoxylate hydrate 3b. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.47−7.25 (m, 5H), 5.20 (s, 2H), 4.92 (s, 1H).
Compound SI-2
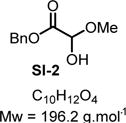 |
SI-2 was prepared by adding methanol (1.0 equiv) in the CDCl3 solution of the crude reaction mixture of benzyl glyoxylate hydrate 3b. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.44−7.30 (m, 5H), 5.34−5.15 (m, 2H), 4.90 (d, J = 11.3 Hz, 1H), 3.80 (d, J = 11.2 Hz, 1H), 3.47 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 169.2, 134.8, 128.7, 128.6, 93.4, 67.8, 55.4. HRMS (DART) m/z: [M + NH4]+ calcd for C10H16NO4, 214.1074; found, 214.1072 (−0.9 ppm). SMILES: OC(O)C(OCC1=CC=CC=C1)=O.
N-Fmoc-Gly(α-Cl)-OBn (6b)
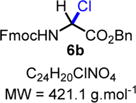 |
Protected α-chloroglycine benzyl ester 6b was synthesized using the following procedure: In a flame-dried scintillation vial, a mixture of 9-fluorenylmethyl carbamate (2b) (120 mg, 0.50 mmol, 1.0 equiv), benzyl glyoxylate hydrate 3b (109 mg, 0.60 mmol, 1.2 equiv), acetyl chloride (107 μL, 1.50 mmol, 3.0 equiv), and acetic acid (3 μL, 0.05 mmol, 10 mol %) was stirred under argon in anhydrous chloroform (5.0 mL) for 24 h at 60 °C. The reaction mixture was cooled to rt and directly evaporated under vacuum to obtain the desired product 6b as a white solid (211 mg, 0.50 mmol, quant yield). Compound 6b could be stored in an amber glass container at rt for 7 days in a desiccator without major decomposition (<5–10%). Rf = 0.30 (EtOAc/hexanes 30:70; UV active, stains green-yellow with vanillin). Caution! Compound 6b is not stable on silica gel; it hydrolyzes back to the corresponding hemiaminal, which can be observed by TLC at the Rf value mentioned above. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.78 (d, J = 7.6 Hz, 2H), 7.57 (s, 2H), 7.45−7.30 (m, 10H), 6.22 (s, 1H), 5.30 (s, 2H), 4.48 (d, J = 6.4 Hz, 2H), 4.24 (t, J = 6.4 Hz, 1H). SMILES: [H][C@](Cl)(C(OCC1=CC=CC=C1)=O) NC(OCC2C(C=CC=C3)=C3C4=C2C=CC=C4)=O.
N-Cbz(α-Cl)-OBn (6c)
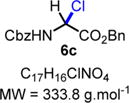 |
Protected α-chloroglycine benzyl ester 6c was synthesized using the following procedure: In a flame-dried scintillation vial, a mixture of benzyl carbamate 2a (76 mg, 0.50 mmol, 1.0 equiv), benzyl glyoxylate hydrate 3b (109 mg, 0.60 mmol, 1.2 equiv), acetyl chloride (107 μL, 1.50 mmol, 3.0 equiv) and acetic acid (3 μL, 0.05 mmol, 10 mol %) was stirred under argon in anhydrous chloroform (5.0 mL) for 16 h at 60 °C. The reaction mixture was cooled to rt and directly evaporated under vacuum to obtain the desired product 6c as a white solid (167 mg, 0.50 mmol, quant yield). Compound 6c can be stored in an amber glass container at rt for 7 days in a desiccator without major decomposition <5–10%). Rf = 0.25 (EtOAc/hexanes 30:70; UV active, stains green-yellow with vanillin) Caution! Compound 6c is not stable on silica gel; it hydrolyzes back to the corresponding hemiaminal, which can be observed by TLC at the Rf mentioned above. 1H NMR (400 MHz, CD3CN): δ (ppm) 7.48−7.26 (m, 10H), 7.05 (bs, 1H), 6.25 (d, J = 10.4 Hz, 1H), 5.25 (dd, J = 12.3 Hz, 2H), 5.15 (s, 2H). 13C NMR (100 MHz, CD3Cl3): δ (ppm) 165.8, 153.9, 135.3, 134.3, 128.9, 128.8, 128.7, 128.7, 128.7, 128.6, 128.5, 128.4, 68.7, 68.1, 63.4. SMILES: [H][C@](Cl)(C(OCC)=O)NC(OCC1C(C=CC=C2)=C2C3=C1C=CC=C3)=O.
Ethyl 2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-2-(2,4-dimethoxyphenyl)acetate (8g)
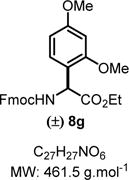 |
Product 8g was synthesized following General Procedure A.
Steps 1/2. Solvent: chloroform (5.0 mL) at 60 °C for 12 h.
Step 3. 1,3-Dimethoxybenzene (52 mg, 0.37 mmol, 1.5 equiv) as the nucleophile at 60 °C for 36 h.
The crude reaction mixture was purified by flash chromatography using an isocratic solvent system of EtOAc/hexanes (15:85) to deliver the desired product 8g (10:1 rr) as a white solid (141 mg, 0.31 mmol, 61% yield). Rf = 0.25 (ethyl acetate/hexanes 75:25; UV active, stains pink-red in vanillin). Mp = 82–85 °C. IR (neat) νmax = 659, 694, 728, 740, 758, 789, 828, 884, 935, 985, 1020, 1043, 1086, 1103, 1120, 1160, 1181, 1207, 1242, 1260, 1296, 1320, 1446, 1507, 1533, 1589, 1614, 1685, 1732, 3320 cm−1. 1H NMR (400 MHz, CD3CN): δ (ppm) 7.85 (d, J = 7.5 Hz, 2H), 7.68 (d, J = 7.4 Hz, 2H), 7.43 (t, J = 7.5 Hz, 2H), 7.39−7.30 (m, 2H), 7.18 (d, J = 8.3 Hz, 1H), 6.57 (d, J = 1.8 Hz, 1H), 6.52 (d, J = 8.3 Hz, 1H), 6.30 (d, J = 8.1 Hz, 1H), 5.38 (d, J = 8.4 Hz, 1H), 4.40−4.28 (m, 2H), 4.25 (t, J = 6.6 Hz, 1H), 4.14 (q, J = 7.1 Hz, 2H), 3.81 (s, 6H), 1.16 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CD3CN): δ (ppm) 171.4, 161.5, 158.4, 156.1, 144.4, 144.3, 141.4, 130.5, 127.9, 127.4, 125.4, 120.2, 118.0, 104.8, 98.9, 66.6, 61.4, 55.6, 55.3, 54.0, 47.2, 13.7. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C27H27NO6Na, 484.1731; found, 484.1723 (−1.6 ppm). SMILES: O=C(OCC1C2=C(C=CC=C2)C3=C1C=CC=C3)N[C@H]-(C(OCC)=O)C4=C(OC)C=C(OC)C=C4.
