

Abstract
Microglia are the main resident macrophage population of the CNS and perform numerous functions required for CNS development, homeostasis, immunity, and repair. Many lines of evidence also indicate that dysregulation of microglia contributes to the pathogenesis of neurodegenerative and behavioral diseases. These observations provide a compelling argument to more clearly define the mechanisms that control microglia identity and function in health and disease. In this Review, we present a conceptual framework for how different classes of transcription factors interact to select and activate regulatory elements that control microglia development and their responses to internal and external signals. We then describe functions of specific transcription factors in normal and pathological contexts and conclude with a consideration of open questions to be addressed in the future.
Introduction
Microglia are tissue-resident macrophages that perform CNS-specific functions (1). They derive from a unique lineage of erythromyeloid precursors (EMPs) in the yolk sac and fetal liver (2). EMPs infiltrate the brain during early development, differentiate into microglia, and maintain their population by self-renewal (3). Microglia distribute themselves throughout the CNS (4) and continuously scan their surroundings (5). Microglia share many traits with other subsets of tissue-resident macrophages, including dependence on the CSF1 receptor (CSF1R) for differentiation and survival, a requirement for PU.1 as an essential lineage-determining transcription factor (LDTF), the ability to efficiently phagocytose tissue debris, and the ability to quickly trigger an inflammatory response following detection of pathogens or tissue damage (6). In addition to responding to injury and infection, microglia carry out functions that are specific to the CNS environment, including secretion of neurotrophic factors and developmental refinement of synaptic networks (7, 8). Upon activation, microglia can acquire a range of phenotypes that can either contribute to disease progression or ameliorate it (9). Although dysregulated microglia are implicated in the pathogenesis of several neurodegenerative and psychiatric conditions (10), mechanisms controlling developmental, homeostatic, and pathogenic programs of microglia gene expression remain poorly understood.
Insights into the transcriptional regulation of cell type–specific functions can be obtained by analysis of transcriptional regulatory elements (11). Promoters provide the obligatory transcriptional start sites necessary for RNA synthesis and are often sites of signal-dependent regulation (Figure 1). However, they are primarily occupied by broadly expressed transcription factors (TFs) such as SP1 and GABP, and by themselves are insufficient to confer the specific regulatory control necessary to generate cell type–specific programs of gene expression. This additional information is provided by distal regulatory elements called enhancers (12). Enhancers represent the most numerous binding sites for LDTFs and signal-dependent transcription factors (SDTFs), and are major sites for the integration of internal and external signals. Enhancers exhibit distinctive patterns of modifications to adjacent histones that can be detected by ChIP sequencing (ChIP-Seq), and these patterns can be used to putatively classify enhancers as inactive, primed, or active (13).
Figure 1. Enhancers and promoters interact to confer cell type–specific expression and response profiles.
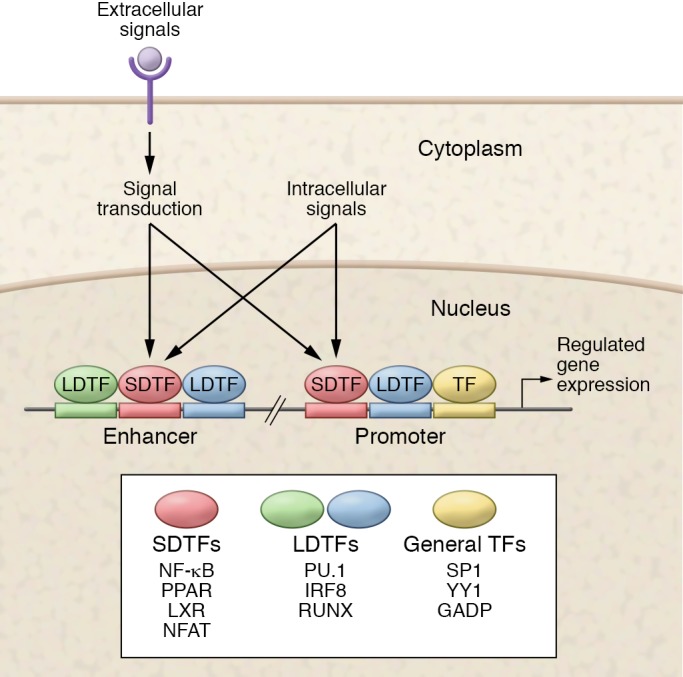
Promoters are primarily occupied by general sequence-specific transcription factors (TFs), but are also important sites of action of lineage-determining TFs (LDTFs) and signal-dependent TFs (SDTFs). Enhancers are primarily selected by LDTFs and are usually the most numerous binding sites for SDTFs. External and internal signals converge on SDTFs to regulate enhancer and promoter function.
In vitro studies of elicited peritoneal macrophages provided the basis for a collaborative/hierarchical model of enhancer selection and activation involving interactions between LDTFs and SDTFs (14–16). According to this model, the initial steps of enhancer selection are driven by collaborative interactions of LDTFs and other “pioneer” factors that recognize factor-specific DNA sequences in closed chromatin and generate nucleosome-free regions (17). The selection of enhancers is thought to be a collaborative process requiring interactions between multiple factors that recognize closely spaced arrangements of corresponding recognition motifs (18). Initial occupancy of enhancers by LDTFs and their collaborative partners can result in histone modifications associated with a primed state of activity. The transition from an inactive or primed state to an active enhancer can be induced by SDTFs (19, 20) (Figure 2) that are activated in response to stimuli through cell signaling pathways that often originate at cell surface receptors. These SDTFs mainly bind to enhancers previously established by LDTFs (15, 16, 21, 22). This process is frequently hierarchical such that SDTF binding is dependent on LDTF binding, whereas loss of the SDTF does not influence the binding of the LDTF (14, 21, 23). The observation that SDTFs are directed to a predetermined set of cell-specific enhancers at least partly explains how a broadly expressed TF can regulate specific transcriptional responses in different cell types. Importantly, the distinction between LDTFs and SDTFs is not always clear cut, as some TFs that regulate the ontogeny of a cell type can also serve as mediators for particular environmental stimuli. In this Review, we examine microglia development and function in the context of this general model, with a focus on roles of the major LDTFs and SDTFs that are expressed in microglia.
Figure 2. Different classes of transcription factors interact and regulate cellular identity.

The top of the figure illustrates the initial steps of enhancer selection in closed chromatin consisting of regularly positioned nucleosomes. Green and blue shading represents closely spaced binding sites for LDTFs. Below, collaborative interactions between LDTFs generate primed enhancers, characterized by a nucleosome-free region and monomethylated H3K4 (H3K4me1). At the bottom of the figure, active SDTFs localize to primed enhancers, resulting in recruitment of coactivators and RNA polymerase II and generation of enhancer RNAs (eRNAs).
