

Abstract
Increasing evidence demonstrates that lysine acetylation is involved in Mycobacterium tuberculosis (Mtb) virulence and pathogenesis. However, previous investigations in Mtb have only monitored acetylation at lysine residues using selected reference strains. We analyzed the global Nε- and O-acetylation of three Mtb isolates: two lineage 7 clinical isolates and the lineage 4 H37Rv reference strain. Quantitative acetylome analysis resulted in identification of 2490 class-I acetylation sites, 2349 O-acetylation and 141 Nε-acetylation sites, derived from 953 unique proteins. Mtb O-acetylation was thereby significantly more abundant than Nε-acetylation. The acetylated proteins were found to be involved in central metabolism, translation, stress responses, and antimicrobial drug resistance. Notably, 261 acetylation sites on 165 proteins were differentially regulated between lineage 7 and lineage 4 strains. A total of 257 acetylation sites on 161 proteins were hypoacetylated in lineage 7 strains. These proteins are involved in Mtb growth, virulence, bioenergetics, host–pathogen interactions, and stress responses. This study provides the first global analysis of O-acetylated proteins in Mtb. This quantitative acetylome data expand the current understanding regarding the nature and diversity of acetylated proteins in Mtb and open a new avenue of research for exploring the role of protein acetylation in Mtb physiology.
Keywords: Mycobacterium tuberculosis, lineage 7, post-translational modifications, acetylome, Nε-acetylation, O-acetylation
Graphical Abstract
INTRODUCTION
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB) in humans. TB is one of the top 10 causes of mortality worldwide and the leading cause of deaths from an infectious disease, leading to 1.8 million deaths and 10.4 million new cases in 2015.1 Due to the increasing prevalence of antimicrobial drug resistance and challenges in vaccine development, it is crucial to understand fundamental aspects of Mtb biology to achieve future elimination of this disease.
Nε-acetylation, which is acetylation at the ε-amine of lysine (K) residues, is an abundant and evolutionarily conserved post-translational modification (PTM) that regulates a broad range of functions in bacteria, including motility and chemotaxis, transcription, metabolism, DNA metabolism, siderophore biosynthesis, and stress responses.2–5 Increasing evidence supports the presence and role of lysine acetylation in mycobacteria.6–12 In Mtb, lysine acetylation presumably confers protein stability and compartmentalization, thereby modulating diverse cellular processes.13–15 Acetylation of Mtb histone-like nucleoid protein MtHU modulates DNA binding and genome organization.16 It has also been shown that reversible lysine acetylation regulates fatty acid metabolism in Mtb and acetate and propionate metabolism in M. smegmatis.6,8 Furthermore, Liu et al. have demonstrated the regulatory role of lysine acetylation in the immunogenicity of the secreted protein HspX in Mtb.17
Lysine acetylation is modulated via both enzymatic and nonenzymatic mechanisms (reviewed in ref 5). The enzymatic mechanism is regulated by the opposing actions of acetyltransferases and deacetylases. Transfer of an acetyl group from acetyl-CoA (AcCoA) to an Nε-lysine has been thought to occur enzymatically through lysine acetyltransferases, generating acetylated K. Although a plethora of Mtb acetyltransferases and deacetylases are predicted to be encoded by the Mtb genome,18 only a few of them are characterized to date. Recently, Lee et al. assigned acetyltransferase activity to the Mtb Rv2170 protein that acetylates lysine residues in isocitrate dehydrogenase, leading to a reduction in its enzymatic activity.19 Other characterized K acetyltransferases in Mtb include Rv0998 and Rv3423.1, which acetylate different target proteins.10,20 However, studies have demonstrated that lysine acetylation can also occur nonenzymatically in bacteria and mitochondria, with the secondary metabolite acetyl-phosphate (AcP) and AcCoA serving as the acetyl group donor.21–24 Nonenzymatic acetylation has been shown to occur via direct interaction of the target protein and AcCoA, which is favored by high pH and high AcCoA concentrations such as those in mitochondria.25,26 Protein deacetylases in Mtb include Rv1151c, the Mtb homologue of cobB, which has been shown to deacetylate and regulate the activity of Mtb acetyl-CoA synthase (ACS).2,7,17 Deletion of deacetylases affects Mtb colony morphology and biofilm formation as well as stress responses.17
Mukherjee et al. discovered that YopJ, a secreted virulence factor from Yersinia pestis, acetylates and inhibits kinase activation in the host by blocking phosphorylation.27 This is the first report regarding the presence and function of O-acetylation, which is acetylation at the –OH group of serine (S) and threonine (T) by a bacterial acetyltransferase. The function of YopJ within Y. pestis itself is not known. However, there is no YopJ ortholog in Mtb. Protein acetylation by itself is known to have a regulatory effect. Current evidence suggests that O-acetylation becomes more important when acetylation takes place on kinase substrates, which is known to regulate myriads of signaling pathways in Mtb.28,29 The transfer of an acetyl group to the –OH group of serine30 and glycoconjugates (peptidoglycans)31,32 has also been reported.
Despite phenotypic variability between strains of Mtb, most of the PTM studies performed to date have used the reference laboratory strain H37Rv as a model organism.15,17,33 Furthermore, bacterial acetylome studies have been limited to the analysis of lysine residues. O-acetylation of proteins has been shown to be involved in regulating key functions in eukaryotes.27 The delineation of such regulatory mechanisms not only will lead to a better understanding of Mtb basic biology and the discovery of potential new drug targets but may also facilitate the development of new vaccines and diagnostic tools.
In this study, we analyzed the global Nε- and O-acetylome of two Mtb lineage 7 clinical isolates and the lineage 4 reference strain H37Rv using nanoscale liquid chromatography coupled to tandem mass spectrometry (nano LC–MS/MS). The aim of this study is to define the global Nε- and O-acetylome profile of Mtb and to predict its possible contribution to the fitness and survival of Mtb using lineage 7 and lineage 4 strains as relevant model organisms.
EXPERIMENTAL PROCEDURES
Mycobacterial Strains and Growth Conditions
Mtb lineage 7 strains (L7-35 and L7-28) and the lineage 4 reference strain (H37Rv) were inoculated onto Middlebrook 7H10 plates in triplicates and incubated in a humidified 37 °C, 5% CO2 incubator. After 32 days, the cells were harvested and transferred to 50 mL Falcon tubes. The cell pellets were resuspended in 30 mL phosphate-buffered saline (PBS) containing, 10 mM PO43−, 137 mM NaCl, and 2.7 mM KCl, pH 7.4, and centrifuged at 3900 rpm for 20 min at 4 °C. The cell pellets were resuspended in 1 mL of PBS, transferred into 2 mL screw capped tubes (Sarstedt, Nümbrecht, Germany), and heat-inactivated at 80 °C for 90 min. Culturing and processing of the Mtb samples prior to heat-inactivation were conducted in a biosafety level 3 facility at Oslo University Hospital, Norway. The heat-inactivated Mtb samples were stored at −20 °C until preparation for MS analysis.
Preparation of Mtb Cell Lysate
The inactivated cell pellets in lysis buffer containing 2% SDS, 10 mM Tris-HCl (pH 7.5), 10 mM DTT, EDTA free protease inhibitor cocktail (Roche), and PhosStop (Roche) were transferred into Lysing Matrix B tubes (Roche, US) and disrupted mechanically by bead beating with a MagNa Lyser for 90 s, speed 6.0 (Roche, US). The lysis procedure was repeated six times with 1 min cooling on ice between each bead beating. The lysate was clarified by centrifugation (15 000g for 15 min) at 21 °C, and the supernatant containing the whole cell lysate proteins was transferred to new 2 mL screw capped microtubes (Sarstedt, Germany). Protein concentration was measured with a Direct Detect infrared spectrometer (DirectDetect, Millipore).
In-Gel Trypsin Digestion
One hundred micrograms of protein dissolved in NuPAGE LDS sample buffer (1×) and NuPAGE sample reducing agent (1×) (Life Technologies) was incubated for 10 min at 70 °C and prefractionated on a 1.0 mm, 4–12% NuPAGE Novex Bis-Tris gel (Life Technologies) at 80 V for 5 min followed by 20 min at 200 V. SDS-PAGE gels were Coomassie stained using a colloidal blue staining kit for NuPAGE according to the manufacturer’s instructions. After staining, each gel lane was divided into six fractions, and each fraction was subjected to in-gel reduction, alkylation, and tryptic digestion.34 Proteins were reduced using 10 mM DTT for 1 h at 56 °C and alkylated with 55 mM iodoacetamide for 1 h at room temperature. The reduced and alkylated peptides were digested with sequence grade trypsin (Promega, 1:100; w/w) for 16 h at 37 °C in 50 mM NH4HCO3. The trypsin digested protein samples were extracted from the gel using acetonitrile (ACN) (50 and 100%), dried by a SpeedVac concentrator (Eppendorf, concentrator 5301), and resuspended using 0.05% trifluoroacetic acid (TFA). The digested protein samples were loaded on to C18 zip-tips activated and equilibrated with 95% ACN/0.1% FA and 0.1% formic acid (FA), respectively. The loaded samples were washed with 0.05% TFA and eluted with 95% ACN/0.1% FA. The eluent was dried using a SpeedVac concentrator, resuspended in 0.1% FA, transferred to autosampler nano LC vials for LC–MS/MS analysis, and stored at −20 °C until they were injected in the LC–MS/MS system.
LC–MS/MS Analysis
Peptide characterization and quantitation were performed by nano LC–MS/MS using a Q Exactive hybrid quadropole-Orbitrap mass spectrometer interfaced with an EASY1000 nanoelectrospray ion source (both from Thermo Scientific).
