

Abstract
The liver is a central fat‐storage organ, making it especially susceptible to steatosis as well as subsequent inflammation and cirrhosis. The mechanisms by which the liver mobilizes stored lipid for energy production, however, remain incompletely defined. The catabolic process of autophagy, a well‐known process of bulk cytoplasmic recycling and cellular self‐regeneration, is a central regulator of lipid metabolism in the liver. In the past decade, numerous studies have examined a selective form of autophagy that specifically targets a unique neutral lipid storage organelle, the lipid droplet, to better understand the function for this process in hepatocellular fatty acid metabolism. In the liver (and other oxidative tissues), this specialized pathway, lipophagy, likely plays as important a role in lipid turnover as conventional lipase‐driven lipolysis. In this review, we highlight several recent studies that have contributed to our understanding about the regulation and effects of hepatic lipophagy. (Hepatology Communications 2017;1:359–369)
Abbreviations
- Atg
autophagy‐related
- ATGL
adipose triglyceride lipase
- CE
cholesterol ester
- CMA
chaperone‐mediated autophagy
- EH
Eps15 homology
- ER
endoplasmic reticulum
- GEF
guanine exchange factor
- GNMT
glycine N‐methyltransferase
- GTPase
guanosine triphosphatase
- HCV
hepatitis C virus
- HSC
hepatic stellate cell
- Hsc70
heat shock cognate 70
- LC3
microtubule‐associated protein 1A/1B‐light chain 3
- LD
lipid droplet
- LRP1
low‐density lipoprotein receptor‐related protein 1
- NAFLD
nonalcoholic fatty liver disease
- siRNA
small interfering RNA
- SOCE
store‐operated Ca2 + entry
- SOD1
superoxide dismutase 1
- TAG
triacylglycerol
Introduction
The liver is a central site for the packaging, redistribution, and processing of fatty acids. As such, liver disease or dysfunction can lead to the progressive accumulation of fat content, a condition (hepatic steatosis) that predisposes individuals to significantly more severe sequelae. This is perhaps best exemplified in the case of nonalcoholic fatty liver disease (NAFLD), a spectrum of diseases ranging in severity from simple steatosis to steatohepatitis or cirrhosis.1 Importantly, it is believed that simple steatosis is completely reversible when patients are placed on an appropriate dietary and weight‐loss regimen.2, 3 As over 25% of the world population is estimated to have some form of fatty liver disease,4 a greater understanding of the natural history of NAFLD is therefore essential.
Under normal conditions, dietary fatty acids released into hepatic portal circulation or those released from the adipose tissue into general circulation can be taken up by the parenchymal cells of the liver, i.e., hepatocytes.5 Within the hepatocytes, fatty acids are quickly esterified to produce triglycerides (triacylglycerols [TAGs]) and cholesterol esters (CEs) in a cytoprotective mechanism that mitigates hepatocytotoxicity of free fatty acids.6, 7 This process of esterification occurs within the endoplasmic reticulum (ER) membrane through the coordinated efforts of multiple ER‐resident enzymes, including diacylglycerol acyltransferases and acylcoenzyme A:cholesterol acyltransferases.8 The net result of these anabolic processes is the deposition of neutral lipids between the ER bilayer within a lens‐like microdomain.7, 9 Recruitment of stabilizing proteins, such as seipin,10, 11, 12 and concomitant further rounds of lipid synthesis13, 14, 15 ultimately cause the outer leaflet of the ER bilayer to distend, resulting in the precipitation of unique spherical cytosolic organelles, i.e., lipid droplets (LDs), that are now known to be key sites of hepatic lipid metabolism.6
The LD represents a cellular organelle of greater complexity than was originally appreciated. Previously thought to represent an inert site of fat sequestration, LDs are now known to have distinct and dynamic proteomes that fluctuate in tandem with the metabolic state of the cell and which may define the composition of the neutral lipid stored within the organelle.16, 17, 18, 19, 20, 21, 22, 23 Significant effort has focused on understanding the biogenesis of these structures to potentially intervene in disorders of lipid dysregulation.7, 24 While substantial inroads have been made toward understanding LD biology (primarily through adipocyte‐centric studies25), the processes of LD catabolism and synthesis in the liver have remained comparatively understudied. Recent studies have begun to draw similarities in lipid metabolism between the two cell types, demonstrating parallels and distinctions between the canonical lipolytic machinery that assists in adipose tissue lipolysis and hepatic cytosolic lipolysis.26, 27, 28
Within the past decade, an alternative mechanism for hepatic LD mobilization, autophagy, has begun to materialize. Canonical macroautophagy is a tightly regulated process that involves the envelopment of cellular material within a double‐membraned structure referred to as an autophagosome.29, 30, 31, 32, 33 The coordination of autophagosome biogenesis is mediated primarily through an intricate network of greater than 30 known factors (autophagy‐related [Atg] proteins), characterized in a host of elegant yeast genetic screens performed over the past 25 years.34 Chief among these factors are Atg7, an E1‐like enzyme, and the E3‐like complex of Atg5‐Atg12‐Atg16, which mediates the conjugation of phosphatidylethanolamine onto the classical autophagosomal marker microtubule‐associated protein 1A/1B‐light chain 3 (LC3; Atg8), forming a lipidated derivative of LC3 (known as LC3‐II).34, 35, 36, 37 Cargo‐laden LC3‐positive autophagosomes are then targeted for fusion with lysosomes, forming hybrid organelles referred to as autolysosomes. This process results in the delivery of hydrolytic enzymes into the autophagosomal lumen and catabolism of cargo into macromolecular substrates that are then made available for biosynthetic and anabolic processes throughout the cell.