Transcriptional control of microglia identity
Microglia derive from a population of EMPs in the extraembryonic yolk sac during the process of primitive hematopoiesis (2, 24). In mice, EMPs exclusively express the TF runt-related TF-1 (RUNX1) between E6.5 and E8.0. Microgliogenesis is crucially dependent on the TFs PU.1 and interferon regulatory factor-8 (IRF8), but not on the TFs inhibitor of DNA binding-2 (ID2); basic leucine zipper TF, ATF-like-3 (BATF3); Krüppel-like factor-4 (KLF4); and MyB (25, 26). In the yolk sac EMPs differentiate into premacrophages (pMacs) that express a core macrophage program that includes the receptors CX3CR1 and CSF1R, as well as the TFs Maf, Batf3, Pparg, Irf8, and zinc finger E-box binding homeobox-2 (Zeb2) (27), a program that persists when these precursors infiltrate various organs to establish resident macrophage populations. Microglia development has been proposed to progress in three developmental stages (28): early microglia (until E14), premicroglia (from E14 to the first weeks after birth), and adult microglia (from a few weeks after birth onward). Early microglia, like EMPs, express genes associated with cell cycle signaling pathways and the TF E2f6 and DNA methyltransferase-1 (Dnmt1), which may help them populate the brain. The genes expressed by premicroglia, including the TFs early growth response-1 (Egr1) and spalt-like TF-1 (Sall1), are associated with neural maturation and synaptic pruning (29, 30). Later, as the brain matures, microglia acquire a surveilling phenotype and express the TFs Jun, Fos, Mef2a, and Mafb, as well as a set of genes that are highly expressed in microglia such as the characteristic purinergic receptor P2RY12, the transmembrane protein TMEM119, and the chemokine receptor CX3CR1 (31–34), the expression of which is largely conserved in humans (35–37) and zebrafish (38).
Compared with monocytes and neutrophils, macrophage enhancers are enriched for a MAF binding motif with Maf and Mafb as the highest expressed corresponding TFs, raising the possibility that these TFs may act collaboratively with PU.1 to specify a general macrophage identity (39). In contrast, microglia enhancers, when compared with other tissue macrophages, were enriched for a MEF2 binding motif and exhibited high expression of the cognate TF Mef2c (39, 40). A comparison with peritoneal macrophages showed that PU.1-bound enhancers in microglia were enriched for PU.1-IRF, HIC2, MEF2, and SMAD binding motifs, implicating these TFs as potential collaborative partners of PU.1. However, enrichment alone cannot establish whether these factors act as LDTFs in collaboration with PU.1 or merely cobind already-established enhancers. To overcome this limitation, experiments exploiting natural genetic variation between two strains of mice were performed, in a manner analogous to a mutagenesis experiment. These experiments showed that disruption of binding sites for STAT3, MAFB, SMAD3, and upstream TF-1 (USF1) affected nearby PU.1 binding specifically in microglia, and these may therefore function as microglia-specific collaborative factors (40).
Systematic analyses of human microglia gene expression from postmortem (36) and surgical tissues (37) indicate broad similarities with mice but also a number of significant differences, including genes implicated in neurodegenerative diseases. Further, age-related changes in gene expression differed significantly between mice and humans (36). Both human and mouse microglia enhancers are enriched for motifs associated with PU.1, IRF, RUNX, MEF2, C/EBP, AP-1, SMAD, and MAF (37), indicating a conserved set of microglia LDTFs. However, Sall2, Sall3, and Smad1 were expressed much more highly in mice, while class II MHC transactivator (CIITA), PPARG, EGR3, and RUNX2 were preferentially expressed in human microglia. These findings suggest that the differences between mouse and human microglia gene expression are mainly driven by species-specific organization of regulatory elements, but some differences also result from differential expression of TFs.
The importance of the brain environment in maintaining microglia identity is suggested by the rapid and extensive changes in gene expression that occurred when they were transferred from the brain to an in vitro environment (32, 37, 40, 41). Substantial subsets of genes associated with risk alleles for neurodegenerative or behavioral diseases exhibited environment-dependent expression (37). Genes that were downregulated in vitro were highly correlated with genes that are upregulated in primitive macrophages following their migration into the fetal brain (28, 37). These changes in gene expression are associated with downregulation of numerous putative microglia LDTFs, such as Sall1 and corresponding alterations in enhancer landscapes. Collectively, these findings provide evidence for an important role of brain-derived signals in controlling a conserved network of signal-dependent and lineage-determining TFs in mouse and human microglia.
LDTFs that establish physiological microglia
Many of the TFs implicated in defining the microglia cell type during development have been shown to play roles in maintaining microglia in a ramified and surveilling state that is characteristic of healthy brain tissue. We discuss three important and well-described transcriptional regulators in more detail below.
PU.1.
PU.1 is a well-characterized LDTF that appears to serve as a master regulator of the myeloid lineage (42). PU.1 is essential for establishing macrophage lineages, drives microgliogenesis, and is a major factor in selecting the microglia enhancer landscape. PU.1-deficient mice exhibit multiple hematopoietic abnormalities and a pronounced depletion of B cell and macrophage populations (43). PU.1 deficiency also ablates microglia in mice (26) and zebrafish (44). Moreover, PU.1 binds to most enhancers in mouse (40) and human microglia (37), highlighting its central role in establishing the microglia enhancer landscape.
SALL1.
SALL1 is a zinc finger transcriptional repressor that belongs to the SALL-like family of TFs that function in tissue morphogenesis (45). SALL1 regulates cortical neurogenesis and laminar fate specification in mice (46). Mutations in SALL1 are associated with Townes-Brocks syndrome, an autosomal dominant developmental disorder in which roughly 10% of patients exhibit neural or behavioral abnormalities (46). Human and mouse microglia uniquely and highly express SALL1 as compared with other macrophage populations, suggesting that it functions as an LDTF (34, 35, 37). Sall1-deficient animals have normal microglia colonization in the brain, but the microglia exhibit an abnormal ameboid morphology (30). Moreover, in the absence of Sall1, microglia are more proinflammatory and phagocytic, and show downregulation of the microglia-specific gene expression signature (29). This altered microglia phenotype has detrimental consequences for neurogenesis and tissue homeostasis. Taken together, these studies suggest that SALL1 inhibits a reactive microglia phenotype, and promotes a physiological surveilling phenotype.
MAFB.