Peptides were injected in triplicates into a precolumn (Acclaim PepMap 100, 75 μm × 2 cm, nanoviper, C18, 3 μm, 100 Å, Thermo Scientific) and separated on an analytical column (PepMap RSLC, C18, 2 μm, 100 Å, 50 μm × 15 cm, Thermo Scientific) with a 75 min solvent gradient at a flow rate of 0.3 μL/min. Gradients from 2 to 30% solvent B for 30 min followed by 30 to 75% solvent B from 30 to 35 min and 75 to 90% solvent B from 35 to 70 min were used. Thereafter, the gradient was kept at 90% solvent B from 70 to 75 min, using 0.1% FA in 3% ACN as solvent A and 0.1% FA in 97% ACN as solvent B (FA: LC–MS grade, Fluka; ACN: LC–MS grade, Merck). The column was operated at 60 °C. The mass spectrometer was operated in data-dependent acquisition mode with automatic switching between MS and MS/MS scans.
The full MS scans were acquired at 70K resolution, with automatic gain control target of 1 × 106 ions, maximum injection time of 200 ms, and a 300–1800 m/z scan range for MS scans. Higher energy collision dissociation (HCD) was used for peptide fragmentation with the normalized collision energy set to 28. The MS/MS scans were performed using a data-dependent top 10 method at a resolution of 17.5K with an automatic gain control target of 5 × 104 ions at maximum injection time of 100 ms and isolation window of 2.0 m/z units. An underfill ratio of 10% and dynamic exclusion duration of 30 s were applied.
Protein and PTM Identification
Protein and PTM site identification from the raw MS data was performed by using MaxQuant software with an integrated Andromeda search engine (version 1.5.7.4).35,36 The raw mass spectral data were searched against the UniProt Mtb protein database (downloaded from http://www.uniprot.org/ on Jan 15, 2017, UniProt ID: UP000001584, Organism/Taxonomy ID: 83332 and with 3993 protein sequences) concatenated to reverse decoy database and protein sequences for common contaminants.
Trypsin/P was specified as a cleavage enzyme allowing up to two missed cleavages. The “re-quantify” and “match between runs” options were utilized with a retention time alignment window of 3 min. Dependent peptide search, second peptide, LFQ, and iBAQ were enabled. Carbamidomethylation on cysteine was set as the fixed modification, and acetylation on protein N-terminal, conversion of N-terminal glutamine and glutamic acid to pyroglutamic acid, and oxidation on methionine were set as the variable modifications. For the PTM analysis, acetyl (KSTY) was set as the variable modification. Unique and razor peptides were used for the quantification of modified peptides (PTM abundance).
Only peptides with a minimum length of seven amino acids that were detected in at least one or more of the replicates were considered for identification. For protein identification, a minimum of two peptides, of which at least one was unique, was required per protein group. The threshold of protein identifications were determined by false discovery rate (FDR) of 0.01. All other parameters in MaxQuant were set to default values.
All modified peptide spectra were validated by applying a stringent site localization probability ≥ 0.75 and PEP ≤ 0.01 prior to further analysis. PTM site identifications with localization probability < 0.75 and PEP > 0.01, protein groups with matches to proteins from the reversed database, and contaminant protein sequences were removed from the analysis. Modified peptides with quantifiable values in at least five of nine biological replicates in the three strains were considered for label-free relative quantification. Acetylated peptide intensities were used for quantification of PTM abundance.
Bioinformatics Analysis
Statistical Analysis
Bioinformatics analysis was performed using Perseus software (version 1.5.6.0) as previously described.37 The protein group output from MaxQuant was used as the basis for all subsequent statistical and Gene Ontology (GO) enrichment analyses. Following protein identification by a MaxQuant database search, validation for multiple comparisons was corrected using the Benjamini–Hochberg correction.38 For identification of significantly changed acetylation sites between the two Mtb lineages, a two-tailed unpaired Student’s t test with FDR ≤ 0.05 and S0 = 2 was applied.
GO and Pathway Enrichment Analyses of Acetylated Proteins
The GO annotation of identified modified proteins was derived from the DAVID Bioinformatics Resources 6.7 and Gene Ontology Consortium bioinformatics databases.39,40 The proteins were classified by GO annotation based on three terms: molecular function (MF), biological process (BP), and cellular component (CC). Acetylated proteins were classified based on their functional category using TubercuList Mtb database (http://tuberculist.epfl.ch/).41,42 The Kyoto Encyclopedia of Genes and Genomes (KEGG) was utilized to annotate the pathways.43 The enriched GO terms and KEGG pathways provided corresponding information on p-value, count, percentage, and fold enrichment. Any pathway, biological process, or molecular function with a p-value ≤ 0.05 was considered significantly enriched. The ScanProsite web-based tool (http://prosite.expasy.org) and GenomeNet Database Resources (http://www.genome.jp/tools/motif/) were used to identify PROSITE signature profiles and active site motifs that match the sequence of identified acetylated peptides.44
PTM Motif Analysis
Motif-X software version 1.245 (http://motif-x.med.harvard.edu/motif-x.html) was used to analyze the enrichment of amino acid sequence motifs at specific positions of acetyl-31-mers (15 amino acids upstream and downstream of the site) in all peptide sequences. All protein sequences in the database were used as the background database parameter, and other parameters were set to their defaults.
Protein–Protein Interaction Network Analysis
Protein–protein interaction (PPI) networks were generated and analyzed using STRING database version 10 (http://string-db.org/)46 with a high confidence threshold of 0.7. Highly connected clusters were identified using MCODE plug-in toolkit, and the interaction network was visualized using the Cytoscape software (http://www.cytoscape.org) (version 3.5.0).47
Ethical Approval
The study obtained ethical approval from the Regional Committee for Medical Research Ethics in Eastern Norway (REK Øst) and Ethiopian Science and Technology Ministry in Addis Ababa, Ethiopia. Sample collection was conducted after obtaining written informed consent.
RESULTS AND DISCUSSION
First Combined Nε- and O-Acetylome Map of Mtb
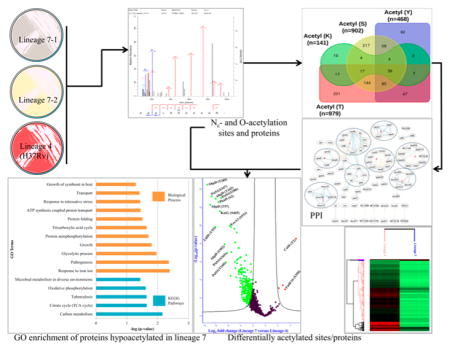
In this study, we analyzed the Nε- and O-acetylome profiles of two Mtb lineage 7 clinical isolate strains and the lineage 4 reference strain H37Rv. A total of 2490 class-I acetylation sites were identified, with 2200 and 2198 acetylation sites in lineage 7 and lineage 4, respectively (Figure 1A,B, Tables 1 and S1–S3). These sites were matched to 1568 proteins (953 unique proteins) (Table S4). A representative spectra of two acetylated peptides from heparin-binding hemagglutinin (HbhA) and conserved hypothetical protein (Rv2020c) are shown in Figure S1. Notably, 2310 (92.77%) sites of the totally identified acetylome were shared between lineage 7 and H37Rv, highlighting the acetylome conservation and consistency of the acetylome data (Figure 1A). Among the 953 unique proteins, 44.28% of the proteins were singly acetylated, whereas the remaining 55.82% of proteins were acetylated at two or more sites (Figure 2C). The most heavily acetylated proteins included the 60 kDa chaperonin 2 GroEL2, mycolipanoate synthase Msl3, polyketide synthase PKS13, ATP synthase subunit beta AtpD, chaperone protein DnaK, and catalase-peroxidase KatG (Figure 2C, Table S4). These heavily acetylated proteins are known to be involved in stress responses, corroborating previous data indicating a role for protein acetylation in stress adaptation in Mtb and other bacteria.17,48,49
Figure 1.

(A) Overall difference and overlap in acetylated sites between Mycobacterium tuberculosis strains of lineage 7 and lineage 4 (H37Rv). (B) Overall difference and overlap in acetylated proteins between four residues, S, T, Y, and K. “n” in (A) and (B) stand for the number of acetylation sites. (C) Distribution of acetylated proteins based on the number of acetylated sites per protein (Tables S1–S3).
Table 1.
Number of Acetylation Sites Per Residue in Mycobacterium tuberculosis Proteins and the Number and Percentage of Unique Proteins Acetylated
| acetylated residues | number of sites | unique proteins | percent (%) |
|---|---|---|---|
| acetyl (lysine, K) | 141 | 109 | 5.66 |
| acetyl (serine, S) | 902 | 568 | 36.22 |
| acetyl (threonine, T) | 979 | 557 | 39.32 |
| acetyl (tyrosine, Y) | 468 | 334 | 18.80 |
| overall unique elements | 2490 | 953 | 23.87 |
Figure 2.

GO enrichment analysis for biological process and KEGG pathways in exclusively O-acetylated Mycobacterium tuberculosis proteins (A) and Nε-acetylated Mtb proteins (B).
