

Abstract
Disorders of protein metabolism are the most common diseases among discovered inherited metabolic disorders. Phenylketonuria (PKU), a relatively common disorder that is responsive to treatment, is an inherited autosomal recessive disorder caused by a deficiency in phenylalanine hydroxylase (PAH) or one of several enzymes mediating biosynthesis or regeneration of the PAH cofactor tetrahydrobiopterin. The objective of this review is to discuss therapeutic strategies that have recently emerged for curing patients with PKU, which have demonstrated promising improvements in managing these patients. Data sourcing included a systematic literature review of PubMed with a focus on emerging knowledge pertaining to this well-studied disease. Recent advances in laboratory diagnosis and therapeutic strategies were described. Collectively, promising and rapid enhancements in neonatal diagnostic technologies and recently emerged therapeutic strategies are paving the way for early diagnosis and treating many inborn errors of metabolism, such as PKU.
Keywords: BH4, gene therapy, molecular diagnosis, phenylalanine hydroxylase, phenylketonuria
Introduction
Inborn errors of metabolism (IEM) involve a heterogeneous group of rare genetic disorders, which are commonly caused by mutant genes, resulting in abnormal proteins that interrupt a metabolic pathway, with clinical manifestations. Almost all known IEMs are inherited in an autosomal-recessive or X-linked-recessive manner. The protein expressed from a defective gene may cause partial deficiency in enzyme activity or a complete loss of function. Moreover, problems in most IEMs result from the toxic effects of accumulated substances, their interference with normal functions, or a reduced capacity for synthesizing essential compounds.
Conventionally, inherited metabolic diseases have been divided into 4 categories: Disorders of amino acid metabolism, carbohydrate metabolism, lysosomal storage, and organic acid metabolism.1 The classification of these diseases has been expanded to include more than 10 categories, based on the recent discovery of several inherited metabolic disorders. The most common diseases among these new categories are disorders of protein metabolism. Remarkably, phenylketonuria (PKU), a treatment-responsive and comparatively common disorder, is considered the most important disease among the known disorders of amino acid metabolism.
In this review, we discuss emerging knowledge on this well-studied disease and describe recent advances in therapeutic strategies.
Phenylalanine (Phe) Metabolism Requires Several Components
Characteristics of Phe and tyrosine (Tyr)
Phe (C9H11NO2) is an essential α-amino acid with a nonpolar side chain. Like all other nonpolar amino acids, Phe is an electrically neutral amino acid that facilitates hydrophobic interactions. As shown in Figure 1, Phe is a precursor of Tyr, highlighting its importance in cellular processes. In contrast to Phe, Tyr is a nonessential α-amino acid with a molecular formula of C9H11NO3. This proteinogenic amino acid is uncharged but has a polar side chain. Tyr is a critical amino acid owing to its various functions. For example, Tyr residues are often phosphorylated on the side chain hydroxyl group, which alters the activity of the target protein. Thus, Tyr phosphorylation is a key step in regulating enzymatic activity and signal transduction. Furthermore, the normal metabolism of Tyr results in its conversion into other compounds, as discussed.
Figure 1.
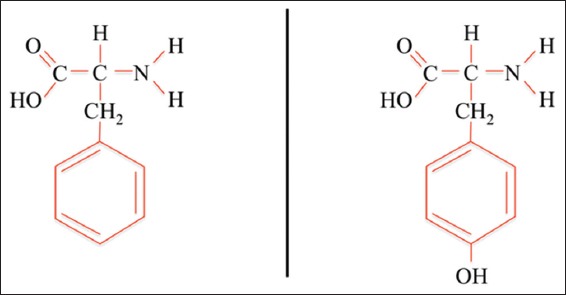
Phenylalanine (left) and tyrosine (right). Tyrosine is generated from phenylalanine by hydroxylation of the phenylalanine benzene ring, catalyzed by phenylalanine hydroxylase
Phenylalanine hydroxylase (PAH)
Phe hydroxylation is catalyzed by PAH, which generates Tyr.2 PAH belongs to a class of monooxygenases named pterin-dependent amino acid hydroxylases; this class contains 3 members (PAH, tryptophan hydroxylase, and tyrosine hydroxylase) that utilize tetrahydrobiopterin 4 (BH4, a pteridine cofactor) and a non-heme iron for catalysis.3
The PAH gene
The human PAH gene (hPAH) is located on chromosome 12q22-12q24, as confirmed by mapping studies.4,5 Kwok et al. isolated full-length hPAH complementary DNA (cDNA) from a human liver cDNA library.6 In addition, digestion of the cDNA clone as a probe for full-length hPAH by several restriction enzymes showed that this gene is comprised of 13 exons and spans 90 kb.7,8 As shown in Figure 2, the entire PAH chromosomal sequence spans 171 kb, including its flanking regions. Within this sequence, approximately 92 kb comprises the un-translated regions (UTRs), with 27 kb in the 5′-UTR, and 65 kb in the 3′-UTR.9
Figure 2.