MAFB is a member of the large MAF subfamily of TFs that binds to the MAF recognition element and forms heterodimers with cMAF, Jun, and Fos. In macrophages, MAFB promotes an antiinflammatory phenotype, and its deletion exacerbates inflammatory and pathological conditions (47, 48). In microglia, expression of MAFB increases during microglia development, and its deletion disrupts gene expression in adult microglia more severely than in premature microglia (28). Motif mutation analysis suggests that MAFB functions as an LDTF in collaboration with PU.1 (40). In microglia, MAFB regulates the expression of immune and viral genes (28), and its expression is downregulated in the SOD1 mouse model of amyotrophic lateral sclerosis (49). In primary microglia, MAFB regulates cell division by dampening the response to granulocyte-macrophage CSF (50). These findings suggest that MAFB helps to maintain a healthy adult microglia phenotype by inhibiting inflammatory and proliferative responses.
Transcriptional control of disease phenotypes
In disease conditions, microglia can adopt various phenotypes that exert beneficial and deleterious effects, including tissue support, excessive pruning of synapses, induction of neuropathic pain, and exacerbation of neurodegeneration (51). New studies are beginning to shed light on how these phenotypes are transcriptionally regulated (Figure 3). The collaborative/hierarchical model suggests that, in disease, a unique combination of pathological and inflammatory factors activates SDTFs that regulate the expression of specific microglia response programs. A major obstacle to understanding the relationships among the different classes of TFs is that the environmental factors and pathological signals are complex and interact on multiple levels. The observation that culturing microglia so that they no longer receive brain-derived signals results in dramatic changes to their enhancer landscape (32, 37, 40) calls into question the extent to which findings about SDTFs obtained in vitro are translatable to microglia in vivo. Furthermore, the frequently used immortalized murine microglia cell line, BV2, only partially recapitulates the response to a simple LPS stimulus observed in primary microglia (52). Given these differences, findings regarding the roles of SDTFs from in vitro studies should be interpreted with caution until they can be confirmed in in vivo conditions. Nevertheless, an increasing number of studies have identified transcriptional regulators of microglia activation and microglia phenotypes in pathological conditions.
Figure 3. Transcription factors that regulate different microglia phenotypes.

Microglia ontogeny is crucially dependent on LDTFs such as PU.1, MAFB, and SALL1 that are essential for the development of the ramified phenotype depicted in the center. Microglia are diverse cells that acquire different functional phenotypes in response to the environment in which they reside. Several transcriptional regulators have been identified that regulate different microglia phenotypes. For example, proinflammatory microglia are regulated by IRF7, RelA, STAT1/3, and FosB. Microglia that mediate neuropathic pain are induced by expression of Irf1, Irf5, and Irf8 in microglia. Microglia that contribute to HIV-associated neurodegeneration are characterized by an induction of p53. Figure adapted with permission from Nature Reviews Neurology (128).
The dynamic interaction between LDTFs and SDTFs in human disease is illustrated by the response of microglia to the mutant form of the huntingtin protein (HTT) responsible for Huntington’s disease (HD) (53). Multiple studies demonstrated increased microglial activation in HD patients, but it remained an open question whether this resulted from the action of mutated HTT (mHTT) in the microglia or represented the response of microglia to mHTT-induced changes in neurons. This was addressed by the expressing of mHTT in BV2 cells, resulting in increased expression of the SDTF NF-κB1 and the proinflammatory cytokines IL-6 and TNF, as well as increased expression and binding of the macrophage LDTFs PU.1 and C/EBP. Neurons that were cocultured with LPS-stimulated microglia expressing mHTT had increased apoptosis compared with neurons cocultured with LPS-stimulated WT microglia.
SDTFs that mediate inflammation
As the resident macrophages of the CNS, microglia are the first to respond to inflammatory insults in the brain, which they do by expressing a wide array of cytokines, chemokines, nitric oxide (NO), and various reactive oxygen species (ROS). Several transcriptional families have been identified whose members regulate the expression of proinflammatory mediators in macrophages and microglia.
NF-κB family.
Members of the NF-κB family are pleiotropic regulators that play key roles in apoptosis and inflammation (54). NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel form homodimers and heterodimers. Without stimulation, NF-κB is sequestered in the cytoplasm. Once activated, it translocates to the nucleus, where it binds to NF-κB binding sites exposed in open chromatin regions and induces the expression of target genes (55). Many studies have examined the role of NF-κB in brain aging and neurodegenerative conditions (54), but, since expression of NF-κB is not limited to microglia, most of these studies have focused on its role in other cell types or its generic expression in the brain.
In microglia, NF-κB is activated by signal transduction pathways responding to the presence of ROS (56), saturated fatty acids (57), α-synuclein (58, 59), and amyloid-β (60). The induced response can be either protective or deleterious depending on the context of the stimulation (61). A small number of studies have experimentally assessed the functional role of NF-κB in microglia. Bacterial LPS is a TLR4 ligand that induces a strong immune response mediated by several TFs, including the NF-κB1/RelA complex. A single intraperitoneal injection of LPS has long-term consequences in microglia and results in blunted expression of proinflammatory cytokines upon LPS restimulation; this attenuation is mediated in part by RelB (62). These observations exemplify how different members of the same NF-κB family can play antagonistic roles in inflammation and mediate changes in the microglia phenotype, and highlight the need for a more thorough understanding of how NF-κB family members regulate microglia phenotypes.
Activator protein-1 family.
The activator protein-1 (AP-1) family of TFs is involved in numerous processes including cell growth, differentiation, apoptosis, and immune activation (63). This family consists of Jun, Fos, activating TF (ATF), and Jun dimerization protein (JDP) subfamilies, and its members can form both homo- and heterodimers. In macrophages, AP-1 members have been described to function as both LDTFs (15) and SDTFs (64). Similarly, in microglia, Jun and Fos are upregulated during microglia development, suggesting that they play roles as LDTFs that regulate microglia identity (28). However, FosB has been suggested to function as an SDTF that regulates excitotoxic microglia activation (65). Constitutive Fosb-knockout mice were relatively resistant to kainate-induced excitotoxicity, with fewer ameboid microglia, and less expression of the proinflammatory cytokines IL-6 and TNF compared with their WT counterparts. Since FosB expression is similar between hippocampal neurons and microglia, it is currently unclear whether FosB ablation directly affects microglia, neurons, or both. More research into the role of AP-1 members in microglia is needed for a better understanding of how these factors regulate both microglia identity and pathological phenotypes.
Interferon regulatory factors.
The interferon regulatory factor (IRF) family consists of nine members that bind to genomic loci known as interferon-sensitive response elements. IRF3, IRF5, and IRF7 mediate type I interferon responses induced by various TLR ligands (66), while IRF1 is essential for the type II interferon response (63). In microglia, IRF8 is a key regulator that appears to function as both an LDTF and an SDTF. IRF8 was recently identified as an essential regulator of tissue macrophage maturation, including microglia (27, 67). Irf8 deletion substantially reduces microglia numbers in mice (26). Microglia-specific disruption of Irf8 resulted in a pronounced reduction in microglia ramification and surface area as well as altered expression of several cell surface markers, indicating increased microglia immune activation (68). In zebrafish, Irf8-null mutants showed a complete absence of microglia (69). IRF8 has also been shown to regulate microglia motility (70).