O-Acetylation at Serine, Threonine, or Tyrosine Represents 94.34% of the Sites Identified on 88.56% of the Acetylated Proteins
Out of a total of 2490 acetylation sites identified, 2349 sites on 844 proteins were found to be O-acetylated. The abundance of acetylation was highest on T residues (39.32%), followed by S (36.39%), Y (18.80%), and K residues (5.66%) (Figure 2B, Table 1). The proportion of O-acetylated residues in Mtb is similar to the phosphorylation profile on the S, T, and Y residues.50 The acetylated proteins identified accounted for 23.87% of the total proteins annotated in Mtb, which may indicate that applying an enrichment-based method would provide an even higher number of O-acetylated proteins than what has been reported for Nε-acetylation.15
Nε-acetylation is the most commonly studied acetylation both in eukaryotes and prokaryotes.4 A recent study on the Mtb acetylome, using an anti-acetyllysine antibody enriched sample, reported a total of 1128 lysine acetylation sites on 658 proteins.15 Although there is no former evidence for the presence of protein O-acetylation in prokaryotes, it is known that O-acetylation is a common modification of bacterial peptidoglycan and other glycoconjugates.31,32 Serine O-acetyltransferase in plants and bacteria plays a role in the biosynthesis of cysteine from serine.30,51 The O-acetyltransferase, N-hydroxyarylamine O-acetyltransferase (NhoA), has been shown to have O-acetyltransferase activity in Salmonella typhimurium.52 On the basis of the evidence discussed so far, including the YopJ O-acetyltransferase, we propose that the mechanism of O-acetylation and deacetylation on S, T, and Y residues in Mtb probably follows a similar pattern as Nε-acetylation involving both enzymatic and nonenzymatic mechanisms and AcCoA as the acetyl group donor. Further investigation is needed to elucidate the responsible mechanisms.
Bioinformatics analysis showed that these proteins share similar functions with Nε-acetylated proteins, as evident from previous reports on lysine acetylation.15,17 Even though there was a relatively low coverage of acetylated proteins in this enrichment-free method, a separate enrichment analysis for 109 lysine acetylated proteins and 840 exclusively O-acetylated proteins provided a distinct profile of biological processes and KEGG pathways associated with a particular acetylation. Exclusively O-acetylated proteins were involved in a broad range of KEGG pathway and biological processes, including fatty acid and carbon metabolism, translation, biosynthesis of secondary metabolites and antibiotics, amino acid metabolism, glyoxylate and dicarboxylate metabolism, and microbial metabolism in diverse environments (Figure 2A). In contrast, proteins acetylated at K residues were found to be involved in limited KEGG pathways and biological processes, primarily translation and the citrate cycle (TCA cycle) (Figure 2B).
These data may indicate that the stoichiometry of O-acetylation is more plentiful than Nε-acetylation. This may support the assumption that O-acetylation is involved in regulating a myriad of biological processes beyond that of Nε-acetylation. Furthermore, the number of O-acetylated peptides significantly outweighs the Nε-acetylated peptides. These frequent occurrences together with their competitive inhibition of phosphorylation may broaden the role of O-acetylated proteins in regulating bacterial physiology.49,53 Further enrichment-dependent methods for each of the four residues is necessary to complete the catalogue of Nε- and O-acetylated proteins and their respective enriched KEGG pathways.
Acetylated Peptides Were Identified Inside or Near PROSITE Signature Motifs
MS-derived sequence windows data was used to search the PROSITE signature motifs, active sites, and domain profiles. Some of the acetylated peptides identified were found near or inside enzyme active site domains (Tables S5-1 and S5-2). The polar residues histidine, cysteine, aspartate, glutamate, arginine, lysine, tyrosine, serine, threonine, asparagine, glutamine, and tryptophan are the most frequent catalytic amino acid residues.54,55 S, T, and Y are the major substrates for protein kinases that are hubs for complex regulatory networks and are involved in blocking phagosome-lysosome fusion, the hallmark of Mtb pathogenesis (reviewed in refs 28 and 29). The interplay between acetylation and phosphorylation of the same residues may have implications in the fine-tuning of certain cellular processes including Mtb pathogenesis. As a result, acetylation of major active site residues, including K, S, T, and Y, may affect protein activity.27,56
Acetylation of K residues found within an enzyme active site neutralizes the positive charge on lysine residues, which results in concomitant alteration in protein activity,57 protein–protein and protein–DNA interactions,58–60 local protein conformation,61 and protein localization.62,63 Protein acetylation increases the net negative charge62,64 on DNA binding proteins that might inhibit their binding capacity of the positively charged lysine residues with the negatively charged phosphate groups on DNA as reviewed by Carabetta et al.5 The inhibitory effect of acetylation on K residues found in the active sites of enzymes ACS, NhoA, adenosylmethionine synthase (MAT), and MbtA is highlighted in this review.
In our study, we found that the pyruvate kinase active site signature (Pyk) was one of the PROSITE motifs identified as acetylated at position K221 (Table S5-2). The lysine residue in the Pyk active site seems to be the acid/base catalyst responsible for the interconversion of pyruvate and enolpyruvate. In accordance with our finding, mutagenesis of the active site K221 of the pyruvate kinase was shown to reduce the activity of this enzyme by a factor of 104 to 105 in Bacillus stearothermophilus.65 Even though the effect of O-acetylation on bacterial enzyme activity has not been investigated to date, it is possible that direct acetylation of such active site residues may modulate, abolish, or induce the enzyme activity49 or interfere with the phosphorylation event.53
Positively Charged Lysine and Arginine Residues Are Enriched toward the N-Terminus of the Acetylation Sites
It is possible that acetylation events follow conserved linear protein sequence motifs similar to the motifs observed in protein phosphorylation. Two conserved putative motifs were identified for acetylated peptides on K residues, namely, RKac and R*Kac, at different abundances (Figure 3A,B).
Figure 3.

(A) Mycobacterium tuberculosis protein acetylation motifs and conservation of acetylation sites. (B) Number of identified peptides contained in each conserved motifs for the four residues.
The acetylation motifs identified in both Nε- and O-acetylated residues were consistent, having the positively charged K and/or arginine (R) residues between −1 and −6 positions to the N-terminus of the acetylated residues and L at −1 or −3 positions in some of the acetylated S and T residues (Figure 3A,B).
Previous studies on lysine acylation showed that the positively charged K and R residues were significantly enriched residues at the C-terminus,15,66 N- and C-terminus,67 and N-terminus49 of the acylation sites. This variation may be attributed to the difference in the methods used in the different studies, for example, in antibodies used for immunoaffinity enrichment.5 These motifs may serve as a recognition signature for putative bacterial acetyltransferases and deacetylases as observed in eukaryotes, or they could be part of an autocatalytic mechanism that facilitates the nonenzymatic acetylation process, thereby regulating substrate specificity, enhancing acetyltransferase activity, and restricting access to nontarget proteins.5,68
Acetylated Peptides Identified Are Involved in Diverse Cellular Processes
Among the 953 acetylated Mtb proteins identified, 78.5% were annotated for KEGG pathways, 92.0% for biological processes, and 94.3% for molecular functions, whereas only 35.1% were annotated for cellular components.
The predicted subcellular location of the acetylated proteins showed that most of the acetylated proteins identified were related to the cytoplasm (60%), while a few proteins were predicted to be membrane associated (20%), in macro-molecular complexes (10%), associated with organelles (8%), or located in the extracellular region (2%) (Figure 4A). The GO analysis of biological processes and molecular functions shows that large numbers of acetylated proteins are enzymes (73%) involved in metabolism (60%) (Figure 4B,C, respectively). Binding proteins are the second largest acetylated protein group in terms of molecular function, accounting for 15% of the total number of acetylated proteins identified (Figure 4B). Proteins associated with cellular processes represented the second largest protein group in terms of biological process, covering 24% of all annotated proteins (Figure 4C).
Figure 4.

Gene Ontology functional classification of the Mycobacterium tuberculosis acetylated proteins identified. (A) Subcellular localization of the acetylated proteins. (B) Classification of the acetylated proteins based on molecular function. (C) Classification of the acetylated proteins based on biological process. (D) Percentage of acetylated proteins within their respective functional category.
The acetylated proteins identified were grouped based on TubercuList functional categories (http://tuberculist.epfl.ch/). The percentage was calculated by dividing the number of acetylated proteins in each group by the total number proteins known to function in a particular category (Figure 4D).41 Proteins involved in information pathways had the most abundant acetylation (105/241, 43.57%), followed by proteins involved in lipid metabolism (106/247, 42.91%) and intermediary metabolism and respiration (373/923, 40.41%).
The GO enrichment analysis and protein–protein interaction analysis (PPI) showed that proteins involved in translation and the structural constituents of ribosomes are the most abundant among the significantly enriched acetylated proteins in terms of biological processes and molecular function, respectively (Figures 5 and S2, Table S6-1). Despite the variations in the methods used and the acetylated residues analyzed, this finding was in agreement with a previous study on lysine acetylation and succinylation.15,17,49
Figure 5.

Gene Ontology enrichment analysis of identified acetylated Mycobacterium tuberculosis proteins based on molecular function, KEGG pathway, and biological process.
It has been shown that lysine acetylation regulates cellular metabolism via different mechanisms such as enzymatic activation or inhibition and by influencing protein stability.17,69,70 We identified eight acetylated enzymes involved in the TCA cycle69,71 and two enzymes, isocitrate lyase (ICL) and malate synthase G (GlcB), involved in the glyoxylate pathway. Both copies of the ICL genes encoding IcL1 and IcL2 are essential for survival of Mtb in vivo.72–74 In E. coli, it has been shown that His356 is one of the catalytic active site residues in ICL.75 Furthermore, Wang et al. have shown that acetylation of AceA (Icl1) with protein acetyltransferase (Pat) or acetylation-mimicking mutations led to a reduction in AceA activity and that its activity was restored by deacetylation via CobB.69 We found that AceAa (Icl2a) and AceAb (Icl2b) were acetylated at two and five sites, respectively. AceAb was acetylated at S355, near the catalytic residue H356, indicating that acetylation of this residue may lead to a conformational change in the protein and therefore affect its enzymatic activity.