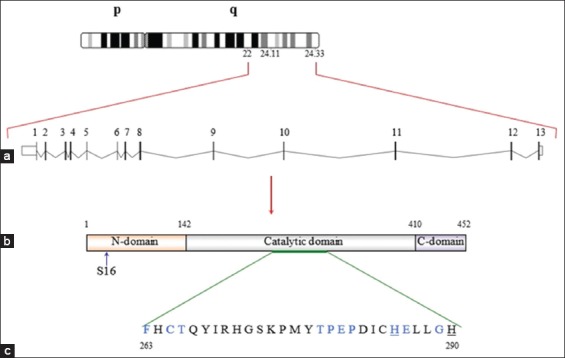
A diagram of the basic structure of the human PAH (hPAH) gene and its encoded protein. (a) The basic structure of the hPAH gene. The gene is located on the long arm of chromosome 12 (102, 836, 326-102, 917, 603) with a genomic size of 79,278 bp. (b) The PAH transcript (length: 4,122 bp) contains 13 exons that encode the 452-amino acid PAH enzyme. (c) The catalytic domain contains a motif of 10 amino acid residues (in blue) and 2 residues (underlined) responsible for BH4 and ferric iron binding, respectively
Structure and function of the PAH enzyme
Homotetrameric and homodimeric forms of eukaryotic PAH have been shown to exist in a pH-dependent equilibrium. Both forms are catalytically active, have different kinetics, and undergo different regulation.10,11 However, the 51.9-kDa PAH monomer protein (452 amino acid residues; National Center of Biotechnology Information Reference Sequence: NP_000268.1) contains 3 domains: An NH2-terminal regulatory domain that represents approximately 26% of the total amino acid residues (117 residues); a catalytic domain that represents 69% of the entire molecule (310 residues); and a COOH-terminal domain containing 25 residues (<6%) responsible for oligomerization (Figure 2).12,13
The NH2-terminal domain of PAH has a βαββαβ topology comprised of an interlocking double-βαβ arrangement, which confers structural flexibility and facilitates the regulatory nature of the domain. Structure-function studies of the human PAH have shown that the activity of the entire enzyme can be affected by alterations in hydrophobic interactions in the NH2-terminal domain.14 Interestingly, the auto regulatory sequence of the NH2-domain (residues 19-33) extends across the catalytic domain at the active site although the regulatory domain is located far from the active site pocket of the catalytic domain.12,14,15 Data from several studies have shown that the conformation of PAH is altered globally by Phe allosteric binding and that a 2-fold increase in Phe affinity, along with elimination of the lag time, is induced by removal of the PAH NH2-terminus.15-17 The serine (Ser) residue at position 16 (Ser16) within the PAH NH2-terminal residues also regulates the enzyme. The Phe concentration required for allosteric activation was reduced by post-transcriptional Ser16 phosphorylation by cyclic adenosine monophosphate-dependent protein kinase.12 Furthermore, data from an in vitro study of human PAH have shown that the basal activity of PAH and its affinity for the substrate Phe were increased by substitution of Ser16 with a polar amino acid having an acidic side chain, such as glutamic acid (Glu) or, to some extent, aspartic acid.18 The NH2-terminus is attached to the catalytic domain through a hinge region (residues 111-117).19 Analysis of the crystal structure of the PAH catalytic domain showed that this region consists of a basket-like structure formed by 13 α-helices and 8 β-strands. The active site within this structure contains a pocket lined by 34 amino acid residues and is situated at the center. Six of these 34 residues are polar, including 3 Glu residues, 2 histidine residues, and 1 Tyr residue; however, most residues are nonpolar (hydrophobic). Pterin- and iron-binding residues are also contained in this region.11,20 The COOH-terminal domain is responsible for oligomerization of PAH monomers.13
With respect to its expression and activity, PAH is distinct among the aromatic amino acid hydroxylases. The central nervous system is the principal site of tryptophan (Trp) and Tyr hydroxylase expression, whereas the liver and kidney are major sites of PAH expression.21 PAH catalyzes catabolic processes, while Trp and Tyr hydroxylases catalyze rate-limiting steps in neurotransmitter and hormone biosynthesis.19
Several biological functions are performed by PAH. For example, PAH plays a critical role in Phe metabolism. Moreover, PAH converts most ingested Phe to Tyr, some of which is utilized for protein synthesis.2 Phe turnover is an alternative pathway through which ingested Phe is converted into phenylpyruvate and finally excreted in the urine. The rate-limiting step, which generates CO2 and water, is also catalyzed by PAH.22
BH4 is an important cofactor
BH4 is an essential, naturally occurring cofactor that plays various roles in human biochemistry. BH4 is biosynthesized de novo from its precursor molecule guanosine triphosphate through Mg2+-, Zn2+-, and nicotinamide adenine dinucleotide phosphate-dependent chemical reactions. As shown in Figure 3, dihydropteridine reductase uses the quinoid form of 7, 8-dihydrobipterin to catalyze the regeneration of BH4.23 Functionally, BH4 acts as a cofactor in the conversion of Phe to Tyr through the catalytic activity of PAH24 and in the production of neurotransmitters. BH4 also serves as a catalyst for nitric oxide synthase during synthesis of the messenger molecule nitric oxide from the amino acid arginine.25 Moreover, BH4 catalyzes the hydroxylation of alkylglycerol by alkylglycerol monooxygenase.26 Thus, insufficient biosynthesis and/or regeneration of this critical cofactor may lead to the development of PKU,27 hereditary progressive dystonia with marked diurnal fluctuation,28 or neurovascular dysfunction.29
Figure 3.
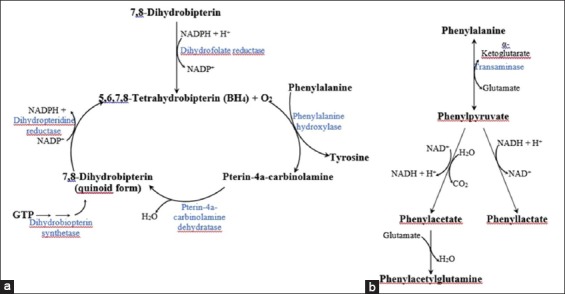
Phenylalanine metabolic pathways. A schematic representation of the normal Phe metabolic pathway is shown in (a). (b) The alternative pathway of Phe metabolism, due to abnormalities in the normal Phe metabolic pathway or an excess of dietary Phe
Normal and abnormal Phe/Tyr metabolism
Figure 3 illustrates the metabolic pathways of Phe. The production of Tyr is the terminal step in the normal metabolism of Phe, catalyzed by PAH. In turn, Tyr participates in a network of biological processes. Many metabolites, including catecholamines, melanin, and thyroid hormones (triiodothyronine and thyroxine), are involved in this network. Transamination eliminates the excess of dietary Phe by converting it into phenylpyruvate. In the context of protein metabolic diseases, however, abnormalities in Phe metabolism and its products may occur due to either a deficiency in the substrate or a defect in PAH or its cofactors.
