

Abstract
Immune responses to allografts represent a major barrier in organ transplantation. Immune tolerance to avoid chronic immunosuppression is a critical goal in the field, recently achieved in the clinic by combining bone marrow transplantation with kidney transplantation following non-myeloablative conditioning. At high levels of chimerism, such protocols can permit central deletional tolerance, yet with a significant risk of graft-versus-host disease (GVHD). In contrast, transient chimerism-based tolerance is devoid of GVHD risk and appears to initially depend on regulatory T cells followed by a gradual, presumably peripheral, clonal deletion of donor-reactive T cells. Here, we review recent mechanistic insights into tolerance and the development of more robust and safer protocols for tolerance induction that will be guided by innovative immune monitoring tools.
Keywords: Tolerance, Mixed chimerism, Clonal deletion, Regulatory T cells
Challenges in translating transplantation tolerance to the clinic
Immune tolerance is a state of unresponsiveness of the immune system to specific donor tissue or cells, in particular in the context of tolerance to self, that prevents autoimmunity. The immune system is educated to discriminate between self and non-self via an intricate set of central and peripheral immune tolerance mechanisms. Regarding T cell tolerance, which is the major emphasis of this article, central tolerance refers to the deletion of reactive clones within the thymus during negative selection. In contrast, peripheral T cell tolerance encompasses several mechanisms that take place outside the thymus, including peripheral deletion, anergy/exhaustion, and the suppressive function of regulatory T cells (Tregs).
In solid organ transplantation, establishing donor-specific immunological tolerance, which avoids the complications of long-term immunosuppression (infections, malignancies, cardiovascular disease, renal failure, etc.), has long been the ultimate goal. Although many approaches have successfully achieved immunological tolerance across MHC barriers in rodents, very few have been translated to humans. The most advanced approach is the addition of hematopoietic cell transplantation (HCT) to kidney allografts in conditioned recipients (referred to henceforth as combined kidney and hematopoietic cell transplantation [CKHCT]), which has yielded very encouraging results. This strategy has offered proof-of-concept that operational tolerance can be induced in humans through mixed chimerism, defined as a state wherein donor and recipient hematopoietic cells coexist at levels sufficient to detect by standard techniques such as flow cytometry (generally 1 to 99%) [1–3]. Toxicities related to the conditioning procedures and the occurrence of opportunistic infections and Graft-versus-Host Disease (GVHD) in some of the protocols indicate a need for further refinements before routine use of these approaches can be considered. Furthermore, achievement of tolerance has not yet been uniform in these protocols. Achievement of this goal will benefit from further mechanistic insights, especially in humans and large animal models.
Also, the lack of reliable biomarkers to identify donor-specific tolerance is a hurdle to structured immunosuppression tapering in clinical transplantation [4, 5]. However, recent advances in identifying biomarkers of transplant tolerance [4, 6] and especially in measuring and tracking the recipient anti-donor alloreactive repertoire [6–8] are likely to facilitate such efforts in the future.
A better grasp of the complex synergistic tolerogenic pathways in rodents and the recent availability of new tools to monitor alloreactivity in humans have helped decipher tolerance mechanisms in clinical settings, paving the way for new protocols that could avoid the need for profound lymphoablation of the recipient. We aim herein to review our current understanding of tolerance mechanisms in clinical and highly relevant preclinical models. We will also discuss some new approaches with potential to further enhance tolerogenic mechanisms in humans based on recent clues gained from experimental models.
I) Tolerance mechanisms associated with sustained mixed chimerism
1) Central tolerance
Sustained mixed hematopoietic chimerism
One of the first avenues explored for sustained tolerance was based on the use of hematopoietic stem cell transfers to achieve durable chimerism. In rodent models, intrathymic deletion of donor-reactive immature T cells is a central feature of this approach for tolerance induction [9]. Indeed, durably engrafted donor hematopoietic stem cells supply circulating T cell/DC precursors that populate the recipient thymus, where they contribute to the pool of thymic dendritic cells (DCs), leading to the deletion of newly-developing donor-reactive T cells [9]. When recipient T cells are globally depleted prior to transplant, this central deletion is the major mechanism both inducing and maintaining tolerance, with no meaningful contribution from regulatory mechanisms [10]. Chimerism in thymic DCs was also detected and correlated with tolerance in chimeric non-human primates [11] and swine [12, 13]. In humans, two clinical CKHCT protocols achieved durable chimerism. Success of one regimen, involving a combination of total lymphoid irradiation and anti-thymocyte globulin, has only been reported for HLA-identical transplants, whereas the other involved extensively HLA-mismatched donors [14, 15]. In the latter, induction of sustained chimerism used the combination of fludarabine, cyclophosphamide and total body irradiation and substantial numbers of donor T cells were administered, with a significant risk of graft-vs-host disease (see below) [14, 15]. Blood T cells from chimeric patients in both studies failed to proliferate in vitro against recipient or donor cells, consistent with (but not specifically indicative of) intrathymic deletion of donor-reactive clones [14, 15]. Other approaches have been successfully used in experimental models to promote central tolerance to an allograft, including thymic transplantation and the transfer of thymus-homing dendritic cell precursors, but their translational potential has yet to be defined (Text Box 1).
Text Box 1. Alternative experimental approaches to induce transplant tolerance through central mechanisms.
Thymus transplantation
An alternative experimental strategy to promote central tolerance involves combining thymus and organ transplantation from the same donor [115, 116]. The powerful tolerance-inducing capacity of this approach was demonstrated in the highly disparate pig-to-mouse [117] xenogeneic combination, and then in humanized mice (i.e. immunodeficient mice reconstituted with human immune cells) after the engraftment of porcine tissue [118, 119]. Vascularized thymic lobe transplantation from juvenile donors to thymectomized young recipients induces T cell tolerance across fully allogeneic barriers in swine [115, 116]. So far in humans, allogeneic thymi have been transplanted, only in the form of cultured thymic tissue, in congenitally athymic infants [120, 121]. Tolerance to simultaneously-grafted parathyroid grafts sharing donor class II HLA alleles [122] suggests the potential of this approach to promote tolerance in humans. Although the deletion of newly-developing thymocytes is a major mechanism by which thymic grafts promote tolerance[123], the generation of Tregs with specificity for the donor is an important mechanism for suppressing non-ablated, pre-existing donor-reactive T cells [118, 124].
Donor antigen-presenting cells homing to the thymus
In addition to the DCs that arise intrathymically from a common T cell/DC precursor, some subsets of thymic DCs originate extrathymically and subsequently colonize the thymus, where they promote tolerance towards antigens loaded in periphery. This includes immature CCR9-expressing plasmacytoid DCs (pDCs) endowed with the ability to home to the thymus, mediate antigen-specific thymocyte deletion [125] and induce regulatory T cells (Tregs) in mice [126]. A similar subset of thymus-resident pDCs, driving the development of Treg, was also identified in human thymi [127]. Importantly, donor-derived thymic DCs injected into the circulation can colonize the thymi of allogeneic mice and prolong skin allograft survival by reshaping the thymocyte repertoire and deleting donor-reactive clones [128]. In addition to these pathways, the direct presentation of donor derived peptide-MHC complexes in the thymus could be promoted by the migration donor-derived exosomes to the thymus, where they coat recipient cells [129]. Crossdressing (i.e. transfer of intact donor peptide-MHC complexes onto recipient antigen-presenting cells) is a phenomenon of unexpectedly large magnitude following organ transplantation [129, 130]. The potential of cross-dressed thymic dendritic cells to mediate central tolerance remains to be addressed.
2) Counteracting Rejection Using Graft-vs-Host Reactivity
Balance between Host-vs-Graft and Graft-vs-Host immune responses
Some allograft types, such as livers and especially intestines, come with high lymphoid cell loads and have the potential to induce GVHD. However, GVH responses are not synonymous with GVHD, as GVH responses confined to the lymphohematopoietic system (Lymphohematopoietic Graft-vs-Host Responses [LGVHR]) can destroy recipient hematopoietic cells without causing GVHD and can balance out host-vs-graft (HvG)-reactive T cells [16–18]. The recent observation that high levels of peripheral blood T cell mixed chimerism occur commonly, without GVHD, in recipients of intestinal allografts, and the association of this chimerism with lack of graft rejection [7] led us to propose that a LGVHR may similarly counteract HvG responses in these patients, promoting hematopoietic chimerism and preventing rejection. In line with this hypothesis, immunosuppression withdrawal in a liver transplant recipient induced the conversion of mixed to full donor chimerism, despite the lack of GVHD [19]. This case report underscores the role of graft-borne GvH-reactive T cells in neutralizing HvG-reactive T cells and in promoting transplant tolerance [19, 20]. Furthermore, we found in intestinal transplant recipients that expanded intra-graft GVH-reactive T cells may have attenuated the HvG response locally, as high GvH/HvG clonal ratios in the graft were associated with slower replacement of graft T cells by the recipient and less rejection [7]. Notably, the expansion of GvH-reactive clones in the graft was found to occur early in association with recipient replacement of graft mucosal antigen-presenting cell populations [7].