Benzyl 2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-2-(2,4-dimethoxyphenyl)acetate (8n)
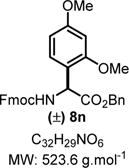 |
Product 8n was synthesized following General Procedure C.
Steps 1/2. Solvent: chloroform (2.5 mL) at 60 °C for 24 h.
Step 3. Dimethoxybenzene (52 mg, 0.37 mmol, 1.5 equiv) as the nucleophile at 40 °C for 60 h.
The crude reaction mixture was purified by flash chromatography using an isocratic solvent system of EtOAc/hexanes (15:85) to deliver the desired product 8n (14:1 rr) as a white solid (92 mg, 0.18 mmol, 71% yield). Rf = 0.30 (EtOAc/hexanes 30:70; UV active, stains pink-red in vanillin). Mp = 75–78 °C. IR (neat) νmax = 659, 694, 727, 739, 757, 790, 985, 1007, 1031, 1043, 1085, 1108, 1120, 1145, 1160, 1180, 1208, 1231, 1260, 1276, 1320, 1450, 1475, 1508, 1533, 1685, 1709, 1732, 2852, 2922 cm−1. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.75 (d, J = 7.5 Hz, 2H), 7.59 (d, J = 7.3 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.32−7.18 (m, 8H), 6.47−6.42 (m, 1H), 6.42 (s, 1H), 5.92 (d, J = 8.7 Hz, 1H), 5.52 (d, J = 8.7 Hz, 1H), 5.22 (d, J = 12.5 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 4.48−4.35 (m, 1H), 4.34−4.26 (m, 1H), 4.22 (t, J = 7.2 Hz, 1H), 3.81 (s, 3H), 3.63 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 171.4, 161.4, 158.2, 156.1, 144.2, 144.1, 141.5, 131.3, 128.6, 128.4, 128.2, 127.9, 127.3, 125.4, 125.4, 118.3, 104.5, 99.2, 67.4, 67.2, 55.7, 55.5, 55.2, 47.4, 30.0. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C32H29NO6Na, 546.1887; found, 546.1879 (−1.5 ppm). SMILES: O=C(N[C@H](C(OCC1=CC=CC=C1)=O)C2=C(OC)C=C(OC)C=C2)OCC3C(C=CC=C4)=C4C5= C3C=CC=C5.
Benzyl 2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-2-(2,4-dimethoxyphenyl)acetate (8o)
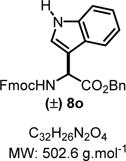 |
Product 8o was synthesized following General Procedure C.
Step 1/2. Solvent: chloroform (5.0 mL) at 60 °C for 24 h.
Step 3. Indole (84 mg, 0.37 mmol, 1.5 equiv) as the nucleophile at −78 °C for 0.5 h.
The crude reaction mixture was purified by flash chromatography using an isocratic solvent system of EtOAc/hexanes (15:85). Another purification was performed by flash chromatography using a solvent system of ether and dichloromethane (2:98) to obtain the pure desired products 8o as a white solid (84 mg, 0.13 mmol, 66% yield). Rf = 0.36 (ethyl acetate/hexanes 30:70; UV active, stains green in vanillin). Mp = 139–142 °C. IR (neat) νmax = 697, 739, 1010, 1045, 1103, 1182, 1216, 1277, 1324, 1450, 1498, 1704, 3400 cm−1. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.20 (s, 1H), 7.73 (dd, J = 17.2, 7.8 Hz, 3H), 7.57 (d, J = 7.4 Hz, 2H), 7.39 (dd, J = 7.4, 6.8 Hz, 3H), 7.29 (s, 8H), 7.14 (dd, J = 15.5, 8.1 Hz, 3H), 5.74 (s, 1H), 5.28−5.10 (m, 2H), 4.50−4.31 (m, 2H), 4.22 (t, J = 7.1 Hz, 1H). 13C NMR (100 MHz, CD3CN): δ (ppm) 171.4, 155.8, 143.8, 141.4, 141.3, 136.4, 135.3, 128.6, 128.4, 128.2, 127.8, 127.2, 125.4, 125.2, 125.2, 123.6, 122.9, 120.5, 120.1, 119.3, 111.6, 110.9, 67.5, 67.3, 51.6, 47.2. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C32H26N2O4Na, 525.1785; found, 525.1779 (+1.1 ppm). SMILES: [H]N1C2C(C=CC=C2)C([C@@H](C(OCC3=CC=CC=C3)=O)NC(OCC4C(C=CC=C5)=C5C6=C4C=CC=C6)=O)=C1.
Ethyl 2-(((Benzyloxy)carbonyl)amino)-2-(5-oxo-2,5-dihydrofur-an-2-yl)acetate (8p)
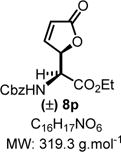 |
Product 8p was synthesized following General Procedure C.
Steps 1/2. Solvent: chloroform (5.0 mL) at 60 °C for 12 h.
Step 3. 2-(Trimethylsiloxy)furan (117 mg, 0.75 mmol, 1.5 equiv) as the nucleophile at 0 °C for 24 h.
The crude reaction mixture was purified by flash chromatography using an isocratic solvent system of CH2Cl2/ether (95:5) to deliver the desired product 8p as a colorless liquid (112 mg, 0.35 mmol, 70% yield). Rf = 0.25 (CH2Cl2/ether 95:5; UV active, stains pink-red in vanillin). IR (neat) νmax = 697, 733, 775, 800, 829, 875, 918, 982, 1026, 1044, 1062, 1100, 1156, 1205, 1248, 1323, 1370, 1455, 1530, 1603, 1699, 1746, 2936, 3311 cm−1. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.49 (dd, J = 5.7, 1.6 Hz, 1H), 7.37−7.28 (m, 5H), 6.13−6.01 (m, 1H), 5.57 (dd, J = 3.6, 1.7 Hz, 1H), 5.48 (bs, 1H), 5.06 (s, 2H), 4.77 (dd, J = 8.1, 2.0 Hz, 1H), 4.30 (q, J = 7.1 Hz, 2H), 1.32 (t, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 172.3, 167.9, 156.1, 152.9, 135.9, 128.6, 128.4, 128.1, 122.9, 82.9, 67.4, 62.9, 54.6, 14.2. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H17NO6Na, 342.0948; found, 342.0951 (+0.9 ppm). SMILES: O=C(C=C1)O[C@@H]1[C@@H](C(OCC)=O)NC(OCC2=CC=CC=C2)=O.