PKU as an Inherited Disorder
PKU is a disorder of Phe metabolism that is mainly caused by deficiencies in PAH or one of the enzymes involved in the synthesis or regeneration of its cofactor BH4, as shown in Figure 3.30 PKU is an inherited autosomal recessive disorder, meaning that the biochemical or clinical features of PKU do not appear in individuals in whom only 1 allele carries the mutation (disease carriers); instead, the mutation must be present in both alleles for PKU to manifest.31
PKU classification
Tolerance to dietary Phe or blood Phe concentrations provides a basis for PKU classification, as summarized in Table 1.31 High blood Phe concentrations (hyperphenylalaninemia [HPA]) have also been demonstrated in approximately 1-2% of patients with PKU and occurs as a result of mutations in 1 of the genes coding for enzymes involved in BH4 biosynthesis or regeneration.32 However, HPA has been reported to be absent in some patients with PKU.33
Table 1.
PKU classification

Incidence
The incidence of PKU varies depending on ethnicity, and 1 in 13,500-19,000 newborns in the United States of America (USA) has PKU,34 whereas the African-American population exhibits PKU at a rate of 1 in 50,000 individuals.35 Finland and Japan have been reported to show the lowest incidence of PKU (<1 in 100,000 and 1 in 108,823 individuals, respectively).36,37 In contrast, high rates of PKU have been reported in populations such as Yemenite Jews in Israel (1 in 5,300 individuals), Scottish populations (1 in 5,300 individuals), Arabic populations (up to 1 in 6,000 individuals), and Czechoslovakian populations (1 in 7,000 individuals). The highest rate of PKU was reported for the Turkish population (1 in 2,600 newborns).38
Diagnosis
Clinical presentation
Infants with PKU are often asymptomatic before consuming food containing Phe and may not be identified by newborn screening,39 explaining the insidious onset of PKU, where symptoms may not appear until early infancy. In general, untreated infants and children with PKU develop a range of impairments, including behavioral, mental, neurological, and physical symptoms. Almost all untreated PKU patients demonstrate behavioral impairments, including aggressiveness, anxiety, hyperactivity, purposeless movements, social withdrawal, and stereotypy. Furthermore, severe mental retardation is the most common outcome and is usually associated with eczema, growth reduction and microcephaly, reduced blond hair, pigmentation of the iris and skin, and a mousy urine odor. The mental retardation observed in patients with PKU is also associated with neurological impairments, such as electroencephalogram abnormalities, epilepsy, gait and tic abnormalities, hyperreflexia, Parkinsonian signs, pyramidal signs, limb spasticity, and tremors. In addition, the brains of untreated PKU patients exhibit disrupted myelination, impaired synaptogenesis, and reduced dendrite arborization.40
Biochemical and molecular genetic diagnosis
PKU diagnosis is performed by detecting an elevated serum Phe concentration; this is the standard method for confirming the positive neonatal screening results.31 Tandem mass spectrometry is one of the most useful laboratory tools for measuring various analytes, including Phe and Tyr, in a single sample.41 PKU diagnosis is also supported by normal or low Tyr levels, in addition to high serum Phe concentrations. Table 1 summarizes the different severities of HPA. All neonates with high serum Phe concentrations should also be evaluated for Pterin disorders by analysis of the urine or blood.32,42 Enzyme activity analysis can be performed to determine the activities of the enzymes involved in BH4 synthesis and regeneration. However, PAH activity cannot be analyzed effectively in the urine or blood because PAH activity is typically only observed in hepatic and renal cells.21,43
PKU diagnosis should be confirmed by molecular analysis. In families with confirmed affected individuals, carriers can be identified by genetic testing. The mutation profile of the gene(s) involved in PKU is limited to certain regions, which are distributed among the structural domains. However, the position and nature of any of these mutations determine the effects of the mutations on enzyme activity, yielding patients with different clinical and biochemical phenotypes.30,44,45
Several approaches have been established for molecular genetics testing. However, variations in the number of mutations within a population have been observed in different countries, and diagnosis based on molecular genetics requires knowledge of the specific mutations present in a certain population. Thus, Sanger sequencing is the method of choice for detecting causative mutations. Because of the large number of known mutations in suspected genes, confirmation of such a diagnosis can be achieved by high-throughput analysis using advanced molecular genetic testing technologies, such as next generation sequencing (NGS). NGS can also be applied as a discovery tool at the exome and genome levels.46-50
Current Strategies for PKU Treatment
PKU is the first metabolic disorder found to cause mental retardation.51 Numerous therapeutic strategies for treating PKU have emerged in recent years.52 In this section, we present current knowledge regarding these therapeutic strategies.
Treatment with a Phe-restricted diet
The management of PKU with dietary habits was established in the mid-1900s.53 Patients with PKU were restricted to diets containing low Phe concentrations for life. Severe mental retardation can be effectively prevented by following this diet plan. However, this approach is also associated with a risk for nutritional deficiencies.54 For example, growth retardation and early-onset osteoporosis can be caused by deficiencies in specific substances, such as minerals or vitamins, in a Phe-restricted diet.55-58 Moreover, huge social and economic burdens are imposed by the Phe-restricted diet.59 In addition, compared with age-matched individuals without PKU, patients with PKU who consume a Phe-restricted diet may not achieve their full neurodevelopment potential.54,60-62 Accordingly, various alternative therapeutic strategies have been proposed for PKU treatment.