As part of the bulk cargo degradation that occurs during macroautophagy, convincing work has shown that LDs can be efficiently incorporated into autophagosomes, ultimately resulting in the release of free fatty acids that can then be used as fuel for mitochondrial β‐oxidation.38 Importantly, a growing body of evidence suggests that LDs can also be selectively recognized (independently from other cytoplasmic structures) by the autophagic machinery (lipophagy)39, 40, 41, 42, 43 to provide a potential mechanism for effectively regulating hepatocellular lipid levels. This review will focus on recent advances made in the fields of lipophagy and hepatic lipid metabolism.
A Key Role for Lipophagy in Hepatic Lipid Metabolism
In 2009, Singh et al.38 provided the first concrete evidence for a connection between autophagy and hepatic lipid metabolism. Treatment of hepatocytes with the lysosomal inhibitor 3‐methyladenine or small interfering RNA (siRNA)‐mediated knockdown of the autophagy regulatory gene Atg5 resulted in increased hepatic TAG content and interfered with subsequent mitochondrial β‐oxidation. Additionally, immunofluorescence imaging revealed that key components of the autophagic machinery (i.e., LC3, a classical marker of autophagic membranes, and lysosomal‐associated membrane protein 1, a lysosomal marker) exhibited a close association with LDs. Using electron microscopy, it was observed that mice fasted for as short as 6 hours had identifiable lipid content enclosed within double‐membrane autophagosomes in hepatocytes, a hallmark of macroautophagic progression. To confirm that these results were representative of hepatic lipophagy in vivo, a hepatocyte‐specific Atg7 knockout mouse was examined and found to have an enlarged liver with increased TAG accumulation following starvation when compared to control mice.38 This study was particularly noteworthy as it was previously assumed that lipid catabolism in the hepatocyte generally mimicked that found in the adipocyte, i.e., using a series of cytoplasmic lipases (i.e., adipose triglyceride lipase [ATGL], hormone‐sensitive lipase, and monoglyceride lipase) that sequentially reduce TAG into component‐free fatty acids for oxidation within mitochondria.
In further support of the Singh et al. study, a subsequent report by Kaushik and Cuervo44 revealed the potential for cooperativity between chaperone‐mediated autophagy (CMA) and macroautophagy in the clearance of hepatic LDs. CMA is a mechanism for the direct internalization of particular protein substrates bearing a canonical KFERQ motif.45, 46, 47 In the case of hepatocytes, the LD‐resident perilipin proteins PLIN2 and PLIN3, which function as barriers against unregulated lipolysis,48, 49, 50, 51 were identified to contain these motifs and shown to directly interact with heat shock cognate 70 (Hsc70), the CMA‐specific chaperone that mediates protein delivery to the lysosome.44, 52 Mutation of this pentapeptide motif in PLIN2 resulted in LD accumulation, suggesting that the removal of the perilipin coat surrounding LDs is a prerequisite for either lipolysis or classical lipophagy.
It now appears evident that coordination and crosstalk exist between the lipolytic and lipophagic pathways. For example, lipase‐driven LD breakdown was recently shown to be partially dependent on autophagy.53 In this study, cold exposure in the mediobasal hypothalamus was observed to drive lipophagy in the liver. The resulting up‐regulation of hepatic lipophagy is coupled to lipolysis as the key cytoplasmic lipases ATGL and hormone‐sensitive lipase were shown to contain motifs that allow for direct interactions with LC3; mutation of these critical motifs was sufficient to ablate ATGL‐driven lipolysis.53 Likewise, ATGL activity may also represent an important upstream checkpoint in the regulation of lipophagy; overexpression of ATGL increases hepatic lipophagy, possibly through a mechanism involving Sirtuin‐1, a nicotinamide adenine dinucleotide‐dependent protein deacetylase.54, 55 Clearly, total organismal LD turnover relies to some extent on the autophagic machinery; therefore, a key area of study for hepatocellular lipid metabolism has subsequently been focused on the regulation of lipophagy.
Regulation of Hepatic Lipophagy
As the processes of cellular lipid metabolism and autophagy are so tightly regulated, it is likely that lipophagy is coordinated at multiple fronts. As outlined below, many candidate regulatory factors may modulate this process either indirectly (as with methionine or calcium signaling) or directly (as in the case of the Rab family of small guanosine triphosphatases [GTPases]).
One example of an enzyme that may indirectly influence hepatic lipophagy is glycine N‐methyltransferase (GNMT). In mice, deficiencies in the expression of GNMT result in increased levels of circulating methionine (as well as its metabolite S‐adenosyl‐L‐methionine).56 This elevation of serum methionine leads to a corresponding reduction in autophagic flux; GNMT‐knockout mice exhibited enhanced methionine levels that resulted in autophagic inhibition due to aberrant methylation of the mammalian target of rapamycin‐interacting protein phosphatase 2A. Lysosomal inhibition in GNMT‐knockout hepatocytes did not result in further increases in cellular TAG levels, indicating that GNMT may play a key role in hepatic lipophagy. The high levels of LD content in individuals with fatty liver and GNMT deficiency may therefore perpetuate an unfavorable cycle whereby liver steatosis is exacerbated by an inability to mobilize LDs via lipophagy.
As with methionine, calcium signaling is another mechanism thought to regulate hepatic lipophagy. It is well established that impaired calcium homeostasis can have broad‐reaching effects resulting in the metabolic dysfunction underlying obesity and diabetes.57 Impairments in a process of cellular Ca2+ influx referred to as store‐operated Ca2+ entry (SOCE) were recently found to result in hepatic lipid accumulation.58 SOCE was shown to be required for proper mitochondrial function, primarily at the level of gene expression, controlling a number of critical factors participating in fatty acid oxidation and components of the electron transport chain. Furthermore, cytosolic lipase expression is impaired in SOCE‐deficient cells; therefore, mice defective in this process of Ca2+ signaling exhibit severe defects in the release of fatty acids from LDs by conventional lipolysis. Interestingly, the cells appear to compensate for the consequent increased intracellular lipid load by up‐regulating the process of lipophagy, potentially a lipotoxicity avoidance mechanism. Fibroblasts isolated from patients with loss‐of‐function mutations in STIM1 or ORAI1, two genes involved in the regulation of SOCE, also exhibited high steady‐state levels of LC3‐II and enhanced levels of autophagic flux. Enhanced autophagic vacuole formation was also observed in the skeletal muscle of SOCE‐deficient mice, reinforcing the importance of Ca2+ signaling on lipophagy not only in the liver but likely in other cell types as well.
