

Abstract
BACKGROUND & AIMS
Induction of non-apoptotic cell death could be an approach to eliminate apoptosis-resistant tumors. We investigated necroptosis-based therapies in mouse models of pancreatic ductal adenocarcinoma cancer (PDAC).
Methods
We screened 273 commercially available kinase inhibitors for cytotoxicity against a human PDAC cell line (PANC1). We evaluated the ability of the aurora kinase inhibitor CCT137690 to stimulate necroptosis in PDAC cell lines (PANC1, PANC2.03, CFPAC1, MiaPaCa2, BxPc3, and PANC02) and the HEK293 cell line, measuring loss of plasma membrane integrity, gain in cell volume, swollen organelles, and cytoplasmic vacuoles. We tested the effects of CCT137690 in colon formation assays, and the effects of the necroptosis (necrostatin-1 and necrosulfonamide), apoptosis, autophagy, and ferroptosis inhibitors. We derived cells from tumors that developed in Pdx1-Cre;K-RasG12D/+;p53R172H/+ (KPC) mice. Genes encoding proteins in cell death pathways were knocked out, knocked down, or expressed from transgenes in PDAC cell lines. Athymic nude or B6 mice were given subcutaneous injections of PDAC cells or tail-vein injections of KPC tumor cells. Mice were given CCT137690 (80 mg/kg) or vehicle and tumor growth was monitored; tumor tissues were collected and analyzed by immunohistochemistry. We compared gene expression levels between human pancreatic cancer tissues (n=130) with patient survival times using the online R2 genomics analysis and visualization platform.
Results
CCT137690 induced necrosis-like death in PDAC cell lines and reduced colony formation; these effects required RIPK1, RIPK3, and MLKL, as well as inhibition of aurora kinase A (AURKA). AURKA interacted directly with RIPK1 and RIPK3 to reduce necrosome activation. AURKA-mediated phosphorylation of glycogen synthase kinase 3 beta (GSK3B) at serine 9 inhibited activation of the RIPK3 and MLKL necrosome. Mutations in AURKA (D274A) or GSK3β (S9A), or pharmacologic inhibitors of RIPK1 signaling via RIPK3 and MLKL, reduced the cytotoxic activity of CCT137690 in PDAC cells. Oral administration of CCT137690 induced necroptosis and immunogenic cell death in subcutaneous and orthotopic tumors in mice, and reduced tumor growth and tumor cell phosphorylation of AURKA and GSK3β. CCT137690 increased survival times of mice with orthotopic KPC PDACs and reduced tumor growth, stroma, and metastasis. Increased expression of AURKA and GSK3β mRNAs associated with shorter survival times of patients with pancreatic cancer.
Conclusions
We identified the aurora kinase inhibitor CCT137690 as an agent that induces necrosis-like death in PDAC cells, via RIPK1, RIPK3, and MLKL. CCT137690 slowed growth of orthotopic tumors from PDAC cells in mice, and expression of AURKA and GSK3β associate with patient survival times. AURKA might be targeted for treatment of pancreatic cancer.
Keywords: regulated cell death, anti-tumor immunity, HMGB1, ATP
Introduction
Pancreatic ductal adenocarcinoma (PDAC), the most common type of pancreatic cancer, is the fourth leading cause of cancer death in the United States. Nowadays, gemcitabine coupled with nab-paclitaxel or FOLFIRINOX is widely accepted as the standard systemic therapy, although it provides limited survival benefit for advanced PDAC patients 1, 2. Intrinsic or acquired resistance to apoptosis is one of the major factors associated with gemcitabine-based therapy failure. This highlights the urgent need to develop approaches to overcome apoptosis resistance or to trigger non-apoptotic forms of programmed cell death in PDAC.
Necroptosis is a caspase-independent form of regulated cell death that is induced by specific stimuli in apoptosis-resistant cells 3, 4. The process of necroptosis involves activation of a series of specific proteins, namely receptor interacting serine/threonine kinase (RIPK)-1, RIPK3, and mixed lineage kinase domain-like (MLKL). MLKL is considered a central executor of necroptosis because it translocates to the plasma membrane and induces its rupture. This process is controlled by RIPK3-mediated phosphorylation of MLKL. Dysregulated necroptosis is implicated in several inflammation-associated diseases, including cancer 5. Necroptosis-based cancer therapy has been proposed as a strategy to eliminate cancer cells when caspases are inhibited or defective 6.
Aurora kinases are a family of three highly homologous serine/threonine kinases, and include three major members, AURKA, AURKB, and AURKC, that play a well-known role in the regulation of cell cycle and division 7. In this study, we screened several kinase inhibitors and determined that CCT137690 exhibits significant anticancer activity in human PDACs via selective inhibition of AURKA and induction of necroptosis. AURKA is a direct negative regulator of necrosome activation. More importantly, oral administration of CCT137690 inhibited tumor growth by induction of necroptosis and immunogenic cell death without major toxicity in three PDAC in vivo models. In addition, increased expression of AURKA in pancreatic cancer tumors in patients correlated with poor overall survival, supporting the notion that AURKA may constitute a relevant therapeutic target for patients with PDAC.