Treatment with a BH4 chaperone
Although the vast majority of patients with PKU are successfully treated through dietary changes,63,64 treatment with a Phe-restricted diet is ineffective in patients with PKU exhibiting BH4 deficiencies. The low activity of a defective enzyme in a metabolic pathway can be increased and the flux can be improved after stabilizing the enzyme conformation with chaperones, and enzyme cofactors can typically function as chaperones.65 Furthermore, BH4 can help stabilize misfolded mutant enzymes and prevent their proteolysis,51 and increase enzyme activity was demonstrated by changing the dose and formulation of BH4. In patients designated as BH4-responsive patients, high BH4 doses correlate with increased PAH activity.52 These patients exhibit improved responses to BH4 therapy and milder disease severity than patients lacking mutations. Thus, the patient’s phenotype can form the basis of his/her responses to BH4 therapy.51
A BH4-loading test was performed to distinguish patients with BH4-related metabolic defects from those with deficiencies in PAH activity.66 Measurement of the enzymatic activity of some recycling enzymes responsible for BH4 could help identify the former group of patients.64 BH4 responses have been found in 10–60% of patients with a deficiency in PAH activity.67-71 Moreover, BH4 is commercially available and is prepared synthetically. In addition, in 2007, sapropterin dihydrochloride was approved in the USA as an adjuvant therapy for PKU. Furthermore, when administered at doses of 5-25 mg/(kg day) for up to 22 weeks, BH4 caused a decrease in the plasma Phe concentration in approximately 32-50% of treated participants, as illustrated by analyzing the effects of cofactor treatment in clinical trials.65
Large neutral amino acid (LNAA) therapy
Another encouraging approach for treating PKU is dietary supplementation with LNAAs. LNAAs can be administered to adult patients with PKU in whom a Phe-restricted diet is not tolerable.72 Four L-type amino acid transporters are present in the transport system through which certain molecules, such as LNAAs, are transported to the brain.73 LNAAs, including the branched-chain amino acids valine, leucine, and isoleucine; the aromatic amino acids Tyr, Trp, and Phe; and some other amino acids, such as threonine, methionine, and His, are transported by these L transporters.74 Thus, to cross the blood–brain barrier, these molecules compete with each other to bind the transporters based on their plasma concentrations. High LNAA supplementation thus inhibits plasma Phe transport and reduces brain Phe concentrations.75 The impact of LNAA supplementation has been shown in multiple studies.76,77
Enzyme therapy
Another approach for treating PKU is enzyme therapy. Irrespective of the disease genotype, according to this approach, alterations in the PAH metabolic phenotype can reduce harmfully high Phe levels in the plasma. Enzyme substitution with Phe ammonia lyase (PAL) or PAH are the 2 available types of enzyme therapy.78 Replacement with PAH-fusion proteins has been shown effective in mouse studies.79 However, PAL-replacement therapy appears to be a more promising approach. In addition to substitution to complement a PAH deficiency, this therapy also converts excess Phe into readily excreted and less toxic products. However, mouse models of PKU have shown that administering injectable or oral version of the PAL enzyme has some limitations.80 For example, injectable PAL activates immune responses, whereas oral administration of PAL is associated with enzyme degradation, thereby reducing the effectiveness of the therapy. Notably, conjugation of PAL with polyethylene glycol (PEG) decreased PAL-induced immune responses.81 Moreover, clinical trials on PKU patients are promising. Phase I and II trials using injectable recombinant PAL conjugated with PEG have shown a reduction in the Phe levels.82,83
Gene therapy
PKU gene therapy has been a major focus of various research groups for the past 2 decades. Profound progress in hepatic gene therapy with adenoviral vectors was demonstrated in PKU mouse models.84,85 However, in contrast to studies of hepatocytes in PKU-model mice, adenovirus-associated vectors could not integrate into the DNA of human hepatocytes, in some studies. While previous studies have not shown success in incorporating the PAH gene into human hepatocytes using adenovirus-associated vectors, other vectors such as lentiviral vectors have demonstrated a stable transduction of the gene of interest in various cell types, which can be used to transduce and stably express PAH in human hepatocytes.
Muscle cells may be attractive targets for gene therapy. Successful gene delivery into murine skeletal muscle cells has been demonstrated in several studies.86,87 Indeed, normal hepatic Phe metabolism has been mimicked in a system involving gene delivery to muscle cells.88 Surprisingly, success regarding other IEMs has been shown in gene therapy trials by exploiting advancements in viral vector design. For example, gene therapy has been shown effective in treating aminoacylase-2 deficiency (known as Canavan disease), alpha 1-antitrypsin deficiency, late infantile variant of neuronal ceroid lipofuscinosis, and familial chylomicronemia syndrome (also known as lipoprotein lipase deficiency).88 Human PKU gene therapy trials may emerge as a result of promising and rapid enhancements in gene therapy studies.
Conclusions
Newborn screening helps in the early establishment of a Phe-restrictive diet and the possibility of avoiding brain damage resulting from HPA in patients with PKU. However, the difficulties of maintaining a strict life-long diet and the occurrence of other complications (despite treatment) make the investigation of new therapeutic strategies of great importance. Moreover, information regarding PKU pathogenesis has increased over the past decade, enabling the development of various novel therapeutic strategies. New treatments, therefore, will eventually become available for addressing the observed HPA in patients with PKU, enabling personalized therapy based on individual genotypes and other specific conditions. Molecular diagnosis by high-throughput sequencing techniques also helps confirm neonatal laboratory-screening results and facilitates the discovery of novel mutations associated with PKU. However, many concerns regarding existing therapeutic strategies need to be addressed and much effort has to be exerted before many of these new therapies can become available for patients. The increasingly competitive pricing and accuracy of NGS for molecular diagnostics has begun to show promise in replacing confirmatory biochemical screening for PKU in newborns in the near future.