In spinal cord microglia, IRF8, in conjunction with IRF1 and IRF5, contributes to the induction of neuropathic pain after peripheral nerve injury (PNI) (71–74). IRF8 expression is markedly upregulated by PNI, which results in hypersensitivity to pain (71). IRF8 is upstream of IRF5, and Irf5-deficient mice exhibited substantial resistance to neuropathic pain (74). In microglia, IRF8 is also upstream of IRF1 and thereby regulates IL-1β expression after PNI (73). Collectively, these studies indicate that IRF8 is both a key regulator of microglia identity and a modulator of a reactive microglia phenotype associated with neuropathic pain.
IRF7 in microglia has been shown to promote both an antiinflammatory phenotype (75) and a proinflammatory phenotype (76). Downregulation of IRF7 by long-term exposure to TGF-β1 or inhibition of IRF7 impairs the ability of microglia to acquire an antiinflammatory phenotype (75), while IFN-β–mediated activation of IRF7 restores the antiinflammatory phenotype. In spinal cord injury, microglia overexpressing IRF7 have decreased proinflammatory activity (75). In contrast, IRF7 promotes LPS-induced inflammation via phosphorylation of STAT1 (76). Reconciliation of these two opposing observations concerning a single TF deserves further attention.
p53 tumor suppressor.
p53 is a well-studied tumor suppressor gene that functions as a complex integrator of signals that regulate DNA damage repair, cell cycle progression, and apoptosis (77). The expression and activity of p53 increases in microglia in response to cellular stress, DNA damage, and oxidative stress (78–80). In postmortem brain tissues from patients with HIV-associated dementia (HAD) elevated levels of p53 were particularly prominent in the microglia (78, 81). HIV-infected p53-deficient neuron/microglia cocultures showed increased neuronal survival compared with WT (78). Microglial p53 activation is detrimental to neuronal synapses during inflammation (82). P53–/– microglia had lower expression of proinflammatory genes and demonstrated increased phagocytosis and expression of tissue support genes in response to IFN-γ (81). p53 may act to downregulate the antiinflammatory transcription factor cMAF through a transcriptional circuit involving TWIST2 and miR-155 (83). Collectively these studies suggest that p53 regulates microglia activity and induces a more reactive inflammatory profile with decreased tissue-supportive functions and phagocytic activity in HAD that might also play a more generic role in regulating microglia in disease.
STAT family.
STAT TFs are pleiotropically expressed and mediate diverse functions such as proliferation, apoptosis, and differentiation in response to cytokines (63). STAT proteins form both homo- and heterodimers and become activated when phosphorylated STAT dimers translocate to the nucleus and bind to genomic elements. The binding pattern of STAT TFs is largely cell type–specific, and individual STAT members often bind in the proximity of cell type–specific TFs (84). Aberrant activation of JAK/STAT signaling in innate immune cells is associated with both multiple sclerosis (MS) and Alzheimer’s disease (AD) (85).
In mice, alterations in the STAT3 binding site affect nearby PU.1 binding in microglia, suggesting that STAT3 functions as an LDTF (40). STAT1 and STAT3 also appear to function as SDTFs that regulate LPS-induced activation (86). In primary mouse microglia, changes in the binding of STAT1, STAT3, and STAT5 in response to LPS were assessed, and a group of STAT-regulated inflammatory genes was identified including the histone 3 lysine-27 (H3K27) demethylase JMJD3. The expression of these putative target genes was only affected if both STAT1 and STAT3 were inhibited simultaneously using siRNA in BV2 cells, suggesting a functional redundancy between these two factors. Knockdown of both STAT1 and STAT3 inhibited the expression of JMJD3 and other inflammatory genes. This evidence suggests that STAT1/STAT3 signaling regulates expression of inflammatory mediators in microglia, possibly via JMJD3.
Antiinflammatory and tissue-supportive mediators
Excessive inflammation in the CNS is detrimental for neuronal health (87). Not surprisingly, several transcriptional mechanisms limit the inflammatory responses of microglia and induce neuroprotective behavior.
MSX family.
The Msh-like homeobox (MSX) genes are part of the homeobox family and are implicated in brain development and neurogenesis (88). In microglia, MSX3 is normally lowly expressed but upregulated during experimental autoimmune encephalomyelitis (EAE) in mice (89). Transplantation of MSX3-overexpressing microglia to EAE mice resulted in milder EAE clinical scores, increased remyelination, and decreased inflammation. ChIP analysis indicated that MSX3 bound at the promoters of Pparg, Stat6, and Jak3, suggesting that it might exert its effect by regulating these genes. This study suggests that MSX3 might play a role in maintaining a tissue-supportive phenotype in microglia.
NR4A2.
Nuclear receptor subfamily 4, group A, member 2 (NR4A2, also known as NURR1) plays an essential role in the generation and maintenance of dopaminergic neurons in the brain (90). Mutations in NR4A2 are associated with familial Parkinson’s disease (PD) (91). In microglia, NR4A2 inhibits LPS-induced expression of proinflammatory cytokines (92). Reduced NR4A2 expression results in exaggerated inflammatory responses in microglia and increases dopaminergic neuron death. NR4A2 exerts an antiinflammatory effect by binding to RelA on inflammatory gene promoters and recruits the CoREST corepressor complex, which clears RelA and represses transcription. Additionally, pharmacological activation of NR4A2 in BV2 cells blocks inflammatory gene expression by inhibiting RelA (93), suggesting that NR4A2 is an antiinflammatory regulator that counteracts RelA-based inflammation in microglia.
Estrogen receptors.
Estrogen receptor-α (ERα) and ERβ are a subclass of the nuclear receptor superfamily that function as estrogen-dependent TFs. Estrogens exert neuroprotective actions by signaling through ERs that are widely distributed in the male and female brain (94). Estradiol is not only a reproductive hormone but also a brain-derived neuroprotective factor in both males and females that enables ERs to coordinate multiple signaling mechanisms that protect the brain from neurodegenerative diseases. In microglia, estrogen signaling was also shown to exert antiinflammatory effects (95). Androstenediol (or 5-androsten-3β,17β-diol) is an estrogenic steroid produced in the brain that binds to ERβ. Androstenediol activation of ERβ suppresses the inflammatory responses of microglia by recruitment of C-terminal binding protein (CtBP) corepressor complexes to AP-1–dependent promoters (96). Reduction of androstenediol or ERβ expression results in exaggerated LPS responses, and administration of androstenediol in vivo prevents EAE. These findings provide evidence for an ERβ signaling pathway that controls inflammatory responses in microglia.