4-Ethyl 1-Methyl 3-(((Benzyloxy)carbonyl)amino)-2,2-dimethyl-succinate (8q)
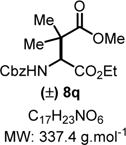 |
Product 8q was synthesized following General Procedure C.
Steps 1/2. Solvent: chloroform (5.0 mL) at 60 °C for 12 h.
Step 3. Trimethyl((3-methylbut-2-en-2-yl)oxy)silane (119 mg, 0.75 mmol, 1.5 equiv) as the nucleophile at 0 °C for 12 h.
The crude reaction mixture was purified by flash chromatography using an isocratic solvent system of hexanes/ethyl acetate (70:30) to deliver the desired product 8q as a colorless liquid (152 mg, 0.45 mmol, 90% yield). Rf = 0.30 (hexanes/ethyl acetate 70:30; UV active). IR (neat) νmax = 683, 702, 729, 757, 787, 824, 848, 875, 919, 983, 1003, 1025, 1046, 1135, 1171, 1207, 1242, 1278, 1332, 1371, 1392, 1455, 1469,1526, 1713, 2982, 3363 cm−1. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.45−7.27 (m, 5H), 5.62 (d, J = 9.5 Hz, 1H), 5.12 (s, 2H), 4.66 (d, J = 9.7 Hz, 1H), 4.16 (ddd, J = 14.2, 7.1, 2.7 Hz, 2H), 3.70 (s, 3H), 1.29 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H), 1.16 (s, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 176.0, 170.6, 156.6, 136.4, 128.8, 128.5, 128.4, 67.5, 61.9, 60.0, 52.4, 45.9, 23.3, 21.6, 14.26. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H23NO6Na, 360.1418; found, 360.1422 (+1.1 ppm). SMILES: O=C(OC)C(C)(C)[C@@H] (C(OCC)=O)NC(OCC1=CC=CC=C1)=O.
Ethyl 2-(((Benzyloxy)carbonyl)amino)-4-oxo-4-phenylbutanoate (8r)
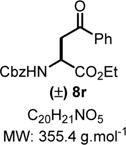 |
Product 8r was synthesized following General Procedure C.
Steps 1/2. Solvent: chloroform (5.0 mL) at 60 °C for 12 h.
Step 3. 1-Phenyl-1-trimethylsiloxyethylene (144 mg, 0.75 mmol, 1.5 equiv) as the nucleophile at −78 °C for 3 h.
The crude reaction mixture was purified by flash chromatography using an isocratic solvent system of hexanes/ethyl acetate (85:15) to deliver the desired product 8r as a colorless liquid (81 mg, 0.23 mmol, 46% yield). Rf = 0.25 (hexanes/ethyl acetate 70:30; UV active). IR (neat) νmax = 690, 879, 1045, 1086, 1217, 1272, 1378, 1450, 1709, 2889, 2973, 3342 cm−1. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.94 (d, J = 7.8 Hz, 2H), 7.63−7.56 (m, 1H), 7.47 (t, J = 7.6 Hz, 2H), 7.38−7.29 (m, 5H), 5.89 (d, J = 8.0 Hz, 1H), 5.16−5.06 (m, 2H), 4.78−4.70 (m, 1H), 4.21 (q, J = 7.0 Hz, 2H), 3.67 (ddd, J = 83.7, 18.2, 4.1 Hz, 2H), 1.23 (t, J = 7.1 Hz, 3H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H22NO5, 356.1492; found, 356.1496 (+1.1 ppm). SMILES: O=C(C1=CC=CC=C1)C[C@@H](C(OCC)=O)-NC(OCC2=CC=CC=C2)=O. The spectral data is consistent with the reported compound in the literature.38
Ethyl 2-(((Benzyloxy)carbonyl)amino)-2-(-2-oxocyclopentyl)-acetate (8s)
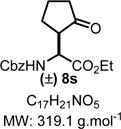 |
Product 8s was synthesized following General Procedure C.
Steps 1/2. Solvent: chloroform (5.0 mL) at 60 °C for 12 h.
Step 3. (Cyclopent-1-en-1-yloxy)trimethylsilane (117 mg, 0.75 mmol, 1.5 equiv) as the nucleophile at 0 °C for 60 h.
The crude reaction mixture was purified by flash chromatography using an isocratic solvent system of hexanes/ethyl acetate (80:20) to deliver the desired product in a mixture of diastereomers (42:58) 8s as a colorless liquid (97 mg, 0.30 mmol, 61% yield). Rf = 0.20 (hexanes/ethyl acetate 70:30; UV active). IR (neat) νmax = 1322, 1370, 1401, 1454, 1520, 1721, 2965, 3340 cm−1. 1H NMR (400 MHz, CD3CN), diastereomers (42:58): δ (ppm) 7.46−7.21 (m, 5H), 6.08−5.83 (m, 1H), 5.08 (dd, J = 8.4, 2.1 Hz, 2H), 4.65−4.47 (m, 1H), 4.23−4.08 (m, 2H), 2.75−2.58 (m, 1H), 2.31−2.18 (m, 1H), 2.11−1.98 (m, 2H), 1.85−1.70 (m, 2H), 1.62 (ddd, J = 23.4, 11.6, 6.7 Hz, 1H), 1.20 (t, J = 9.9 Hz, 3H). 13C NMR (100 MHz, CDCl3), diastereomers (42:58): δ (ppm) 217.7, 217.3, 170.9, 170.5, 156.4, 155.8, 136.2, 136.1, 128.4, 128.4, 128.1, 128.0, 128.0, 127.9, 66.9, 66.9, 61.7, 61.6, 61.6, 53.2, 53.0, 51.6, 50.8, 37.9, 37.7, 25.9, 25.6, 20.5, 20.3, 14.0. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H21NO5Na, 342.1312; found, 342.1318 (+1.8 ppm). SMILES: O=C(CCC1)[C@H]1[C@@H](C(OCC)=O)NC(OCC2=CC=CC=C2)=O.