Lysosomal calcium signaling also appears to be involved in the induction of autophagy via the control of key transcriptional regulators, implying the existence of a Ca2+‐regulated lysosome‐to‐nucleus signaling axis that helps the cell respond appropriately to nutrient demand.59, 60 Transcriptional regulation of lipophagy may thus be mediated by factors such as the transcription factor TFE3, which binds to promoters key to the process of lysosomal biogenesis.61 Lentivirus‐mediated overexpression of this transcriptional regulator in hepatoma cells results in an increased association of LC3 with LDs and reductions in total cellular lipid content.62 It is unknown, however, if similar results are observed in vivo. Similarly, knockout of TFEB, another transcriptional regulator of lysosome biogenesis, also resulted in an accumulation of hepatocellular LDs.63 Notably, overexpression of TFEB was able to rescue both obesity and metabolic syndrome in mice, lending support to the idea that the negative effects of excessive LD accumulation in the liver might be therapeutically addressed by modulation of these key Ca2+‐controlled transcriptional regulators.
Another indirect mechanism for hepatocellular lipophagy regulation relates to the consumption of caffeine. A recent study suggested that caffeine promotes autophagic flux in hepatocytes with a possible preference for LDs; electron microscopy revealed lipid content in the autophagosomes and autolysosomes of hepatocytes from caffeine‐treated mice.64, 65 Ultimately, the fatty acids liberated as a result of caffeine‐stimulated lipophagy undergo β‐oxidation within mitochondria, resulting in elevated levels of serum β‐hydroxybutyrate, an effect that was inhibited when the lysosomal inhibitor chloroquine was co‐administered to mice. The mechanisms whereby caffeine may exert these effects, however, remain undefined.
Other factors have recently been shown to participate in the regulation of hepatic lipophagy. For example, liver‐specific knockout of low‐density lipoprotein receptor‐related protein 1 (LRP1) in mice showed that, following a 24‐hour fast, a nearly 7‐fold increase in hepatic TAG accumulation was observed as compared to wild‐type mice.66 An accumulation of the autophagic cargo adapter p62 was also noted when isolated LRP–/– hepatocytes were incubated in the presence of palmitate, suggesting that global autophagy is impaired when this receptor is perturbed. However, no accumulation of LC3 was observed, suggesting that LRP1 is not involved in the biogenesis of autophagic membranes but rather that lysosome‐mediated degradation of LDs had been compromised.
A similar phenomenon was observed in the livers of fasted mice with a global knockout of superoxide dismutase 1 (SOD1).67 SOD1 converts superoxide (generated from reactive oxygen species) to hydrogen peroxide as a means of alleviating oxidative stress. Kurahashi et al.67 found that hepatocytes from SOD1‐knockout mice exhibited a ballooned phenotype. Following a 48‐hour fast, elevated levels of PLIN2, a marker of enhanced LD content, were also observed. Accumulation of LC3‐II in the fasted knockout cells was greater than that observed for fasted wild‐type cells, as was p62. Taken together, the above studies reaffirm that hepatic lipophagy can be regulated in a number of different ways. As such, identifying factors that might mediate the targeted control of this process is attractive from a therapeutic standpoint.
Role for the Rab GTPases in Hepatic Lipophagy
The identification of direct modulators of hepatocellular lipophagy has been the subject of intense recent focus. Promising examples of lipophagy‐regulating proteins are the small Rab GTPases, part of the Ras superfamily of monomeric G proteins best known as master coordinators of membrane trafficking events.68 Rab GTPases function as molecular switches that transition between GTP‐bound (“active”) and guanosine diphosphate‐bound (“inactive”) states with the assistance of guanine exchange factors (GEFs) and GTPase‐activating proteins, respectively. All Rab proteins undergo prenylation at C‐terminal cysteine residues; this is required for the Rabs to be inserted into target membranes. Once recruited to the site of action, active Rab GTPases interact with specific effector proteins, thus regulating vesicle formation, movement, tethering, and fusion.68, 69 Because lipophagy is a process that relies on coordinated membrane trafficking events, it is not surprising that numerous Rabs have been identified as part of the LD proteome.18, 20, 22, 70 However, the roles played by many of the LD‐associated Rab GTPases remain poorly defined. Below, we discuss a subset of Rab proteins (Rab7, Rab10, Rab18, and Rab32) that have been routinely found in LD proteomic screens and that are likely to play a key role in the lipophagic process.
Rab7 MEDIATES THE RECRUITMENT OF MULTIVESICULAR BODIES AND LYSOSOMES TO LDs DURING LIPOPHAGY
Rab7 is found to reside predominantly on multivesicular bodies and late endosomes and is implicated in cargo trafficking to later endocytic compartments as well as in lysosome biogenesis.71 Rab7 is also involved in the maturation of autophagic vesicles, making it an attractive candidate to facilitate LD interactions with degradative organelles and thus promote LD breakdown.72 Recently, Schroeder and coworkers73 discovered a central role for Rab7 in mediating hepatocellular lipophagy. In their study, the authors found that nutrient deprivation activates Rab7 on LDs and vesicular structures that intimately associate with LDs during starvation. It appears that active Rab7 recruits and mediates the association of multivesicular bodies and lysosomes with LDs. This role of Rab7 is essential for starvation‐induced lipophagy as loss of Rab7 function after siRNA‐mediated knockdown or expression of a dominant‐negative mutant impaired LD breakdown.73 In a follow‐up study, it was observed that chronic ethanol exposure inhibits Rab7 activity, disrupts degradative compartment morphology and motility, and causes reduced LD turnover during the process of lipophagy.74 These findings provided some additional insights into how alcohol might exacerbate the progression of fatty liver.
