

Abstract
Purpose
To explore the effect of the CCL2 and CCR2 system on the activation and migration of microglia and monocytes in light-induced photoreceptor apoptosis.
Methods
At 1 day, 3 days, 7 days, and 14 days after light exposure, OX42 and ED1 immunostaining were used to label the activation and migration of microglia and monocytes. Double immunostaining of CCL2 with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), OX42, or glial fibrillary acidic protein (GFAP) was applied to explore the relationships among CCL2, apoptotic photoreceptors, activated microglia and monocytes, and macroglial cells (Müller cells and astrocytes). Real-time PCR was used to evaluate the mRNA levels of retinal CCL2 and CCR2 and the proinflammatory factors interleukin (IL)-1 beta and tumor necrosis factor (TNF)-alpha.
Results
Real-time PCR analyses showed that CCL2 and CCR2 expression gradually increased after light exposure and peaked at 3 days, coinciding with the infiltration of OX42-positive cells and the expression of IL-1 beta and TNF-alpha in the outer retina. Double immunostaining of CCL2 with TUNEL revealed that CCL2 was expressed robustly in about 30% of the apoptotic photoreceptors at the early stage. As degeneration progressed, immunostaining of CCL2 with OX42 showed that activated and migrated microglia and monocytes expressed CCL2. At the late stage, Müller cells became the main source of CCL2, which was illustrated by CCL2 immunostaining with GFAP.
Conclusions
Light exposure led to apoptosis of photoreceptors, which expressed CCL2, accelerating an inflammation-mediated cascade by activating and attracting microglia and monocytes and promoting their secretion of CCL2 in the injured position.
Introduction
Photoreceptor degeneration is an important characteristic of age-related macular degeneration (AMD) and retinitis pigmentosa (RP). As a defensive reaction against various stress signals, inflammation is a self-sacrificial process [1]. Inflammation has been shown to be actively involved in the pathogenesis of photoreceptor degenerative diseases, although the retina is considered an immune-privileged organ [2-4]. Macroglial cells (Müller cells and astrocytes) and microglial cells are two basic types of glial cells in the mammalian retina. Regarded as the major resident immune cells in the retina [5], resting microglia play an important role in the immune surveillance and host defense under physiologic conditions [6]. Upon injury or disease, microglia and monocytes are activated. Many neurotoxic factors are secreted, such as interleukin (IL)-1 beta and tumor necrosis factor (TNF)-alpha, contributing to neurodegeneration [7]. In a model of light-induced photoreceptor degeneration, our previous work showed that retinal microglia and monocytes are activated and recruited to the area of photoreceptor death, along with upregulation of proinflammatory factor IL-1 beta [8]. However, the process by which microglia and monocytes are recruited to the injury-specific region after light-induced photoreceptor apoptosis remains unknown.
CC chemokines, which regulate monocyte/macrophage activation and recruitment, are involved in the pathogenesis of immune-mediated inflammation [9]. Studies have shown that several CC chemokines increase significantly after inflammation-associated degeneration. In particular, the chemokine CCL2 is the most well characterized [10,11]. Elevated expression levels of CCL2 and its receptor CCR2 have been found in acute and chronic multiple sclerosis plaques [11], Alzheimer disease [12], and experimental allergic encephalomyelitis and anterior uveitis [13]. These findings indicate that the CCL2 system is required in lesion formation by inducing the expression of proinflammatory factors and recruiting macrophages. Furthermore, Rutar et al. [14] demonstrated that monocyte recruitment after light-induced cell death is correlated with the spatial-temporal expression of CCL2 by Müller cells; and targeted suppression of CCL2 in Müller cells inhibits the recruitment of monocytes/microglia [15]. However, the cellular localization of the expression of CCL2 and its receptor CCR2 in the retina after light-induced photoreceptor degeneration and the relationship among photoreceptor apoptosis, the expression pattern of CCL2, and infiltration of microglia and monocytes need to be further explored.
In the current study, the expression pattern of CCL2 in a rat photic injury model was analyzed to delineate the role of CCL2 in the activation and migration of microglia and monocytes in light-induced photoreceptor degeneration. The results showed that CCL2 was expressed only by astrocytes under normal conditions; while in the early stage of photoreceptor injury, the apoptotic photoreceptors and the migrated microglia and monocytes secreted abundant CCL2, which was in concert with the upregulation of IL-1 beta and TNF-alpha. The overall expression pattern of CCL2 correlated closely with the migration and activation of microglia and monocytes. At the late stage of photic injury, instead of microglia and monocytes and photoreceptors, Müller cells became a rich source of CCL2. In particular, the spatial-temporal expression of CCL2 in the light-induced injured retina was demonstrated to reveal the important role of the CCL2 system in the neuro-glia interaction in light-induced photoreceptor degeneration.
Methods
Animals and light exposure
All animal experiments were implemented with a methodology similar to the Statement by the Association for Research in Vision and Ophthalmology Statement for Use of Animals in Research. The subjects were Sprague Dawley rats weighing 200–250 g and aged 4–6 weeks. All rats were kept in a cage with a 12 h:12 h light-dark cycle. After adaption for 24 h in the dark, the rats were maintained at 24 °C and exposed to bright blue light at about 2,500 lux measured with a numerical luminometer (JC07-TES-1334A, Beixin, China). All rats were subjected to the conditions above and returned to the cage after bright blue light exposure.