Materials and Methods
Antibodies and reagents
The antibodies to c-PARP (#9544), p-MLKL (Ser358, #91689), MLKL (human specific, #14993), MLKL (mouse specific, #28640), p-H3 (Ser10, #3377), GSK3β (#9315), p-GSK3β (Ser9, #9336), RIPK3 (#13526), p-AURKA(Thr288)/AURKB(Thr232)/AURKC(Thr198) (#2914), AURKA (human specific, #14475), Flag (#14793), and HA (#3724) were purchased from Cell Signaling Technology. The antibodies to GAPDH (#G8795) and β-actin (#A5441) were purchased from Sigma Aldrich. The antibodies to RIPK1 (#NB100-56160), LC3 (#NB100-2220), HMGB1 (#H00003146-M08), and AURKA (#NBP2-36737) were purchased from Novus Biologicals. The antibodies to GPX4 (#ab125066) and p-MLKL (Ser345, # ab196436) were purchased from Abcam. The antibody to p-AURKA (Thr288, # GTX55002) was purchased from GeneTex Inc. The antibody to TNFR1 (#MAB225R) was purchased from R&D Systems. CCT137690 (#S2744), ferrostatin-1 (#S7243), AR-A014418 (#S7435), staurosporine (#S1421), erastin (#S7242), and kinase inhibitor library (#L1200) were obtained from Selleck Chemicals. Necrostatin-1 (#N9037) and gemcitabine (#G6423) were purchased from Sigma Aldrich. Z-VAD-FMK (#219007) and XXVI (#361569) were purchased from EMD Millipore. Chloroquine (#193919) was purchased from MP Biomedicals. Necrosulfonamide (#5025) and recombinant human TNF (#210-TA-005) were purchased from R&D Systems.
Cell lines
The PDAC cell lines (PANC1, PANC2.03, CFPAC1, MiaPaCa2, BxPc3, and PANC02) and human embryonic kidney 293 (HEK293) cell line were obtained from American Type Culture Collection (ATCC) or the National Cancer Institute of USA. The mouse PDAC cell line KPC was derived from tumors from KPC mice (Pdx1-Cre;K-RasG12D/+;p53R172H/+) as previously described 8. RIPK1−/−, RIPK3−/− and MLKL−/− mouse embryonic fibroblasts (MEFs) were a gift from Dr. Douglas R. Green. The ATG5−/− MEFs were a gift from Dr. Noboru Mizushima. ATG7−/− MEFs were a gift from Dr. Masaaki Komatsu. BAX−/−/BAK−/− MEFs were purchased from ATCC. Normal human pancreatic duct epithelial cells (HPDEs) were purchased from Applied Biological Materials Inc. All cells were mycoplasma-free and authenticated using Short Tandem Repeat DNA Profiling Analysis.
Animal models
All animal experiments were approved by the Institutional Animal Care and Use Committees and performed in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (http://www.aaalac.org).
To generate murine subcutaneous tumors, 5×106 PANC1, 1×106 PANC02, or 1×106 KPC cells in 100 μl phosphate buffered saline (PBS) were injected subcutaneously to the right of the dorsal midline in six- to eight-week-old athymic nude or B6 mice (Charles River, USA). To generate orthotopic tumors, 1× 106 KPC cells in 10 l PBS were surgically implanted into the tail of the pancreas in B6 mice. Pdx1-Cre and K-rasG12D/+ transgenic mice on B6 background were received from the MMHCC/NCI Mouse Repository. These mice were crossed to generate KC animals as we previously described 9.
Full Methods are available in the SUPPLEMENTAL MATERIALS AND METHODS.
Results
Anticancer activity of CCT137690 in PDAC cell lines
To identify compounds with anticancer activity against PDAC, we used a human PDAC cell line (PANC1) to screen 273 compounds from a commercially available library of kinase inhibitors. In the primary cytotoxicity assays using a single concentration, we identified the following top five kinase inhibitors: 1) NVP-BGT226: a dual phosphoinositide 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor; 2) IMD0354: an IκB kinase β (IKKβ) inhibitor; 3) CCT137690: an aurora kinase inhibitor; 4) PF-03814735: an aurora kinase inhibitor; and 5) SNS-314: an aurora kinase inhibitor (Fig. 1A, 1B, and Table S1). NVP-BGT226 10, IMD0354 11, PF-03814735 12, and SNS-314 13 have previously been reported to cause growth inhibition or cell death in PDAC cells. We therefore focused on the study of CCT137690 for the following experiments due to its previously unidentified role in PDAC treatment. CCT137690 induced necrosis-like death in PANC1 cells, as confirmed by live cell imaging analysis (Video S1). In addition to PANC1, CCT137690 dose-dependently killed other human PDAC cell lines, including PANC2.03, BxPc3, CFPAC1, and MiaPaCa2 in vitro (Fig. 1C). In contrast, normal HPDEs were resistance to CCT137690 treatment (Fig. 1C). Colony formation assays confirmed that the reproductive integrity of the PDAC cells after CCT137690 treatment was significantly reduced (Fig. 1D and 1E). Altogether, these results suggest that CCT137690 has anticancer activity in human PDAC cells.