Rab10 MEDIATES AUTOPHAGIC ENGULFMENT OF LDs DURING LIPOPHAGY
As with Rab7, Rab10 is a Rab GTPase that repeatedly appears in LD proteomic screens. Rab10 has been previously implicated in basolateral endocytic recycling, insulin‐dependent glucose transporter type 4 trafficking to the plasma membrane, and in the regulation of ER tubule dynamics and morphology.75, 76, 77 Li et al.78 reported a novel function for Rab10 in regulating LD breakdown during lipophagy. These findings suggest that Rab10 is activated during nutrient starvation or autophagy‐inducing conditions, ultimately promoting its recruitment to membranous structures surrounding the LD surface that are positive for LC3, Atg16L1, and lysosomal‐associated membrane protein 1, thus likely representing nascent autophagic structures. Once Rab10 is activated on autophagic membranes, it binds its effector protein (endocytic adaptor Eps15 homology [EH] domain binding protein 1) as well as the membrane remodeling adenosine triphosphatase EH domain containing 2 protein. The trimeric complex extends the autophagic membranes around the LD surface, eventually resulting in complete LD engulfment within an autophagosome. The function of Rab10 lies, therefore, in promoting the autophagic engulfment of LDs during lipophagic progression. Indeed, the disruption of Rab10 function by siRNA‐mediated knockdown or expression of a dominant‐negative mutant results in LD accumulation. Interestingly, loss of Rab7 significantly impaired the downstream recruitment of Rab10 to the autophagic membranes surrounding the LD, while localization of Rab7 was not impacted by the absence of Rab10, suggesting that these two lipophagy regulators do not play overlapping roles.78
Rab18: AN EXCLUSIVE LD RESIDENT Rab GTPase
In contrast to Rab7 and Rab10, which associate with LDs as well as with lysosomal and autophagic membranes, respectively, Rab18 is thought to exist solely as an LD resident protein. Two independent groups have simultaneously reported that Rab18 directly associates with the LD monolayer in adipocyte and nonadipocyte cell models.79, 80 Using both immunofluorescence and immunogold electron microscopy, they showed that fluorescently labeled wild‐type or constitutively active (but not dominant‐negative) Rab18 mutants localize directly on the LD surface. Both groups also noted a close apposition of the ER network around the LDs in Rab18‐expressing cells, proposing that this Rab GTPase may regulate LD–ER associations. Despite its discovery on the LDs more than a decade ago, the role of Rab18 has not been completely established. The observation that Rab18 connects LDs with the ER raises a question whether Rab18 could be involved in regulating LD formation/growth at the ER. Stimulation of adipocytes with either insulin or β‐adrenergic agents increased Rab18 recruitment to the LD surface, suggestive of a potential role in mediating adipocellular lipogenic and lipolytic functions.79, 81 Perhaps the most convincing evidence for the role of Rab18 in LD homeostasis comes from patients with Warburg micro syndrome, a neurological disorder caused by mutations in Rab18.82 Treatment of fibroblasts isolated from these patients with oleic acid for 18 or 24 hours resulted in accumulation of larger LDs compared to control fibroblasts.83 Most recently, Li et al.84 discovered that transport protein particle II is recruited to the LD surface after lipid loading where it functions as an LD‐associated GEF for Rab18. Interestingly, the consequences for the loss of transport protein particle II GEF activity were comparable to those observed after Rab18 inhibition resulting in impaired lipolysis and accumulation of enlarged LDs. Further studies will prove informative in clarifying the role of Rab18 in LD biology and whether it plays any specific role in hepatic lipophagy.
A POTENTIAL ROLE FOR Rab32 IN THE REGULATION OF LIPID STORAGE DURING AUTOPHAGY
Rab32 has been shown to localize to mitochondria where it functions as an A‐kinase anchoring protein and regulates mitochondrial dynamics.85 It also participates in melanosome biogenesis by regulating post‐Golgi trafficking of melanogenic enzymes.86 The first evidence for the potential role of Rab32 in regulating LD storage came from studies done in larval adipose tissue of Drosophila. Wang et al.87 found that expression of dominant‐negative or loss‐of‐function mutants of Rab32 resulted in a reduction in both LD size and total TAG levels. The authors further reported that Rab32 localized to ring‐like structures that were positive for LysoTracker (lysosome) and LC3 markers. Intriguingly, loss of Rab32 function resulted in impaired autophagy, suggesting that Rab32 may exert its role in LD storage by regulating lipophagic LD breakdown. Such a proposed role for Rab32 is not surprising as it has been implicated previously in the formation of autophagosomes.88 Consistent with the study performed using Drosophila, Li et al.89 showed that depletion of Rab32 in mammalian hepatocyte cell models also results in reduced intracellular LD levels. Although still preliminary, these studies suggest an interesting avenue for further investigations into the role of Rab32 in mediating LD breakdown.
Other Roles for Hepatic Lipophagy
Similar to the effects of caffeine (as described above), acute exposures to alcohol appear to accelerate hepatic autophagy.90 The macroautophagy induced by ethanol appears to exhibit a particular selectivity toward mitochondria as well as LDs, indicating that mitophagy and lipophagy are up‐regulated as part of a comprehensive hepatoprotective response to acute ethanol exposure. This may perhaps be mediated through the activation of transcription factors, such as FoxO3a.91 Any potentially beneficial function of autophagy in response to short‐term alcohol intake may be a double‐edged sword, however; the chronic consumption of alcohol appears to be especially detrimental to lipophagy. Livers from patients with a history of chronic alcohol abuse are often identifiable as having microvesicular steatosis, a histologic observation that can indicate the potential for cirrhotic progression.92, 93 Chronic ethanol exposure may interfere with the nuclear localization of transcriptional lipophagic regulators, such as TFEB (as above),94 or cause the harmful modulation of key mediators of LD interactions with the lipophagic machinery (as with Rab7).74 Together, these data serve as strong evidence that lipophagy normally serves a hepatoprotective role in response to alcohol consumption. Disruption of this pathway over time, however, may provide an explanation for the prevalence of steatosis that persists with chronic alcohol abuse.