6-Benzyl 1-Methyl-5-(((benzyloxy)carbonyl)amino)-(E)-hex-2-enedioate (8t)
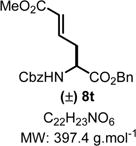 |
Product 8t was synthesized following General Procedure C.
Steps 1/2. Solvent: chloroform (5.0 mL) at 60 °C for 12 h.
Step 3. (E)-((1-Methoxybuta-1,3-dien-1-yl)oxy)trimethylsilane39 (129 mg, 0.75 mmol, 1.5 equiv) as the nucleophile at −78 °C for 3 h.
The crude reaction mixture showing one regioisomer was purified by flash chromatography using an isocratic solvent system of hexanes/ethyl acetate (80:20) to deliver the desired product 8t as a colorless liquid (109 mg, 0.27 mmol, 55% yield). Rf = 0.30 (hexanes/ethyl acetate 70:30; UV active). IR (neat) νmax = 696, 737, 775, 844, 910, 977, 1044, 1082, 1173, 1265, 1340, 1386, 1435, 1455, 1523, 1660, 1715, 2951, 3344 cm−1. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.42−7.28 (m, 10H), 6.92−6.68 (m, 1H), 5.83 (dd, J = 15.6, 0.9 Hz, 1H), 5.69 (d, J = 7.9 Hz, 1H), 5.17 (dd, J = 23.7, 12.2 Hz, 2H), 5.10 (s, 2H), 4.57 (dd, J = 13.2, 6.4 Hz, 1H), 3.69 (d, J = 0.9 Hz, 3H), 2.75 (dt, J = 12.6, 6.1 Hz, 1H), 2.68−2.58 (m, 1H). 13C NMR (100 MHz, CDCl3): δ (ppm) 170.9, 166.1, 155.7, 142.2, 136.1, 134.9, 128.6, 128.6, 128.5, 128.5, 128.4, 128.2, 128.0, 124.6, 67.5, 67.0, 52.9, 51.5, 34.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C22H23NO6Na, 420.1418; found, 420.1418 (0 ppm). SMILES: O=C(N[C@H](C(OCC1=CC=CC=C1)=O)C/C=C/C(OC)=O)OCC2=CC=CC=C2.
Ethyl 2-(((Benzyloxy)carbonyl)amino)-2-(4-ethoxy-2-oxocyclo-hex-3-en-1-yl)acetate (8u)
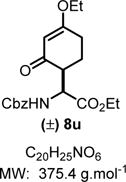 |
Product 8u was synthesized following General Procedure C.
Steps 1/2. Solvent: chloroform (5.0 mL) at 60 °C for 12 h.
Step 3. ((5-Ethoxycyclohexa-1,5-dien-1-yl)oxy)trimethylsilane40 (159 mg, 0.75 mmol, 1.5 equiv) as the nucleophile at −20 °C for 12 h.
The crude reaction mixture was purified by flash chromatography using an isocratic solvent system of hexanes/ethyl acetate (70:30) to deliver the mixture of two diastereomers (1:1) of 8u as a colorless liquid (129 mg, 0.37 mmol, 74% yield). The two diasteromers were separated on preparative TLC (same plates were run 3 times) using a solvent system of hexanes and ethy acetate (80:20) to obtain the two pure diasteromers in a 1:1 ratio. Rf = 0.20 (hexanes/ethyl acetate 70:30; UV active). IR (neat) νmax = 697, 738, 776, 819, 851, 892, 1026, 1051, 1191, 1328, 1379, 1454, 1499, 1600, 1645, 1716, 2938, 3348 cm−1. syn-Diastereomer: 1H NMR (400 MHz, CD3CN) δ ppm 7.55−7.22 (m, 5H), 5.62 (d, J = 9.4 Hz, 1H), 5.27 (s, 1H), 5.20−5.00 (m, 2H), 4.44 (dd, J = 9.6, 3.0 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.98−3.81 (m, 2H), 3.20−3.03 (m, 1H), 2.63 (ddd, J = 17.5, 12.6, 4.7 Hz, 1H), 2.37 (ddd, J = 17.7, 4.9, 2.2 Hz, 1H), 2.08−1.97 (m, 1H), 1.83 (ddd, J = 25.9, 12.9, 5.0 Hz, 1H), 1.30 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CD3CN) δ ppm 198.5, 179.2, 172.1, 157.7, 138.1, 129.4, 128.9, 128.6, 102.7, 67.3, 65.7, 62.0, 55.2, 49.2, 29.6, 25.4, 14.4, 14.4. anti-Diastereomer: 1H NMR (400 MHz, CD3CN) δ ppm 7.48−7.22 (m, 5H), 5.98 (d, J = 7.7 Hz, 1H), 5.29 (d, J = 1.2 Hz, 1H), 5.19−5.00 (m, 2H), 4.60 (dd, J = 9.3, 4.2 Hz, 1H), 4.11 (q, J = 7.0 Hz, 2H), 4.02−3.85 (m, 2H), 2.76 (dt, J = 12.2, 4.8 Hz, 1H), 2.61−2.47 (m, 1H), 2.41−2.32 (m, 1H), 2.08−1.97 (m, 2H), 1.30 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CD3CN) δ ppm 197.9, 178.9, 172.5, 157.1, 138.1, 129.4, 128.9, 128.7, 102.7, 67.2, 65.6, 62.0, 54.9, 48.9, 29.5, 24.7, 14.4. HRMS (ESI-TOF) m/z [M − H]− calcd for C20H25NO6 374.1609, found 374.1608 (−0.3 ppm). SMILES: O=C(C=C1O)CC[C@H]1[C@@H](C(OCC)=O)NC(OCC2=CC=CC=C2)=O.