Nuclear respiratory factor 2.
Nuclear respiratory factor 2 (NRF2, also referred to as nuclear factor 2 erythroid 2 like 2 [NFE2L2]) regulates an antioxidant response to the oxidative damage that is characteristic of many neurodegenerative conditions (97). In macrophages, NRF2 expression represses inflammation not only by regulating the antioxidative defense (98), but also by directly inhibiting the recruitment of RNA polymerase II to Il6 and Tnf (99). Many studies suggest that NRF2 activation is involved in neuroprotection (97) by regulating antioxidative proteins such as heme oxygenase-1 (HO-1). In microglia, NRF2 has been demonstrated to counteract inflammation and neurodegeneration in the tau pathology model for AD (100), the MPTP model for PD (101), hematoma (102), and LPS-induced inflammation (103). Stressed neurons signal microglia through fractalkine to induce the NRF2-mediated antioxidative response (100). These findings were corroborated in human AD brain tissues, suggesting that this mechanism might be an adaptive tissue-restorative response to pathological conditions. Besides regulating the antioxidative response, NRF2 has also been implicated in regulation of phagocytic clearance, a process that is essential for the uptake of blood cells in hematoma, via upregulation of CD36 (102). Collectively, these results indicate that NRF2 dampens inflammation through mediating the antioxidative response and facilitating phagocytosis, thereby positioning it as a crucial regulator of a microglia tissue-restorative phenotype.
SDTFs that regulate cholesterol and lipid metabolism
Lipids and cholesterol derivatives are essential for the CNS, where they function as key components of myelin sheets, synapses, dendrites, and intercellular signaling molecules (104, 105). Defects in cholesterol metabolism are clearly described in neurodegenerative diseases (106). The PPAR and liver X receptor/retinoid X receptor (LXR/RXR) families play key roles at the crossroads of lipid and cholesterol metabolism with immune responses in tissue macrophages (107). In microglia, much less is known about these factors since, to date, no studies have experimentally targeted members of the LXR/RXR or the PPAR family to microglia in vivo.
PPARs.
The PPAR family of nuclear receptor TFs consists of three members: PPARα, PPARδ, and PPARγ. PPARs function as lipid sensors and are involved in fatty acid metabolism (108). In adipose-resident macrophages, members of the PPAR family are implicated in suppressing fatty acid–induced inflammation associated with obesity and metabolic syndrome by increasing fatty acid oxidation (109). PPARγ is a central switch that inhibits the expression of proinflammatory mediators (110) and induces a wound healing phenotype (111, 112). However, the role of PPARs in microglia and neuroinflammation is still poorly understood. A rapidly growing number of studies have investigated the role of PPARγ agonists in neurodegenerative conditions such as AD, PD, HD, and amyotrophic lateral sclerosis, but these findings have not conclusively shown a protective effect (113). However, in AD mice, a PPARγ agonist did show beneficial effects by counteracting neurodegeneration, lowering amyloid-β deposits, and inhibiting neuroinflammation (114). It has been suggested that the beneficial effects of PPARγ agonist are produced by inhibition of the proinflammatory activity of microglia and promotion of their phagocytic activity (115–117).
LXRs and RXRs.
Liver X receptors (LXRs) and retinoid X receptors (RXRs) are nonsteroid nuclear receptors that play key roles in cholesterol homeostasis and inflammation (118, 119). The LXR isoforms LXRα and LXRβ can form dimers with any of the three RXR isoforms (RXRα, RXRβ, and RXRγ) and are activated in the presence of cholesterol derivatives such as oxysterols. In macrophages, the majority of LXR binding sites are found near PU.1-bound primed enhancers (15). LXRs have antiinflammatory effects and modify macrophage phagocytic capability (120). Age-related neurodegenerative conditions have been linked to the disruption of cholesterol metabolism and LXR signaling (118). Likewise, LXR agonists have been shown to produce cognitive improvement in AD mouse models (121–123), but the mechanisms are not well understood. It has been suggested that the protective effects of LXR agonists are achieved by promotion of an increase in phagocytosis of amyloid-β by microglia (117), by inhibition of microglia nitric oxide synthetase (NOS) activity (124), or by attenuation of the microglia inflammatory response (125).
Conclusions and future perspectives
Many transcriptional regulators that mediate microglia in health and disease have been identified, but much remains unknown about their functional consequences, how they guide microglia phenotypic diversity, the effects of natural genetic variation, and their responses to specific signals in the local microenvironment.
One challenge is that many studies are purely descriptive or are performed on cultured microglia. The recent development of effective and inducible drivers of Cre-recombinase such as Cx3cr1-CreER (126) and Sall1-CreER (29) enables microglia-targeted knockout and overexpression of genes of interest. These tools could not only provide stronger and more detailed experimental evidence but also give qualitatively new insights into the functional roles of TFs in regulating microglia in vivo.
A further challenge is to untangle the interrelationships between transcriptional regulators and the microglia enhancer landscape, which is required to understand the molecular mechanisms underlying their functional roles in microglia. With the increasing adoption of high-throughput technologies, it is now possible to map the binding patterns of different TFs in the context of the enhancer landscape. Additionally, new techniques that unravel the 3D structure of chromatin, such as Hi-C, can be used to determine enhancer-promoter interactions, and help to address whether particular TFs regulate specific classes of enhancers.
Naturally occurring variation in noncoding regions of the genome can have modulatory effects on gene regulation or enhancer formation and could have considerable importance for understanding the involvement of such genetic variation in the roles of microglia in neurodegenerative diseases. Relatively large numbers of human microglia samples are necessary to determine whether disease-associated SNPs act on microglia expression and/or enhancer profiles. Therefore, the effect of natural genetic variation on microglia expression and enhancer landscapes remains largely unexplored.
Another major aim for future research will be to identify the specific factors of the local CNS microenvironment, such as TGF-β or amyloid-β, that either help to maintain microglial physiological identity or are responsible for disease-specific responses. Additionally, further identification of the TFs through which these signals are transduced to produce the microglia-specific patterns of gene expression remains an ongoing subject of investigation.
Microglia can exhibit a diverse range of phenotypes in response to their surroundings, but these phenotypes are much more complex than the simple M1/M2 dichotomy (127) originally proposed for macrophages. Nor are these phenotypic states easily classified on a one-disease/one-phenotype basis. The proper framework for cataloging microglia diversity across pathological conditions, brain regions, local microenvironment, and developmental stages remains elusive. New technologies such as single-cell RNA sequencing and mass cytometry should facilitate a framework for microglia phenotype classification.
Finally, the degree to which findings about transcriptional regulation in mice and zebrafish translate to humans is critical for insight into the pathophysiology of neurodegenerative and neuropsychiatric diseases and is still largely unaddressed. Further studies of human microglia are essential to identify the strengths and weaknesses of animal and cell culture models.