References
- 1.Das SK. Inborn errors of metabolism: Challenges and management. Indian J Clin Biochem. 2013;28:311–3. doi: 10.1007/s12291-013-0371-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaufman S. The phenylalanine hydroxylating system from mammalian liver. Adv Enzymol Relat Areas Mol Biol. 1971;35:245–319. doi: 10.1002/9780470122808.ch6. [DOI] [PubMed] [Google Scholar]
- 3.Fitzpatrick PF. Tetrahydropterin-dependent amino acid hydroxylases. Annu Rev Biochem. 1999;68:355–81. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 4.Lidsky AS, Law ML, Morse HG, Kao FT, Rabin M, Ruddle FH, et al. Regional mapping of the phenylalanine hydroxylase gene and the phenylketonuria locus in the human genome. Proc Natl Acad Sci U S A. 1985;82:6221–5. doi: 10.1073/pnas.82.18.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lidksy AS, Robson KJ, Thirumalachary C, Barker PE, Ruddle FH, Woo SL. The PKU locus in man is on chromosome 12. Am J Hum Genet. 1984;36:527–33. [PMC free article] [PubMed] [Google Scholar]
- 6.Kwok SC, Ledley FD, DiLella AG, Robson KJ, Woo SL. Nucleotide sequence of a full-length complementary DNA clone and amino acid sequence of human phenylalanine hydroxylase. Biochemistry. 1985;24:556–61. doi: 10.1021/bi00324a002. [DOI] [PubMed] [Google Scholar]
- 7.Güttler F, Woo SL. Molecular genetics of PKU. J Inherit Metab Dis. 1986;9(Suppl1):58–68. doi: 10.1007/BF01800859. [DOI] [PubMed] [Google Scholar]
- 8.Konecki DS, Schlotter M, Trefz FK, Lichter-Konecki U. The identification of two mis-sense mutations at the PAH gene locus in a Turkish patient with phenylketonuria. Hum Genet. 1991;87:389–93. doi: 10.1007/BF00197153. [DOI] [PubMed] [Google Scholar]
- 9.Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat. 2007;28:831–45. doi: 10.1002/humu.20526. [DOI] [PubMed] [Google Scholar]
- 10.Bjørgo E, de Carvalho RM, Flatmark T. A comparison of kinetic and regulatory properties of the tetrameric and dimeric forms of wild-type and Thr427-->Pro mutant human phenylalanine hydroxylase: Contribution of the flexible hinge region Asp425-Gln429 to the tetramerization and cooperative substrate binding. Eur J Biochem. 2001;268:997–1005. doi: 10.1046/j.1432-1327.2001.01958.x. [DOI] [PubMed] [Google Scholar]
- 11.Erlandsen H, Patch MG, Gamez A, Straub M, Stevens RC. Structural studies on phenylalanine hydroxylase and implications toward understanding and treating phenylketonuria. Pediatrics. 2003;112:1557–65. [PubMed] [Google Scholar]
- 12.Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD, et al. Structural basis of autoregulation of phenylalanine hydroxylase. Nat Struct Biol. 1999;6:442–8. doi: 10.1038/8247. [DOI] [PubMed] [Google Scholar]
- 13.Fusetti F, Erlandsen H, Flatmark T, Stevens RC. Structure of tetrameric human phenylalanine hydroxylase and its implications for phenylketonuria. J Biol Chem. 1998;273:16962–7. doi: 10.1074/jbc.273.27.16962. [DOI] [PubMed] [Google Scholar]
- 14.Carluccio C, Fraternali F, Salvatore F, Fornili A, Zagari A. Structural features of the regulatory ACT domain of phenylalanine hydroxylase. PLoS One. 2013;8:e79482. doi: 10.1371/journal.pone.0079482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Dangott LJ, Fitzpatrick PF. Regulation of phenylalanine hydroxylase: Conformational changes upon phenylalanine binding detected by hydrogen/deuterium exchange and mass spectrometry. Biochemistry. 2010;49:3327–35. doi: 10.1021/bi1001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daubner SC, Hillas PJ, Fitzpatrick PF. Expression and characterization of the catalytic domain of human phenylalanine hydroxylase. Arch Biochem Biophys. 1997;348:295–302. doi: 10.1006/abbi.1997.0435. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Ilangovan U, Daubner SC, Hinck AP, Fitzpatrick PF. Direct evidence for a phenylalanine site in the regulatory domain of phenylalanine hydroxylase. Arch Biochem Biophys. 2011;505:250–5. doi: 10.1016/j.abb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miranda FF, Teigen K, Thórólfsson M, Svebak RM, Knappskog PM, Flatmark T, et al. Phosphorylation and mutations of Ser(16) in human phenylalanine hydroxylase. Kinetic and structural effects. J Biol Chem. 2002;277:40937–43. doi: 10.1074/jbc.M112197200. [DOI] [PubMed] [Google Scholar]
- 19.Flydal MI, Martinez A. Phenylalanine hydroxylase: Function, structure, and regulation. IUBMB Life. 2013;65:341–9. doi: 10.1002/iub.1150. [DOI] [PubMed] [Google Scholar]
- 20.Hufton SE, Jennings IG, Cotton RG. Structure and function of the aromatic amino acid hydroxylases. Biochem J. 1995311:353–66. doi: 10.1042/bj3110353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lichter-Konecki U, Hipke CM, Konecki DS. Human phenylalanine hydroxylase gene expression in kidney and other nonhepatic tissues. Mol Genet Metab. 1999;67:308–16. doi: 10.1006/mgme.1999.2880. [DOI] [PubMed] [Google Scholar]