Role of GVHR in clinical mixed chimerism protocols
The perennial challenge in clinical HCT has been the reliance on GVH reactivity both to counterbalance HVG reactivity and to mediate graft-vs-tumor (GVT) effects, as this GVH reactivity is often associated with GVHD, especially when HLA barriers are traversed. GVH reactivity can be controlled as a beneficial (mediating GVT effects) LGVHR that does not migrate to the epithelial GVH target tissues but promotes full donor chimerism, if inflammatory stimuli associated with conditioning have subsided by the time when GVH-reactive T cells are administered in a delayed donor lymphocyte infusion [16, 17, 21]. We have aimed to develop clinical protocols that are completely devoid of GVHD risk, which we consider unacceptable when HCT is added to an organ transplant solely for the purpose of tolerance induction (ie in the absence of malignancy or any other indication for HCT). Our transient chimerism protocol was first shown to meet this criterion in recipients of HLA-mismatched transplants for malignancy [22] before being adapted for the induction of renal allograft tolerance across HLA barriers in the absence of malignancy [1–3]. These protocols include peri-transplant treatment with a depleting anti-CD2 antibody that eliminates donor, in addition to recipient, T cells, thereby avoiding any GVHR from the HCT and permitting robust maintenance of recipient hematopoietic stem cells (HSCs) (Figure 1). In contrast, other clinical tolerance induction protocols that are associated with sustained donor chimerism may rely on GVH reactivity to achieve this outcome (Figure 1) [2, 14, 15]. In one study, the full donor chimerism achieved in most patients is not in keeping with the maintenance of recipient HSCs that would be expected following non-myeloablative conditioning [2, 14, 15]. Despite full donor leukocyte chimerism, however, when donor and recipient blood types were mismatched, most of the patients retained the recipient’s blood type [14]. Recipient HSCs may survive without contributing to chimerism when their progeny are destroyed by LGVHR, consistent with the immunoprotection of the HSC niche that has recently been described [23]. The observation of persistent recipient erythropoiesis in the presence of full donor leukocyte chimerism in humans similarly suggests the persistence of recipient HSCs whose HLA-expressing progeny are destroyed by a GVHR while HLA antigen-deficient erythrocytes survive. [14] Unfortunately, this has been associated with significant clinical GVHD in several cases, resulting in the death of one of them (Ildstad et al, Abstract 450.1, 2016 congress of The Transplantation Society, Hong-Kong). In another CKHCT protocol, efforts to extend tolerance induction from the HLA-matched to the HLA-mismatched setting utilized a dose-escalation of donor T cells. While more sustained mixed chimerism in HLA haplotype-matched recipients of this protocol was achieved with a 50-fold increase in the donor T cell dose compared to that in HLA-matched recipients (50 vs 1×10ˆ6/kg), successful immunosuppression withdrawal has not yet been reported [15]. Collectively, these studies emphasize that GVHR may serve as a potent contributor to the induction of durable chimerism but is associated with GVHD risk (Figure 1). Using an approach of ex vivo donor T cell depletion combined with in vivo recipient T cell depletion, we have previously obtained proof-of-principle in hematologic malignancy patients that durable mixed chimerism can be achieved across HLA haplotype barriers without relying on GVH reactivity [22]. However, the anti-CD2 mAb needed for this regimen subsequently became unavailable, 2 of 4 patients lost chimerism, and the protocol has not been evaluated in patients who had not had previous cytoreductive therapy for lymphoma. Thus, the reliable achievement of durable chimerism across HLA barriers without risk of GVHD in humans remains elusive. Given the ample demonstrations that life-long mixed chimerism can be achieved without relying on GVH reactivity in animal models, we believe this remains an attainable and desirable goal in humans. As is discussed below, Tregs have the potential to advance this goal.
Figure 1. Mechanisms involved in clinical chimerism-based regimens for inducing tolerance across HLA barriers.
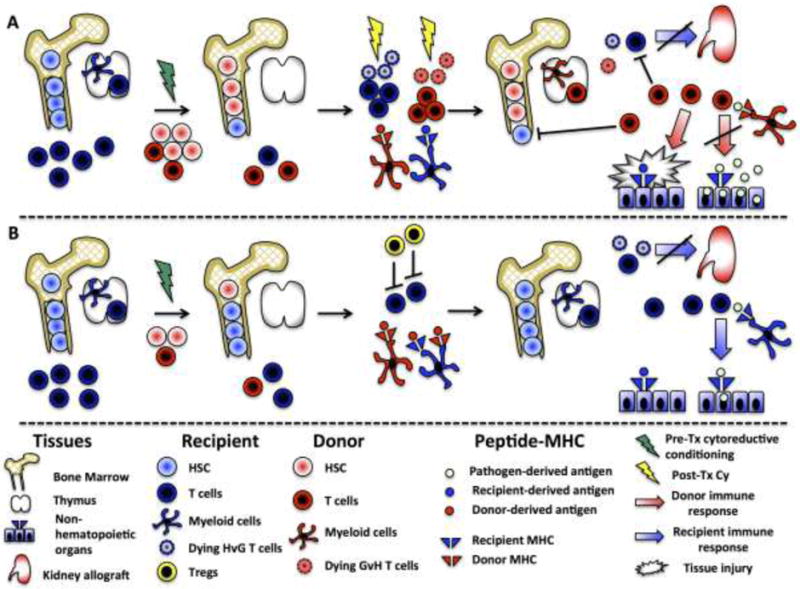
(A) Sustained full chimerism:, A large number of donor CD34+ HSCs (up to 17×106/kg) and donor T cells (3.8×106/kg) plus “facilitating cells” are administered to the recipients along with the kidney allograft after non-myeloablative conditioning (green lightning symbol). Graft-vs-Host-reactivity drives the expansion of donor T cells that destroy recipient T cells and hematopoietic cells, creating HSC niche space in bone marrow. The GVH response is partly attenuated by post-transplant cyclophosphamide treatment (yellow lightning). Durably engrafted donor HSCs supply the thymus with T-cell precursors and dendritic cells progenitors that induce central donor-specific tolerance. These conditions allow sustained full chimerism. Donor-derived T cells can mediate GVHD, yet fail to eliminate infectious organisms from recipient’s tissues, as post-transplant pathogen-specific immune responses are restricted to donor-MHC.
(B) Transient mixed chimerism: Unfractionated bone marrow and kidney allograft are transplanted following non-myeloablative donor and recipient T cell-depleting induction (green lightning). Early Treg expansion, driven by lymphopenia and relative Treg sparing by the conditioning regimen combined with the presence of donor antigen, prevents the activation of HvG-reactive T cells and creates a microenvironmentthat supports peripheral deletion of donor-reactive T cells. In contrast to full chimeras, individuals with mixed or transient chimerism preserve efficient anti-infectious immune responses.
Abbreviations: Cy, cyclophosphamide; GvH, Graft-vs-Host; HSC, hematopoietic stem cells; HvG, Host-vs-Graft; Tregs, regulatory T cells;
II) Tolerance mechanisms associated with transient mixed chimerism
For clarity, tolerance mechanisms will be discussed separately in this section, although growing evidence suggests that they are intimately interconnected. Indeed, the ultimate stage of T cell exhaustion is physical deletion [24], while recent data support the role of Tregs in promoting CD8+ T cell exhaustion [25]. Furthermore, apoptotic body uptake by phagocytic cells can induce local TGF-β secretion, which in turn promotes the generation and intra-graft expansion of Tregs [26].
1) Peripheral deletion
Evidence for peripheral deletion in clinical CKHCT protocol
In mice achieving mixed chimerism and donor-specific tolerance with costimulation blockade, specific deletion of T cells expressing donor-reactive TCR occurs in the periphery [27–30]. Moreover, in humans and in non-human primates with transient chimerism and long-term tolerance after CKHCT, thymic deletion of donor-reactive T cells is unlikely to be the major mechanism of tolerance. However, proliferation and cytotoxicity assays demonstrate donor-specific hyporesponsiveness in these patients [1, 6, 31]. Functional evidence suggested that regulatory mechanisms played an early, but not a long-term, role in this systemic hyporesponsiveness to the donor [31]. Together, these findings suggested that peripheral mechanisms control the anti-donor response, but did not indicate whether the clones persist as quiescent T cells (anergic, exhausted) or become progressively deleted. To address this important issue, we established a method for identifying and tracking the donor-reactive TCR repertoire [6]. Host-vs-Graft (HvG)-reactive clones were identified via pre-transplant mixed lymphocyte reaction (MLR, where recipient anti-donor T cells are identified by dilution of the dye CFSE), followed by high-throughput sequencing of TCRβ CDR3 regions and comparison to unstimulated pre-transplant T cells to identify MLR-expanded clones as donor-reactive. Pre-defined HvG and non-HvG clones (those that were detected in the unstimulated pool and did not expand in MLR) could then be quantified in post-transplant blood [6] and biopsy samples [7]. This method allowed demonstration of gradual deletion of the clones with the strongest alloreactivity in tolerant patients, suggesting peripheral deletion as a significant tolerance mechanism (Figure 1). Importantly, such deletion did not occur in the one patient who failed tolerance induction and rejected the graft upon immunosuppression withdrawal [6]. Remarkably, this non-tolerant patient exhibited complete donor-specific unresponsiveness in all in vitro assays, suggesting that the donor-specific T cells were “anergic” under the conditions of these assays, but that this anergy did not translate to a robustly tolerant state in vivo. These sobering findings reinforce the unreliability of functional in vitro assays in identifying a tolerant state and suggest that tracking of donor-specific TCRs may have potential to distinguish tolerant from non-tolerant patients. This possibility deserves further exploration in view of the need for reliable predictors of “spontaneous” allograft tolerance that would permit weaning of immunosuppression in appropriate patients without increasing the risk of graft rejection.