TUNEL
Immediate cell death was detected with use of a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining detection kit (R&D Systems, Minneapolis, MN), according to the manufacturer’s protocol. TUNEL-positive cells were defined as cells with green color staining under a confocal laser microscope (TCS SP2; Leica Microsystems, Bensheim, Germany). The number of TUNEL-positive cells in the outer nuclear layer (ONL) was counted.
Immunohistochemistry
After removal of the cornea and lens, the eyecups were fixed with 4% paraformaldehyde for 2 h. The eyecups were kept in different levels of sucrose solutions (20–30% in phosphate buffer) and maintained at 4 °C. Optimal cutting temperature compound was added, and the samples were frozen by liquid nitrogen. Ten-micrometer sections were cut using a cryostat (Bright Instruments Ltd., Huntingdon, UK). Sections within approximately 1 mm of the optic nerve head were recovered on gelatin-coated slides. These slides were air-dried, followed by incubation with blocking buffer (5% goat serum) at room temperature for 1 h. The sections were washed three times with 0.01 M PBS (1X; 120 mM NaCl, 20 mM KCl, 10 mM NaPO4, 5 mM KPO4, pH 7.4). after incubation. Incubation with the following primary antibodies was performed: monoclonal mouse anti-rat OX42 antibody (1:100; Abcam, Cambridge, MA), monoclonal mouse anti-rat ED1 antibody (1:100; Millipore, Millipore, MA), polyclonal rabbit anti-rat CCL2 antibody (1:100; Abcam), and rabbit anti-mouse CCR2 antibody (1:100; Abcam). For double staining, the sections were incubated with the following primary antibody pairs: CCL2 at 1:100 and OX42 at 1:100, CCL2 at 1:100 and mouse anti-rat glial fibrillary acidic protein (GFAP) at 1:100, and CCL2 at 1:100 and TUNEL reaction solution. Afterward, the sections were washed three times with 0.01 M PBS and incubated with two kinds of secondary antibodies for 45 min. The first one was conjugated to Alexa 488 (1:1,000; Invitrogen, Carlsbad, CA), and the second one was conjugated to Alexa 555 (1:1,000; Invitrogen). The temperature of the incubation process was maintained at 37 °C.
For immunostaining of retinal flat mounts, the retinas were dissected out after 4% paraformaldehyde fixation. The retinas were kept in 0.3% Triton X-100 in PBS to maintain their permeability. One hour later, they were washed three times with PBS and incubated with two kinds of primary antibodies (CCL2 at 1:100 and OX42 at 1:100; CCL2 at 1:100 and GFAP at 1:100). Afterward, they were washed three times with 0.01 M PBS and incubated with two kinds of secondary antibodies for 45 min, Alexa 488 (1:1,000; Invitrogen) and Alexa 555 (1:1,000; Invitrogen). The temperature of the incubation process was maintained at 37 °C. The sections or retinas were mounted with antifade mounting medium after washing with PBS and detected with a confocal laser microscope (TCS SP2; Leica Microsystems, Bensheim, Germany).
Quantitative real-time PCR
After bright blue light exposure, the retinas were extracted and frozen by liquid nitrogen until the time of the experiment. As described previously, the total RNA was extracted from the retinas and quantified. According to the manufacturer’s suggestion, the first-strand cDNA was compounded from total RNA through the use of reverse transcriptase and oligo(dT) primer. A 20-µl mixture was prepared from 500 nM primer, 1 µl of cDNA, and 1× PCR mix (iQ SYBR Green Supermix; Bio-Rad, Hercules, CA). This mixture was the base for the PCRs, and the following PCR parameters were applied in a thermocycler: 95 °C for 5 min followed by 50 cycles at 95 °C for 15 s, 65 °C for 15 s, and 72 °C for 15 s. The fluorescence threshold (Ct) was calculated. The lack of nonspecific products was indicated by the melting-point curves and electrophoresis in 3% agarose gels. GAPDH was regarded as a standard of mRNA expression. A comparison Ct method was used. The primer sequences used are shown in Table 1.
Table 1. The primer sequences used were as follows.
| Gene | Primer (5′-3′) |
|---|---|
| CCL2 |
F: AAGAAGCTGTAGTATTTGTCACCAAGCTCA |
| R: CATCAGGTACGATCCAGGCT |
|
| CCR2 |
F: CGCAGAGTTGACAAGTTGTG |
| R: CGCAGAGTTGACAAGTTGTG |
|
| IL-1beta |
F: ATGCACCTGTACGATCACTGA |
| R: ACAAAGGACATGGAGAACACC |
|
| TNF-alpha |
F: GAGGCAATAGGTTTTGAGGGCCAT |
| R: GGGACACACAAGCATCAAG |
Western blotting
RIPA buffer was used as the medium for the retinas and contained the following ingredients: 1% Triton X-100, 5% sodium dodecyl sulfate (SDS), 5% deoxycholic acid, 0.5 M Tris–HCl pH 7.5, 10% glycerol, 1 mM ethylene diamine tetraacetic acid, 1 mM phenylmethylsulfonyl fluoride, 5 × 10–12 μg/ml aprotinin, 1 × 10–12 μg/ml leupeptin, 1 × 10–12 μg/ml pepstatin, 200 mM sodium orthovanadate, and 200 mM sodium fluoride.