- 22.Kaufman S. A model of human phenylalanine metabolism in normal subjects and in phenylketonuric patients. Proc Natl Acad Sci U S A. 1999;96:3160–4. doi: 10.1073/pnas.96.6.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thöny B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347:1–16. [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufman S. A new cofactor required for the enzymatic conversion of phenylalanine to tyrosine. J Biol Chem. 1958;230:931–9. [PubMed] [Google Scholar]
- 25.Tejero J, Stuehr D. Tetrahydrobiopterin in nitric oxide synthase. IUBMB Life. 2013;65:358–65. doi: 10.1002/iub.1136. [DOI] [PubMed] [Google Scholar]
- 26.Tietz A, Lindberg M, Kennedy EP. A new pteridine-requiring enzyme system for the oxidation of glyceryl ethers. J Biol Chem. 1964;239:4081–90. [PubMed] [Google Scholar]
- 27.Scriver CR, Clow CL. Phenylketonuria and other phenylalanine hydroxylation mutants in man. Annu Rev Genet. 1980;14:179–202. doi: 10.1146/annurev.ge.14.120180.001143. [DOI] [PubMed] [Google Scholar]
- 28.Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain Dev. 2011;33:195–201. doi: 10.1016/j.braindev.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Wu G, Meininger CJ. Nitric oxide and vascular insulin resistance. Biofactors. 2009;35:21–7. doi: 10.1002/biof.3. [DOI] [PubMed] [Google Scholar]
- 30.Blau NT, Cotton RG, Hyland K. Disorders of tetrahydrobiopterin and related biogenic amines. In: Scriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001. pp. 1725–76. [Google Scholar]
- 31.Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376:1417–27. doi: 10.1016/S0140-6736(10)60961-0. [DOI] [PubMed] [Google Scholar]
- 32.Thöny B, Blau N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum Mutat. 2006;27:870–8. doi: 10.1002/humu.20366. [DOI] [PubMed] [Google Scholar]
- 33.Bonafé L, Thöny B, Penzien JM, Czarnecki B, Blau N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet. 2001;69:269–77. doi: 10.1086/321970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.National Institutes of Health Consensus Development Panel. National institutes of health consensus development conference statement: Phenylketonuria: Screening and management, October 16-18 2000. Pediatrics. 2001;108:972–82. doi: 10.1542/peds.108.4.972. [DOI] [PubMed] [Google Scholar]
- 35.Hofman KJ, Steel G, Kazazian HH, Valle D. Phenylketonuria in U.S. blacks: Molecular analysis of the phenylalanine hydroxylase gene. Am J Hum Genet. 1991;48:791–8. [PMC free article] [PubMed] [Google Scholar]
- 36.Guldberg P, Henriksen KF, Sipilä I, Güttler F, de la Chapelle A. Phenylketonuria in a low incidence population: Molecular characterisation of mutations in Finland. J Med Genet. 1995;32:976–8. doi: 10.1136/jmg.32.12.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tada K, Tateda H, Arashima S, Sakai K, Kitagawa T, Aoki K, et al. Follow-up study of a nation-wide neonatal metabolic screening program in Japan. A collaborative study group of neonatal screening for inborn errors of metabolism in Japan. Eur J Pediatr. 1984;142:204–7. doi: 10.1007/BF00442450. [DOI] [PubMed] [Google Scholar]
- 38.Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: An inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31–41. [PMC free article] [PubMed] [Google Scholar]
- 39.Yang L, Mao H, Yang R. Delays in referral, and parents refusing treatment for children with PKU. J Med Screen. 2011;18:214. doi: 10.1258/jms.2011.011133. [DOI] [PubMed] [Google Scholar]
- 40.Walter JH, Lachmann RH, Burgard P. Hyperphenylalaninaemia. In: Saudubray JM, Van den Berghe G, Walter JH, editors. Inborn Metabolic Diseases. Berlin: Springer; 2012. pp. 251–64. [Google Scholar]
- 41.Chace DH, Millington DS, Terada N, Kahler SG, Roe CR, Hofman LF. Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin Chem. 1993;39:66–71. [PubMed] [Google Scholar]
- 42.Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104(Suppl):S2–9. doi: 10.1016/j.ymgme.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 43.Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, et al. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet Med. 2014;16:188–200. doi: 10.1038/gim.2013.157. [DOI] [PubMed] [Google Scholar]
- 44.Guldberg P, Rey F, Zschocke J, Romano V, François B, Michiels L, et al. AEuropean multicenter study of phenylalanine hydroxylase deficiency: Classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet. 1998;63:71–9. doi: 10.1086/301920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zschocke J. Phenylketonuria mutations in Europe. Hum Mutat. 2003;21:345–56. doi: 10.1002/humu.10192. [DOI] [PubMed] [Google Scholar]
- 46.Park J, Jang W, Chae H, Kim Y, Chi HY, Kim M, et al. Comparison of targeted next-generation and sanger sequencing for the BRCA1 and BRCA2 mutation screening. Ann Lab Med. 2016;36:197–201. doi: 10.3343/alm.2016.36.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matthijs G, Souche E, Alders M, Corveleyn A, Eck S, Feenstra I, et al. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet. 2016;24:2–5. doi: 10.1038/ejhg.2015.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sikkema-Raddatz B, Johansson LF, de Boer EN, Almomani R, Boven LG, van den Berg MP, et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat. 2013;34:1035–42. doi: 10.1002/humu.22332. [DOI] [PubMed] [Google Scholar]