Apoptotic pathways and targeted therapeutics
Deletion of donor-reactive T cells can occur at different stages of development, and through different mechanisms. Understanding which mechanisms promote tolerance induction has important implications for the design of immunosuppressive drug combinations that might promote the establishment of tolerance (Figure 2). While calcineurin inhibitors (CNI), unlike mTOR inhibitors, were shown to block Activation-Induced Cell Death (AICD) and thereby prevent tolerance induction in a costimulatory blockade-based tolerance model, this conclusion was drawn from experiments using one CNI, cyclosporine, but not tacrolimus [32]. Mice constitutively expressing Bcl-xL were also shown to be resistant to induction of transplantation tolerance through costimulatory blockade [33] and to induction of mixed chimerism with costimulatory blockade [34]. Bcl-xL and Bcl-2 hamper intrinsic cell death by maintaining mitochondrial integrity, but provide little protection from the death receptor (FAS and TRAIL)-induced cell-extrinsic apoptosis pathway (often referred to as AICD) [35]. In a mixed chimerism-based model using costimulatory blockade, constitutive Bcl-xL expression impaired the early deletion of donor-reactive T cells and prevented the induction of donor-specific tolerance [34], whereas FasL was dispensable for CD4 cell tolerance [36]. A Bcl-2/Bcl-xL inhibitor (ABT-737), combined with co-stimulatory blockade and donor bone marrow cells, induces complete peripheral deletion of alloreactive T cells, allowing mixed chimerism induction without cytoreductive conditioning [37]. Furthermore, Bim, a key player in the intrinsic apoptosis pathway, is required for induction of mixed chimerism without lymphoablation [37]. Collectively, these data suggest that the intrinsic apoptosis pathway may be more important than the extrinsic pathway in peripheral deletion in these models. Importantly, while both cyclosporine and tacrolimus interfere with calcineurin-dependent T cell activation, only cyclosporine and not tacrolimus inhibits the mitochondrial permeability transition pore, an endpoint to the intrinsic apoptotic pathway (Figure 2) [38]. Consistently, in pig models, tacrolimus was found to better facilitate the induction of tolerance than cyclosporine [39]. Cyclosporine derivatives (such as Debio-025 or NIM811) lacking anticalcineurin activity but inhibiting mitochondrial permeability could be useful in investigating the separate roles of the two apoptosis pathways.
Figure 2. Approaches to inducing exhaustion and death of HvG-reactive T cells.
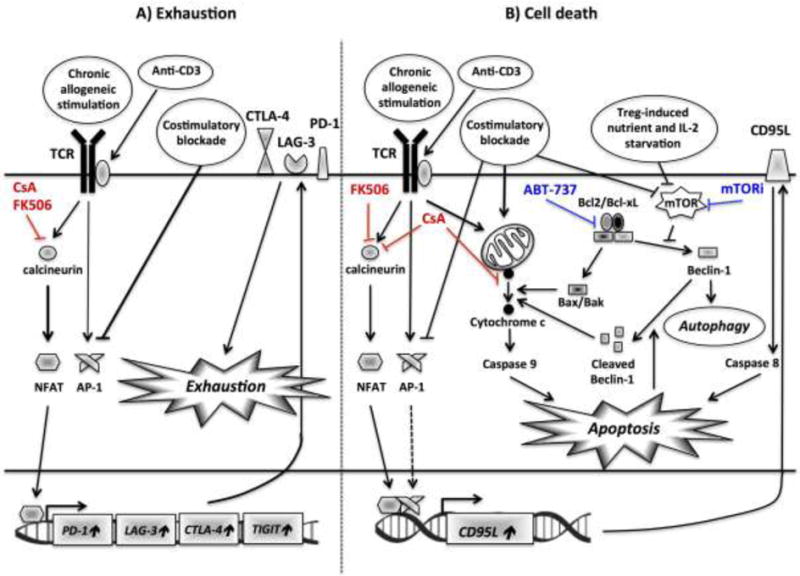
A) Prolonged inefficient antigenic stimulation or anti-CD3 therapy results in altered balance between NFAT and AP-1, inducing an exhaustion program, as defined by the expression of several inhibitory cell surface receptors, including PD1, LAG3, TIM3, TIGIT and CTLA-4 and a hyporesponsive state. Both CsA and FK506 inhibit NFAT-dependent T-cell exhaustion.
B) The two main apoptosis pathways, namely extrinsic (caspase 8-dependent) and intrinsic (caspase 9-dependent), involved in the peripheral deletion of HvG T cells following tolerance-inducing protocols are depicted. The complex crosstalk between autophagy and mitochondrial apoptosis is illustrated. Autophagy is initiated in response to metabolic cues (nutrient deprivation) and/or costimulatory blockade, both sensed by mTOR. If this process is insufficient, it triggers cytochrome c release and mitochondrial apoptosis. Furthermore, caspase activity hampers Beclin-1-dependent autophagy, fueling a positive amplification loop. Although both calcineurin inhibitors (CsA and FK506) block NFAT-dependent activation-cell death (extrinsic), CsA inhibits, unlike FK506, mitochondria depolarization and cytochrome c release. On the other hand, mitochondrial apoptosis is enhanced by mTOR and Bcl2/Bcl-xL (ABT-737) inhibition.
Abbreviations: AP-1, activator protein 1; CsA, cyclosporin A; TCLA-4, cytotoxic T lymphocyte-associated protein 4; LAG3, lymphocyte-activation gene 3; mTORi, mammalian target of rapamycin inhibitor; NFAT, nuclear factor of activated T-cells; PD-1, programmed cell death protein 1; TCR, T cell receptor;
Targeted memory T cell depletion
Unlike laboratory mice [40], patients already harbor a large repertoire of memory T cells at the time of transplantation. Heterologous immunity and CD8 T-cell crossreactivity constitute a significant barrier to immunologic tolerance in large outbred animals and humans. For example, up to 45% of anti-CMV T cell clones have been reported to be alloreactive [41]. Generally, achieving mixed chimerism in non-human primates (NHP) is far more challenging than in rodents, apparently due to the presence of abundant crossreactive memory T cells [42]. NHPs with the greatest frequency of pre-transplant donor-reactive memory T cells fail to become tolerant after non-myeloablative CKHCT [43]. In addition, viral or bacterial infections to which humans may be exposed can prevent or even break established tolerance following transplantation [44, 45]. Memory T cells are less dependent on costimulatory signals and are more resistant to suppression by regulatory T cells (Tregs). Interestingly, the type of pathogen involved in the generation of crossreactive CD8+ memory T cells can determine the nature of the dominant memory T cell subset, whose susceptibility to costimulation/integrin blockade varies greatly [46]. Central memory T cells are more sensitive [46], possibly because of lower Bcl-2 expression upon cytokine stimulation, than effector memory T cells [47]. Attempts to eliminate donor-reactive memory T cells have yielded very promising results in NHPs [48, 49]. Both alefacept, a fusion protein that antagonizes LFA-3/CD2, and a humanized anti-CD8 monoclonal antibody successfully restrain the expansion of memory T cells after kidney transplantation and allow tolerance induction via a delayed mixed chimerism protocol [48, 49]. Also, the use of a Bcl-2/Bcl-XL inhibitor, currently developed in clinical oncology, restores costimulation blockade efficiency in mice whose immune system has been primed by donor cells and allows mixed chimerism-based donor-specific tolerance across memory T cell barriers [47]. However, the model of adoptive transfer of memory T cells used in these studies may be less challenging than induction of tolerance in a native host with alloreactive memory T and B cells, in which induction of mixed chimerism is inhibited by B cell-dependent T cell memory, even after exhaustive T cell depletion and costimulatory blockade [50].
Skewing the balance toward regulation
A critical goal of tolerogenic protocols is to tip the balance toward suppression by targeting effector cells while sparing Tregs [51]. Recent studies demonstrate that Mcl-1 rather than Bcl-2 expression is critical for the survival and fitness of Foxp3-expressing Tregs, in contrast to naive, effector and memory T cells [52]. Building on this insight into murine Treg biology, Cippa et al demonstrated that this differential effect of selective Bcl-2/Bcl-xL inhibition creates a more favorable environment for Tregs, which are required for tolerance induction by ABT-737 (Figure 3) [53].
Figure 3. Pharmacological and biological agents with potential to tip the balance toward tolerance.