Ethyl 3-Hydroxy-6-oxopiperidine-2-carboxylate (12)
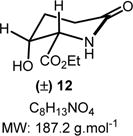 |
To a solution of 8p (96 mg, 0.3 mmol, 1.0 equiv) in ethanol (10.0 mL) was added 10% Pd/C (30 mg, 0.03 mmol, 0.1 equiv). The reaction mixture was hydrogenated at rt under hydrogen pressure (balloon: 1.0 atm). The starting material 8p was consumed after 3 h, as indicated by TLC. The hydrogen balloon was removed, and the reaction mixture was stirred another 12 h at rt until full consumption of the intermediate (Rf = 0.1 in ethyl acetate/methanol 90:10; stains yellow in ninhydrin). The reaction mixture was then filtered through Celite and washed with ethanol (5.0 mL). The filtrate was evaporated under vacuum to obtain 12 in pure form (55 mg, 0.29 mmol, 98% yield) as a pale yellow liquid. Rf = 0.2 (methanol/dichloromethane 5:95; not UV active, stains yellow-brown in a ninhydrin solution). IR (neat) νmax = 860, 881, 939, 968, 1024, 1072, 1177, 1445, 1574, 1650, 1731, 1773, 2981, 3368 cm−1. 1H NMR (400 MHz, CDCl3): δ (ppm) 4.94 (td, J = 7.2, 3.1 Hz, 1H), 4.23 (qd, J = 7.1, 1.5 Hz, 2H), 3.47 (d, J = 3.1 Hz, 1H), 2.74−2.62 (m, 1H), 2.53 (ddd, J = 17.9, 10.3, 7.9 Hz, 1H), 2.40−2.27 (m, 2H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 177.1, 172.5, 80.8, 61.8, 57.6, 28.6, 24.2, 14.2. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C8H14NO4, 188.0917; found, 188.0919 (+1.1 ppm). SMILES: O=C1N[C@]([H])(C(OCC)=O)[C@@](O)([H])CC1.
Ethyl 2-Amino-4-phenylbutanoate (13)
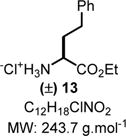 |
To a solution of 8r (71 mg, 0.2 mmol, 1.0 equiv) in ethanol (5.0 mL) was added 1 N HCl (200 μL) followed by 10% Pd/C (20 mg, 0.02 mmol, 0.1 equiv). The mixture was hydrogenated at 55 °C under hydrogen pressure (balloon: 1.0 atm). The reaction was completed after 3 h as determined by TLC, and the mixture was then filtered through Celite and washed with ethanol (5.0 mL). The filtrate was evaporated under vacuum to obtain the crude reaction mixture as a solid. The solid was suspended in diethyl ether (3.0 mL), sonicated for 5 min, and finally filtered to obtain 13 (41 mg, 0.17 mmol, 89% yield) as a white solid. Rf = 0.1 (ethyl acetate/methanol 90:10; UV active, stains yellow in ninhydrin). IR (neat) νmax = 672, 702, 737, 759, 981, 1019, 1033, 1062, 1084, 1104, 1156, 1199, 1238, 1266, 1377, 1454, 1497, 1752, 2843, 2866, 2973 cm−1. 1H NMR (400 MHz, D2O): δ (ppm) 7.33−7.23 (m, 2H), 7.19 (d, J = 7.5 Hz, 3H), 4.16−4.06 (m, 2H), 3.99 (t, J = 6.3 Hz, 1H), 2.76−2.54 (m, 2H), 2.27−2.03 (m, 2H), 1.16 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, D2O): δ (ppm) 170.1, 139.8, 128.8, 128.4, 126.6, 63.5, 52.3, 31.3, 30.2, 13.1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H18NO2, 208.1332; found, 208.1331 (−0.5 ppm). SMILES: [NH3+]C(C(OCC)=O)CCC1=CC=CC=C1.[Cl−].
2-Amino-6-ethoxy-6-oxohexanoic Acid (14)
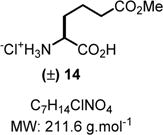 |
To a solution of 8t (80 mg, 0.2 mmol, 1.0 equiv) in ethanol (5.0 mL) was added HCl (1 N, 200 μL) followed by 10% Pd/C (20 mg, 0.02 mmol, 0.1 equiv). The mixture was hydrogenated at rt under hydrogen pressure (balloon: 1.0 atm). When the reaction was completed as determined by TLC, the mixture was filtered through Celite and washed with ethanol (5.0 mL). The filtrate was evaporated under vacuum to obtain the crude reaction mixture. The solid was suspended in diethyl ether (3.0 mL), sonicated for 5.0 min, and finally filtered to obtain 14 in pure form (36 mg, 0.17 mmol, 83% yield) as a white solid. Rf = 0.35 (n-butanol/acetic acid/water 3:1:1 (by volume); stains yellow-brown in ninhydrin). IR (neat) νmax = 739, 804, 827, 888, 943, 985, 1093, 1141, 1183, 1203, 1220, 1260, 1352, 1437, 1481, 1602, 1727, 2982 cm−1. 1H NMR (400 MHz, D2O): δ (ppm) 3.95 (t, J = 6.2 Hz, 1H), 3.71 (s, 3H), 2.49 (t, J = 7.2 Hz, 2H), 2.04−1.85 (m, 2H), 1.83−1.64 (m, 2H). 13C NMR (100 MHz, D2O): δ (ppm) 176.2, 172.0, 52.6, 52.2, 32.7, 29.0, 19.7. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C7H14NO4, 176.0917; found, 176.0918 (+0.6 ppm). SMILES: [NH3+]C(C(O)=O)CCCC(OC)=O.[Cl−].
Supplementary Material
Acknowledgments
The authors thank Ms. M. J. Gosselin (FAU) for her initial work on the scope of the Friedel–Crafts reaction. This research was supported by the National Institutes of Health (R15GM116025).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b01274.
Empirical determination of the E value of iminium, overall order of a Friedel–Crafts reaction, kinetic profiling, copies of 1H and 13C NMR spectra for all new compounds (PDF)
ORCID
Stéphane P. Roche: 0000-0002-3019-2168
Notes
The authors declare no competing financial interest.