The tissue extracts were kept on ice for 10 min before centrifugation (10,000 ×g for 25 min at 4 °C). The total protein content of the extracts was evaluated with the bicinchoninic acid assay. The retina extracts were stored in 5 × buffer (60 mM Tris–HCl pH 7.4, 25% glycerol, 2% SDS, 14.4 mM 2-mercaptoethanol, and 0.1% bromophenol blue). The buffer was boiled for 5 min and resolved by electrophoresis in a 12% SDS-polyacrylamide gel. Proteins were transferred on a nitrocellulose membrane, and Ponceau S was used to stain the blots. The protein bands were detected such that equal loading of protein and uniform transfer could be guaranteed. The blots were blocked in TBST buffer (20 mM Tris–HCl pH 7.6, 137 mM NaCl, and 0.1% Tween-20). At the beginning of this process, 5% nonfat dry milk was added to the buffer, and the blocking process lasted for 45 min. The blots were probed with polyclonal rabbit anti-rat CCL2 antibody (1:1,000; Abcam) or polyclonal rabbit anti-rat CCR2 antibody (1:1,000; Abcam) for 24 h, followed by the horseradish peroxidase–conjugated goat anti-rabbit secondary antibody (1:10,000 dilution). X-ray film and the chemiluminescence system were used to detect the antibodies. An ImageMaster Video Documentation System (GE Healthcare / Amersham Pharmacia) was used to measure the signal intensity. The optical densities (mean ± standard deviation) for each sample were measured in three separate blots.
Data analysis
The mRNA and protein levels of target molecules from different groups were compared. The expression changes were relative to a control. One-way ANOVA was used, followed by post-hoc multiple comparison. A p value of less than 0.05 was considered statistically significant.
Results
Migration and activation of microglia and monocytes in light-induced photoreceptor degeneration
In healthy retinas, a few OX42-positive cells (known as retinal microglia) with slim processes were seen in the inner retina (Figure 1A). A small number of OX42-positive cells appeared in the ONL after light exposure, increased gradually at 1 day (Figure 1B), and peaked at 3 days (Figure 1C) in the subretinal space. The OX42-positive cells underwent transformation from resting ramified to activated amoeboid at 3 days and 7 days after exposure to light (Figure 1C,D).
Figure 1.

Microglia/monocyte activation and infiltration in light-induced photoreceptor degeneration. A, E: In healthy retinas, OX42-positive microglia were seen only in the retinal ganglion cells and the inner plexiform layer with long, slim processes, while no ED1-positive monocytes were seen in the retina. B, F: One day after exposure to light, a small number of monocytes/microglia began to infiltrate into the outer nuclear layer (ONL) and subretinal space. C: The number of OX42-positive cells peaked at 3 days, with activated amoeboid morphology. D, H: At 7 days, many OX42-positive cells remained in the thinned ONL and subretinal space, while the number of ED1-positive cells in the outer retina peaked at that time.
There were no ED1-positive cells (known as possible infiltrated macrophages) in the healthy retina (Figure 1E). At 1 day after light exposure (Figure 1F), a small number of ED1-positive cells appeared in the ONL. The ED1-positive cells increased in the subretinal space at 3 days (Figure 1G) and peaked at 7 days (Figure 1H).
Overall expression of CCL2 and CCR2 in the retina after light exposure
CCL2 immunoreactivity was localized in the ganglion cell layer in healthy retinas (Figure 2A). One day after light exposure, some CCL2 expression was present in the ONL (Figure 2B), and it began to increase at 3 days (Figure 2C). At 7 days, CCL2 expression was present in radially oriented processes traversing the inner retina (Figure 2D). At 14 days, CCL2 continued to be expressed in the radially oriented processes (data not shown). There was no immunoreactivity to CCR2 in the healthy retinas (Figure 2E). In the experimental groups, some CCR2 was present in the ganglion cell layer at 1 day after light exposure (Figure 2F), and it increased further from the inner nuclear layer to the ONL at 3 days (Figure 2G). Seven days after light exposure, immunoreactivity to CCR2 was localized in the ONL (Figure 2H).
Figure 2.

Expression of CCL2 and CCR2 in the retina after extensive light exposure. A: CCL2 was scarcely expressed in the retinal ganglion cells (RGCs) of a healthy retina. B, C: At 1 day after light exposure, there was a small amount of CCL2 expression in the outer nuclear layer (ONL), which increased in the RGCs and the inner nuclear layer (INL) at 3 days. D: At 7 days, CCL2 was observed in cells with radially oriented processes (yellow arrows) throughout the RGCs. E: CCR2 was scarcely expressed in a healthy retina. F, G: At 1 day, CCR2 was observed in the RGCs of the retina, and it was expressed robustly in the INL and the ONL at 3 days after light exposure. H: CCR2 was richly expressed in the ONL at 7 days.
Representative localization of CCL2 in the retina after light exposure
Double immunofluorescence staining was performed on the retinas with antibodies to GFAP and CCL2. Based on the robust expression of CCL2 in the ONL after light exposure, combined TUNEL and immunofluorescence staining were performed to determine the relationship between CCL2 and apoptotic photoreceptors. Colocalization of CCL2 and GFAP was observed in the healthy retinal slice (Figure 3A–C) and the flat mount (Figure 3D–F), indicating that astrocytes might be the main source of CCL2 in the healthy retina. One day after light exposure, increased expression of CCL2 was observed in the ONL (Figure 3G). Double immunostaining of CCL2 with TUNEL staining revealed that some of the TUNEL-positive cells were also CCL2-positive. In the vision field, the number of CCL2-positive cells was found to be 20, and the number of TUNEL-positive cells was 63, indicating that about 31.74% of the apoptotic photoreceptors secreted CCL2 after light-induced injury (Figure 3G–L). Double immunostaining of CCL2 with OX42 in the retinal slice at 3 days further revealed colocalization of CCL2 and OX42 in the ONL (Figure 4A–F), demonstrating that some migrated microglia also expressed CCL2, which was confirmed by the result in the retinal flat mount (Figure 4G–L). The activation of microglia was relieved at 7 days, as indicated by the immunostaining of OX42 in the flat mount (Figure 5D–F), and there was no colocalization of CCL2 and OX42 compared with the colocalization at 3 days (Figure 5A–C), signifying that the migrated microglia did not express CCL2 in the late phase of light-induced injury. Seven days after light exposure, the fluorescent markers showed that CCL2 colocalized with the GFAP-immunoreactive Müller cell processes (Figure 5G–L) and astrocytes (Figure 5M,O).