Lipophagy does not appear to be restricted to metabolism of triglyceride‐enriched LDs alone; indeed, it also appears to serve an important role in the breakdown of sterol ester‐enriched LDs. A number of disorders result in the accumulation of free cholesterol. Hepatic bile acid metabolism may therefore be another mechanism by which lipophagy is regulated. For example, elevated levels of cholesterol 7α‐hydroxylase together with decreased bile acids resulting from cholestyramine feeding were recently shown to result in increased levels of hepatic autophagy. This was shown by Wang et al.95 to promote the lipophagy‐mediated hydrolysis of CEs and the transport of lysosomal cholesterol to the ER.
Additionally, the process of lipophagy plays an important role in more than just the physiology of hepatocytes; another important liver‐resident cell type that uses lipophagy is the hepatic stellate cell (HSC). In their quiescent state, HSCs are a principal site of storage for retinol ester‐enriched LDs and vitamin A within the liver. As a consequence of tissue damage, HSCs become activated and in this process lose LDs and promote the deposition of extracellular matrix. This loss of LDs is known to partially rely on the lipophagic turnover of LDs.96, 97 Stimulation of liver injury with CCl4, a compound that promotes fibrosis, was found to increase the autophagic turnover of LDs in HSCs.97 Stellate cell‐specific inhibition of Atg7 expression was also found to attenuate liver fibrosis in vivo.
In addition, lipophagy appears to be central to the replication and function of some viruses. Severe infections with dengue virus often have hepatic involvement. Studies have reported that this particular virus is especially dependent on an up‐regulation of autophagy to drive cellular LD turnover and promote replication.98 Mechanistically, it was recently shown that dengue virus can stimulate lipophagy in HepG2 hepatoma cells via activation of 5′ adenosine monophosphate‐activated kinase.99 This activation subsequently results in inhibition of mammalian target of rapamycin and consequently increased autophagy. siRNA‐mediated silencing of 5′ adenosine monophosphate‐activated kinase resulted in a diminished capacity for catabolism of LDs and suppressed the replication of dengue virus and production of infectious virus particles. Like dengue virus, both hepatitis B100 and hepatitis C (HCV)101 viruses up‐regulate host cell autophagy as a replicative strategy. In particular, HCV appears to hijack lipophagy in order to specifically promote LD formation and accumulation as a mechanism for staging viral assembly. Virally encoded structural (core) and nonstructural (NS5A) proteins are embedded on the LD surface, allowing for efficient HCV virion production.102, 103, 104 HCV also appears to target another host cell lipophagy‐related factor, the immunity‐related GTPase family M protein, in order to promote efficient replication.105, 106 Reduced immunity‐related GTPase family M expression is capable of not only interfering with autophagic progression but also negatively impacting viral replication, suggesting an important role for this lipophagy‐related gene in the pathogenesis of hepatitis viruses.
Conclusions
During the past decade, significant progress has been made toward understanding hepatocellular lipophagy (Fig. 1). Key factors and pathways linked to the regulation of this process are being identified but are at an early stage for therapeutic intervention of complex diseases, such as NAFLD. Outstanding questions will no doubt focus on further defining the crosstalk between lipases and the core lipophagic machinery. Additionally, it will be important to reconcile studies supporting a catabolic role for lipophagy with other reports suggesting that inhibition of the autophagic machinery actually reduces hepatic steatosis (see, e.g., Kim et al.107 and Shibata et al.108). The basis for these divergent findings is currently unclear but may be derived from the variety of models, contexts of the inhibition (i.e., whole‐animal or tissue‐specific knockouts), and methods of quantification used in the evaluation of hepatic steatosis.109 Furthermore, despite progress made in understanding the interactions between organelles, such as mitochondria and the autophagic machinery, little is known regarding the LD‐specific proteins that guide the selective process of lipophagy. Identification of these factors will no doubt fuel a better understanding of the mechanisms underlying LD metabolism in the liver. Finally, it will be important to clarify the relative contributions played by various forms of lysosome‐mediated breakdown (i.e., conventional macrolipophagy versus chaperone‐mediated lipophagy and microlipophagy) in the context of hepatic lipid metabolism.
Figure 1.
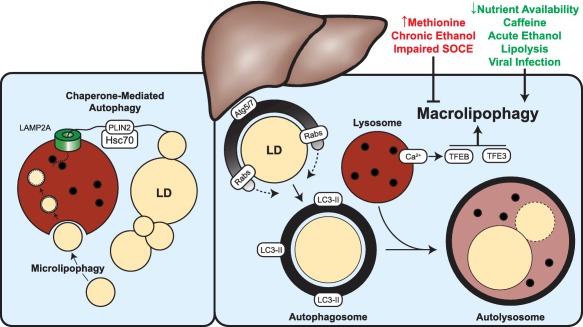
Hepatic lipophagy can be regulated at many different levels. The machinery associated with CMA appears to prime LDs for further processing in an Hsc70‐ and LAMP2A‐dependent manner. LD‐resident perilipin proteins (e.g., PLIN2) appear to be selectively recognized by the Hsc70 chaperone for targeted degradation within the lysosome, exposing the surface of the LD and rendering it susceptible to further processing by cytosolic lipases or alternative components of the autophagic machinery. Microlipophagy, a poorly understood mechanism of direct LD engulfment within the lysosome, may also play an important role in mediating the clearance of accumulated LDs. Classical macrolipophagy (or more generally, lipophagy) is a highly regulated process involving the recruitment of autophagic membranes directly to the LD, which ultimately results in the enclosure of the LD within an autophagosome. Subsequent fusion of an autophagosome with a degradative lysosome results in the formation of a hybrid structure, the autolysosome, in which LDs are ultimately catabolized. As discussed in this review, the initiation and progression of macrolipophagy can be positively and negatively influenced by numerous intracellular and extracellular factors. Abbreviation: Lamp2A, lysosomal‐associated membrane protein 2A.