Acknowledgments
We thank Leslie Van Ael for her assistance with manuscript and figure preparation. IRH is supported by the Dutch MS Research Foundation and the Gemmy and Mibeth Tichelaar Foundation. DS was supported by a UCSD institutional predoctoral training grant (T32DK007541). CKG is supported by the Ben and Wanda Hildyard Chair in Hereditary Diseases. We thank Alexi Nott, Johannes Schlachetzki, and Claudia Han for proofreading the manuscript.
Version 1. 07/31/2017
Electronic publication
Version 2. 09/01/2017
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2017;127(9):3220–3229.https://doi.org/10.1172/JCI90604.
Contributor Information
Inge R. Holtman, Email: irholtman@gmail.com.
Dylan Skola, Email: dskola@ucsd.edu.
References
- 1.Masuda T, Prinz M. Microglia: a unique versatile cell in the central nervous system. ACS Chem Neurosci. 2016;7(4):428–434. doi: 10.1021/acschemneuro.5b00317. [DOI] [PubMed] [Google Scholar]
- 2.Hoeffel G, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity. 2015;42(4):665–678. doi: 10.1016/j.immuni.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402. doi: 10.1146/annurev-immunol-032713-120240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry VH, Hume DA, Gordon S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience. 1985;15(2):313–326. doi: 10.1016/0306-4522(85)90215-5. [DOI] [PubMed] [Google Scholar]
- 5.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 6.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell. 2014;158(1):15–24. doi: 10.1016/j.cell.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. 2015;36(10):605–613. doi: 10.1016/j.it.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eggen BJ, Raj D, Hanisch UK, Boddeke HW. Microglial phenotype and adaptation. J Neuroimmune Pharmacol. 2013;8(4):807–823. doi: 10.1007/s11481-013-9490-4. [DOI] [PubMed] [Google Scholar]
- 10.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777–783. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 11.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16(3):144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinz S, et al. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503(7477):487–492. doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghisletti S, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32(3):317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Zaret KS, Watts J, Xu J, Wandzioch E, Smale ST, Sekiya T. Pioneer factors, genetic competence, and inductive signaling: programming liver and pancreas progenitors from the endoderm. Cold Spring Harb Symp Quant Biol. 2008;73:119–126. doi: 10.1101/sqb.2008.73.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyes J, Felsenfeld G. Tissue-specific factors additively increase the probability of the all-or-none formation of a hypersensitive site. EMBO J. 1996;15(10):2496–2507. [PMC free article] [PubMed] [Google Scholar]
- 19.Samstein RM, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151(1):153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trompouki E, et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell. 2011;147(3):577–589. doi: 10.1016/j.cell.2011.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaikkonen MU, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51(3):310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barish GD, et al. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24(24):2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mullen AC, et al. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011;147(3):565–576. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 26.Kierdorf K, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16(3):273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 27.Mass E, et al. Specification of tissue-resident macrophages during organogenesis. Science. 2016;353(6304):aaf4238. doi: 10.1126/science.aaf4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matcovitch-Natan O, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353(6301):aad8670. doi: 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- 29.Buttgereit A, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. 2016;17(12):1397–1406. doi: 10.1038/ni.3585. [DOI] [PubMed] [Google Scholar]
- 30.Koso H, et al. Conditional rod photoreceptor ablation reveals Sall1 as a microglial marker and regulator of microglial morphology in the retina. Glia. 2016;64(11):2005–2024. doi: 10.1002/glia.23038. [DOI] [PubMed] [Google Scholar]
- 31.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butovsky O, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hickman SE, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16(12):1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89(1):37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galatro TF, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci. doi: 10.1038/nn. [published online ahead of print July 3, 2017]. https://doi.org/10.1038/nn.4597. [DOI] [PubMed] [Google Scholar]
- 37.Gosselin DS, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. doi: 10.1126/science. [published online ahead of print May 25, 2017]. https://doi.org/10.1126/science.aal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oosterhof N, et al. Identification of a conserved and acute neurodegeneration-specific microglial transcriptome in the zebrafish. Glia. 2017;65(1):138–149. doi: 10.1002/glia.23083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavin Y, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gosselin D, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159(6):1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA. Diverse requirements for microglial survival, specification, and function revealed by defined-medium cultures. Neuron. 2017;94(4):759–773.e8. doi: 10.1016/j.neuron.2017.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nerlov C, Graf T. PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 1998;12(15):2403–2412. doi: 10.1101/gad.12.15.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKercher SR, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15(20):5647–5658. [PMC free article] [PubMed] [Google Scholar]
- 44.Herbomel P, Thisse B, Thisse C. Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev Biol. 2001;238(2):274–288. doi: 10.1006/dbio.2001.0393. [DOI] [PubMed] [Google Scholar]
- 45.de Celis JF, Barrio R. Regulation and function of Spalt proteins during animal development. Int J Dev Biol. 2009;53(8–10):1385–1398. doi: 10.1387/ijdb.072408jd. [DOI] [PubMed] [Google Scholar]
- 46.Harrison SJ, Nishinakamura R, Jones KR, Monaghan AP. Sall1 regulates cortical neurogenesis and laminar fate specification in mice: implications for neural abnormalities in Townes-Brocks syndrome. Dis Model Mech. 2012;5(3):351–365. doi: 10.1242/dmm.002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cuevas VD, et al. MAFB determines human macrophage anti-inflammatory polarization: relevance for the pathogenic mechanisms operating in multicentric carpotarsal osteolysis. J Immunol. 2017;198(5):2070–2081. doi: 10.4049/jimmunol.1601667. [DOI] [PubMed] [Google Scholar]
- 48.Hasegawa H, et al. The role of macrophage transcription factor MafB in atherosclerotic plaque stability. Atherosclerosis. 2016;250:133–143. doi: 10.1016/j.atherosclerosis.2016.05.021. [DOI] [PubMed] [Google Scholar]
- 49.Butovsky O, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77(1):75–99. doi: 10.1002/ana.24304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koshida R, Oishi H, Hamada M, Takahashi S. MafB antagonizes phenotypic alteration induced by GM-CSF in microglia. Biochem Biophys Res Commun. 2015;463(1–2):109–115. doi: 10.1016/j.bbrc.2015.05.036. [DOI] [PubMed] [Google Scholar]