Figure 3.

Localization of CCL2 in a healthy retina and apoptotic photoreceptors by double immunofluorescence staining. A, D: In a healthy retinal slice and flat mount, CCL2 was mainly expressed in the retinal ganglion cell layer. B, E: Glial fibrillary acidic protein (GFAP) staining was localized in the astrocytes. C, F: The merged images revealed that CCL2 was expressed by astrocytes in a healthy retina. G: At 1 day after light exposure, CCL2 was expressed robustly in the outer nuclear layer (ONL). H: Large numbers of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)–positive cells were observed in the ONL. I: The merged image shows that some of the CCL2 expression was colocalized in TUNEL-positive cells. J, K, L: Amplified images of G, H, and I, respectively.
Figure 4.

Localization of CCL2 in activated microglia and monocytes by double immunofluorescence staining. A, B: At 3 days after light exposure, in the retinal slice, CCL2 expression was observed in the outer nuclear layer (ONL), and many OX42-positive cells were activated and migrated into the ONL and subretinal space. C: The merged image shows that CCL2 was colocalized in those activated and migrated microglia and monocytes. D, E, F: Amplified images of A, B, and C, respectively. G, H: In the retinal flat mount, CCL2 was expressed richly in the outer retina, where OX42-positive cells were found. I: The merged image shows that CCL2 was colocalized in those microglia and monocytes. J, K, L: Amplified images of G, H, and I, respectively.
Figure 5.

Localization of CCL2 in the late stage of photic injury by double immunofluorescence staining. A, B, C: In the flat mount, CCL2 was colocalized in OX42-positive cells with amoeboid morphology at 3 days after light exposure. D, E, F: At 7 days, no expression of CCL2 was observed in OX42-positive cells. G: However, expression of CCL2 was observed in cells with radially oriented processes throughout the inner nuclear layer. H: Strong immunoreactivity to glial fibrillary acidic protein (GFAP) appeared from the internal limiting membrane to the inner plexiform layer at 7 days. I: The merged image shows that CCL2 was colocalized in GFAP-positive Müller glia. J, K, L: Amplified images of G, H, and I, respectively. M, N, O: Immunofluorescent staining in the retinal flat mount confirmed the observation in the retinal slice.
Patterns of CCL2, CCR2, IL-1 beta, and TNF-alpha expression in the retina after light exposure
The levels of retinal CCL2 and CCR2 mRNA at various time points after exposure to light were examined. CCL2 and CCR2 were detected as early as 1 day after light exposure, and the patterns of gene expression were similar. CCL2 and CCR2 were significantly upregulated and maximized at the peak of 3 days (Figure 6A,B). To determine whether the inflammation correlates with the presence of CC chemokines, the expression levels of the proinflammatory factors IL-1 beta and TNF-alpha were examined. Both factors showed a gradually increasing trend that peaked on the third day after light exposure, coinciding with the expression of CCL2 and CCR2 (Figure 6C,D).
Figure 6.
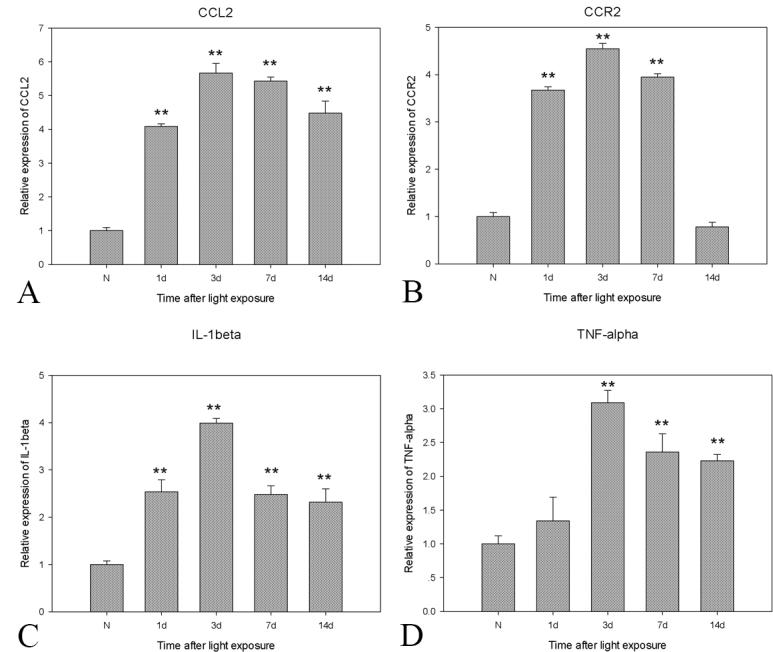
Real-time PCR of CCL2 and CCR2 and IL-1 beta and TNF-alpha expression after extensive light exposure. A, B: The mRNA of CCL2 and CCR2 began to increase as early as 1 day, peaked at 3 days, and slowly decreased. C, D: The expression pattern of interleukin (IL)-1 beta and tumor necrosis factor (TNF)-alpha was consistent with the patterns of CCL2 and CCR2. GAPDH expression served as the loading control. The mean ± standard deviation of data from three independent experiments is shown, **, p<0.01.