References
- 1.(a) Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Science. 1995;268:726. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]; (b) Marahiel MA, Stachelhaus T, Mootz HD. Chem Rev. 1997;97:2651. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]; (c) Walsh CT. Science. 2004;303:1805. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]; (d) Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; (e) Walsh CT, O’Brien RV, Khosla C. Angew Chem, Int Ed. 2013;52:7098. doi: 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Kaneko I, Kamoshida K, Takahashi S. J Antibiot. 1989;42:236. doi: 10.7164/antibiotics.42.236. [DOI] [PubMed] [Google Scholar]; (b) Kleinkauf H, Dohren H. Eur J Biochem. 1990;192:1. doi: 10.1111/j.1432-1033.1990.tb19188.x. [DOI] [PubMed] [Google Scholar]; (c) Boman HG. Annu Rev Immunol. 1995;13:61. doi: 10.1146/annurev.iy.13.040195.000425. [DOI] [PubMed] [Google Scholar]; (d) Vertesy L, Aretz W, Knauf M, Markus A, Vogel M, Wink J. J Antibiot. 1999;52:374. doi: 10.7164/antibiotics.52.374. [DOI] [PubMed] [Google Scholar]; (e) Chiu H-T, Hubbard BK, Shah AN, Eide J, Fredenburg RA, Walsh CT, Khosla C. Proc Natl Acad Sci U S A. 2001;98:8548. doi: 10.1073/pnas.151246498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Soloshonok VA, Izawa K. Asymmetric synthesis and application of α-amino acids. Oxford University Press; Washington, DC: 2009. [Google Scholar]; (b) Blaskovich MA. Handbook on syntheses of amino acids: general routes for the syntheses of amino acids. Oxford University Press; Oxford, New York: 2010. [Google Scholar]
- 4.(a) Maruoka K, Ooi T. Chem Rev. 2003;103:3013–3028. doi: 10.1021/cr020020e. [DOI] [PubMed] [Google Scholar]; (b) Lygo B, Andrews BI. Acc Chem Res. 2004;37:518–525. doi: 10.1021/ar030058t. [DOI] [PubMed] [Google Scholar]; (c) O’Donnell MJ. Acc Chem Res. 2004;37:506–517. doi: 10.1021/ar0300625. [DOI] [PubMed] [Google Scholar]
- 5.(a) Liang J, Ruble JC, Fu GC. J Org Chem. 1998;63:3154. [Google Scholar]; Trost BM, Ariza X. J Am Chem Soc. 1999;121:10727. [Google Scholar]; (b) Cabrera S, Reyes E, Aleman J, Milelli A, Kobbelgaard S, Joergensen KA. J Am Chem Soc. 2008;130:12031. doi: 10.1021/ja804567h. [DOI] [PubMed] [Google Scholar]; (c) Terada M, Tanaka H, Sorimachi K. J Am Chem Soc. 2009;131:3430. doi: 10.1021/ja8090643. [DOI] [PubMed] [Google Scholar]; (d) Avila EP, Justo RMS, Goncalves VP, Pereira AA, Diniz R, Amarante GW. J Org Chem. 2015;80:590. doi: 10.1021/jo5024975. [DOI] [PubMed] [Google Scholar]
- 6.(a) Lee EC, Fu GC. J Am Chem Soc. 2007;129:12066. doi: 10.1021/ja074483j. [DOI] [PubMed] [Google Scholar]; (b) Liu B, Zhu S-F, Zhang W, Chen C, Zhou Q-L. J Am Chem Soc. 2007;129:5834. doi: 10.1021/ja0711765. [DOI] [PubMed] [Google Scholar]
- 7.For selected examples of the Strecker reaction, see:; (a) Krueger CA, Kuntz KW, Dzierba CD, Wirschun WG, Gleason JD, Snapper ML, Hoveyda AH. J Am Chem Soc. 1999;121:4284. [Google Scholar]; (b) Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN. Nature. 2009;461:968. doi: 10.1038/nature08484. and references cited herein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For selected examples of the Mannich reaction, see:; (a) Hagiwara E, Fujii A, Sodeoka M. J Am Chem Soc. 1998;120:2474. [Google Scholar]; (b) Ferraris D, Young B, Dudding T, Lectka T. J Am Chem Soc. 1998;120:4548. [Google Scholar]; (c) Juhl K, Gathergood N, Jørgensen KA. Angew Chem, Int Ed. 2001;40:2995. doi: 10.1002/1521-3773(20010817)40:16<2995::AID-ANIE2995>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]; (d) Córdova A, Notz W, Zhong G, Betancort JM, Barbas CF., III J Am Chem Soc. 2002;124:1842. doi: 10.1021/ja017270h. [DOI] [PubMed] [Google Scholar]
- 9.(a) Petasis NA, Zavialov IA. J Am Chem Soc. 1997;119:445. [Google Scholar]; (b) Beenen MA, Weix DJ, Ellman JA. J Am Chem Soc. 2006;128:6304–6305. doi: 10.1021/ja060529h. [DOI] [PubMed] [Google Scholar]
- 10.(a) Knowles WS. Acc Chem Res. 1983;16:106. [Google Scholar]; (b) Noyori R, Ikeda T, Ohkuma T, Widhalm M, Kitamura M, Takaya H, Akutagawa S, Sayo N, Saito T. J Am Chem Soc. 1989;111:9134. [Google Scholar]; (c) Burk MJ, Feaster JE, Nugent WA, Harlow RL. J Am Chem Soc. 1993;115:10125. [Google Scholar]; (d) van den Berg M, Minnaard AJ, Schudde EP, van Esch J, de Vries AHM, de Vries JG, Feringa BL. J Am Chem Soc. 2000;122:11539. [Google Scholar]
- 11.For selected examples of the Friedel–Crafts reaction, see:; (a) Johannsen M. Chem Commun. 1999;2233 [Google Scholar]; (b) Saaby S, Fang X, Gathergood N, Jørgensen KA. Angew Chem, Int Ed. 2000;39:4114. doi: 10.1002/1521-3773(20001117)39:22<4114::aid-anie4114>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; (c) Uraguchi D, Sorimachi K, Terada M. J Am Chem Soc. 2004;126:11804. doi: 10.1021/ja046185h. [DOI] [PubMed] [Google Scholar]; (d) Shirakawa S, Berger R, Leighton JL. J Am Chem Soc. 2005;127:2858. doi: 10.1021/ja042522a. [DOI] [PubMed] [Google Scholar]
- 12.For a discussion on protecting groups compatibility in peptide synthesis and α-iminoglycinate functionalization, see:; Nakamura Y, Matsubara R, Kiyohara H, Kobayashi S. Org Lett. 2003;5:2481. doi: 10.1021/ol034717d. [DOI] [PubMed] [Google Scholar]