ACS is another protein acetylated at three sites and involved in the synthesis of AcCoA, a key intermediate in energy metabolism and an acetyl group donor in protein acetylation (Table S3). ACS was the first enzyme in Mtb shown to be regulated by reversible post-translational acetylation via cAMP-dependent protein acetyltransferase.11,12 The acetylation status of ACS determines the activity that might influence the availability of the acetyl donor, AcCoA, and the metabolic state of the cell.76
The glyoxylate cycle is another pathway enriched by acetylated proteins. When the TCA cycle is downregulated upon oxygen and nutrient/glucose depletion, replenishment of TCA cycle intermediates is achieved via the glyoxylate cycle using the AcCoA from fatty acid β-oxidation as a carbon source for subsequent metabolic pathways in the synthesis of biomolecules (glucose, amino acids, DNA, and RNA).77–79 Protein acetylation has been shown to regulate the activity of enzymes controlling the direction of glycolysis versus gluconeo-genesis and the branching between the TCA cycle and glyoxylate cycle by Pat and deacetylase.69
Fatty acid metabolism was one of the pathways identified by KEGG pathway enrichment analysis in this study. In addition to being a source of AcCoA, fatty acids are an integral component of the Mtb cell wall and known to be related to Mtb pathogenicity.80 It has been shown that reversible protein acetylation can regulate the activity of a number of fatty-acid-CoA ligases in Mtb.6,70 These findings suggest that acetylation may play a role in the regulation of various cellular processes in Mtb. Additional functional studies are needed to validate these claims.
Very Few Mtb Proteins Involved in Genome Maintenance Are Acetylated
Most components involved in DNA repair, recombination, and replication (3R components) were not acetylated even though they are located in the cytosol among core metabolic enzymes. The only 3R components found to be acetylated were DNA gyrases TopA, GyrA, and GyrB, single-stranded binding protein SSB, nucleotide excision repair DNA damage sensor UvrA, and recombination factors RecB and RecF. Ku ligase and DnaA were previously found to be acetylated in M. smegmatis and E. coli, respectively.81,82 Thus, along with all surface components, most 3R enzymes were found to be constituents of the nonacetylated complement of Mtb cells (Table S4).
Proteins Involved in Antimicrobial Drug Resistance Are Acetylated in Mtb
Several bacterial species, including Mtb, alter their proteins involved in drug resistance or drug targets, which in turn decreases or blocks the affinity for drug binding without affecting normal activity.49 PTMs may alter the net charge on the protein, conformation, interaction, and activity, thereby modulating the bacterial response to drugs. Acetylation of kinase substrates might alter the signaling pathways that lead to drug resistance/sensitivity. Seven Mtb proteins associated with isoniazide (INH) resistance, including KatG, InhA, NdhA KasA, AhpC, FadE24, and AcpM, were acetylated (Table S4).49,83 KatG, a catalase-peroxidase enzyme, is responsible for peroxidative activation of the prodrug INH and acts as a virulence factor to protect against oxidative stress.84 Mutations at KatG positions S315T and R463L have been shown to diminish its capacity to activate INH and confer INH resistance to Mtb.85 An acylation study showed that succinylation of KatG at K310 near the S315T mutation assists the enzyme in retaining its native antioxidant activity, while the INH activating property was reduced by almost 30% and the minimum inhibitory concentration of bacteria increased up to 200-fold.49 Among the eight differentially acetylated sites on KatG, S465 was found near the natural mutation R463L. Acetylation of this residue may lead to a conformational change in the protein and therefore affect its activity.
Serine/threonine protein kinases (STPKs) and two-component signal transduction systems are key regulators of metabolic processes, including transcription, cell development, stress response, virulence, host–pathogen interactions, and drug resistance.86,87 Four of the 11 Mtb STPKs, PknD, PknK, PknG, and PknH, were acetylated at various residues. OpcA and Wag31 have been shown to be upregulated in INH-resistant Mtb strains;49,88 OpcA and Wag31 are involved in peptidoglycan biosynthesis and oxidative stress responses.89,90 Another protein, PpiA, is involved in cationic antimicrobial peptide (CAMP) resistance.91 MurF is a protein involved in cell wall synthesis and implicated in vancomycin resistance in Mtb.92 Thus, acetylation of a protein in an active site residue or anywhere in the protein sequence may alter the its function in various modes.
InhA, NADH-dependent enoyl-ACP reductase, is a major enzyme involved in the biosynthesis of mycolic acids. Mutation at InhA I74T has been associated with resistance to ethambutol (EMB), INH, rifampicin (RMP), and streptomycin (SM).93 We identified an acetylation site on InhA at position T79, which is only five amino acids away from the natural mutation I74T. Other acetylated proteins with a role in resistance to first-line anti-TB drugs include RpoB, EmbR, PhoP, FabG1, and RpsL.83,94
Mutations in the genes encoding DNA gyrase subunits, gyrA and gyrB, are the most common mechanisms for acquiring fluoroquinolone (FQ) resistance in Mtb.95 The most frequent FQ resistance-associated mutations, termed the quinolone resistance-determining region, resides between codons 74 and 113 in gyrA and between 461 and 538 in gyrB.95–97 We found two acetylation sites at T500 and S473, which are located within the quinolone resistance-determining region of GyrB and may play a role in drug resistance, DNA replication, and Mtb survival. Another acetylated proteins involved in drug resistance is enhanced intracellular survival (Eis), an acetyl-transferase that confers resistance to kanamycin by modifying the drug.98,99
Proteins Associated with Virulence, Growth, and Stress Responses Are Differentially Acetylated between Lineage 7 and Lineage 4 Strains
From a total of 2490 acetylation sites identified on 953 proteins, 1085 sites on 506 proteins were eligible for quantification. We found that 261 acetylation sites on 165 proteins were differentially acetylated between lineage 7 and lineage 4 at S0 = 2 and FDR ≤ 0.05 (Figure 6A,B). Interestingly, 257 sites on 161 proteins involved in Mtb growth and virulence were hypoacetylated in lineage 7 with fold changes between 4.2 and 628.4. Only four sites on four proteins were significantly hyperacetylated in lineage 7 strains. Lineage 7 is a recently identified lineage of Mtb, characterized by slow-growth and reduced virulence phenotypes.100,101 The GO enrichment analysis of 161 proteins hypoacetylated in lineage 7 revealed that pathogenesis, growth, glycolysis, response to iron ion, response to nitrosative stress, and protein folding were among the significantly enriched biological processes (Figure 7A,B). Carbon metabolism, TCA cycle, oxidative phosphorylation, and microbial metabolism in diverse environments were some of the significantly enriched pathways from these hypoacetylated proteins (Figure 7B, Table S6-2).
Figure 6.

(A) Hierarchical clustering of differentially acetylated proteins between lineage 7 and lineage 4 strains of Mycobacterium tuberculosis (Mtb). (B) Volcano plot of differentially acetylated proteins between lineage 7 and H37Rv (S0 = 2, FDR ≤ 0.05; −log Student’s t test p-value on the Y-axis and Student’s t test difference on the X-axis). Red: hyperacetylation; green: hypoacetylation.
Figure 7.

(A) Protein–protein interaction network of differentially acetylated protein groups involved in (1) ESX-3 secretion, (2) host–pathogen interactions, (3) glycolysis, (4) TCA cycle, (5) fatty acid metabolism, (6) stress response, (7) transcription and translation, and (8) ATP synthase. (B) GO enrichment analysis of proteins hypoacetylated in lineage 7.
A total of 71 acetylated sites on 69 proteins and 109 acetylated sites on 93 proteins were exclusively identified in Mtb lineage 7 and lineage 4, respectively. GO enrichment analysis of the exclusively identified acetylated proteins showed a strain-specific enrichment of biological processes. Carbon and fatty acid metabolism were enriched in lineage 7 (Figure S3), whereas translation and amino acid metabolism were enriched in lineage 4 (H37Rv) (Figure S3). Enrichment of carbon and fatty acid metabolism in proteins exclusively identified in lineage 7 might be considered a compensatory mechanism for energy generation.
A number of enzymes involved in carbon metabolism, fatty acid metabolism, stress response, growth, virulence, and the Esx-3 secretion system were hypoacetylated in lineage 7 (Figure 7A,B, Tables 2 and S7). The dihydrolipoyllysine-residue acetyltransferase component of the pyruvate dehydrogenase complex (DlaT) and aconitate hydratase A (AcnA) are two proteins involved in the TCA cycle. Both enzymes were found to be hyperacetylated at lysine residues in lineage 4 strains. AcnA was acetylated at K273 position with fold change of 7.83, whereas DlaT was acetylated at two positions, K273 and K287, with fold changes of 8.27 and 6.66, respectively. Glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) and enolase (Eno) are two hypoacetylated enzymes involved in glycolytic pathway. Eno was acetylated at five sites with fold changes between −8.14 and 15.59, whereas GAPDH was acetylated at position S259 with a fold change of 7.03 (Tables 2 and S7).
Table 2.