To determine whether mRNA expression correlates with the production of chemokine proteins, western blotting was used to assay CCL2 and CCR2, during the course of the light-induced injury. The CCL2 protein level was correlated with the mRNA expression (Figure 7A,B), with its peak at 3 days. However, a few differences were found in terms of the expression pattern of CCR2 protein compared with CCR2 mRNA. The overall trend was similar, while the maximum expression was found at 7 days after light exposure (Figure 7A,C).
Figure 7.

Western blot analysis of CCL2 and CCR2 expression after extensive light exposure. A, B: CCL2 protein expression began to increase at 1 day and peaked at 3 days. A, C: CCR2 protein expression began to increase at 3 days and peaked at 7 days after light exposure. Actin protein expression served as the loading control. The mean ± standard deviation of data from three independent experiments is shown, *,p<0.05; **,p<0.01.
Comparative changes in OX42-positive and ED1-positive cells in the outer retina with CCL2 and CCR2 and IL-1 beta and TNF-alpha expression
The number of OX42-positive cells in the outer retina increased gradually at 1 day and peaked at 3 days after light exposure (Figure 8A), following the peak of the TUNEL-positive cells (our previously published data) [8] and coinciding with the peak of CCL2 and CCR2 and IL-1 beta and TNF-alpha expression (Figure 8A,B). The number of ED1-positive cells in the outer retina increased slowly at 1 day and peaked at 7 days after light exposure (Figure 8B), which was slightly delayed compared with CCL2 and CCR2 and IL-1 beta and TNF-alpha expression (Figure 8A,B). The number of OX42-positive cells was greater than the number of ED1-positive cells at various time points, except 7 days after light exposure (Figure 8A,B).
Figure 8.

Comparative changes in microglia/monocyte infiltration, as well as CCL2 and CCR2 and IL-1 beta and TNF-alpha expression. A: The number of OX42-positive cells increased rapidly at as early as 1 day after light exposure and peaked at 3 days. B: The increasing number of ED1-positive cells in the outer retina occurred just following the infiltration of OX42-positive cells and peaked at 7 days. A, B: The expression patterns of CCL2 and CCR2 and interleukin (IL)-1 beta and tumor necrosis factor (TNF)-alpha were consistent with the infiltration of OX42-positive cells, with the peak at 3 days.
Discussion
Previous studies by our laboratory [8,16] and others [17,18] have shown that microglia and monocytes are activated and migrate to the outer retina in the photic injury model, indicating its importance in photoreceptor apoptosis. To explore the relationship between microglia/monocyte infiltration and photoreceptor apoptosis, the current study focused on the expression pattern of the chemokine CCL2 in light-induced photoreceptor apoptosis. The spatial-temporal expression of CCL2 was examined during the progression of photoreceptor apoptosis, and its relationship with photoreceptor apoptosis, microglia/monocyte infiltration, and upregulation of proinflammatory factors was analyzed.
Intense blue light exposure was first used to induce apoptosis of photoreceptors such that approximately 30% of the apoptotic photoreceptors began to express CCL2. Second, the robust recruitment of microglia and monocytes to the outer retina occurred upon photoreceptor apoptosis, which was correlated with the upregulation of the proinflammatory factors IL-1 beta and TNF-alpha. Double immunofluorescence staining revealed that CCL2 was expressed mainly by infiltrated microglia and monocytes. Third, our data further showed that Müller cells, taking the place of apoptotic photoreceptors and microglia and monocytes, became a rich source of CCL2 at the late stage of photic injury, indicating a possible important role of CCL2 in Müller cell–associated inflammatory signaling pathways.
Photic injury shares an important feature with human retinal dystrophies: the loss of visual cells. Two classes of photic injury have been proposed [19]. Class I damage occurs when long-term exposure to light at 380–410 nm with a low irradiance damages photoreceptor cells [19]. Class II damage occurs after short-term (<4 h) exposure to light at 470 nm with a high irradiance damages the photoreceptors and RPE cells [19]. In this study, 24-h exposure to blue light at 400 nm with 2,500 lux was designed to mimic class I damage. Retinal degeneration caused by photic injury proceeds faster than inherited retinal degeneration [20]. The results demonstrated that the disappearance of most of the photoreceptors was found at 7 days after light exposure, which was similar to the late phase of human retinal degenerative disease. In addition, the apoptosis of photoreceptors was synchronized after light exposure. This facilitated the detection of biochemical markers, assessment of the behavioral change of retinal microglia, and analysis of the molecular network involved in the process [21].
Chemokines constitute a class of chemoattractant cytokines, which are known as C, CC, CXC, and CX3C according to the systematic nomenclature [22]. The CC-chemokine subfamily is basically constituted by monocyte chemoattractant proteins [23]. MCP-1, the first to be discovered, was recently reclassified as CCL2 [24]. Recent evidence has shown that CCL2 appears to be an important mediator of the inflammatory response in many neurodegenerative brain diseases [25]. In acute neuroinflammation, CCL2 is considered a detrimental factor in stroke and excitotoxic injury [26,27]. An elevated level of CCL2 also has been detected in chronic multiple sclerosis plaques [25]. In the retina, CCL2 expression is low in healthy young animals but increases with age and stress levels in animal models of glaucoma [28], experimental autoimmune uveitis [29], diabetic retinopathy [30], and age-related macular degeneration [31,32]. The current investigation verified the upregulation of CCL2 and its receptor CCR2 in light-induced photoreceptor degeneration and revealed that the expression pattern of CCL2 and CCR2 was consistent with the infiltration of microglia and monocytes. Upregulation of the chemokine CCL2 might have an important role in mediating local neuroinflammatory responses driven by activated microglia and monocytes.