- 13.For the use of N-acylimino ester generated from α-bromoglycine esters in metal-catalyzed Mannich reactions, see:; (a) Kobayashi S, Kitagawa H, Matsubara R. J Comb Chem. 2001;3:401. doi: 10.1021/cc010009c. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi S, Matsubara R, Kitagawa H. Org Lett. 2002;4:143. doi: 10.1021/ol017062u. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi S, Matsubara R, Nakamura Y, Kitagawa H, Sugiura M. J Am Chem Soc. 2003;125:2507. doi: 10.1021/ja0281840. [DOI] [PubMed] [Google Scholar]; (d) Nakamura Y, Matsubara R, Kiyohara H, Kobayashi S. Org Lett. 2003;5:2481. doi: 10.1021/ol034717d. [DOI] [PubMed] [Google Scholar]; (e) Matsubara R, Nakamura Y, Kobayashi S. Angew Chem, Int Ed. 2004;43:1679. doi: 10.1002/anie.200353237. [DOI] [PubMed] [Google Scholar]; and in the organocatalyzed Mannich reaction, see:; (f) Poulsen TB, Alemparte C, Saaby S, Bella M, Jørgensen KA. Angew Chem, Int Ed. 2005;44:2896. doi: 10.1002/anie.200500144. [DOI] [PubMed] [Google Scholar]
- 14.(a) Fini F, Sgarzani V, Pettersen D, Herrera RP, Bernardi L, Ricci A. Angew Chem, Int Ed. 2005;44:7975. doi: 10.1002/anie.200502646. [DOI] [PubMed] [Google Scholar]; (b) Palomo C, Oiarbide M, Laso A, López R. J Am Chem Soc. 2005;127:17622. doi: 10.1021/ja056594t. [DOI] [PubMed] [Google Scholar]; (c) Song J, Shih HW, Deng L. Org Lett. 2007;9:603. doi: 10.1021/ol062837q. [DOI] [PubMed] [Google Scholar]; (d) Gianelli C, Sambri L, Carlone A, Bartoli G, Melchiorre P. Angew Chem, Int Ed. 2008;47:8700. doi: 10.1002/anie.200803819. [DOI] [PubMed] [Google Scholar]; (e) Takada K, Tanaka S, Nagasawa K. Synlett. 2009;2009:1643. [Google Scholar]; (f) Galzerano P, Agostino D, Bencivenni G, Sambri L, Bartoli G, Melchiorre P. Chem - Eur J. 2010;16:6069. doi: 10.1002/chem.200903217. [DOI] [PubMed] [Google Scholar]; (g) Gao J, Chuan Y, Li J, Xie F, Peng Y. Org Biomol Chem. 2012;10:3730. doi: 10.1039/c2ob00049k. [DOI] [PubMed] [Google Scholar]
- 15.For asymmetric Mannich-3CRs, see:; (a) Córdova A. Chem -Eur J. 2004;10:1987–1997. doi: 10.1002/chem.200305646. [DOI] [PubMed] [Google Scholar]; (b) Hamashima Y, Sasamoto N, Hotta D, Somei H, Umebayashi N, Sodeoka M. Angew Chem, Int Ed. 2005;44:1525. doi: 10.1002/anie.200462202. [DOI] [PubMed] [Google Scholar]; (c) Enders D, Grondal C, Vrettou M, Raabe G. Angew Chem, Int Ed. 2005;44:4079. doi: 10.1002/anie.200500810. [DOI] [PubMed] [Google Scholar]; (d) Ramasastry SSV, Zhang H, Tanaka F, Barbas CF., III J Am Chem Soc. 2007;129:288. doi: 10.1021/ja0677012. [DOI] [PubMed] [Google Scholar]; For asymmetric Mannich-3CRs, see:; (e) Ishitani H, Komiyama S, Hasegawa Y, Kobayashi S. J Am Chem Soc. 2000;122:762. [Google Scholar]; (f) Pan SC, List B. Org Lett. 2007;9:1149. doi: 10.1021/ol0702674. [DOI] [PubMed] [Google Scholar]; For asymmetric Petasis-3CR, see:; (g) Lou S, Schaus SE. J Am Chem Soc. 2008;130:6922. doi: 10.1021/ja8018934. [DOI] [PMC free article] [PubMed] [Google Scholar]; For asymmetric Friedel–Crafts-3CRs; (h) Saaby S, Bayon P, Aburel PS, Jorgensen KA. J Org Chem. 2002;67:4352. doi: 10.1021/jo0256787. [DOI] [PubMed] [Google Scholar]; (i) Kang Q, Zhao Z-A, You S-L. Tetrahedron. 2009;65:1603. [Google Scholar]
- 16.For multicomponent syntheses of α-amino esters, using a Petasis reaction, see:; (a) Petasis NA, Goodman A, Zavialov IA. Tetrahedron. 1997;53:16463. [Google Scholar]; Using a Barbier reaction:; (b) Le Gall E, Pignon A. J Chem Educ. 2012;89:1190. [Google Scholar]; (c) Bisai A, Singh VK. Tetrahedron. 2012;68:3480. [Google Scholar]; Using a Mannich reaction:; (d) Akiyama T, Takaya J, Kagoshima H. Adv Synth Catal. 2002;344:338. [Google Scholar]; (e) Hamashima Y, Sasamoto N, Hotta D, Somei H, Umebayashi N, Sodeoka M. Angew Chem, Int Ed. 2005;44:1525. doi: 10.1002/anie.200462202. [DOI] [PubMed] [Google Scholar]; Using a Friedel–Crafts reaction:; (f) Bensel N, Pevere V, Desmurs JR, Wagner A, Mioskowski C. Tetrahedron Lett. 1999;40:879. [Google Scholar]; (g) Grundmann P, Fessner W-D. Adv Synth Catal. 2008;350:1729. [Google Scholar]; (h) Csütörtöki R, Szatmári I, Mándi A, Kurtán T, Fülöp F. Synlett. 2011;2011:1940. and references cited therein. [Google Scholar]
- 17.Wasa M, Liu RY, Roche SP, Jacobsen EN. J Am Chem Soc. 2014;136:12872. doi: 10.1021/ja5075163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roche SP, Samanta SS, Gosselin MMJ. Chem Commun. 2014;50:2632. doi: 10.1039/c3cc48884e. [DOI] [PubMed] [Google Scholar]
- 19.For the synthesis of α-chloroglycines, see:; (a) Mooiweer HH, Hiemstra H, Speckamp WN. Tetrahedron. 1989;45:4627. [Google Scholar]; (b) Mooiweer HH, Ettema KWA, Hiemstra H, NicoSpeckamp W. Tetrahedron. 1990;46:2991. [Google Scholar]; (c) Williams RM, Aldous DJ, Aldous SC. J Org Chem. 1990;55:4657. [Google Scholar]; (d) Kobayashi S, Matsubara R, Nakamura Y, Kitagawa H, Sugiura M. J Am Chem Soc. 2003;125:2507. doi: 10.1021/ja0281840. [DOI] [PubMed] [Google Scholar]; (e) Hafez AM, Dudding T, Wagerle TR, Shah MH, Taggi AE, Lectka T. J Org Chem. 2003;68:5819. doi: 10.1021/jo034150e. [DOI] [PubMed] [Google Scholar]
- 20.Xu F, Devine P. Org Process Res Dev. 2010;14:666. [Google Scholar]
- 21.See the Supporting Information for complete experimental details.