Author names in bold designate shared co‐first authorship.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supported by the National Institutes of Health (T32DK007352 [R.J.S.], 5R37DK044650 [M.A.M.], and 5R01AA020735 [C.A.C. and M.A.M.]) and the Minnesota Partnership for Biotechnology and Medical Genomics (MNP #16.28 [R.J.S. and M.A.M.]).
Potential conflict of interest: Nothing to report.
REFERENCES
- 1. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA 2015;313:2263‐2273. [DOI] [PubMed] [Google Scholar]
- 2. Harrison SA, Fecht W, Brunt EM, Neuschwander‐Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: a randomized, prospective trial. Hepatology 2009;49:80‐86. [DOI] [PubMed] [Google Scholar]
- 3. Wong VW, Chan RS, Wong GL, Cheung BH, Chu WC, Yeung DK, et al. Community‐based lifestyle modification programme for non‐alcoholic fatty liver disease: a randomized controlled trial. J Hepatol 2013;59:536‐542. [DOI] [PubMed] [Google Scholar]
- 4. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 5. Bradbury MW. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: possible role in steatosis. Am J Physiol Gastrointest Liver Physiol 2006;290:G194‐G198. [DOI] [PubMed] [Google Scholar]
- 6. Walther TC, Farese RV Jr. Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 2012;81:687‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilfling F, Haas JT, Walther TC, Farese RV Jr. Lipid droplet biogenesis. Curr Opin Cell Biol 2014;29:39‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buhman KK, Chen HC, Farese RV Jr. The enzymes of neutral lipid synthesis. J Biol Chem 2001;276:40369‐40372. [DOI] [PubMed] [Google Scholar]
- 9. Khandelia H, Duelund L, Pakkanen KI, Ipsen JH. Triglyceride blisters in lipid bilayers: implications for lipid droplet biogenesis and the mobile lipid signal in cancer cell membranes. PLoS One 2010;5:e12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pagac M, Cooper DE, Qi Y, Lukmantara IE, Mak HY, Wu Z, et al. SEIPIN regulates lipid droplet expansion and adipocyte development by modulating the activity of glycerol‐3‐phosphate acyltransferase. Cell Rep 2016;17:1546‐1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salo VT, Belevich I, Li S, Karhinen L, Vihinen H, Vigouroux C, et al. Seipin regulates ER‐lipid droplet contacts and cargo delivery. EMBO J 2016;35:2699‐2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang H, Becuwe M, Housden BE, Chitraju C, Porras AJ, Graham MM, et al. Seipin is required for converting nascent to mature lipid droplets. Elife 2016;5.pii:e16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fei W, Shui G, Zhang Y, Krahmer N, Ferguson C, Kapterian TS, et al. A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLoS Genet 2011;7:e1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krahmer N, Guo Y, Wilfling F, Hilger M, Lingrell S, Heger K, et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab 2011;14:504‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell 2013;24:384‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3‐L1 adipocytes. J Biol Chem 2004;279:46835‐46842. [DOI] [PubMed] [Google Scholar]
- 17. Cermelli S, Guo Y, Gross SP, Welte MA. The lipid‐droplet proteome reveals that droplets are a protein‐storage depot. Curr Biol 2006;16:1783‐1795. [DOI] [PubMed] [Google Scholar]
- 18. Hodges BD, Wu CC. Proteomic insights into an expanded cellular role for cytoplasmic lipid droplets. J Lipid Res 2010;51:262‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang L, Ding Y, Chen Y, Zhang S, Huo C, Wang Y, et al. The proteomics of lipid droplets: structure, dynamics, and functions of the organelle conserved from bacteria to humans. J Lipid Res 2012;53:1245‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krahmer N, Hilger M, Kory N, Wilfling F, Stoehr G, Mann M, et al. Protein correlation profiles identify lipid droplet proteins with high confidence. Mol Cell Proteomics 2013;12:1115‐1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Currie E, Guo X, Christiano R, Chitraju C, Kory N, Harrison K, et al. High confidence proteomic analysis of yeast LDs identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation. J Lipid Res 2014;55:1465‐1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan SA, Wollaston‐Hayden EE, Markowski TW, Higgins L, Mashek DG. Quantitative analysis of the murine lipid droplet‐associated proteome during diet‐induced hepatic steatosis. J Lipid Res 2015;56:2260‐2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsieh K, Lee YK, Londos C, Raaka BM, Dalen KT, Kimmel AR. Perilipin family members preferentially sequester to either triacylglycerol‐specific or cholesteryl‐ester‐specific intracellular lipid storage droplets. J Cell Sci 2012;125:4067‐4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thiam AR, Farese RV, Jr. , Walther TC. The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol 2013;14:775‐786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette‐Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte‐specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem 1991;266:11341‐11346. [PubMed] [Google Scholar]
- 26. Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP. Hepatic lipid droplet biology: getting to the root of fatty liver. Hepatology 2015;62:964‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 2011;53:116‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner‐Gruenberger R, Riederer M, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004;306:1383‐1386. [DOI] [PubMed] [Google Scholar]
- 29. Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, et al. Mammalian autophagy: how does it work? Annu Rev Biochem 2016;85:685‐713. [DOI] [PubMed] [Google Scholar]
- 30. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010;221:3‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009;43:67‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 2015;16:461‐472. [DOI] [PubMed] [Google Scholar]
- 33. Noda NN, Inagaki F. Mechanisms of Autophagy. Annu Rev Biophys 2015;44:101‐122. [DOI] [PubMed] [Google Scholar]
- 34. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011;27:107‐132. [DOI] [PubMed] [Google Scholar]