Numerous studies have shown that photoreceptor apoptosis occurs in light-induced photoreceptor degeneration, whereas activated microglia and monocytes infiltrate into the necrotic lesions [8,17,18]. CCL2 has been proposed to be the factor that initiates the reactive sequelae of microglia. Our previous study [33] showed the effective expression of green fluorescent protein–tagged CCR2 in primary microglia with the use of lentiviral vector enhanced CCL2-mediated microglial recruitment. In addition, a substantial decrease in monocyte recruitment after experimental retinal detachment was found in CCL2-deficient knockout mice [34]. However, there has been no direct identification of the relationship among the specific expression of CCL2, apoptotic photoreceptors, and migrated microglia and monocytes. Our data demonstrated that intense light exposure caused significant photoreceptor apoptosis that peaked at 1 day, the early stage of photic injury (our previously published study) [8], and that CCL2 was expressed in some of those apoptotic photoreceptors. Photoreceptor apoptosis induced infiltration of microglia and monocytes that underwent significant transformation, from resting ramified to activated amoeboid. CCL2 was expressed richly in the infiltrated microglia and monocytes, as shown by double immunostaining in the retinal slice and flat mount preparations. Such expression is consistent with upregulation of the proinflammatory factors IL-1 beta and TNF-alpha, which are proapoptotic and associated with the progression of retinal degeneration. Several studies, including our published work, have indicated that IL-1 beta is secreted by activated microglia under pathological conditions [8,35,36]. Zeng et al. [37] showed that activated microglia in the ONL are the major source of TNF-alpha, whereas diseased and apoptotic photoreceptor cells can be another source. It is possible that factors released by apoptotic photoreceptors attract microglia and that the microglia secrete more factors and amplify the inflammation-associated injury. The current investigation is the first to show the specific distribution of CCL2 in apoptotic photoreceptors as early as 24 h after light exposure. Together, these results demonstrate that CCL2 amplifies the microglia/monocyte-mediated inflammatory response and that it might be a key factor in the crosstalk network between apoptotic photoreceptors and activated microglia and monocytes.
There is some controversy regarding the precise distribution and cellular localization of CCL2 expression after light-induced damage. Zhang et al. considered that Müller cells or RPE cells might be the source [18]. Several in vitro studies have demonstrated that cultured RPE and microglial cells express CCL2 in response to stimulatory cytokines [31,38]. In addition, Rutar et al. [14] showed the preferential distribution of CCL2 mRNA in Müller cells in the region of the incipient lesion as early as 12 h after light exposure. However, in the present study, the expression pattern of CCL2 was changed, followed by the progression of photoreceptor degeneration. In the early phase, CCL2 protein expression was detected in some of the apoptotic photoreceptors, indicating that increased CCL2 in dying photoreceptors attracted microglia toward the outer retina. Accumulated microglia phagocytosed the debris of apoptotic photoreceptors and secreted CCL2, which may have self-amplified pathological processes. In the late phase, the expression of CCL2 was colocalized and correlated with the expression of GFAP. As we know, Müller cells do not express GFAP in the healthy retina [39]. However, the de novo expression of GFAP is a general response to injury and inflammation, which is known as “glial stress” [40]. Reactive Müller cells proliferate, communicate with other retinal cell types, and play an important role in neuron–glia and glia–glia interactions [41]. In a Müller cell–microglial cell coculture system, the levels of CCL2 and CCL3 were elevated in the Müller cells, suggesting that Müller cells may be able to amplify inflammatory responses in bidirectional communication with microglia [42]. Meanwhile, responding Müller cells may be able to transmit inflammatory signals to adjacent Müller cells or across multiple retinal layers by their radial processes [43]. In the current study, in the late phase of photic injury, CCL2 was expressed in Müller glia with a spatial distribution pattern. It is reasonable to propose that CCL2 might also participate in the inflammatory signaling pathways that regulate neuron–glia and glia–glia crosstalk in photoreceptor degeneration.
In conclusion, this study demonstrated that intensive light exposure led to apoptosis of photoreceptors, which expressed CCL2 to activate and attract microglia and monocytes to the injured site; the infiltrated microglia and monocytes self-secreted CCL2 to amplify and accelerate a microglia/monocyte-mediated neuroinflammatory cascade, which is detrimental to photoreceptor degeneration. Müller glia became a rich source of CCL2 in the late stage of photic injury, indicating the possible role of CCL2 in glial stress-associated inflammatory signaling pathways. The potential modulation of the endogenous CCL2 system might provide a promising pathway to regulate microglia/monocyte activation in retinal degenerative disease.
Acknowledgments
Supported by research grants from Chinese Natural Science Foundation (NO. 81570855), Chinese National Natural Science Foundation (NO. 81,570,854), Chinese National Natural Science Foundation for Young Scholars (NO. 81500723). Chinese National Natural Science Foundation for Young Scholars (NO.81400410).