List of Proteins Hypoacetylated in Mycobacterium tuberculosis Lineage 7 Strains
| protein name | gene ID | acetylation | fold change |
|---|---|---|---|
| alkyl hydroperoxide reductase AhpD | Rv2159c | T285 | −628.43 |
| alkyl hydroperoxide reductase AhpD | Rv2159c | T57 | −450.62 |
| phosphate-binding protein PstS 1 | pstS1 | S347 | −388.45 |
| putative L-lactate dehydrogenase | lldD | Y55 | −258.83 |
| alkyl hydroperoxide reductase AhpD | Rv2159c | T165 | −246.35 |
| serine/threonine-protein kinase PknD | pknD | S2 | −179.07 |
| catalase-peroxidase | katG | S465 | −153.46 |
| phosphate-binding protein PstS 1 | pstS1 | S186 | −130.58 |
| alkyl hydroperoxide reductase AhpD | Rv2159c | S302 | −89.98 |
| phosphate-binding protein PstS 1 | pstS1 | T160 | −78.49 |
| phosphate-binding protein PstS 1 | pstS1 | S365 | −68.62 |
| ESX-3 secretion system protein EccA3 | eccA3 | S533 | −49.83 |
| ESAT-6-like protein EsxO | esxO | T2 | −46.69 |
| ESX-3 secretion system protein EccC3 | eccC3 | T891 | −45.42 |
| isoniazid-induced protein IniB | iniB | S20 | −42.15 |
| mycolipanoate synthase | msl3 | Y1279 | −30.14 |
| phosphate-binding protein PstS 1 | pstS1 | T328 | −28.94 |
| probable CDP-diacylglycerol pyrophosphatase | cdh | Y84 | −28.23 |
| phosphate-binding protein PstS 1 | pstS1 | K324 | −27.53 |
| chaperone protein DnaK | dnaK | Y106 | −24.68 |
| nitrate reductase alpha subunit | narG | T2 | −23.58 |
| ferritin BfrB | bfrB | Y49 | −18.81 |
| probable thiol peroxidase | tpx | T13 | −18.61 |
| cytochrome BD ubiquinol oxidase subunit I | cydA | S307 | −18.27 |
| ESAT-6-like protein EsxO | esxO | Y65 | −18.09 |
| alpha-crystallin | hspX | T101 | −18.03 |
| ESX-3 secretion system protein EccC3 | eccC3 | T726 | −17.80 |
| mycolipanoate synthase | msl3 | S2016 | −16.50 |
| ESX-3 secretion system protein EccC3 | eccC3 | T176 | −16.15 |
| enolase | eno | K335 | −15.59 |
| ESX-3 secretion system protein EccA3 | eccA3 | T433 | −15.40 |
| mycolipanoate synthase | msl3 | T1259 | −14.74 |
| ESX-3 secretion system protein EccC3 | eccC3 | S125 | −14.54 |
| long-chain-fatty-acid-CoA ligase FadD15 | fadD15 | T160 | −14.35 |
| enolase | eno | S39 | −14.15 |
| probable thiol peroxidase | tpx | T98 | −13.50 |
| enolase | eno | S198 | −13.30 |
| alpha-crystallin | hspX | S91 | −13.27 |
Energy metabolism is associated with growth and virulence in intracellular bacteria, including Mtb.102,103 Central carbon metabolism uses different carbon sources to generate the building blocks, cofactors, and energy for cell growth. Wang et al. have shown that enzymes involved in Salmonella enterica central carbon metabolism are regulated by reversible lysine acetylation, involving protein acetyltransferase (Pat) and deacetylases (CobB).69 In S. enterica, acetylation of GAPDH has been shown to favor the glycolytic pathway while inhibiting gluconeogenesis by more than 30%, whereas deacetylation of GAPDH by the sirtiun CobB stimulates gluconeogenesis and inhibits the glycolytic pathway.69 Furthermore, it has been shown that S. enterica deficient in cobB (with high acetylation) grow faster than the wild-type cells in minimal glucose medium but grow slower than the wild type in minimal citrate medium. This indicates the importance of protein acetylation in regulating bacterial growth.69 The detailed mechanism of the regulation needs to be investigated. The activity of pyruvate dehydrogenase, one of the hypoacetylated enzymes involved in the TCA cycle, is reduced by lysine acetylation in eukaryotes,104,105 and this might also be true in bacteria. ACS was found to be hypoacetylated in lineage 7 strains with a fold change of 5.44. AcCoA is the substrate for the glyoxylate cycle.106 The glyoxylate cycle has been shown to be upregulated during Mtb growth arrest and involved in Mtb persistence.107,108 Moreover, previous works indicated the possible role of ACS in controlling the expression and/or activity of the glyoxylate cycle in E. coli.109,110 Therefore, the acetylation status of Mtb ACS might have a role in modulating Mtb physiology and persistence.11,106 Mtb utilizes lactate as a potential carbon and energy source inside macrophages.111 L-lactate dehydrogenase (LldD), an enzyme involved in the oxidation of lactate, was hypoacetylated in lineage 7 with a fold change of −258.83.
AcCoA can be generated from different substrates, including glucose, fatty acids, amino acids, and citrate. Once generated, it is used in ATP synthesis via the TCA cycle and the synthesis of fatty acids, amino acids, and other metabolites (reviewed in ref 76). In addition, AcCoA is an acetyl group donor for protein acetylation. Thus, acetylation of enzymes involved in AcCoA synthesis, ACS, may also modulate all processes involving AcCoA. These processes also consume AcCoA and thereby alter the availability of AcCoA in the cell and the metabolic state of the cell.
In addition to the enzymes involved in TCA and glyoxylate cycles, a number of long-chain fatty-acid-CoA ligases associated with fatty acid metabolism were hypoacetylated in Mtb lineage 7 strains with fold changes ranging from 4.54 to 14.35 (Figure 7A, Tables 2 and S5). Enzymes involved in fatty acid β-oxidation were hypoacetylated in lineage 7 (Tables 2 and S7). Fatty acid metabolism, both synthesis and catabolism, is an important cellular process for Mtb fitness and survival. More importantly, fatty acids and their derivatives are an integral component of the cell wall complex and implicated in Mtb pathogenicity, fitness, and survival.17,80,112 Mtb utilizes fatty acids as principal source of energy during dormancy and reactivation.103,113 Survival of Mtb inside the phagolysosome depends on the pathogen’s ability to synthesize virulence factors (proteins) and other biomolecules in a glucose-limited stress environment.79 Reversible acetylation has been shown to modulate the activity of several fatty-acid-CoA ligases.70 Polyketide synthase (PKS13) was hypoacetylated in lineage 7 at six sites with fold changes ranging from −3.89 to −5.98. PKS13 is an enzyme involved in the final steps of mycolic acid biosynthesis. Mycolic acids are an integral component of Mtb cell wall and known to be related to its pathogenicity.80
ESX-3 secretion system components were found to be highly acetylated in H37Rv. Esx-3 is implicated in essential physiologic processes and metal homeostasis and is crucial for Mtb growth in vivo and in vitro.114 The phosphate-binding protein PstS 1 is involved in inorganic phosphate uptake and its disruption has been shown to be associated with decreased virulence and attenuated growth.115 PstS 1 was hypoacetylated at six sites in lineage 7 with fold changes between −388.45 and −27.53.
Other groups of differentially acetylated proteins involved in stress response, virulence, and pathogenesis include chaperon proteins (DnaK, HspX), oxidoreductases (AhpD, Tpx, and KatG), ESX-1 secretion-associated proteins (EsxB, EspI, EspR, and EspF), mammalian cell entry proteins (Mce1B, Mce1F, and Mce1C), STPKs (PknD and PknH), and drug resistance-associated proteins (RpoB, RpoC, and IniB) (Tables 2 and S7). These proteins are indispensable for Mtb in signal transduction mechanisms that lead to bacterial adaptation to its environment,87 detoxification, and drug resistance85,116 or are involved in the entry and survival of the pathogen inside macrophages.117 Although the exact mechanism remains unknown, acetylation of enzymes involved in Mtb fitness and survival may lead to a change in the net charge of the protein, altering its stability and compartmentalization, bringing a conformational change, and/or blocking kinase substrates, and may thus modulate activity.
CONCLUSIONS
This study generated the first extensive data on O-acetylation in Mtb. We identified a total of 2490 acetylation sites on 953 Mtb unique proteins involved in a wide variety of fundamental cellular processes. These acetylated proteins are involved in Mtb core metabolic processes, bioenergetics, virulence, and drug resistance. Our findings provide novel insight into a range of functions predicted to be regulated by Nε- and O-acetylation in Mtb. The stoichiometry of Mtb O-acetylation is significantly higher than that of Nε-acetylation. This fact may in turn support the assumption that O-acetylation is involved in a broader range of processes than the Nε-acetylated counterparts. Due to the limited number of functional studies on the impact of acetylation conducted to date, the contribution of lysine acetylation in Mtb drug resistance is not fully understood. This study provides new knowledge on acetylated proteins and acetylation sites found near naturally occurring mutations that are known to be involved in Mtb drug resistance, and it may be used as a basis for further functional studies. Differentially acetylated proteins involved in Mtb virulence, energy metabolism/growth, and stress responses were uniformly biased toward being hypoacetylated in lineage 7 strains. This may lead to a metabolic state that makes one strain better fitted to a certain environment compared to the other. The real impact (inhibition/activation) of acetylation on protein activity needs to be further investigated through functional studies.
This MS-based prediction focusing on the nature and role of protein acetylation in Mtb should be verified by using both in vivo and in vitro functional studies, which can provide a proof of concept for the role of acetylation in Mtb physiology. Future studies could be performed by analyzing deletion mutants (i.e., acetylase or deacetylase) or substitution mutants at specific acetylation sites. Deletion of either the acetyltransferases or deacetylases can be used to unveil the role of these enzymes in the acetylation process, while point mutations are used to assess the functional role of a specific acetylation site. Finally, modification-specific antibodies could be generated to permit the use of immunoaffinity-enriched samples for nano LC–MS/ MS analysis, which may enable the identification of large numbers of acetylated proteins to establish the complete Mtb O-acetylome.