- 22.Claridge TDW. In: High Resolution NMR Techniques in Organic Chemistry. Baldwin JE, Williams RM, editors. Vol. 19. Pergamon Press; Oxford: 1999. p. 114. [Google Scholar]
- 23.The addition of MS 3 Å (1 g/mol) to the reaction mixture drastically decreases the reaction rate, probably due to the sequestration of HCl in the sieves. Also, α-chloroglycine ester 6a was found to be unstable upon silica gel chromatography or exposure to water, leading in both cases to the spontaneous formation of α-hydroxyglycine ester 5a.
- 24.(a) Williams RM, Aldous DJ, Aldous SC. J Org Chem. 1990;55:4657. [Google Scholar]; (b) Coqueron P-Y, Didier C, Ciufolini MA. Angew Chem, Int Ed. 2003;42:1411. doi: 10.1002/anie.200390363. [DOI] [PubMed] [Google Scholar]; (c) Hafez AM, Dudding T, Wagerle TR, Shah MH, Taggi AE, Lectka T. J Org Chem. 2003;68:5819. doi: 10.1021/jo034150e. [DOI] [PubMed] [Google Scholar]; (d) Kobayashi S, Matsubara R, Nakamura Y, Kitagawa H, Sugiura M. J Am Chem Soc. 2003;125:2507. doi: 10.1021/ja0281840. [DOI] [PubMed] [Google Scholar]; (e) Zhang J, Ciufolini MA. Org Lett. 2009;11:2389. doi: 10.1021/ol900455m. [DOI] [PubMed] [Google Scholar]
- 25.Using excess ethyl glyoxylate 3a, or in a “true” multicomponent direct reaction between arene and ethyl glyoxylate, mono- and bis-aryl Friedel–Crafts products 3a are observed.
- 26.(a) Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]; (b) Mayr H. Angew Chem, Int Ed. 2011;50:3612. [Google Scholar]
- 27.Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 28.Ammer J, Nolte C, Mayr H. J Am Chem Soc. 2012;134:13902. doi: 10.1021/ja306522b. [DOI] [PubMed] [Google Scholar]
- 29.Gotta MF, Mayr H. J Org Chem. 1998;63:9769. [Google Scholar]
- 30.Lakhdar S, Mayr H. Chem Commun. 2011;47:1866. doi: 10.1039/c0cc04295a. [DOI] [PubMed] [Google Scholar]
- 31.Lakhdar S, Westermaier M, Terrier F, Goumont R, Boubaker T, Ofial AR, Mayr H. J Org Chem. 2006;71:9088. doi: 10.1021/jo0614339. [DOI] [PubMed] [Google Scholar]
- 32.Mayr H, Bug T, Gotta MF, Hering N, Irrgang B, Janker B, Kempf B, Loos R, Ofial AR, Remennikov G, Schimmel H. J Am Chem Soc. 2001;123:9500. doi: 10.1021/ja010890y. [DOI] [PubMed] [Google Scholar]
- 33.(a) Mayr H, Bug T, Gotta MF, Hering N, Irrgang B, Janker B, Kempf B, Loos R, Ofial AR, Remennikov G, Schimmel H. J Am Chem Soc. 2001;123:9500. doi: 10.1021/ja010890y. [DOI] [PubMed] [Google Scholar]; (b) Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]; (c) Lakhdar S, Westermaier M, Terrier F, Goumont R, Boubaker T, Ofial AR, Mayr H. J Org Chem. 2006;71:9088. doi: 10.1021/jo0614339. [DOI] [PubMed] [Google Scholar]; (d) Nigst TA, Westermaier M, Ofial AR, Mayr H. Eur J Org Chem. 2008;2008:2369. [Google Scholar]; (e) Ammer J, Nolte C, Mayr H. J Am Chem Soc. 2012;134:13902. doi: 10.1021/ja306522b. [DOI] [PubMed] [Google Scholar]
- 34.Roos EC, Hiemstra H, Speckamp WN, Kaptein B, Kamphuis J, Schoemaker HE. Synlett. 1992;1992:451. [Google Scholar]
- 35.Examples of third-order kinetics in Friedel–Crafts:; (a) Choi SU, Brown HC. J Am Chem Soc. 1963;85:2596. [Google Scholar]; (b) DeHaan FP, Delker GL, Covey WD, Ahn J, Cowan RL, Fong CH, Kim GY, Kumar A, Roberts MP. J Org Chem. 1986;51:1587. [Google Scholar]; (c) DeHaan FP, Chan WH, Chang J, Ferrara DM, Wainschel LA. J Org Chem. 1986;51:1591. [Google Scholar]
- 36.For the salt effect, see:; (a) Fainberg AH, Winstein S. J Am Chem Soc. 1956;78:2763. [Google Scholar]; (b) McLennan DJ. Acc Chem Res. 1976;9:281. and references cited herein. [Google Scholar]
- 37.(a) Royer J, Oudeyer S, Dudot B. Heterocycles. 2005;65:823. [Google Scholar]; (b) Landelle G, Claraz A, Oudeyer S, Levacher V. Tetrahedron Lett. 2012;53:2414. [Google Scholar]
- 38.Yang C-F, Shen C, Wang J-Y, Tian S-K. Org Lett. 2012;14:3092. doi: 10.1021/ol301180z. [DOI] [PubMed] [Google Scholar]
- 39.The starting material was prepared using the following protocol:; Ratjen L, García-García P, Lay F, Beck ME, List B. Angew Chem, Int Ed. 2011;50:754. doi: 10.1002/anie.201005954. [DOI] [PubMed] [Google Scholar]
- 40.The starting material was prepared using the following protocol:; Perkins JR, Carter RG. J Am Chem Soc. 2008;130:3290. doi: 10.1021/ja7113486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.