- 51.Gertig U, Hanisch UK. Microglial diversity by responses and responders. Front Cell Neurosci. 2014;8:101. doi: 10.3389/fncel.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das A, et al. Transcriptome sequencing reveals that LPS-triggered transcriptional responses in established microglia BV2 cell lines are poorly representative of primary microglia. J Neuroinflammation. 2016;13(1):182. doi: 10.1186/s12974-016-0644-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crotti A, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17(4):513–521. doi: 10.1038/nn.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lanzillotta A, et al. NF-κB in innate neuroprotection and age-related neurodegenerative diseases. Front Neurol. 2015;6:98. doi: 10.3389/fneur.2015.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhatt D, Ghosh S. Regulation of the NF-κB-mediated transcription of inflammatory genes. Front Immunol. 2014;5:71. doi: 10.3389/fimmu.2014.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rivas-Arancibia S, Zimbrón LF, Rodríguez-Martínez E, Maldonado PD, Borgonio Pérez G, Sepúlveda-Parada M. Oxidative stress-dependent changes in immune responses and cell death in the substantia nigra after ozone exposure in rat. Front Aging Neurosci. 2015;7:65. doi: 10.3389/fnagi.2015.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Z, et al. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-κB signalling. Br J Nutr. 2012;107(2):229–241. doi: 10.1017/S0007114511002868. [DOI] [PubMed] [Google Scholar]
- 58.Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJ, Anthony DC. The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation. 2011;8:166. doi: 10.1186/1742-2094-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoenen C, et al. Alpha-synuclein proteins promote pro-inflammatory cascades in microglia: stronger effects of the A53T mutant. PLoS One. 2016;11(9):e0162717. doi: 10.1371/journal.pone.0162717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen J, et al. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280(48):40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- 61.von Bernhardi R, Tichauer JE, Eugenín J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J Neurochem. 2010;112(5):1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x. [DOI] [PubMed] [Google Scholar]
- 62.Schaafsma W, et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav Immun. 2015;48:205–221. doi: 10.1016/j.bbi.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 63.Kaminska B, Mota M, Pizzi M. Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim Biophys Acta. 2016;1862(3):339–351. doi: 10.1016/j.bbadis.2015.10.026. [DOI] [PubMed] [Google Scholar]
- 64.Fontana MF, Baccarella A, Pancholi N, Pufall MA, Herbert DR, Kim CC. JUNB is a key transcriptional modulator of macrophage activation. J Immunol. 2015;194(1):177–186. doi: 10.4049/jimmunol.1401595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nomaru H, et al. Fosb gene products contribute to excitotoxic microglial activation by regulating the expression of complement C5a receptors in microglia. Glia. 2014;62(8):1284–1298. doi: 10.1002/glia.22680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao GN, Jiang DS, Li H. Interferon regulatory factors: at the crossroads of immunity, metabolism, and disease. Biochim Biophys Acta. 2015;1852(2):365–378. doi: 10.1016/j.bbadis.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 67.Hagemeyer N, et al. Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for Irf8 in macrophage maturation. EMBO J. 2016;35(16):1730–1744. doi: 10.15252/embj.201693801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Minten C, Terry R, Deffrasnes C, King NJ, Campbell IL. IFN regulatory factor 8 is a key constitutive determinant of the morphological and molecular properties of microglia in the CNS. PLoS ONE. 2012;7(11):e49851. doi: 10.1371/journal.pone.0049851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shiau CE, Kaufman Z, Meireles AM, Talbot WS. Differential requirement for irf8 in formation of embryonic and adult macrophages in zebrafish. PLoS One. 2015;10(1):e0117513. doi: 10.1371/journal.pone.0117513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Masuda T, et al. IRF8 is a transcriptional determinant for microglial motility. Purinergic Signal. 2014;10(3):515–521. doi: 10.1007/s11302-014-9413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Masuda T, et al. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep. 2012;1(4):334–340. doi: 10.1016/j.celrep.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Akagi T, et al. Interferon regulatory factor 8 expressed in microglia contributes to tactile allodynia induced by repeated cold stress in rodents. J Pharmacol Sci. 2014;126(2):172–176. doi: 10.1254/jphs.14143SC. [DOI] [PubMed] [Google Scholar]
- 73.Masuda T, et al. Transcription factor IRF1 is responsible for IRF8-mediated IL-1β expression in reactive microglia. J Pharmacol Sci. 2015;128(4):216–220. doi: 10.1016/j.jphs.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 74.Masuda T, et al. Transcription factor IRF5 drives P2X4R+-reactive microglia gating neuropathic pain. Nat Commun. 2014;5:3771. doi: 10.1038/ncomms4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen M, et al. Chronic exposure to TGFβ1 regulates myeloid cell inflammatory response in an IRF7-dependent manner. EMBO J. 2014;33(24):2906–2921. doi: 10.15252/embj.201489293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tanaka T, Murakami K, Bando Y, Yoshida S. Interferon regulatory factor 7 participates in the M1-like microglial polarization switch. Glia. 2015;63(4):595–610. doi: 10.1002/glia.22770. [DOI] [PubMed] [Google Scholar]
- 77.Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- 78.Garden GA, et al. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004;18(10):1141–1143. doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- 79.Davenport CM, Sevastou IG, Hooper C, Pocock JM. Inhibiting p53 pathways in microglia attenuates microglial-evoked neurotoxicity following exposure to Alzheimer peptides. J Neurochem. 2010;112(2):552–563. doi: 10.1111/j.1471-4159.2009.06485.x. [DOI] [PubMed] [Google Scholar]
- 80.Aloi MS, Su W, Garden GA. The p53 transcriptional network influences microglia behavior and neuroinflammation. Crit Rev Immunol. 2015;35(5):401–415. doi: 10.1615/CritRevImmunol.v35.i5.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jayadev S, et al. Transcription factor p53 influences microglial activation phenotype. Glia. 2011;59(10):1402–1413. doi: 10.1002/glia.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jebelli J, Hooper C, Pocock JM. Microglial p53 activation is detrimental to neuronal synapses during activation-induced inflammation: Implications for neurodegeneration. Neurosci Lett. 2014;583:92–97. doi: 10.1016/j.neulet.2014.08.049. [DOI] [PubMed] [Google Scholar]