References
- 1.Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp Mol Med. 2006;38:333–47. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
- 2.Hughes EH, Schlichtenbrede FC, Murphy CC, Sarra GM, Luthert PJ, Ali RR, Dick AD. Generation of activated sialoadhesin-positive microglia during retinal degeneration. Invest Ophthalmol Vis Sci. 2003;44:2229–34. doi: 10.1167/iovs.02-0824. [DOI] [PubMed] [Google Scholar]
- 3.Ardeljan CP, Ardeljan D, Abu-Asab M, Chan CC. Inflammation and Cell Death in Age-Related Macular Degeneration: An Immunopathological and Ultrastructural Model. J Clin Med. 2014;3:1542–60. doi: 10.3390/jcm3041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sennlaub F, Auvynet C, Calippe B, Hu SJ, Dominguez E, Camelo S, Levy O, Guyon E, Saederup N, Charo IF, Rooijen NV, Nandrot E, Bourges JL, Behar-Cohen F, Sahel JA, Guillonneau X, Raoul W, Combadiere C. CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med. 2013;5:1775–93. doi: 10.1002/emmm.201302692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: just bystander or target for therapy? Prog Retin Eye Res. 2015;45:30–57. doi: 10.1016/j.preteyeres.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. 2007;81:1345–51. doi: 10.1189/jlb.0207114. [DOI] [PubMed] [Google Scholar]
- 7.Li L, Eter N, Heiduschka P. The microglia in healthy and diseased retina. Exp Eye Res. 2015;136:116–30. doi: 10.1016/j.exer.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 8.Ni YQ, Xu GZ, Hu WZ, Shi L, Qin YW, Da CD. Neuroprotective effects of naloxone against light-induced photoreceptor degeneration through inhibiting retinal microglial activation. Invest Ophthalmol Vis Sci. 2008;49:2589–98. doi: 10.1167/iovs.07-1173. [DOI] [PubMed] [Google Scholar]
- 9.Baggiolini M, Dahinden CA. CC chemokines in allergic inflammation. Immunol Today. 1994;15:127–133. doi: 10.1016/0167-5699(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–83. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- 11.McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. 1998;86:20–9. doi: 10.1016/s0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- 12.Prat E, Baron P, Meda L, Scarpini E, Galimberti D, Ardolino G, Catania A, Scarlato G. The human astrocytoma cell line U373MG produces monocyte chemotactic protein (MCP)-1 upon stimulation with beta-amyloid protein. Neurosci Lett. 2000;283:177–80. doi: 10.1016/s0304-3940(00)00966-6. [DOI] [PubMed] [Google Scholar]
- 13.Adamus G, Machnicki M, Amundson D, Adlard K, Offner H. Similar pattern of MCP-1 expression in spinal cords and eyes of Lewis rats with experimental autoimmune encephalomyelitis associated anterior uveitis. J Neurosci Res. 1997;50:531–8. doi: 10.1002/(SICI)1097-4547(19971115)50:4<531::AID-JNR4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 14.Rutar M, Natoli R, Valter K, Provis JM. Early focal expression of the chemokine Ccl2 by Müller cells during exposure to damage-inducing bright continuous light. Invest Ophthalmol Vis Sci. 2011;52:2379–88. doi: 10.1167/iovs.10-6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutar M, Natoli R, Provis JM. Small interfering RNA-mediated suppression of Ccl2 in Müller cells attenuates microglial recruitment and photoreceptor death following retinal degeneration. J Neuroinflammation. 2012;9:221. doi: 10.1186/1742-2094-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang X, Ni Y, Liu T, Ren H, Xu G. Inhibition of LPS-induced retinal microglia activation by naloxone does not prevent photoreceptor death. Inflammation. 2013;36:42–52. doi: 10.1007/s10753-012-9518-6. [DOI] [PubMed] [Google Scholar]
- 17.Santos AM, Martin-Oliva D, Ferrer-Martin RM, Tassi M, Calvente R, Sierra A, Carrasco MC, Marín-Teva JL, Navascués J, Cuadros MA. Microglial response to light-induced photoreceptor degeneration in the mouse retina. J Comp Neurol. 2010;518:477–92. doi: 10.1002/cne.22227. [DOI] [PubMed] [Google Scholar]
- 18.Zhang C, Shen JK, Lamn TT, Zeng HY, Chiang SK, Yang F, Tso MO. Activation of microglia and chemokines in light-induced retinal degeneration. Mol Vis. 2005;11:887–95. [PubMed] [Google Scholar]
- 19.Wu J, Seregard S, Algvere PV. Photochemical damage of the retina. Surv Ophthalmol. 2006;51:461–81. doi: 10.1016/j.survophthal.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Wu J, Seregard S, Algvere PV. Photochemical damage of the retina. Surv Ophthalmol. 2006;51:461–81. doi: 10.1016/j.survophthal.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Wenzel A, Grimm C, Samardzija M, Remé CE.Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res 200524275–306.Epub 2004 Nov 11 [DOI] [PubMed] [Google Scholar]
- 22.Bajetto A, Bonavia R, Barbero S, Boddeke HW. Chemokines and their receptors in the central nervous system. Front Neuroendocrinol. 2001;22:147–84. doi: 10.1006/frne.2001.0214. [DOI] [PubMed] [Google Scholar]