- 35. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000;19:5720‐5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 2005;169:425‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004;432:1032‐1036. [DOI] [PubMed] [Google Scholar]
- 38. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011;7:279‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu K, Czaja M. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 2013;20:3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol 2012;2012:282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang CW. Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta 2016;1861:793‐805. [DOI] [PubMed] [Google Scholar]
- 43. Weidberg H, Shvets E, Elazar Z. Lipophagy: selective catabolism designed for lipids. Dev Cell 2009;16:628‐630. [DOI] [PubMed] [Google Scholar]
- 44. Kaushik S, Cuervo AM. Degradation of lipid droplet‐associated proteins by chaperone‐mediated autophagy facilitates lipolysis. Nat Cell Biol 2015;17:759‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arias E, Cuervo AM. Chaperone‐mediated autophagy in protein quality control. Curr Opin Cell Biol 2011;23:184‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996;273:501‐503. [DOI] [PubMed] [Google Scholar]
- 47. Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 1990;15:305‐309. [DOI] [PubMed] [Google Scholar]
- 48. Hickenbottom SJ, Kimmel AR, Londos C, Hurley JH. Structure of a lipid droplet protein; the PAT family member TIP47. Structure 2004;12:1199‐1207. [DOI] [PubMed] [Google Scholar]
- 49. Jiang HP, Harris SE, Serrero G. Molecular cloning of a differentiation‐related mRNA in the adipogenic cell line 1246. Cell Growth Differ 1992;3:21‐30. [PubMed] [Google Scholar]
- 50. Jiang HP, Serrero G. Isolation and characterization of a full‐length cDNA coding for an adipose differentiation‐related protein. Proc Natl Acad Sci USA 1992;89:7856‐7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kimmel AR, Brasaemle DL, McAndrews‐Hill M, Sztalryd C, Londos C. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT‐family of intracellular lipid storage droplet proteins. J Lipid Res 2010;51:468‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70‐kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989;246:382‐385. [DOI] [PubMed] [Google Scholar]
- 53. Martinez‐Lopez N, Garcia‐Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab 2016;23:113‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khan SA, Sathyanarayan A, Mashek MT, Ong KT, Wollaston‐Hayden EE, Mashek DG. ATGL‐catalyzed lipolysis regulates SIRT1 to control PGC‐1alpha/PPAR‐alpha signaling. Diabetes 2015;64:418‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sathyanarayan A, Mashek MT, Mashek DG. ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep 2017;19:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zubiete‐Franco I, García‐Rodríguez JL, Martínez‐Uña M, Martínez‐Lopez N, Woodhoo A, Juan VG, et al. Methionine and S‐adenosylmethionine levels are critical regulators of PP2A activity modulating lipophagy during steatosis. J Hepatol 2016;64:409‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Arruda AP, Hotamisligil GS. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab 2015;22:381‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maus M, Cuk M, Patel B, Lian J, Ouimet M, Kaufmann U, et al. Store‐operated Ca2 + entry controls induction of lipolysis and the transcriptional reprogramming to lipid metabolism. Cell Metab 2017;25:698‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Medina DL, Di Paola S, #Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015;17:288‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012;31:1095‐1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Martina JA, Diab HI, Lishu L, Jeong‐A L, #Patange S, Raben N, et al. The nutrient‐responsive transcription factor TFE3 promotes autophagy, #lysosomal |biogenesis, and clearance of cellular debris. Sci Signal 2014;7:ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xiong J, Wang K, He J, Zhang G, Zhang D, Chen F. TFE3 alleviates hepatic steatosis through autophagy‐induced lipophagy and PGC1α‐mediated fatty acid β‐oxidation. Int J Mol Sci 2016;17:387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. TFEB controls cellular lipid metabolism through a starvation‐induced autoregulatory loop. Nat Cell Biol 2013;15:647‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ding WX. Drinking coffee burns hepatic fat by inducing lipophagy coupled with mitochondrial β‐oxidation. Hepatology 2014;59:1235‐1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy‐lysosomal pathway in mice. Hepatology 2014;59:1366‐1380. [DOI] [PubMed] [Google Scholar]
- 66. Hamlin AN, Basford JE, Jaeschke A, Hui DY. LRP1 protein deficiency exacerbates palmitate‐induced steatosis and toxicity in hepatocytes. J Biol Chem 2016;291:16610‐16619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kurahashi T, Hamashima S, Shirato T, Lee J, Homma T, Kang ES, et al. An SOD1 deficiency enhances lipid droplet accumulation in the fasted mouse liver by aborting lipophagy. Bioch Biophys Res Commun 2015;467:866‐871. [DOI] [PubMed] [Google Scholar]
- 68. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 2009;10:513‐525. [DOI] [PubMed] [Google Scholar]
- 69. Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev 2011;91:119‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fujimoto Y, Itabe H, Sakai J, Makita M, Noda J, Mori M, et al. Identification of major proteins in the lipid droplet‐enriched fraction isolated from the human hepatocyte cell line HuH7. Biochim Biophys Acta 2004;1644:47‐59. [DOI] [PubMed] [Google Scholar]
- 71. Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell 2000;11:467‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004;117:4837‐4848. [DOI] [PubMed] [Google Scholar]
- 73. Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015;61:1896‐1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schulze RJ, Rasineni K, Weller SG, Schott MB, Schroeder B, Casey CA, et al. Ethanol exposure inhibits hepatocyte lipophagy by inactivating the small guanosine triphosphatase Rab7. Hepatol Commun 2017;1:140‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen CC, Schweinsberg PJ, Vashist S, Mareiniss DP, Lambie EJ, Grant BD. RAB‐10 is required for endocytic recycling in the Caenorhabditis elegans intestine. Mol Biol Cell 2006;17:1286‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. English AR, Voeltz GK. Rab10 GTPase regulates ER dynamics and morphology. Nat Cell Biol 2013;15:169‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE. Rab10 in insulin‐stimulated GLUT4 translocation. Biochem J 2008;411:89‐95. [DOI] [PubMed] [Google Scholar]