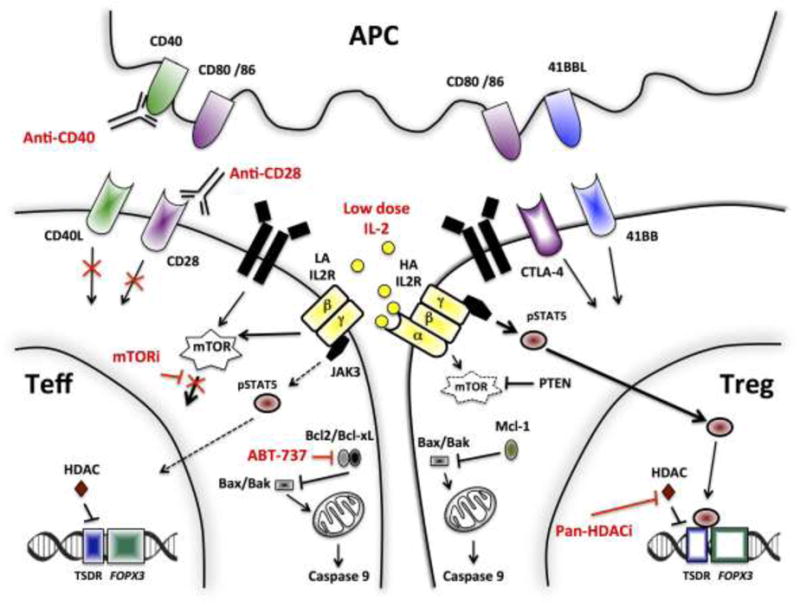
Low-dose IL-2 selectively activates high-affinity IL2R-expressing Tregs, but has limited effect on Teffs. IL2R signaling relies mostly on the Jak3/STAT5 pathway in Tregs. In contrast, the PI3K/Akt/mTOR pathway is highly dispensable for the generation, expansion and survival of Tregs, yet not of Teff. Hence, mTORi favors Tregs over Teffs. Similarly, the critical role of anti-apoptotic Bcl2/Bcl-xL genes in hindering mitochondrial apoptosis in Teffs, but not Tregs, makes the former more vulnerable to Bcl2/Bcl-xL inhibition. Exposure of FOXP3+ Treg cells to pan-HDACi promotes the stability and function of Tregs, through acetylation of the FOXP3 gene. Tregs are unaffected by anti-CD40 antibody as they do not upregulate CD40L expression.
Abbreviations: HA IL2R, high affinity IL-2 receptor; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; LA IL2R, low affinity IL-2 receptor; mTORi, mammalian target of rapamycin inhibitor; NFAT, nuclear factor of activated T-cells; PD-1, programmed cell death protein 1; PTEN, phosphatase and tensin homolog; TCR, T cell receptor; Teff, effector T cells; Treg, regulatory T cell; TSDR, Treg-specific demethylated region.
2) Anergy and exhaustion
Anergy and T cell exhaustion are two states of qualitative impairment of T cell function [54]. The former may occur quite quickly after TCR stimulation when the integration of costimulation signals results in an inhibitory signal, leading to the inability of T cells to produce IL-2 or proliferate [54]. There are several forms of anergy, some of which may be reversed by exogenous IL-2. In contrast, CD8+ T cell exhaustion requires chronic stimulation resulting from an overwhelming antigen load and/or prolonged exposure to an environment that is suboptimal for their response, such as lack of CD4+ T cell help and high level of inhibitory signals [24, 55]. The ability to reverse T cell exhaustion with checkpoint inhibitors has provided a major advance in cancer immunotherapy [54, 56]. A handful of key transcription factors are involved in shaping the transcriptomic landscape of exhausted T cells, including eomesodermin (Eomes), T-box 21 (Tbet), PR domain zinc protein 1 (Blimp1), forkhead box O1 (Foxo1), nuclear factor of activated T cells (NFAT) and von Hippel-Lindau tumor suppressor (VHL). Interestingly, the core transcriptional program of exhausted CD8+ T cells only partly overlaps with that of exhausted CD4+ T cells [57]. T cell exhaustion has been identified as one of the main immune escape mechanisms in the settings of chronic infection and malignancies [24, 55]. The limited studies of exhaustion in clinical transplantation include studies in HCV-infected liver transplant recipients [55]. Chronic exposure to alloantigens and the blunting of the immune response by immunotherapies provides the conditions that promote T cell exhaustion, which is therefore likely to be a mechanism involved in other tolerance settings [55].
Administration of IL-2 breaks transient mixed-chimerism-based tolerance in NHP and leads to rapid allograft rejection. In this setting, IL-2 disrupts peripheral tolerance, resulting in expansion of CD8+ alloreactive memory T cells [58]. This finding suggests that alloreactive T cells persist in the anergic (or suppressed) state, and can be reactivated upon IL-2 injection. Different forms of anergy are suggested by the observation of decreased but persisting donor-specific clones 2 years after transplantation in tolerant CKBMT patients [6]. These clones did not cause rejection, in contrast to those in a patient who failed tolerance induction and showed no reduction in donor-reactive T cell clones following the transplant, yet showed donor-specific anergy under in vitro allostimulation conditions [6].
Studies in mice demonstrate that both CD4+ and CD8+ donor-reactive T cells from recipients of allogeneic BMT with anti-CD40L are donor-unresponsive before being deleted. Tolerance of CD8 + T cells may involve exhaustion, as multiple inhibitory pathways, including PD-1 [59], LAG-3, CTLA-4 and TGF-β, are required, whereas exhaustion-associated molecules are not required for CD4 tolerance [60]. Notably, in the same model, NFAT activation is critical in tolerizing CD8 alloreactive T cells, yet is completely dispensable for the induction of CD4 tolerance [61]. Recent data shed light on the molecular mechanisms linking costimulatory blockade, NFAT and exhaustion in CD8+ T cells (Figure 2) [57]. Nuclear translocation and activation of NFAT controls T cell fate decisions in a manner that depends on complexes with other transcription factor partners, such as AP-1 [57]. TCR activation along with costimulation blockade profoundly unbalances the equilibrium between NFAT and AP-1, resulting in monomeric NFAT that preferentially activates a transcriptomic program driving T cell exhaustion (Figure 2) [57]. Together, these results suggest that T cell exhaustion contributes to the state of CD8+ T cell hypofunction and deletion observed in the model of mixed chimerism induced by anti-CD40L blockade.
Reduced expression of the AP-1 complex was also noticed in tolerant T cells infiltrating islet allografts after anti-CD3 antibody [62]. Moreover, CD8+ T cells isolated from tolerated islet allografts failed to produce IFNγ in response to donor antigens [62]. Single cell PCR of individual graft-infiltrating CD8+ T cells revealed high expression of the inhibitory receptors LAG-3, TGF-β-induced PD-1, PDL-1, as well as increased expression of Eomes in tolerant compared to control CD8+ T cells [62], highly suggestive of T cell exhaustion [55]. Furthermore, downregulated expression of hypoxia-inducible factor HIFα in tolerant CD8+ T cells [62] is aligned with the low metabolic requirement and the high expression of VHL in exhausted T cells [63]. Similar findings in type I diabetic patients treated with the anti-CD3 monoclonal antibody teplizumab demonstrate the relevance of these new insights into peripheral CD8 tolerance [64]. A population of hypoproliferative EOMES+TIGIT+KLRG1+ CD8+ T cells is readily identified in the blood of patients with the greatest response to anti-CD3 treatment [64].
3) Regulatory cells
Regulatory T cells (Tregs)
Mounting evidence implicates FOXP3-expressing Tregs in clinical transplant tolerance induction. The rare kidney transplant recipients who spontaneously develop a state of operational tolerance demonstrate an increased frequency of highly suppressive memory Tregs with a fully demethylated FOXP3 Treg-Demethylated Region (TSDR), a hallmark epigenetic change of stable Tregs [65]. Similarly, marked enrichment of FOXP3-expressing Tregs during the early post-transplant course has been observed following the non-myeloablative anti-CD2 mAb-based CKHCT protocol that leads to transient chimerism [31, 66]. Consistently, a recent study suggested donor-specific FOXP3+ Treg expansion in tolerant NHPs receiving a T cell depletion/costimulatory blockade-based transient chimerism CKHCT regimen [67]. Mouse models have decisively established a causal link between accumulations of donor-reactive FOXP3+ Tregs and transplant tolerance through a variety of experimental approaches, including targeted deletion of FOXP3-expressing Tregs, which disrupted well-established tolerance [68, 69]. Importantly, Tregs have the capacity to elicit infectious tolerance, permitting spreading of tolerance toward a broad diversity of donor antigens [69].
Numerous strategies that promote Treg expansion or stability are being implemented in the clinic (Figure 3). Dramatic Treg expansion in vivo has been observed upon low-dose subcutaneous IL-2 treatment in patients with chronic GVHD [74]. However, excessive IL-2 may activate alloreactive effector T cells (Teff), preventing or breaking transplant tolerance, as recently described in NHPs [58]. In this respect, close monitoring of downstream targets of the IL-2R pathway (e.g. phosphorylated Stat5) in Tregs and Teff may help to adjust IL-2 doses [75]. Further, the development of an IL-2 mutant that fails to recruit pro-inflammatory cells expressing low-affinity IL2 receptor (i.e. composed of the βγ IL-2 R chains only) is currently underway and appears promising (patent n° US2014/0286898). Histone deacetylases (HDACs) play a critical role in regulating gene expression through epigenetic changes. Pan-HDAC inhibition in BMT patients was associated with a reduced rate of acute GVHD [76], along with marked enrichment of circulating Tregs with highly demethylated TSDR [77]. HDAC-selective targeting (such as HDAC-6 or 9) has a safer toxicity profile and has yielded promising results in experimental transplant models [78]. Moreover, HDAC inhibition elicits a synergistic effect with mTOR inhibition in promoting transplant tolerance [79]. The favorable effect of mTOR inhibitors on Treg generation and expansion has been extensively documented and reviewed elsewhere [80].
Costimulatory blockade targeting the CD40/CD40L and CD28/CD80/CD86 pathways has long been used to promote peripheral tolerance in rodent models and has significantly improved mixed-chimerism-based tolerance in monkeys by reducing the need for cytoreduction [81]. Fc-silent forms of anti-CD40 (CFZ533) [82, 83] and anti-CD40L (BMS-986004) [84] antibodies, which lack the side effects of the respective full-length antibodies (B lymphopenia and thromboembolic events), are currently being developed for clinical applications in transplantation. Growing evidence suggests that costimulation through CD40L is highly dispensable in human Tregs, in contrast to conventional T cells, and that CD40/CD40L blockade may tip the balance toward suppression. Indeed, gene expression profiles revealed marked down-regulation of CD40L in human Tregs compared to non-Tregs [70]. Moreover, human Tregs fail to up-regulate CD40L upon activation, in contrast to conventional CD4+ T cells [85]. Also of interest, blocking anti-CD28 antibody treatment was associated with an increased Treg frequency in large animal models [86], and was extremely potent in preventing alloantibody development by blocking follicular T-helper cell activation [87], while preserving CTLA-4 dependent T follicular regulatory T cell function [88].