- 49.de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 50.De Leeneer K, Hellemans J, Steyaert W, Lefever S, Vereecke I, Debals E, et al. Flexible, scalable, and efficient targeted resequencing on a benchtop sequencer for variant detection in clinical practice. Hum Mutat. 2015;36:379–87. doi: 10.1002/humu.22739. [DOI] [PubMed] [Google Scholar]
- 51.Bélanger-Quintana A, Burlina A, Harding CO, Muntau AC. Up to date knowledge on different treatment strategies for phenylketonuria. Mol Genet Metab. 2011;104(Suppl):S19–25. doi: 10.1016/j.ymgme.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Spronsen FJ, Enns GM. Future treatment strategies in phenylketonuria. Mol Genet Metab. 2010;99(Suppl 1):S90–5. doi: 10.1016/j.ymgme.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 53.Woolf LI. Excretion of conjugated phenylacetic acid in phenylketonuria. Biochem J. 1951;49:9–10. [PubMed] [Google Scholar]
- 54.Enns GM, Koch R, Brumm V, Blakely E, Suter R, Jurecki E. Suboptimal outcomes in patients with PKU treated early with diet alone: Revisiting the evidence. Mol Genet Metab. 2010;101:99–109. doi: 10.1016/j.ymgme.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 55.Arnold GL, Vladutiu CJ, Kirby RS, Blakely EM, Deluca JM. Protein insufficiency and linear growth restriction in phenylketonuria. J Pediatr. 2002;141:243–6. doi: 10.1067/mpd.2002.126455. [DOI] [PubMed] [Google Scholar]
- 56.Barat P, Barthe N, Redonnet-Vernhet I, Parrot F. The impact of the control of serum phenylalanine levels on osteopenia in patients with phenylketonuria. Eur J Pediatr. 2002;161:687–8. doi: 10.1007/s00431-002-1091-9. [DOI] [PubMed] [Google Scholar]
- 57.Acosta PB, Yannicelli S, Singh R, Mofidi S, Steiner R, DeVincentis E, et al. Nutrient intakes and physical growth of children with phenylketonuria undergoing nutrition therapy. J Am Diet Assoc. 2003;103:1167–73. doi: 10.1016/S0002-8223(03)00983-0. [DOI] [PubMed] [Google Scholar]
- 58.Dobbelaere D, Michaud L, Debrabander A, Vanderbecken S, Gottrand F, Turck D, et al. Evaluation of nutritional status and pathophysiology of growth retardation in patients with phenylketonuria. J Inherit Metab Dis. 2003;26:1–11. doi: 10.1023/a:1024063726046. [DOI] [PubMed] [Google Scholar]
- 59.Simon E, Schwarz M, Roos J, Dragano N, Geraedts M, Siegrist J, et al. Evaluation of quality of life and description of the sociodemographic state in adolescent and young adult patients with phenylketonuria (PKU) Health Qual Life Outcomes. 2008;6:25. doi: 10.1186/1477-7525-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gassió R, Artuch R, Vilaseca MA, Fusté E, Boix C, Sans A, et al. Cognitive functions in classic phenylketonuria and mild hyperphenylalaninaemia: Experience in a paediatric population. Dev Med Child Neurol. 2005;47:443–8. doi: 10.1017/s0012162205000861. [DOI] [PubMed] [Google Scholar]
- 61.Waisbren SE, Noel K, Fahrbach K, Cella C, Frame D, Dorenbaum A, et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: A systematic literature review and meta-analysis. Mol Genet Metab. 2007;92:63–70. doi: 10.1016/j.ymgme.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 62.Gentile JK, Ten Hoedt AE, Bosch AM. Psychosocial aspects of PKU: Hidden disabilities-a review. Mol Genet Metab. 2010;99(Suppl 1):S64–7. doi: 10.1016/j.ymgme.2009.10.183. [DOI] [PubMed] [Google Scholar]
- 63.Blaskovics ME, Schaeffler GE, Hack S. Phenylalaninaemia. Differential diagnosis. Arch Dis Child. 1974;49:835–43. doi: 10.1136/adc.49.11.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis. 2009;32:333–42. doi: 10.1007/s10545-009-1067-2. [DOI] [PubMed] [Google Scholar]
- 65.Lichter-Konecki U. Neurotransmission and neurotoxicity (phenylketonuria and dopamine) In: Lee B, Scaglia F, editors. Inborn Errors of Metabolism: From Neonatal Screening to Metabolic Pathways. New York: Oxford University Press; 2014. pp. 241–70. [Google Scholar]
- 66.Ponzone A, Guardamagna O, Spada M, Ferraris S, Ponzone R, Kierat L, et al. Differential diagnosis of hyperphenylalaninaemia by a combined phenylalanine-tetrahydrobiopterin loading test. Eur J Pediatr. 1993;152:655–61. doi: 10.1007/BF01955242. [DOI] [PubMed] [Google Scholar]
- 67.Bernegger C, Blau N. High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: A study of 1,919 patients observed from 1988 to 2002. Mol Genet Metab. 2002;77:304–13. doi: 10.1016/s1096-7192(02)00171-3. [DOI] [PubMed] [Google Scholar]
- 68.Muntau AC, Röschinger W, Habich M, Demmelmair H, Hoffmann B, Sommerhoff CP, et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med. 2002;347:2122–32. doi: 10.1056/NEJMoa021654. [DOI] [PubMed] [Google Scholar]
- 69.Matalon R, Michals-Matalon K, Koch R, Grady J, Tyring S, Stevens RC. Response of patients with phenylketonuria in the US to tetrahydrobiopterin. Mol Genet Metab. 2005;86(Suppl 1):S17–21. doi: 10.1016/j.ymgme.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 70.Zurflüh MR, Zschocke J, Lindner M, Feillet F, Chery C, Burlina A, et al. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat. 2008;29:167–75. doi: 10.1002/humu.20637. [DOI] [PubMed] [Google Scholar]