- 83.Su W, et al. The p53 transcription factor modulates microglia behavior through microRNA-dependent regulation of c-Maf. J Immunol. 2014;192(1):358–366. doi: 10.4049/jimmunol.1301397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kang K, Robinson GW, Hennighausen L. Comprehensive meta-analysis of Signal Transducers and Activators of Transcription (STAT) genomic binding patterns discerns cell-specific cis-regulatory modules. BMC Genomics. 2013;14:4. doi: 10.1186/1471-2164-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yan Z, Gibson SA, Buckley JA, Qin H, Benveniste EN. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol. doi: 10.1016/j.clim.2016.09.014. [published online ahead of print October 3, 2016]. http://doi.org/10.1016/j.clim.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Przanowski P, et al. The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia. J Mol Med. 2014;92(3):239–254. doi: 10.1007/s00109-013-1090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10(4):217–224. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- 88.Ramos C, Robert B. msh/Msx gene family in neural development. Trends Genet. 2005;21(11):624–632. doi: 10.1016/j.tig.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 89.Yu Z, et al. MSX3 switches microglia polarization and protects from inflammation-induced demyelination. J Neurosci. 2015;35(16):6350–6365. doi: 10.1523/JNEUROSCI.2468-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zetterström RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276(5310):248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- 91.Le WD, et al. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2003;33(1):85–89. doi: 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- 92.Saijo K, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137(1):47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De Miranda BR, et al. The Nurr1 activator 1,1-bis(3’-indolyl)-1-(p-chlorophenyl)methane blocks inflammatory gene expression in BV-2 microglial cells by inhibiting nuclear factor κB. Mol Pharmacol. 2015;87(6):1021–1034. doi: 10.1124/mol.114.095398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Arevalo MA, Azcoitia I, Garcia-Segura LM. The neuroprotective actions of oestradiol and oestrogen receptors. Nat Rev Neurosci. 2015;16(1):17–29. doi: 10.1038/nrn3856. [DOI] [PubMed] [Google Scholar]
- 95.Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglial activation. Endocrinology. 2000;141(10):3646–3656. doi: 10.1210/endo.141.10.7693. [DOI] [PubMed] [Google Scholar]
- 96.Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145(4):584–595. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang Y, et al. An overview of the molecular mechanisms and novel roles of Nrf2 in neurodegenerative disorders. Cytokine Growth Factor Rev. 2015;26(1):47–57. doi: 10.1016/j.cytogfr.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 98.Li N, et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173(5):3467–3481. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 99.Kobayashi EH, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lastres-Becker I, et al. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain. 2014;137(Pt 1):78–91. doi: 10.1093/brain/awt323. [DOI] [PubMed] [Google Scholar]
- 101.Rojo AI, Innamorato NG, Martín-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010;58(5):588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 102.Zhao X, et al. Cleaning up after ICH: the role of Nrf2 in modulating microglia function and hematoma clearance. J Neurochem. 2015;133(1):144–152. doi: 10.1111/jnc.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Park SY, Jin ML, Ko MJ, Park G, Choi YW. Anti-neuroinflammatory effect of Emodin in LPS-stimulated microglia: involvement of AMPK/Nrf2 activation. Neurochem Res. 2016;41(11):2981–2992. doi: 10.1007/s11064-016-2018-6. [DOI] [PubMed] [Google Scholar]
- 104.Bozek K, et al. Organization and evolution of brain lipidome revealed by large-scale analysis of human, chimpanzee, macaque, and mouse tissues. Neuron. 2015;85(4):695–702. doi: 10.1016/j.neuron.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 105.Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6(4):254–264. doi: 10.1007/s13238-014-0131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Petrov AM, Kasimov MR, Zefirov AL. Brain cholesterol metabolism and its defects: linkage to neurodegenerative diseases and synaptic dysfunction. Acta Naturae. 2016;8(1):58–73. [PMC free article] [PubMed] [Google Scholar]
- 107.Kidani Y, Bensinger SJ. Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol Rev. 2012;249(1):72–83. doi: 10.1111/j.1600-065X.2012.01153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23(7):351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 109.Namgaladze D, Brüne B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim Biophys Acta. 2016;1861(11):1796–1807. doi: 10.1016/j.bbalip.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 110.Barlic J, Zhang Y, Foley JF, Murphy PM. Oxidized lipid-driven chemokine receptor switch, CCR2 to CX3CR1, mediates adhesion of human macrophages to coronary artery smooth muscle cells through a peroxisome proliferator-activated receptor gamma-dependent pathway. Circulation. 2006;114(8):807–819. doi: 10.1161/CIRCULATIONAHA.105.602359. [DOI] [PubMed] [Google Scholar]
- 111.Chen H, et al. Macrophage peroxisome proliferator-activated receptor γ deficiency delays skin wound healing through impairing apoptotic cell clearance in mice. Cell Death Dis. 2015;6:e1597. doi: 10.1038/cddis.2014.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bashir S, Sharma Y, Elahi A, Khan F. Macrophage polarization: the link between inflammation and related diseases. Inflamm Res. 2016;65(1):1–11. doi: 10.1007/s00011-015-0874-1. [DOI] [PubMed] [Google Scholar]
- 113.Skerrett R, Pellegrino MP, Casali BT, Taraboanta L, Landreth GE. Combined liver X receptor/peroxisome proliferator-activated receptor γ agonist treatment reduces amyloid β levels and improves behavior in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2015;290(35):21591–21602. doi: 10.1074/jbc.M115.652008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pérez MJ, Quintanilla RA. Therapeutic actions of the thiazolidinediones in Alzheimer’s disease. PPAR Res. 2015;2015:957248. doi: 10.1155/2015/957248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci. 2012;32(30):10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32(48):17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Savage JC, et al. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J Neurosci. 2015;35(16):6532–6543. doi: 10.1523/JNEUROSCI.4586-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Courtney R, Landreth GE. LXR regulation of brain cholesterol: from development to disease. Trends Endocrinol Metab. 2016;27(6):404–414. doi: 10.1016/j.tem.2016.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gabbi C, Warner M, Gustafsson JÅ. Action mechanisms of liver X receptors. Biochem Biophys Res Commun. 2014;446(3):647–650. doi: 10.1016/j.bbrc.2013.11.077. [DOI] [PubMed] [Google Scholar]
- 120.Spann NJ, Glass CK. Sterols and oxysterols in immune cell function. Nat Immunol. 2013;14(9):893–900. doi: 10.1038/ni.2681. [DOI] [PubMed] [Google Scholar]
- 121.Cramer PE, et al. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335(6075):1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Skerrett R, Malm T, Landreth G. Nuclear receptors in neurodegenerative diseases. Neurobiol Dis. 2014;72 Pt A:104–116. doi: 10.1016/j.nbd.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sandoval-Hernández AG, Restrepo A, Cardona-Gómez GP, Arboleda G. LXR activation protects hippocampal microvasculature in very old triple transgenic mouse model of Alzheimer’s disease. Neurosci Lett. 2016;621:15–21. doi: 10.1016/j.neulet.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 124.Secor McVoy JR, Oughli HA, Oh U. Liver X receptor-dependent inhibition of microglial nitric oxide synthase 2. J Neuroinflammation. 2015;12:27. doi: 10.1186/s12974-015-0247-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cui W, Sun Y, Wang Z, Xu C, Peng Y, Li R. Liver X receptor activation attenuates inflammatory response and protects cholinergic neurons in APP/PS1 transgenic mice. Neuroscience. 2012;210:200–210. doi: 10.1016/j.neuroscience.2012.02.047. [DOI] [PubMed] [Google Scholar]
- 126.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–991. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- 128.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]