As an alternative approach to in vivo Treg promotion, Tregs can be manufactured as exvivo cellular products for tolerance purpose. However, regulatory T cell therapy according to Good Manufacturing Practices (GMP) faces several challenges, including the isolation of a pure population that remains stable upon expansion, the issue of antigen specificity and the number of cells requested to obtain a therapeutic effect [89]. Concerns about Treg stability [90, 91] have converged on the viewpoint that Tregs can be very stable as long as they are fully committed, according to the so called “heterogeneity model” [92]. Consistently, one year after the infusion of deuterium-labeled highly-purified CD4+CD25highCD127low Tregs to humans, deuterium was only detected within the Treg compartment, demonstrating the lack of leakage into other lineages [93]. Polyclonal Treg infusion has been implemented in clinical trials as GVHD prophylaxis (ClinicalTrials.gov: NCT00602693) and in new onset Type 1 diabetes patients (ClinicalTrials.gov: NCT01210664), with excellent safety profiles [93–95]. Both polyclonal and donor-specific Treg infusion are being evaluated in several clinical organ transplant trials (reviewed in reference [89]). The addition of polyclonal Tregs to donor bone marrow allowed achievement of stable chimerism and tolerance in the absence of cytoreductive conditioning in a rodent model [96]. In efforts to convert a transient chimerism protocol to durable chimerism without relying on GVH reactivity in a preclinical model, we are exploring the addition of expanded polyclonal recipient Treg infusion in NHP and have indeed observed prolongation of mixed chimerism in association with more robust allograft tolerance. Whereas the original NHP transient chimerism CKHCT protocol relies on the presence of a donor kidney in the early post-transplant period to promote tolerance, the prolonged chimerism achieved by the addition of expanded recipient Tregs allows acceptance of a donor kidney grafted as late as 4 months after the BMT, indicating a more robust form of tolerance than that induced by BMT alone [97]. The addition of donor-reactive Treg-enriched recipient cells to liver transplants dramatically increased the success rate of immunosuppressive drug weaning in a pilot study (clinical trial registration number: UMIN-000015789) [98]. Allospecific Tregs offer several advantages over polyclonal Tregs, including greater efficiency in preventing rejection [99] and less potential for toxicity due to global immunosuppression. However, the very low frequency of allospecific Tregs represents a substantial barrier for clinical translation [99]. To tackle this challenge, a recent study used an allogeneic HLA-targeting chimeric antigen receptors to redirect human Treg specificity toward the allograft, with very promising preclinical results [100].
Regulatory B cells
A B cell-associated gene expression and flow cytometric signature was identified in two different cohorts of spontaneously operational tolerant kidney transplant recipients and in CKHCT recipients in whom mixed chimerism-based tolerance was successfully achieved [101]. However, these initial studies did not control for the fact that tolerant patients were off immunosuppression while non-tolerant subjects were treated with drugs that could have reduced B cell numbers and/or function. A recent reanalysis revealed that much of this signature was indeed attributable to immunosuppressive drugs used in the non-tolerant subjects [102]. However, consistent with a possible role for B cells in tolerance, B cells from tolerant patients produce IL-10, express inhibitory receptors [103], and restrain T cell proliferation in a granzyme B-dependent manner [104]. In the murine BMT/anti-CD154 model, recipient B cells were required for the tolerization of recipient peripheral CD8 T cells, but this effect did not require IL-10 production or APC function of the B cells [29, 105], suggesting that B cells promote tolerance through additional, unknown mechanisms.
4) Intra-kidney graft immunomodulatory mechanisms
Studies attempting to induce tolerance to different organs via transient mixed chimerism have emphasized the critical role of the kidney allograft itself [42]. The presence of a co-transplanted kidney allograft was found to be required for long-term tolerance of heart allografts following transient chimerism induction in large animal models [42]. The critical role of the kidney was compellingly demonstrated by the prompt rejection of co-transplanted heart after kidney allograft removal [42]. Moreover, the re-transplantation of a tolerated class I MHC-mismatched kidney allograft plus lymphocytes from the tolerant animal leads to acceptance of a donor-matched naive kidney allograft with systemic donor-specific hyporesponsiveness [106, 107]. This “kidney effect” might be related to its ability to establish an immunologic environment conducive to tolerance. Transforming growth factor-β and indoleamine 2,3-dioxygenase are abundantly produced by renal parenchymal cells. Furthermore, the PD1 ligand PDL-1, whose role is increasingly evident in transplant immunology [108], is strongly inducible by tubular epithelial cells upon alloimmune attack [109]. This finding may explain the observation that T cells from CKHCT patients were unresponsive to donor renal tubular epithelial cells [110] and is reminiscent of the instrumental role of the PD1/PDL1 axis in spontaneous liver allograft acceptance in mice [111]. Moreover, accumulation of FOXP3+ Tregs in tolerated grafts has been extensively reported in mice [62, 112], and the graft itself was found to be a major site for suppression [69]. Similarly, FOXP3+ T cells are detected in biopsies from tolerant CKHCT recipients with transient chimerism [1, 66].
Concluding remarks
Two recent studies have provided proof of concept that transplant tolerance can be induced in humans across MHC barriers through CKHCT. However, the safety profile of a regimen that is associated with full donor chimerism raises concerns. In the other protocol, which is associated with transient chimerism, avoidance of an early engraftment syndrome (i.e. capillary leak syndrome occurring at the time of initial hematopoietic recovery) is a desirable goal. To this end, approaches to making chimerism more durable and less dependent on the kidney itself would allow extension of tolerance to less tolerogenic organs. Ongoing clinical and preclinical studies are moving toward achievement of both of these goals. Preclinical studies suggest several promising approaches for enhancing peripheral tolerance via deletion, exhaustion and suppression, that may allow further reductions in the need for lymphoablation and cytoreduction in future protocols. Murine studies demonstrating the potency of multiple parallel tolerance mechanisms in promoting robust transplantation tolerance [113] and particularly the combination of central deletion and regulation that is achieved with incomplete lymphodepletion and Treg-promoting mixed chimerism regimens [30, 114] are useful in guiding these approaches. Together, major new insights into transplant tolerance mechanisms, along with the emergence of new immunotherapies and tools for immune monitoring, open up new avenues for developing clinical tolerance protocols with reduced risks and greater efficacy (see Outstanding Questions).
Outstanding questions.
Does the ability of an immune system to be reset for transplant tolerance depend on recipient’s age (thymus output, repertoire diversity, immune senescence)?
Is there a level of heterologous memory T cell response that could not be overcome by targeted lymphodepletion?
Is anti-HLA pre-sensitization an insurmountable hurdle to induce mixed chimerism-based tolerance?
Can recurrent autoimmunity be prevented by tolerance regimens that allow allograft acceptance in patients with underlying autoimmune disease?
Can clinical transplant tolerance resist immune-triggering events such as infections and vaccinations?
Can new drugs and cells being used in rodent models be extended to achieve robust tolerance in primates?
Can regulatory T cells be used in combination with minimal host conditioning and donor hematopoietic cell transplantation to achieve durable chimerism without a graft-vs-host response in humans?
Would thymus transplant be sufficient to induce tolerance toward donor antigens recognized both through direct and indirect presentation pathways?
Can alloreactive TCR repertoire tracking provide a biomarker for tolerance and rejection?
What is the role of direct vs semidirect vs indirect alloreactivity in late graft loss?
Since the prevention of donor-specific antibody generation requires tolerization of indirect allorecognition, reliable tools are needed to monitor this response.
Can exosomes that promote semi-direct allorecognition participate in tolerance induction?
Trends.
A current clinical tolerance protocol achieving sustained full chimerism across HLA barriers in humans likely depends on some level of Graft-versus-Host reactivity that counteracts the Host-versus-Graft response. This is associated with a significant risk of life-threatening GVHD.
In contrast, tolerance induction through transient mixed chimerism involves sparing and early expansion of regulatory T cells and progressive deletion of donor-specific T cells in the periphery.
A better understanding of the molecular pathways involved in peripheral tolerance is paving the way for refined tolerance-inducing protocols that will use biological and pharmacological agents to synergistically achieve tolerance without relying on graft-vs-host reactivity.
Regulatory T cell therapy, T cell pro-apoptotic agents, and costimulatory blockers are entering into the clinic and have the potential to make chimerism-based tolerance strategies safer and more robust.
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award #P01 AI045897, #P01 AI106697, # UM1 AI109565 and #R56AI122332, by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award # UC4DK104207 and # R01 DK103585, by the Heart, Lung and Blood Institute of the National Institutes of Health under Award # NIH P01HL018646, by the Office of the Director of the National Institutes of Health Award # NIH R01 OD017949, and by Department of Defense grant # W81XWH-15-1-0234. J.Z. was supported by a Columbia University Schaefer award, a Fulbright fellowship, la Fondation pour la Recherche Médicale and the Emmanuel Boussard’s foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Defense. We thank Ms. TeShima Brennen for expert assistance with manuscript preparation.