Supplementary Material
Acknowledgments
Funding
Funding was received from Research Council of Norway (RCN) FRIMEDBIO project no. 204747 and RCN GLOB-VAC project nos. 234506 to T.T. and 192468 to C.H.-H. and from Norwegian South-Eastern Health Authority project 2013080 to S.A.Y. and T.T.
We thank Tahira Riaz for acquiring the MS data. We also thank the patients for consenting to participate in the study and selected health care facilities in the Amhara Region, Ethiopia, for facilitating the study. We are grateful to the Armauer Hansen Research Institute (AHRI), Addis Ababa, Ethiopia, and the Norwegian Institute of Public Health for facilitating the transfer of lineage 7 strains for WGS at Oslo University Hospital.
ABBREVIATIONS
- Mtb
Mycobacterium tuberculosis
- PTM
post-translational modification
- MS
mass spectrometry
- GO
Gene Ontology
- FDR
false discovery rate
- PEP
posterior error probability
- KEGG
Kyoto Encyclopedia of Genes and Genomes
Footnotes
Author Contributions
T.T., A.G.B., and S.A.Y. conceived the study and study design. S.A.Y. collected the lineage 7 isolates. A.G.B. performed the MS sample preparation as well as performed bioinformatics and statistical analyses. A.G.B. and T.T. evaluated and interpreted the data and drafted the paper. All authors edited and approved the final manuscript.
Notes
The authors declare no competing financial interest.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE118 partner repository with the data set identifier PXD006630.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteo-me.7b00429.
Representative spectra of Nε- and O-acetylated peptides in Mtb (Figure S1); protein–protein interaction network analysis of Nε- and O-acetylated proteins in Mtb (Figure S2); GO enrichment analysis of acetylated proteins exclusively identified in Mycobacterium tuberculosis strains of lineage 7 and lineage 4 (Figure S3) (PDF)
List of class-I acetylation sites identified in H37Rv (Table S1); list of class-I acetylation sites identified in lineage 7 (Table S2); list of total identified class-I acetylation sites (Table S3); list of 953 identified acetylated proteins and number of acetylation sites per protein (Table S4); PROSITE patterns of acetylated proteins (Table S5-1); list of identified PROSITE profiles of acetylated peptides (Table S5-2); Gene Ontology enrichment analysis for Nε- and O-acetylated proteins using the DAVID classification system (Table S6-1); Gene Ontology enrichment analysis for differentially acetylated proteins between lineage 7 and lineage 4 (H37Rv) using the DAVID classification system (Table S6-2); list of differentially acetylated proteins in lineage 7 versus lineage 4 (Table S7) (XLSX)
References
- 1.Global Tuberculosis Report 2016. World Health Organization; Geneva: 2016. [accessed May 15, 2017]. http://www.who.int/tb/publications/global_report/en/ [Google Scholar]
- 2.Cain JA, Solis N, Cordwell SJ. Beyond gene expression: the impact of protein post-translational modifications in bacteria. J Proteomics. 2014;97:265–86. doi: 10.1016/j.jprot.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Vergnolle O, Xu H, Tufariello JM, Favrot L, Malek AA, Jacobs WR, Jr, Blanchard JS. Post-translational Acetylation of MbtA Modulates Mycobacterial Siderophore Biosynthesis. J Biol Chem. 2016;291:22315–22326. doi: 10.1074/jbc.M116.744532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ouidir T, Kentache T, Hardouin J. Protein lysine acetylation in bacteria: Current state of the art. Proteomics. 2016;16:301–9. doi: 10.1002/pmic.201500258. [DOI] [PubMed] [Google Scholar]
- 5.Carabetta VJ, Cristea IM. The regulation, function, and detection of protein acetylation in bacteria. J Bacteriol. 2017;199:e00107–17. doi: 10.1128/JB.00107-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nambi S, Gupta K, Bhattacharyya M, Ramakrishnan P, Ravikumar V, Siddiqui N, Thomas AT, Visweswariah SS. Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J Biol Chem. 2013;288:14114–24. doi: 10.1074/jbc.M113.463992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu J, Deng JY, Li R, Wei H, Zhang Z, Zhou Y, Zhang Y, Zhang XE. Cloning and characterization of NAD-dependent protein deacetylase (Rv1151c) from Mycobacterium tuberculosis. Biochemistry (Moscow) 2009;74:743–8. doi: 10.1134/s0006297909070062. [DOI] [PubMed] [Google Scholar]
- 8.Hayden JD, Brown LR, Gunawardena HP, Perkowski EF, Chen X, Braunstein M. Reversible acetylation regulates acetate and propionate metabolism in Mycobacterium smegmatis. Microbiology. 2013;159:1986–1999. doi: 10.1099/mic.0.068585-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee HJ, Lang PT, Fortune SM, Sassetti CM, Alber T. Cyclic AMP regulation of protein lysine acetylation in Mycobacterium tuberculosis. Nat Struct Mol Biol. 2012;19:811–8. doi: 10.1038/nsmb.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nambi S, Basu N, Visweswariah SS. cAMP-regulated protein lysine acetylases in mycobacteria. J Biol Chem. 2010;285:24313–23. doi: 10.1074/jbc.M110.118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H, Hegde SS, Blanchard JS. Reversible Acetylation and Inactivation of Mycobacterium tuberculosis Acetyl-CoA Synthetase Is Dependent on cAMP. Biochemistry. 2011;50:5883–5892. doi: 10.1021/bi200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li R, Gu J, Chen P, Zhang Z, Deng J, Zhang X. Purification and characterization of the acetyl-CoA synthetase from Mycobacterium tuberculosis. Acta Biochim Biophys Sin. 2011;43:891–899. doi: 10.1093/abbs/gmr076. [DOI] [PubMed] [Google Scholar]
- 13.van Els CA, Corbiere V, Smits K, van Gaans-van den Brink JA, Poelen MC, Mascart F, Meiring HD, Locht C. Toward Understanding the Essence of Post-Translational Modifications for the Mycobacterium tuberculosis Immunoproteome. Front Immunol. 2014;5:361. doi: 10.3389/fimmu.2014.00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okkels LM, Müller EC, Schmid M, Rosenkrands I, Kaufmann SH, Andersen P, Jungblut PR. CFP10 discriminates between nonacetylated and acetylated ESAT-6 of Mycobacterium tuberculosis by differential interaction. Proteomics. 2004;4:2954–2960. doi: 10.1002/pmic.200400906. [DOI] [PubMed] [Google Scholar]
- 15.Xie L, Wang X, Zeng J, Zhou M, Duan X, Li Q, Zhang Z, Luo H, Pang L, Li W, Liao G, Yu X, Li Y, Huang H, Xie J. Proteome-wide lysine acetylation profiling of the human pathogen Mycobacterium tuberculosis. Int J Biochem Cell Biol. 2015;59:193–202. doi: 10.1016/j.biocel.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh S, Padmanabhan B, Anand C, Nagaraja V. Lysine acetylation of the Mycobacterium tuberculosis HU protein modulates its DNA binding and genome organization. Mol Microbiol. 2016;100:577–88. doi: 10.1111/mmi.13339. [DOI] [PubMed] [Google Scholar]
- 17.Liu F, Yang M, Wang X, Yang S, Gu J, Zhou J, Zhang XE, Deng J, Ge F. Acetylome analysis reveals diverse functions of lysine acetylation in Mycobacterium tuberculosis. Mol Cell Proteomics. 2014;13:3352–66. doi: 10.1074/mcp.M114.041962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 19.Lee W, VanderVen BC, Walker S, Russell DG. Novel protein acetyltransferase, Rv2170, modulates carbon and energy metabolism in Mycobacterium tuberculosis. Sci Rep. 2017;7:72. doi: 10.1038/s41598-017-00067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jose L, Ramachandran R, Bhagavat R, Gomez RL, Chandran A, Raghunandanan S, Omkumar RV, Chandra N, Mundayoor S, Kumar RA. Hypothetical protein Rv3423.1 of Mycobacterium tuberculosis is a histone acetyltransferase. FEBS J. 2016;283:265–281. doi: 10.1111/febs.13566. [DOI] [PubMed] [Google Scholar]
- 21.Weinert BT, Iesmantavicius V, Wagner SA, Scholz C, Gummesson B, Beli P, Nystrom T, Choudhary C. Acetyl-Phosphate Is a Critical Determinant of Lysine Acetylation in E. coli. Mol Cell. 2013;51:265–272. doi: 10.1016/j.molcel.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, Lima BP, Scholle M, Mrksich M, Anderson WF, Gibson BW, Schilling B, Wolfe AJ. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One. 2014;9:e94816. doi: 10.1371/journal.pone.0094816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosono S, Tamura M, Suzuki S, Kawamura Y, Yoshida A, Nishiyama M, Yoshida M. Changes in the Acetylome and Succinylome of Bacillus subtilis in Response to Carbon Source. PLoS One. 2015;10:e0131169. doi: 10.1371/journal.pone.0131169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paik WK, Pearson D, Lee HW, Kim S. Nonenzymatic acetylation of histones with acetyl-CoA. Biochim Biophys Acta, Nucleic Acids Protein Synth. 1970;213:513–22. doi: 10.1016/0005-2787(70)90058-4. [DOI] [PubMed] [Google Scholar]
- 25.Wagner GR, Payne RM. Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288:29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baeza J, Smallegan MJ, Denu JM. Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem Biol. 2015;10:122–8. doi: 10.1021/cb500848p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science. 2006;312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- 28.Cousin C, Derouiche A, Shi L, Pagot Y, Poncet S, Mijakovic I. Protein-serine/threonine/tyrosine kinases in bacterial signaling and regulation. FEMS Microbiol Lett. 2013;346:11–9. doi: 10.1111/1574-6968.12189. [DOI] [PubMed] [Google Scholar]