- 78. Li Z, Schulze RJ, Weller SG, Krueger EW, Schott MB, Zhang X, et al. A novel Rab10‐EHBP1‐EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci Adv 2016;2:e1601470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Martin S, Driessen K, Nixon SJ, Zerial M, Parton RG. Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J Biol Chem 2005;280:42325‐42335. [DOI] [PubMed] [Google Scholar]
- 80. Ozeki S, Cheng J, Tauchi‐Sato K, Hatano N, Taniguchi H, Fujimoto T. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum‐derived membrane. J Cell Sci 2005;118:2601‐2611. [DOI] [PubMed] [Google Scholar]
- 81. Pulido MR, Diaz‐Ruiz A, Jimenez‐Gomez Y, Garcia‐Navarro S, Gracia‐Navarro F, Tinahones F, et al. Rab18 dynamics in adipocytes in relation to lipogenesis, lipolysis and obesity. PLoS One 2011;6:e22931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bem D, Yoshimura S, Nunes‐Bastos R, Bond FC, Kurian MA, Rahman F, et al. Loss‐of‐function mutations in RAB18 cause Warburg micro syndrome. Am J Hum Genet 2011;88:499‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liegel RP, Handley MT, Ronchetti A, Brown S, Langemeyer L, Linford A, et al. Loss‐of‐function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans. Am J Hum Genet 2013;93:1001‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li C, Luo X, Zhao S, Siu GK, Liang Y, Chan HC, et al. COPI‐TRAPPII activates Rab18 and regulates its lipid droplet association. EMBO J 2017;36:441‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Alto NM, Soderling J, Scott JD. Rab32 is an A‐kinase anchoring protein and participates in mitochondrial dynamics. J Cell Biol 2002;158:659‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wasmeier C, Romao M, Plowright L, Bennett DC, Raposo G, Seabra MC. Rab38 and Rab32 control post‐Golgi trafficking of melanogenic enzymes. J Cell Biol 2006;175:271‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang C, Liu Z, Huang X. Rab32 is important for autophagy and lipid storage in Drosophila. PLoS One 2012;7:e32086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hirota Y, Tanaka Y. A small |GTPase, #human Rab32, is required for the formation of autophagic vacuoles under basal conditions. Cell Mol Life Sci 2009;66:2913‐2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li Q, Wang J, Wan Y, Chen D. Depletion of Rab32 decreases intracellular lipid accumulation and induces lipolysis through enhancing ATGL expression in hepatocytes. Biochem Biophys Res Commun 2016;471:492‐496. [DOI] [PubMed] [Google Scholar]
- 90. Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol‐induced hepatotoxicity and steatosis in mice. Gastroenterology 2010;139:1740‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ni HM, Du K, You M, Ding WX. Critical role of FoxO3a in alcohol‐induced autophagy and hepatotoxicity. Am J Pathol 2013;183:1815‐1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995;346:987‐990. [DOI] [PubMed] [Google Scholar]
- 93. Yeh MM, Brunt EM. Pathological features of fatty liver disease. Gastroenterology 2014;147:754‐764. [DOI] [PubMed] [Google Scholar]
- 94. Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM Jr. Acute and chronic ethanol administration differentially modulate hepatic autophagy and transcription factor EB. Alcohol Clin Exp Res 2015;39:2354‐2363. [DOI] [PubMed] [Google Scholar]
- 95. Wang Y, Ding Y, Li J, Chavan H, Matye D, Ni HM, et al. Targeting the enterohepatic bile acid signaling induces hepatic autophagy via a CYP7A1‐AKT‐mTOR axis in mice. Cell Mol Gastroenterol Hepatol 2016;3:245‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Thoen LF, Guimaraes EL, Dolle L, Mannaerts I, Najimi M, Sokal E, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol 2011;55:1353‐1360. [DOI] [PubMed] [Google Scholar]
- 97. Hernandez‐Gea V, Ghiassi‐Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012;142:938‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Heaton NS, Randall G. Dengue virus‐induced autophagy regulates lipid metabolism. Cell Host Microbe 2010;8:422‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jordan TX, Randall G. Dengue virus activates the AMP kinase‐mTOR axis to stimulate a proviral lipophagy. J Virol 2017;91.pii:e02020‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A 2010;107:4383‐4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 2009;106:14046‐14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Harris C, Herker E, Farese RV Jr, Ott M. Hepatitis C virus core protein decreases lipid droplet turnover: a mechanism for core‐induced steatosis. J Biol Chem 2011;286:42615‐42625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Herker E, Ott M. Unique ties between hepatitis C virus replication and intracellular lipids. Trends Endocrinol Metab 2011;22:241‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 2007;9:1089‐1097. [DOI] [PubMed] [Google Scholar]
- 105. Gregoire IP, Richetta C, Meyniel‐Schicklin L, Borel S, Pradezynski F, Diaz O, et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog 2011;7:e1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lin Y‐C, Chang P‐F, Lin H‐F, Liu K, Chang M‐H, Ni Y‐H. Variants in the autophagy‐related gene IRGM confer susceptibility to non‐alcoholic fatty liver disease by modulating lipophagy. J Hepatol 2016;65:1209‐1216. [DOI] [PubMed] [Google Scholar]
- 107. Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 2013;19:83‐92. [DOI] [PubMed] [Google Scholar]
- 108. Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, et al. The MAP1‐LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 2009;382:419‐423. [DOI] [PubMed] [Google Scholar]
- 109. Cingolani F, Czaja MJ. Regulation and functions of autophagic lipolysis. Trends Endocrinol Metab 2016;27:696‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]