Glossary
- Activation-induced cell death (AICD)
AICD is programmed cell death is the process by which T cells undergo apoptosis following activation in a manner controlled through the interaction of death factors (FASL and TRAIL) and their receptors. AICD, also known as the extrinsic apoptosis pathway, involves a death-inducing signaling complex and caspase 8
- Central tolerance
Central tolerance is the mechanism by which newly developing T cells are rendered nonreactive to self antigens, as the result of the intra-thymic deletion of those with the greatest reactivity to self
- Exhaustion
T-cell exhaustion is a state of T cell dysfunction, defined by poor effector function, a hallmark transcriptomic pattern, and sustained expression of inhibitory receptors. It usually results from chronic antigen stimulation in a suboptimal environment
- Heterologous immunity
Heterologous immunity refers to the phenomenon by which T cells that were generated during an earlier infection can recognize and mount a memory T cell response against allogeneic antigens through TCR crossreactivity
- Intrinsic apoptosis pathway
The intrinsic pathway of apoptosis (also named the mitochondrial pathway) gets activated in response to various types of intracellular stress, including nutrient starvation. Mitochondrial outer membrane permeabilization and cytochrome-c release are key points of no return in the intrinsic apoptosis pathway, leading to caspase 9 activation
- Lymphohematopoietic graft-versus-host response (LGVHR)
LGVHR refers to graft-vs-host reactivity that eliminates recipient lymphohematopoietic cells without trafficking to epithelial GVHD target tissues (skin, intestines and liver) and therefore without causing clinical GVHD
- Mixed or Full Chimerism
Mixed hematopoietic chimerism refers to a state wherein recipient and donor hematopoietic cells coexist in an individual, with a donor cell frequency ranging from >1% to <100%. Full chimerism denotes donor reconstitution close to 100%
- Peripheral tolerance
Peripheral tolerance refers to the mechanisms that take place outside of the thymus to prevent mature T lymphocytes from mounting an efficient immune response against a given set of antigens. Mechanisms include anergy, exhaustion, ignorance, deletion and suppression
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kawai T, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. The New England journal of medicine. 2008;358(4):353–61. doi: 10.1056/NEJMoa071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leventhal J, et al. Chimerism and tolerance without GVHD or engraftment syndrome in HLA-mismatched combined kidney and hematopoietic stem cell transplantation. Sci Transl Med. 2012;4(124):124ra28. doi: 10.1126/scitranslmed.3003509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scandling JD, et al. Tolerance and chimerism after renal and hematopoietic-cell transplantation. N Engl J Med. 2008;358(4):362–8. doi: 10.1056/NEJMoa074191. [DOI] [PubMed] [Google Scholar]
- 4.DeWolf S, Shen Y, Sykes M. A New Window into the Human Alloresponse. Transplantation. 2016;100(8):1639–49. doi: 10.1097/TP.0000000000001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menon MC, Murphy B, Heeger PS. Moving Biomarkers toward Clinical Implementation in Kidney Transplantation. J Am Soc Nephrol. 2017;28(3):735–747. doi: 10.1681/ASN.2016080858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris H, et al. Tracking donor-reactive T cells: Evidence for clonal deletion in tolerant kidney transplant patients. Science translational medicine. 2015;7(272):272ra10. doi: 10.1126/scitranslmed.3010760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuber J, et al. Bidirectional intragraft alloreactivity drives the repopulation of human intestinal allografts and correlates with clinical outcome. Sci Immunol. 2016;1(4) doi: 10.1126/sciimmunol.aah3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeWolf S, Sykes M. Alloimmune T cells in transplantation. J Clin Invest. 2017 doi: 10.1172/JCI90595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomita Y, Khan A, Sykes M. Role of intrathymic clonal deletion and peripheral anergy in transplantation tolerance induced by bone marrow transplantation in mice conditioned with a nonmyeloablative regimen. J Immunol. 1994;153(3):1087–98. [PubMed] [Google Scholar]
- 10.Khan A, Tomita Y, Sykes M. Thymic dependence of loss of tolerance in mixed allogeneic bone marrow chimeras after depletion of donor antigen. Peripheral mechanisms do not contribute to maintenance of tolerance. Transplantation. 1996;62(3):380–7. doi: 10.1097/00007890-199608150-00014. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, et al. Mixed allogeneic chimerism and renal allograft tolerance in cynomolgus monkeys. Transplantation. 1995;59(2):256–62. [PubMed] [Google Scholar]
- 12.Fuchimoto Y, et al. Mixed chimerism and tolerance without whole body irradiation in a large animal model. J Clin Invest. 2000;105(12):1779–89. doi: 10.1172/JCI8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horner BM, et al. Predictors of organ allograft tolerance following hematopoietic cell transplantation. Am J Transplant. 2006;6(12):2894–902. doi: 10.1111/j.1600-6143.2006.01563.x. [DOI] [PubMed] [Google Scholar]
- 14.Leventhal JR, et al. Immune reconstitution/immunocompetence in recipients of kidney plus hematopoietic stem/facilitating cell transplants. Transplantation. 2015;99(2):288–98. doi: 10.1097/TP.0000000000000605. [DOI] [PubMed] [Google Scholar]
- 15.Scandling JD, et al. Chimerism, graft survival, and withdrawal of immunosuppressive drugs in HLA matched and mismatched patients after living donor kidney and hematopoietic cell transplantation. Am J Transplant. 2015;15(3):695–704. doi: 10.1111/ajt.13091. [DOI] [PubMed] [Google Scholar]
- 16.Chakraverty R, et al. An inflammatory checkpoint regulates recruitment of graft-versus-host reactive T cells to peripheral tissues. J Exp Med. 2006;203(8):2021–31. doi: 10.1084/jem.20060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sykes M, Sheard MA, Sachs DH. Graft-versus-host-related immunosuppression is induced in mixed chimeras by alloresponses against either host or donor lymphohematopoietic cells. The Journal of experimental medicine. 1988;168(6):2391–6. doi: 10.1084/jem.168.6.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sykes M, Sheard MA, Sachs DH. Effects of T cell depletion in radiation bone marrow chimeras. II. Requirement for allogeneic T cells in the reconstituting bone marrow inoculum for subsequent resistance to breaking of tolerance. J Exp Med. 1988;168(2):661–73. doi: 10.1084/jem.168.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander SI, et al. Chimerism and tolerance in a recipient of a deceased-donor liver transplant. The New England journal of medicine. 2008;358(4):369–74. doi: 10.1056/NEJMoa0707255. [DOI] [PubMed] [Google Scholar]
- 20.Zuber J, et al. Macrochimerism in Intestinal Transplantation: Association With Lower Rejection Rates and Multivisceral Transplants, Without GVHD. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2015 doi: 10.1111/ajt.13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelot MR, et al. Lymphohematopoietic graft-vs.-host reactions can be induced without graft-vs.-host disease in murine mixed chimeras established with a cyclophosphamide-based nonmyeloablative conditioning regimen. Biol Blood Marrow Transplant. 1999;5(3):133–43. doi: 10.1053/bbmt.1999.v5.pm10392959. [DOI] [PubMed] [Google Scholar]
- 22.Spitzer TR, et al. Nonmyeloablative haploidentical stem-cell transplantation using anti-CD2 monoclonal antibody (MEDI-507)-based conditioning for refractory hematologic malignancies. Transplantation. 2003;75(10):1748–51. doi: 10.1097/01.TP.0000064211.23536.AD. [DOI] [PubMed] [Google Scholar]
- 23.Fujisaki J, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474(7350):216–9. doi: 10.1038/nature10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Penaloza-MacMaster P, et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med. 2014;211(9):1905–18. doi: 10.1084/jem.20132577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perruche S, et al. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med. 2008;14(5):528–35. doi: 10.1038/nm1749. [DOI] [PubMed] [Google Scholar]
- 27.Wekerle T, et al. Extrathymic T cell deletion and allogeneic stem cell engraftment induced with costimulatory blockade is followed by central T cell tolerance. J Exp Med. 1998;187(12):2037–44. doi: 10.1084/jem.187.12.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurtz J, et al. Mechanisms of early peripheral CD4 T-cell tolerance induction by anti-CD154 monoclonal antibody and allogeneic bone marrow transplantation: evidence for anergy and deletion but not regulatory cells. Blood. 2004;103(11):4336–43. doi: 10.1182/blood-2003-08-2642. [DOI] [PubMed] [Google Scholar]
- 29.Fehr T, et al. Rapid deletional peripheral CD8 T cell tolerance induced by allogeneic bone marrow: role of donor class II MHC and B cells. J Immunol. 2008;181(6):4371–80. doi: 10.4049/jimmunol.181.6.4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Domenig C, et al. Roles of deletion and regulation in creating mixed chimerism and allograft tolerance using a nonlymphoablative irradiation-free protocol. J Immunol. 2005;175(1):51–60. doi: 10.4049/jimmunol.175.1.51. [DOI] [PubMed] [Google Scholar]
- 31.Andreola G, et al. Mechanisms of donor-specific tolerance in recipients of haploidentical combined bone marrow/kidney transplantation. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11(6):1236–47. doi: 10.1111/j.1600-6143.2011.03566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, et al. Blocking both signal 1 and signal 2 of T-cell activation prevents apoptosis of alloreactive T cells and induction of peripheral allograft tolerance. Nat Med. 1999;5(11):1298–302. doi: 10.1038/15256. [DOI] [PubMed] [Google Scholar]
- 33.Wells AD, et al. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat Med. 1999;5(11):1303–7. doi: 10.1038/15260. [DOI] [PubMed] [Google Scholar]