- 71.Karacic I, Meili D, Sarnavka V, Heintz C, Thöny B, Ramadza DP, et al. Genotype-predicted tetrahydrobiopterin (BH4)-responsiveness and molecular genetics in Croatian patients with phenylalanine hydroxylase (PAH) deficiency. Mol Genet Metab. 2009;97:165–71. doi: 10.1016/j.ymgme.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 72.van Spronsen FJ, de Groot MJ, Hoeksma M, Reijngoud DJ, van Rijn M. Large neutral amino acids in the treatment of PKU: From theory to practice. J Inherit Metab Dis. 2010;33:671–6. doi: 10.1007/s10545-010-9216-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vogel KR, Arning E, Wasek BL, Bottiglieri T, Gibson KM. Non-physiological amino acid (NPAA) therapy targeting brain phenylalanine reduction: Pilot studies in PAHENU2 mice. J Inherit Metab Dis. 2013;36:513–23. doi: 10.1007/s10545-012-9524-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yano S, Moseley K, Azen C. Large neutral amino acid supplementation increases melatonin synthesis in phenylketonuria: A new biomarker. J Pediatr. 2013;162:999–1003. doi: 10.1016/j.jpeds.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pietz J, Kreis R, Rupp A, Mayatepek E, Rating D, Boesch C, et al. Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. J Clin Invest. 1999;103:1169–78. doi: 10.1172/JCI5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matalon R, Michals-Matalon K, Bhatia G, Grechanina E, Novikov P, McDonald JD, et al. Large neutral amino acids in the treatment of phenylketonuria (PKU) J Inherit Metab Dis. 2006;29:732–8. doi: 10.1007/s10545-006-0395-8. [DOI] [PubMed] [Google Scholar]
- 77.Sanjurjo P, Aldamiz L, Georgi G, Jelinek J, Ruiz JI, Boehm G. Dietary threonine reduces plasma phenylalanine levels in patients with hyperphenylalaninemia. J Pediatr Gastroenterol Nutr. 2003;36:23–6. doi: 10.1097/00005176-200301000-00007. [DOI] [PubMed] [Google Scholar]
- 78.Sarkissian CN, Shao Z, Blain F, Peevers R, Su H, Heft R, et al. Adifferent approach to treatment of phenylketonuria: Phenylalanine degradation with recombinant phenylalanine ammonia lyase. Proc Natl Acad Sci U S A. 1999;96:2339–44. doi: 10.1073/pnas.96.5.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eavri R, Lorberboum-Galski H. A novel approach for enzyme replacement therapy. The use of phenylalanine hydroxylase-based fusion proteins for the treatment of phenylketonuria. J Biol Chem. 2007;282:23402–9. doi: 10.1074/jbc.M703367200. [DOI] [PubMed] [Google Scholar]
- 80.Gámez A, Wang L, Straub M, Patch MG, Stevens RC. Toward PKU enzyme replacement therapy: PEGylation with activity retention for three forms of recombinant phenylalanine hydroxylase. Mol Ther. 2004;9:124–9. doi: 10.1016/j.ymthe.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 81.Sarkissian CN, Gámez A. Phenylalanine ammonia lyase, enzyme substitution therapy for phenylketonuria, where are we now? Mol Genet Metab. 2005;86(Suppl 1):S22–6. doi: 10.1016/j.ymgme.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 82.Longo N, Harding CO, Burton BK, Grange DK, Vockley J, Wasserstein M, et al. Phase 1 trial of subcutaneous rAvPAL-PEG in subjects with phenylketonuria. Lancet. 2014;384:37. doi: 10.1016/S0140-6736(13)61841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blau N, Longo N. Alternative therapies to address the unmet medical needs of patients with phenylketonuria. Expert Opin Pharmacother. 2015;16:791–800. doi: 10.1517/14656566.2015.1013030. [DOI] [PubMed] [Google Scholar]
- 84.Harding CO, Gillingham MB, Hamman K, Clark H, Goebel-Daghighi E, Bird A, et al. Complete correction of hyperphenylalaninemia following liver-directed, recombinant AAV2/8 vector-mediated gene therapy in murine phenylketonuria. Gene Ther. 2006;13:457–62. doi: 10.1038/sj.gt.3302678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ding Z, Georgiev P, Thöny B. Administration-route and gender-independent long-term therapeutic correction of phenylketonuria (PKU) in a mouse model by recombinant adeno-associated virus 8 pseudotyped vector-mediated gene transfer. Gene Ther. 2006;13:587–93. doi: 10.1038/sj.gt.3302684. [DOI] [PubMed] [Google Scholar]
- 86.Ding Z, Harding CO, Rebuffat A, Elzaouk L, Wolff JA, Thöny B. Correction of murine PKU following AAV-mediated intramuscular expression of a complete phenylalanine hydroxylating system. Mol Ther. 2008;16:673–81. doi: 10.1038/mt.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rebuffat A, Harding CO, Ding Z, Thöny B. Comparison of adeno-associated virus pseudotype 1, 2, and 8 vectors administered by intramuscular injection in the treatment of murine phenylketonuria. Hum Gene Ther. 2010;21:463–77. doi: 10.1089/hum.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alexander IE, Cunningham SC, Logan GJ, Christodoulou J. Potential of AAV vectors in the treatment of metabolic disease. Gene Ther. 2008;15:831–9. doi: 10.1038/gt.2008.64. [DOI] [PubMed] [Google Scholar]