- 29.Chao J, Wong D, Zheng X, Poirier V, Bach H, Hmama Z, Av-Gay Y. Protein kinase and phosphatase signaling in Mycobacterium tuberculosis physiology and pathogenesis. Biochim Biophys Acta, Proteins Proteomics. 2010;1804:620–7. doi: 10.1016/j.bbapap.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Denk D, Bock A. L-cysteine biosynthesis in Escherichia coli: nucleotide sequence and expression of the serine acetyltransferase (cysE) gene from the wild-type and a cysteine-excreting mutant. Microbiology. 1987;133:515–25. doi: 10.1099/00221287-133-3-515. [DOI] [PubMed] [Google Scholar]
- 31.Anonsen JH, Borud B, Vik A, Viburiene R, Koomey M. Structural and genetic analyses of glycan O-acetylation in a bacterial protein glycosylation system: evidence for differential effects on glycan chain length. Glycobiology. 2017;27:888–899. doi: 10.1093/glycob/cwx032. [DOI] [PubMed] [Google Scholar]
- 32.Moynihan PJ, Clarke AJ. O-Acetylated peptidoglycan: controlling the activity of bacterial autolysins and lytic enzymes of innate immune systems. Int J Biochem Cell Biol. 2011;43:1655–9. doi: 10.1016/j.biocel.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 33.Yang M, Wang Y, Chen Y, Cheng Z, Gu J, Deng J, Bi L, Chen C, Mo R, Wang X, Ge F. Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis. Mol Cell Proteomics. 2015;14:796–811. doi: 10.1074/mcp.M114.045922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2007;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 35.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 36.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 37.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–40. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 38.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–84. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 39.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 41.Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList–10 years after. Tuberculosis (Oxford, U K) 2011;91:1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Camus JC, Pryor MJ, Medigue C, Cole ST. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology. 2002;148:2967–2973. doi: 10.1099/00221287-148-10-2967. [DOI] [PubMed] [Google Scholar]
- 43.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou MF, Schwartz D. Biological sequence motif discovery using motif-x. Current Protoc Bioinform. 2011:13.15.1–13.15.24. doi: 10.1002/0471250953.bi1315s35. [DOI] [PubMed] [Google Scholar]
- 46.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma Q, Wood TK. Protein acetylation in prokaryotes increases stress resistance. Biochem Biophys Res Commun. 2011;410:846–51. doi: 10.1016/j.bbrc.2011.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie L, Liu W, Li Q, Chen S, Xu M, Huang Q, Zeng J, Zhou M, Xie J. First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. J Proteome Res. 2015;14:107–19. doi: 10.1021/pr500859a. [DOI] [PubMed] [Google Scholar]
- 50.Fortuin S, Tomazella GG, Nagaraj N, Sampson SL, Gey van Pittius NC, Soares NC, Wiker HG, de Souza GA, Warren RM. Phosphoproteomics analysis of a clinical Mycobacterium tuberculosis Beijing isolate: expanding the mycobacterial phosphoproteome catalog. Front Microbiol. 2015;6:6. doi: 10.3389/fmicb.2015.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saito K, Yokoyama H, Noji M, Murakoshi I. Molecular cloning and characterization of a plant serine acetyltransferase playing a regulatory role in cysteine biosynthesis from watermelon. J Biol Chem. 1995;270:16321–6. doi: 10.1074/jbc.270.27.16321. [DOI] [PubMed] [Google Scholar]
- 52.Saito K, Yamazoe Y, Kamataki T, Kato R. Mechanism of activation of proximate mutagens in Ames’ tester strains: The acetyl-CoA dependent enzyme in Salmonella typhimurium TA98 deficient in TA981, 8-DNP6 catalyzes DNA-binding as the cause of mutagenicity. Biochem Biophys Res Commun. 1983;116:141–147. doi: 10.1016/0006-291x(83)90392-3. [DOI] [PubMed] [Google Scholar]
- 53.Mukherjee S, Hao YH, Orth K. A newly discovered post-translational modification–the acetylation of serine and threonine residues. Trends Biochem Sci. 2007;32:210–6. doi: 10.1016/j.tibs.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 54.Holliday GL, Mitchell JB, Thornton JM. Understanding the functional roles of amino acid residues in enzyme catalysis. J Mol Biol. 2009;390:560–77. doi: 10.1016/j.jmb.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 55.Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol. 2002;324:105–21. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 56.Thao S, Escalante-Semerena JC. Control of protein function by reversible Nvarepsilon-lysine acetylation in bacteria. Curr Opin Microbiol. 2011;14:200–4. doi: 10.1016/j.mib.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang T, Wang S, Lin Y, Xu W, Ye D, Xiong Y, Zhao S, Guan KL. Acetylation negatively regulates glycogen phosphorylase by recruiting protein phosphatase 1. Cell Metab. 2012;15:75–87. doi: 10.1016/j.cmet.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 59.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 60.Thao S, Chen CS, Zhu H, Escalante-Semerena JC. Nε–lysine acetylation of a bacterial transcription factor inhibits its DNA-binding activity. PLoS One. 2010;5:e15123. doi: 10.1371/journal.pone.0015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howe FS, Boubriak I, Sale MJ, Nair A, Clynes D, Grijzenhout A, Murray SC, Woloszczuk R, Mellor J. Lysine acetylation controls local protein conformation by influencing proline isomerization. Mol Cell. 2014;55:733–44. doi: 10.1016/j.molcel.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A. 2012;109:10558–63. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ishfaq M, Maeta K, Maeda S, Natsume T, Ito A, Yoshida M. Acetylation regulates subcellular localization of eukaryotic translation initiation factor 5A (eIF5A) FEBS Lett. 2012;586:3236–41. doi: 10.1016/j.febslet.2012.06.042. [DOI] [PubMed] [Google Scholar]
- 64.Shaw BF, Schneider GF, Bilgicer B, Kaufman GK, Neveu JM, Lane WS, Whitelegge JP, Whitesides GM. Lysine acetylation can generate highly charged enzymes with increased resistance toward irreversible inactivation. Protein Sci. 2008;17:1446–1455. doi: 10.1110/ps.035154.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sakai H. Mutagenesis of the active site lysine 221 of the pyruvate kinase from Bacillus stearothermophilus. J Biochem. 2005;137:141–5. doi: 10.1093/jb/mvi027. [DOI] [PubMed] [Google Scholar]
- 66.Pan J, Ye Z, Cheng Z, Peng X, Wen L, Zhao F. Systematic analysis of the lysine acetylome in Vibrio parahemolyticus. J Proteome Res. 2014;13:3294–302. doi: 10.1021/pr500133t. [DOI] [PubMed] [Google Scholar]
- 67.Pan J, Chen R, Li C, Li W, Ye Z. Global Analysis of Protein Lysine Succinylation Profiles and Their Overlap with Lysine Acetylation in the Marine Bacterium Vibrio parahemolyticus. J Proteome Res. 2015;14:4309–18. doi: 10.1021/acs.jproteome.5b00485. [DOI] [PubMed] [Google Scholar]
- 68.Berndsen CE, Denu JM. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr Opin Struct Biol. 2008;18:682–9. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–7. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guan KL, Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci. 2011;36:108–16. doi: 10.1016/j.tibs.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–4. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr, Russell DG. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 73.Zu Bentrup KH, Miczak A, Swenson DL, Russell DG. Characterization of Activity and Expression of Isocitrate Lyase in Mycobacterium avium and Mycobacterium tuberculosis. J Bacteriol. 1999;181:7161–7167. doi: 10.1128/jb.181.23.7161-7167.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–44. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rehman A, Mcfadden BA. The Consequences of Replacing Histidine 356 in Isocitrate Lyase fromEscherichia coli. Arch Biochem Biophys. 1996;336:309–315. doi: 10.1006/abbi.1996.0562. [DOI] [PubMed] [Google Scholar]
- 76.Shi L, Tu BP. Protein Acetylation as a Means to Regulate Protein Function in Tune with Metabolic State. Biochem Soc Trans. 2014;42:1037–1042. doi: 10.1042/BST20140135. [DOI] [PubMed] [Google Scholar]
- 77.Wayne LG, Lin KY. Glyoxylate metabolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic conditions. Infect Immun. 1982;37:1042–1049. doi: 10.1128/iai.37.3.1042-1049.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dunn MF, Ramirez-Trujillo JA, Hernandez-Lucas I. Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology. 2009;155:3166–3175. doi: 10.1099/mic.0.030858-0. [DOI] [PubMed] [Google Scholar]
- 79.Lorenz MC, Fink GR. Life and death in a macrophage: role of the glyoxylate cycle in virulence. Eukaryotic Cell. 2002;1:657–62. doi: 10.1128/EC.1.5.657-662.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guenin-Mace L, Simeone R, Demangel C. Lipids of pathogenic Mycobacteria: contributions to virulence and host immune suppression. Transboundary Emerging Dis. 2009;56:255–68. doi: 10.1111/j.1865-1682.2009.01072.x. [DOI] [PubMed] [Google Scholar]