- 23.Andjelkovic AV, Pachter JS. Characterization of binding sites for chemokines MCP-1 and MIP-1alpha on human brain microvessels. J Neurochem. 2000;75:1898–1906. doi: 10.1046/j.1471-4159.2000.0751898.x. [DOI] [PubMed] [Google Scholar]
- 24.Bacon K, Baggiolini M, Broxmeyer H, Horuk R, Lindley I, Mantovani A, Maysushima K, Murphy P, Nomiyama H, Oppenheim J, Rot A, Schall T, Tsang M, Thorpe R, Van Damme J, Wadhwa M, Yoshie O, Zlotnik A, Zoon K. IS/WHO Subcommittee on Chemokine Nomenclature. Chemokine/chemokine receptor nomenclature. J Interferon Cytokine Res. 2002;22:1067–8. doi: 10.1089/107999002760624305. [DOI] [PubMed] [Google Scholar]
- 25.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–15. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 26.Chen D, Ding Y, Schroppel B, Zhang N, Fu S, Chen D, Zhang H, Bromberg JS. Differential chemokine and chemokine receptor gene induction by ischemia,alloantigen, and gene transfer in cardiac grafts. Am J Transplant. 2003;3:1216–29. doi: 10.1046/j.1600-6143.2003.00207.x. [DOI] [PubMed] [Google Scholar]
- 27.Sheehan JJ, Zhou C, Gravanis I, Rogove AD, Wu YP, Bogenhagen DF, Tsirka SE. Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J Neurosci. 2007;27:1738–45. doi: 10.1523/JNEUROSCI.4987-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiu K, Yeung SC, So KF, Chang RC. Modulation of morphological changes of microglia and neuroprotection by monocyte chemoattractant protein-1 in experimental glaucoma. Cell Mol Immunol. 2010;7:61–8. doi: 10.1038/cmi.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuaillon N, Shen DF, Berger RB, Lu B, Rollins BJ, Chan CC. MCP-1 expression in endotoxin-induced uveitis. Invest Ophthalmol Vis Sci. 2002;43:1493–8. [PubMed] [Google Scholar]
- 30.Rangasamy S, McGuire PG, Franco Nitta C, Monickaraj F, Oruganti SR, Das A. Chemokine mediated monocyte trafficking into the retina: role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS One. 2014;9:e108508. doi: 10.1371/journal.pone.0108508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falk MK, Singh A, Faber C, Nissen MH, Hviid T, Sørensen TL. CX3CL1/CX3CR1 and CCL2/CCR2 chemokine/chemokine receptor complex in patients with AMD. PLoS One. 2014;9:e112473. doi: 10.1371/journal.pone.0112473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raoul W, Auvynet C, Camelo S, Guillonneau X, Feumi C, Combadière C, Sennlaub F. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J Neuroinflammation. 2010;7:87. doi: 10.1186/1742-2094-7-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang XS, Ni YQ, Liu TJ, Zhang M, Ren H, Jiang R, Huang X, Xu GZ. CCR2 overexpression promotes the efficient recruitment of retinal microglia in vitro. Mol Vis. 2012;18:2982–92. [PMC free article] [PubMed] [Google Scholar]
- 34.Nakazawa T, Hisatomi T, Nakazawa C, Noda K, Maruyama K, She H, Matsubara A, Miyahara S, Nakao S, Yin Y, Benowitz L, Hafezi-Moghadam A, Miller JW. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci USA. 2007;104:2425–30. doi: 10.1073/pnas.0608167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang AL, Yu AC, Lau LT, Lee C. Wu le M, Zhu X, Tso MO. Minocycline inhibits LPS-induced retinal microglia activation. Neurochem Int. 2005;47:152–8. doi: 10.1016/j.neuint.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 36.Streit WJ, Conde JR, Harrison JK. Chemokines and Alzheimer’s disease. Neurobiol Aging. 2001;22:909–13. doi: 10.1016/s0197-4580(01)00290-1. [DOI] [PubMed] [Google Scholar]
- 37.Zeng HY, Zhu XA, Zhang C, Yang LP, Wu LM, Tso MO. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in rd mice. Invest Ophthalmol Vis Sci. 2005;46:2992–9. doi: 10.1167/iovs.05-0118. [DOI] [PubMed] [Google Scholar]
- 38.Elner SG, Strieter R, Elner VM, Rollins BJ, Del Monte MA, Kunkel SL. Monocyte chemotactic protein gene expression by cytokinetreated human retinal pigment epithelial cells. Lab Invest. 1991;64:819–25. [PubMed] [Google Scholar]
- 39.Ekström P, Sanyal S, Narfström K, Chader GJ, van Veen T. Accumulation of glial fibrillary acidic protein in Müller radial glia during retinal degeneration. Invest Ophthalmol Vis Sci. 1988;29:1363–71. [PubMed] [Google Scholar]
- 40.Eisenfeld AJ, Bunt-Milam AH, Sarthy PV. Müller cell expression of glial fibrillary acidic protein after genetic and experimental photoreceptor degeneration in the rat retina. Invest Ophthalmol Vis Sci. 1984;25:1321–8. [PubMed] [Google Scholar]
- 41.Abcouwer SF. Müller Cell-Microglia Cross Talk Drives Neuroinflammation in Diabetic Retinopathy. Diabetes. 2017;66:261–3. doi: 10.2337/dbi16-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang M, Ma W, Zhao L, Fariss RN, Wong WT. Adaptive Müller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J Neuroinflammation. 2011;8:173. doi: 10.1186/1742-2094-8-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newman EA. Propagation of intercellular calcium waves in retinal astrocytes and Müller cells. J Neurosci. 2001;21:2215–23. doi: 10.1523/JNEUROSCI.21-07-02215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]