- 34.Wekerle T, et al. Peripheral deletion after bone marrow transplantation with costimulatory blockade has features of both activation-induced cell death and passive cell death. J Immunol. 2001;166(4):2311–6. doi: 10.4049/jimmunol.166.4.2311. [DOI] [PubMed] [Google Scholar]
- 35.Tang Q, et al. CD28/B7 regulation of anti-CD3-mediated immunosuppression in vivo. J Immunol. 2003;170(3):1510–6. doi: 10.4049/jimmunol.170.3.1510. [DOI] [PubMed] [Google Scholar]
- 36.Kurtz J, et al. Lack of role for CsA-sensitive or Fas pathways in the tolerization of CD4 T cells via BMT and anti-CD40L. Am J Transplant. 2003;3(7):804–16. doi: 10.1034/j.1600-6143.2003.00128.x. [DOI] [PubMed] [Google Scholar]
- 37.Cippa PE, et al. Targeting apoptosis to induce stable mixed hematopoietic chimerism and long-term allograft survival without myelosuppressive conditioning in mice. Blood. 2013;122(9):1669–77. doi: 10.1182/blood-2012-09-453944. [DOI] [PubMed] [Google Scholar]
- 38.Merlini L, et al. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc Natl Acad Sci U S A. 2008;105(13):5225–9. doi: 10.1073/pnas.0800962105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Utsugi R, et al. Induction of transplantation tolerance with a short course of tacrolimus (FK506): I. Rapid and stable tolerance to two-haplotype fully mhc-mismatched kidney allografts in miniature swine. Transplantation. 2001;71(10):1368–79. doi: 10.1097/00007890-200105270-00003. [DOI] [PubMed] [Google Scholar]
- 40.Steinert EM, et al. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell. 2015;161(4):737–49. doi: 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amir AL, et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood. 2010;115(15):3146–57. doi: 10.1182/blood-2009-07-234906. [DOI] [PubMed] [Google Scholar]
- 42.Sachs DH, Kawai T, Sykes M. Induction of tolerance through mixed chimerism. Cold Spring Harb Perspect Med. 2014;4(1):a015529. doi: 10.1101/cshperspect.a015529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nadazdin O, et al. Host alloreactive memory T cells influence tolerance to kidney allografts in nonhuman primates. Sci Transl Med. 2011;3(86):86ra51. doi: 10.1126/scitranslmed.3002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed EB, et al. IL-6 induced by Staphylococcus aureus infection prevents the induction of skin allograft acceptance in mice. Am J Transplant. 2011;11(5):936–46. doi: 10.1111/j.1600-6143.2011.03476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang T, et al. Infection with the intracellular bacterium, Listeria monocytogenes, overrides established tolerance in a mouse cardiac allograft model. Am J Transplant. 2010;10(7):1524–33. doi: 10.1111/j.1600-6143.2010.03066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Badell IR, et al. Pathogen Stimulation History Impacts Donor-Specific CD8(+) T Cell Susceptibility to Costimulation/Integrin Blockade-Based Therapy. Am J Transplant. 2015;15(12):3081–94. doi: 10.1111/ajt.13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cippa PE, et al. Bcl-2 inhibition to overcome memory cell barriers in transplantation. Am J Transplant. 2014;14(2):333–42. doi: 10.1111/ajt.12554. [DOI] [PubMed] [Google Scholar]
- 48.Lee S, et al. Alefacept promotes immunosuppression-free renal allograft survival in nonhuman primates via depletion of recipient memory T cells. Am J Transplant. 2013;13(12):3223–9. doi: 10.1111/ajt.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamada Y, et al. Overcoming memory T-cell responses for induction of delayed tolerance in nonhuman primates. Am J Transplant. 2012;12(2):330–40. doi: 10.1111/j.1600-6143.2011.03795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levesque V, et al. B-cell-dependent memory T cells impede nonmyeloablative mixed chimerism induction in presensitized mice. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11(11):2322–31. doi: 10.1111/j.1600-6143.2011.03683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.You S. Differential sensitivity of regulatory and effector T cells to cell death: a prerequisite for transplant tolerance. Front Immunol. 2015;6:242. doi: 10.3389/fimmu.2015.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pierson W, et al. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3(+) regulatory T cells. Nat Immunol. 2013;14(9):959–65. doi: 10.1038/ni.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gabriel SS, et al. Distinctive Expression of Bcl-2 Factors in Regulatory T Cells Determines a Pharmacological Target to Induce Immunological Tolerance. Front Immunol. 2016;7:73. doi: 10.3389/fimmu.2016.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35(2):51–60. doi: 10.1016/j.it.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanchez-Fueyo A, Markmann JF. Immune Exhaustion and Transplantation. Am J Transplant. 2016;16(7):1953–7. doi: 10.1111/ajt.13702. [DOI] [PubMed] [Google Scholar]
- 56.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–76. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martinez GJ, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. 2015;42(2):265–78. doi: 10.1016/j.immuni.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamada Y, et al. Repeated Injections of IL-2 Break Renal Allograft Tolerance Induced via Mixed Hematopoietic Chimerism in Monkeys. Am J Transplant. 2015;15(12):3055–66. doi: 10.1111/ajt.13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haspot F, et al. Peripheral deletional tolerance of alloreactive CD8 but not CD4 T cells is dependent on the PD-1/PD-L1 pathway. Blood. 2008;112(5):2149–55. doi: 10.1182/blood-2007-12-127449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lucas CL, et al. LAG-3, TGF-beta, and cell-intrinsic PD-1 inhibitory pathways contribute to CD8 but not CD4 T-cell tolerance induced by allogeneic BMT with anti-CD40L. Blood. 2011;117(20):5532–40. doi: 10.1182/blood-2010-11-318675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fehr T, et al. A CD8 T cell-intrinsic role for the calcineurin-NFAT pathway for tolerance induction in vivo. Blood. 2010;115(6):1280–7. doi: 10.1182/blood-2009-07-230680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baas M, et al. TGFbeta-dependent expression of PD-1 and PD-L1 controls CD8(+) T cell anergy in transplant tolerance. Elife. 2016;5:e08133. doi: 10.7554/eLife.08133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doedens AL, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14(11):1173–82. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Long SA, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci Immunol. 2016;1(5) doi: 10.1126/sciimmunol.aai7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braza F, et al. Central Role of CD45RA-Foxp3hi Memory Regulatory T Cells in Clinical Kidney Transplantation Tolerance. J Am Soc Nephrol. 2015;26(8):1795–805. doi: 10.1681/ASN.2014050480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sprangers B, et al. Origin enriched regulatory T cells in patients receiving combined kidney/bone marrow transplantation to induce transplantation tolerance. Am J Transplant. 2017 doi: 10.1111/ajt.14251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hotta K, et al. Induced regulatory T cells in allograft tolerance via transient mixed chimerism. JCI Insight. 2016;1(10) doi: 10.1172/jci.insight.86419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu M, et al. Infiltrating Foxp3(+) regulatory T cells from spontaneously tolerant kidney allografts demonstrate donor-specific tolerance. Am J Transplant. 2013;13(11):2819–30. doi: 10.1111/ajt.12445. [DOI] [PubMed] [Google Scholar]
- 69.Kendal AR, et al. Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J Exp Med. 2011;208(10):2043–53. doi: 10.1084/jem.20110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferraro A, et al. Interindividual variation in human T regulatory cells. Proc Natl Acad Sci U S A. 2014;111(12):E1111–20. doi: 10.1073/pnas.1401343111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arvey A, et al. Genetic and epigenetic variation in the lineage specification of regulatory T cells. Elife. 2015;4:e07571. doi: 10.7554/eLife.07571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Coghill JM, et al. CC chemokine receptor 8 potentiates donor Treg survival and is critical for the prevention of murine graft-versus-host disease. Blood. 2013;122(5):825–36. doi: 10.1182/blood-2012-06-435735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plitas G, et al. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity. 2016;45(5):1122–1134. doi: 10.1016/j.immuni.2016.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koreth J, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365(22):2055–66. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsuoka K, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med. 2013;5(179):179ra43. doi: 10.1126/scitranslmed.3005265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Choi SW, et al. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. 2014;15(1):87–95. doi: 10.1016/S1470-2045(13)70512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choi SW, et al. Histone deacetylase inhibition regulates inflammation and enhances Tregs after allogeneic hematopoietic cell transplantation in humans. Blood. 2015;125(5):815–9. doi: 10.1182/blood-2014-10-605238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Zoeten EF, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. 2011;31(10):2066–78. doi: 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tao R, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13(11):1299–307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 80.Zuber J, et al. Harnessing regulatory T cells for transplant tolerance in the clinic through mTOR inhibition: myth or reality? Curr Opin Organ Transplant. 2011;16(6):606–13. doi: 10.1097/MOT.0b013e32834c237a. [DOI] [PubMed] [Google Scholar]
- 81.Kawai T, et al. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6(2):114. doi: 10.1038/72162. [DOI] [PubMed] [Google Scholar]
- 82.Cordoba F, et al. A novel, blocking, Fc-silent anti-CD40 monoclonal antibody prolongs nonhuman primate renal allograft survival in the absence of B cell depletion. Am J Transplant. 2015;15(11):2825–36. doi: 10.1111/ajt.13377. [DOI] [PubMed] [Google Scholar]