- 81.Zhou Y, Chen T, Zhou L, Fleming J, Deng J, Wang X, Wang L, Wang Y, Zhang X, Wei W, Bi L. Discovery and characterization of Ku acetylation in Mycobacterium smegmatis. FEMS Microbiol Lett. 2015;362:fnu051. doi: 10.1093/femsle/fnu051. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Q, Zhou A, Li S, Ni J, Tao J, Lu J, Wan B, Li S, Zhang J, Zhao S, et al. Reversible lysine acetylation is involved in DNA replication initiation by regulating activities of initiator DnaA in Escherichia coli. Sci Rep. 2016;6:30837. doi: 10.1038/srep30837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith T, Wolff KA, Nguyen L. Pathogenesis of Mycobacterium tuberculosis and its Interaction with the Host Organism. Springer; 2012. Molecular biology of drug resistance in Mycobacterium tuberculosis; pp. 53–80. [Google Scholar]
- 84.Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol. 2004;52:1291–302. doi: 10.1111/j.1365-2958.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- 85.Cohen T, Becerra MC, Murray MB. Isoniazid resistance and the future of drug-resistant tuberculosis. Microb Drug Resist. 2004;10:280–5. doi: 10.1089/mdr.2004.10.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gotoh Y, Eguchi Y, Watanabe T, Okamoto S, Doi A, Utsumi R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr Opin Microbiol. 2010;13:232–9. doi: 10.1016/j.mib.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 87.Prisic S, Husson RN. Mycobacterium tuberculosis Serine/Threonine Protein Kinases. Microbiol Spectrum. 2014;2:1–26. doi: 10.1128/microbiolspec.MGM2-0006-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jhingan GD, Kumari S, Jamwal SV, Kalam H, Arora D, Jain N, Kumaar LK, Samal A, Rao KV, Kumar D, Nandicoori VK. Comparative Proteomic Analyses of Avirulent, Virulent, and Clinical Strains of Mycobacterium tuberculosis Identify Strain-specific Patterns. J Biol Chem. 2016;291:14257–73. doi: 10.1074/jbc.M115.666123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mukherjee P, Sureka K, Datta P, Hossain T, Barik S, Das KP, Kundu M, Basu J. Novel role of Wag31 in protection of mycobacteria under oxidative stress. Mol Microbiol. 2009;73:103–19. doi: 10.1111/j.1365-2958.2009.06750.x. [DOI] [PubMed] [Google Scholar]
- 90.Kang CM, Nyayapathy S, Lee JY, Suh JW, Husson RN. Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology. 2008;154:725–735. doi: 10.1099/mic.0.2007/014076-0. [DOI] [PubMed] [Google Scholar]
- 91.Unal CM, Steinert M. Microbial peptidyl-prolyl cis/trans isomerases (PPIases): virulence factors and potential alternative drug targets. Microbiol Mol Biol Rev. 2014;78:544–71. doi: 10.1128/MMBR.00015-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Munshi T, Gupta A, Evangelopoulos D, Guzman JD, Gibbons S, Keep NH, Bhakta S. Characterisation of ATP-dependent Mur ligases involved in the biogenesis of cell wall peptidoglycan in Mycobacterium tuberculosis. PLoS One. 2013;8:e60143. doi: 10.1371/journal.pone.0060143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tseng ST, Tai CH, Li CR, Lin CF, Shi ZY. The mutations of katG and inhA genes of isoniazid-resistant Mycobacterium tuberculosis isolates in Taiwan. J Microbiol Immunol Infect. 2015;48:249–55. doi: 10.1016/j.jmii.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 94.Ramaswamy SV, Reich R, Dou SJ, Jasperse L, Pan X, Wanger A, Quitugua T, Graviss EA. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2003;47:1241–50. doi: 10.1128/AAC.47.4.1241-1250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chien JY, Chiu WY, Chien ST, Chiang CJ, Yu CJ, Hsueh PR. Mutations in gyrA and gyrB among Fluoroquinolone-and Multidrug-Resistant Mycobacterium tuberculosis Isolates. Anti-microb Agents Chemother. 2016;60:2090–6. doi: 10.1128/AAC.01049-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang JY, Lee LN, Lai HC, Wang SK, Jan IS, Yu CJ, Hsueh PR, Yang PC. Fluoroquinolone resistance in Mycobacterium tuberculosis isolates: associated genetic mutations and relationship to antimicrobial exposure. J Antimicrob Chemother. 2007;59:860–5. doi: 10.1093/jac/dkm061. [DOI] [PubMed] [Google Scholar]
- 97.Pitaksajjakul P, Wongwit W, Punprasit W, Eampokalap B, Peacock S, Ramasoota P. Mutations in the gyrA and gyrB genes of fluoroquinolone-resistant Mycobacterium tuberculosis from TB patients in Thailand. Southeast Asian J Trop Med Public Health. 2005;36:228–237. [PubMed] [Google Scholar]
- 98.Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S. Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc Natl Acad Sci U S A. 2011;108:9804–8. doi: 10.1073/pnas.1105379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsodikov OV, Green KD, Garneau-Tsodikova S. A random sequential mechanism of aminoglycoside acetylation by Mycobacterium tuberculosis Eis protein. PLoS One. 2014;9:e92370. doi: 10.1371/journal.pone.0092370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yimer SA, Norheim G, Namouchi A, Zegeye ED, Kinander W, Tonjum T, Bekele S, Mannsaker T, Bjune G, Aseffa A, Holm-Hansen C. Mycobacterium tuberculosis lineage 7 strains are associated with prolonged patient delay in seeking treatment for pulmonary tuberculosis in Amhara Region, Ethiopia. J Clin Microbiol. 2015;53:1301–9. doi: 10.1128/JCM.03566-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yimer SA, Birhanu AG, Kalayou S, Riaz T, Zegeye ED, Beyene GT, Holm-Hansen C, Norheim G, Abebe M, Aseffa A, Tonjum T. Comparative Proteomic Analysis of Mycobacterium tuberculosis Lineage 7 and Lineage 4 Strains Reveals Differentially Abundant Proteins Linked to Slow Growth and Virulence. Front Microbiol. 2017;8:795. doi: 10.3389/fmicb.2017.00795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eisenreich W, Dandekar T, Heesemann J, Goebel W. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol. 2010;8:401–12. doi: 10.1038/nrmicro2351. [DOI] [PubMed] [Google Scholar]
- 103.Munoz-Elias EJ, McKinney JD. Carbon metabolism of intracellular bacteria. Cell Microbiol. 2006;8:10–22. doi: 10.1111/j.1462-5822.2005.00648.x. [DOI] [PubMed] [Google Scholar]
- 104.Zhang X, Ji R, Liao X, Deng LY, Castillero E, Givens R, George I, Schulze PC. Lysine Acetylation of Pyruvate Dehydrogenase Reduces Enzymatic Activity and Contributes to Impaired Substrate Metabolism in the Failing Myocardium. Circulation. 2014;130:A18978. [Google Scholar]
- 105.Ozden O, Park SH, Wagner BA, Song HY, Zhu Y, Vassilopoulos A, Jung B, Buettner GR, Gius D. SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radical Biol Med. 2014;76:163–172. doi: 10.1016/j.freeradbiomed.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li R, Gu J, Chen P, Zhang Z, Deng J, Zhang X. Purification and characterization of the acetyl-CoA synthetase from Mycobacterium tuberculosis. Acta Biochim Biophys Sin. 2011;43:891–9. doi: 10.1093/abbs/gmr076. [DOI] [PubMed] [Google Scholar]
- 107.McKinney JD, zu Bentrup KH, Muñoz-Elías EJ, Miczak A, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 108.Shi L, Sohaskey CD, Pfeiffer C, Datta P, Parks M, McFadden J, North RJ, Gennaro ML. Carbon flux rerouting during Mycobacterium tuberculosis growth arrest. Mol Microbiol. 2010;78:1199–1215. doi: 10.1111/j.1365-2958.2010.07399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev. 2005;69:12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Contiero J, Beatty C, Kumari S, DeSanti CL, Strohl WR, Wolfe A. Effects of mutations in acetate metabolism on high-cell-density growth of Escherichia coli. J Ind Microbiol Biotechnol. 2000;24:421–430. [Google Scholar]
- 111.Billig S, Schneefeld M, Huber C, Grassl GA, Eisenreich W, Bange FC. Lactate oxidation facilitates growth of Mycobacterium tuberculosis in human macrophages. Sci Rep. 2017;7:6484. doi: 10.1038/s41598-017-05916-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Banerjee R, Vats P, Dahale S, Kasibhatla SM, Joshi R. Comparative genomics of cell envelope components in mycobacteria. PLoS One. 2011;6:e19280. doi: 10.1371/journal.pone.0019280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Daniel J, Sirakova T, Kolattukudy P. An acyl-CoA synthetase in Mycobacterium tuberculosis involved in triacylglycerol accumulation during dormancy. PLoS One. 2014;9:e114877. doi: 10.1371/journal.pone.0114877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen JM. Mycosins of the Mycobacterial Type VII ESX Secretion System: the Glue That Holds the Party Together. mBio. 2016;7:e02062–16. doi: 10.1128/mBio.02062-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peirs P, Lefevre P, Boarbi S, Wang XM, Denis O, Braibant M, Pethe K, Locht C, Huygen K, Content J. Mycobacterium tuberculosis with disruption in genes encoding the phosphate binding proteins PstS1 and PstS2 is deficient in phosphate uptake and demonstrates reduced in vivo virulence. Infection and immunity. 2005;73:1898–1902. doi: 10.1128/IAI.73.3.1898-1902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Colangeli R, Helb D, Sridharan S, Sun J, Varma-Basil M, Hazbon MH, Harbacheuski R, Megjugorac NJ, Jacobs WR, Jr, Holzenburg A, Sacchettini JC, Alland D. The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol Microbiol. 2005;55:1829–40. doi: 10.1111/j.1365-2958.2005.04510.x. [DOI] [PubMed] [Google Scholar]
- 117.Zhang F, Xie JP. Mammalian cell entry gene family of Mycobacterium tuberculosis. Mol Cell Biochem. 2011;352:1–10. doi: 10.1007/s11010-011-0733-5. [DOI] [PubMed] [Google Scholar]
- 118.Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.