- 83.Okimura K, et al. Characterization of ASKP1240, a fully human antibody targeting human CD40 with potent immunosuppressive effects. Am J Transplant. 2014;14(6):1290–9. doi: 10.1111/ajt.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim SC, et al. Fc-Silent Anti-CD154 Domain Antibody Effectively Prevents Nonhuman Primate Renal Allograft Rejection. Am J Transplant. 2017;17(5):1182–1192. doi: 10.1111/ajt.14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schoenbrunn A, et al. A converse 4-1BB and CD40 ligand expression pattern delineates activated regulatory T cells (Treg) and conventional T cells enabling direct isolation of alloantigen-reactive natural Foxp3+ Treg. J Immunol. 2012;189(12):5985–94. doi: 10.4049/jimmunol.1201090. [DOI] [PubMed] [Google Scholar]
- 86.Poirier N, et al. Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci Transl Med. 2010;2(17):17ra10. doi: 10.1126/scitranslmed.3000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ville S, et al. Anti-CD28 Antibody and Belatacept Exert Differential Effects on Mechanisms of Renal Allograft Rejection. J Am Soc Nephrol. 2016;27(12):3577–3588. doi: 10.1681/ASN.2015070774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sage PT, et al. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. 2014;41(6):1026–39. doi: 10.1016/j.immuni.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trzonkowski P, et al. Hurdles in therapy with regulatory T cells. Sci Transl Med. 2015;7(304):304ps18. doi: 10.1126/scitranslmed.aaa7721. [DOI] [PubMed] [Google Scholar]
- 90.Komatsu N, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20(1):62–8. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- 91.Rubtsov YP, et al. Stability of the regulatory T cell lineage in vivo. Science. 2010;329(5999):1667–71. doi: 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miyao T, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36(2):262–75. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 93.Bluestone JA, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7(315):315ra189. doi: 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brunstein CG, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant. 2013;19(8):1271–3. doi: 10.1016/j.bbmt.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brunstein CG, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061–70. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pilat N, et al. Treg-therapy allows mixed chimerism and transplantation tolerance without cytoreductive conditioning. Am J Transplant. 2010;10(4):751–62. doi: 10.1111/j.1600-6143.2010.03018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duran-Struuck R, et al. Effect of ex vivo Expanded Recipient Regulatory T Cells on Hematopoietic Chimerism and Kidney Allograft Tolerance Across MHC Barriers in Cynomolgus Macaques. Transplantation. 2016 doi: 10.1097/TP.0000000000001559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Todo S, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. 2016;64(2):632–43. doi: 10.1002/hep.28459. [DOI] [PubMed] [Google Scholar]
- 99.Sagoo P, et al. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. 2011;3(83):83ra42. doi: 10.1126/scitranslmed.3002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.MacDonald KG, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. 2016;126(4):1413–24. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Newell KA, et al. Longitudinal studies of a B cell-derived signature of tolerance in renal transplant recipients. Am J Transplant. 2015;15(11):2908–20. doi: 10.1111/ajt.13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rebollo-Mesa I, et al. Biomarkers of Tolerance in Kidney Transplantation: Are We Predicting Tolerance or Response to Immunosuppressive Treatment? Am J Transplant. 2016 doi: 10.1111/ajt.13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pallier A, et al. Patients with drug-free long-term graft function display increased numbers of peripheral B cells with a memory and inhibitory phenotype. Kidney Int. 2010;78(5):503–13. doi: 10.1038/ki.2010.162. [DOI] [PubMed] [Google Scholar]
- 104.Chesneau M, et al. Tolerant Kidney Transplant Patients Produce B Cells with Regulatory Properties. J Am Soc Nephrol. 2015;26(10):2588–98. doi: 10.1681/ASN.2014040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mollov JL, et al. Recipient dendritic cells, but not B cells, are required antigen-presenting cells for peripheral alloreactive CD8+ T-cell tolerance. Am J Transplant. 2010;10(3):518–26. doi: 10.1111/j.1600-6143.2009.02967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Okumi M, et al. The induction of tolerance of renal allografts by adoptive transfer in miniature swine. Am J Transplant. 2013;13(5):1193–202. doi: 10.1111/ajt.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Madariaga ML, et al. Kidney-induced cardiac allograft tolerance in miniature swine is dependent on MHC-matching of donor cardiac and renal parenchyma. Am J Transplant. 2015;15(6):1580–90. doi: 10.1111/ajt.13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Riella LV, et al. Role of the PD-1 pathway in the immune response. Am J Transplant. 2012;12(10):2575–87. doi: 10.1111/j.1600-6143.2012.04224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Starke A, et al. Renal tubular PD-L1 (CD274) suppresses alloreactive human T-cell responses. Kidney Int. 2010;78(1):38–47. doi: 10.1038/ki.2010.97. [DOI] [PubMed] [Google Scholar]
- 110.Fudaba Y, et al. Myeloma responses and tolerance following combined kidney and nonmyeloablative marrow transplantation: in vivo and in vitro analyses. Am J Transplant. 2006;6(9):2121–33. doi: 10.1111/j.1600-6143.2006.01434.x. [DOI] [PubMed] [Google Scholar]
- 111.Morita M, et al. PD-1/B7-H1 interaction contribute to the spontaneous acceptance of mouse liver allograft. Am J Transplant. 2010;10(1):40–6. doi: 10.1111/j.1600-6143.2009.02859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Graca L, Cobbold SP, Waldmann H. Identification of regulatory T cells in tolerated allografts. J Exp Med. 2002;195(12):1641–6. doi: 10.1084/jem.20012097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miller ML, et al. Tracking of TCR-Tg T cells reveals multiple mechanisms maintain cardiac transplant tolerance in mice. Am J Transplant. 2016 doi: 10.1111/ajt.13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kurtz J, Wekerle T, Sykes M. Tolerance in mixed chimerism – a role for regulatory cells? Trends Immunol. 2004;25(10):518–23. doi: 10.1016/j.it.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 115.Kamano C, et al. Vascularized thymic lobe transplantation in miniature swine: thymopoiesis and tolerance induction across fully MHC-mismatched barriers. Proc Natl Acad Sci U S A. 2004;101(11):3827–32. doi: 10.1073/pnas.0306666101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tasaki M, et al. Role of bone marrow maturity, insulin-like growth factor 1 receptor and forkhead box protein N1 in thymic involution and rejuvenation. Am J Transplant. 2016 doi: 10.1111/ajt.13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhao Y, et al. Skin graft tolerance across a discordant xenogeneic barrier. Nat Med. 1996;2(11):1211–6. doi: 10.1038/nm1196-1211. [DOI] [PubMed] [Google Scholar]
- 118.Kalscheuer H, et al. Xenograft tolerance and immune function of human T cells developing in pig thymus xenografts. J Immunol. 2014;192(7):3442–50. doi: 10.4049/jimmunol.1302886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nikolic B, et al. Normal development in porcine thymus grafts and specific tolerance of human T cells to porcine donor MHC. J Immunol. 1999;162(6):3402–7. [PubMed] [Google Scholar]
- 120.Markert ML, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood. 2007;109(10):4539–47. doi: 10.1182/blood-2006-10-048652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Markert ML, et al. First use of thymus transplantation therapy for FOXN1 deficiency (nude/SCID): a report of 2 cases. Blood. 2011;117(2):688–96. doi: 10.1182/blood-2010-06-292490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chinn IK, Markert ML. Induction of tolerance to parental parathyroid grafts using allogeneic thymus tissue in patients with DiGeorge anomaly. J Allergy Clin Immunol. 2011;127(6):1351–5. doi: 10.1016/j.jaci.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhao Y, et al. Immune restoration by fetal pig thymus grafts in T cell-depleted, thymectomized mice. J Immunol. 1997;158(4):1641–9. [PubMed] [Google Scholar]
- 124.Rodriguez-Barbosa JI, et al. Enhanced CD4 reconstitution by grafting neonatal porcine tissue in alternative locations is associated with donor-specific tolerance and suppression of preexisting xenoreactive T cells. Transplantation. 2001;72(7):1223–31. doi: 10.1097/00007890-200110150-00007. [DOI] [PubMed] [Google Scholar]
- 125.Hadeiba H, et al. Plasmacytoid dendritic cells transport peripheral antigens to the thymus to promote central tolerance. Immunity. 2012;36(3):438–50. doi: 10.1016/j.immuni.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hadeiba H, et al. CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nat Immunol. 2008;9(11):1253–60. doi: 10.1038/ni.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Martin-Gayo E, et al. Plasmacytoid dendritic cells resident in human thymus drive natural Treg cell development. Blood. 2010;115(26):5366–75. doi: 10.1182/blood-2009-10-248260. [DOI] [PubMed] [Google Scholar]
- 128.Duncan SR, et al. Thymic dendritic cells traffic to thymi of allogeneic recipients and prolong graft survival. J Clin Invest. 2002;109(6):755–64. doi: 10.1172/JCI12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marino J, et al. Donor exosomes rather than passenger leukocytes initiate alloreactive T cell responses after transplantation. Sci Immunol. 2016;1(1) doi: 10.1126/sciimmunol.aaf8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu Q, et al. Donor dendritic cell-derived exosomes promote allograft-targeting immune response. J Clin Invest. 2016;126(8):2805–20. doi: 10.1172/JCI84577. [DOI] [PMC free article] [PubMed] [Google Scholar]