Figure 1. Identification of CCT137690 as a novel anticancer agent limiting PDAC cells.
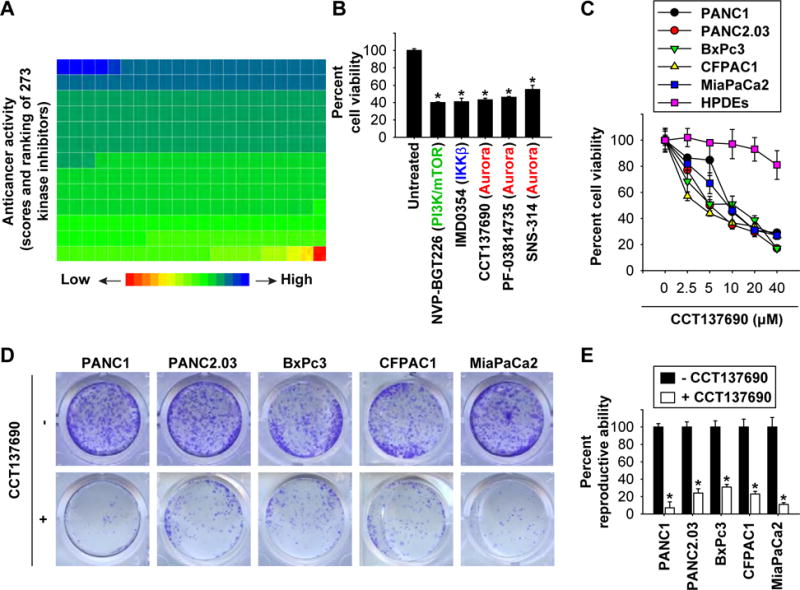
(A, B) PANC1 cells were treated with a kinase inhibitor (10 μM) for 24 hours and then cell viability was assayed. Ranking of the anticancer activity of 273 kinase inhibitors is shown by the heat map; one block represents a kinase inhibitor (A). The top five anti-cancer kinase inhibitors are shown in panel B (n=3, *p < 0.05 versus untreated group). (C) Indicated PDAC or normal HPDE cells were treated with CCT137690 (2.5–40 μM) for 24 hours, and then cell viability was assayed. (D, E) Clonogenic cell survival assay determined the reproductive ability of a cell after treatment with CCT137690 (10 μM) (n=3, *p < 0.05 versus untreated group).
Necroptosis mediates the primary anticancer activity of CCT137690
Evaluating the morphology of CCT137690-treated PANC1 cells, we identified characteristics of necrosis, including loss of plasma membrane integrity, gain in cell volume, swollen organelles, and cytoplasmic vacuoles (Fig. 2A). CCT137690 induced biochemical markers of apoptosis (cleavage of poly (ADP-ribose) polymerase [c-PARP]), autophagy (lipidation of microtubule associated protein 1 light chain 3 to generate the electrophoretically mobile form II [LC3-II]), ferroptosis (degradation of glutathione peroxidase 4 [GPX4]), and necroptosis/necrosis (release of high mobility group box 1 [HMGB1]) (Fig. 2B). These findings suggest that CCT137690 causes a mixed type of cell death. Remarkably, the necroptosis inhibitors necrostatin-1 and necrosulfonamide significantly restored cell viability (Fig. 2C and Fig. S1A) and reduced cell death (Fig. S1B) in PDAC cells (PANC1, PANC2.03, BxPc3, CFPAC1, and MiaPaCa2) following CCT137690 treatment. However, Z-VAD-FMK (an apoptosis inhibitor), chloroquine (an autophagy inhibitor), and ferrostatin-1 (a ferroptosis inhibitor) had no significant effects on cell viability (Fig. 2C and Fig. S1A) and cell death (Fig. S1B) following CCT137690 treatment. As an internal control, ZVAD-FMK (but not necrostatin-1) inhibited the death of PDAC cells induced by the pro-apoptotic agent staurosporine, while ferrostatin-1 (but not necrostatin-1) inhibited ferroptosis induction by erastin (Fig. 2D). These data indicate that the anticancer activity of CCT137690 depends on the induction of necroptosis.
Figure 2. Induction of necroptosis contributes to the anticancer activity of CCT137690.
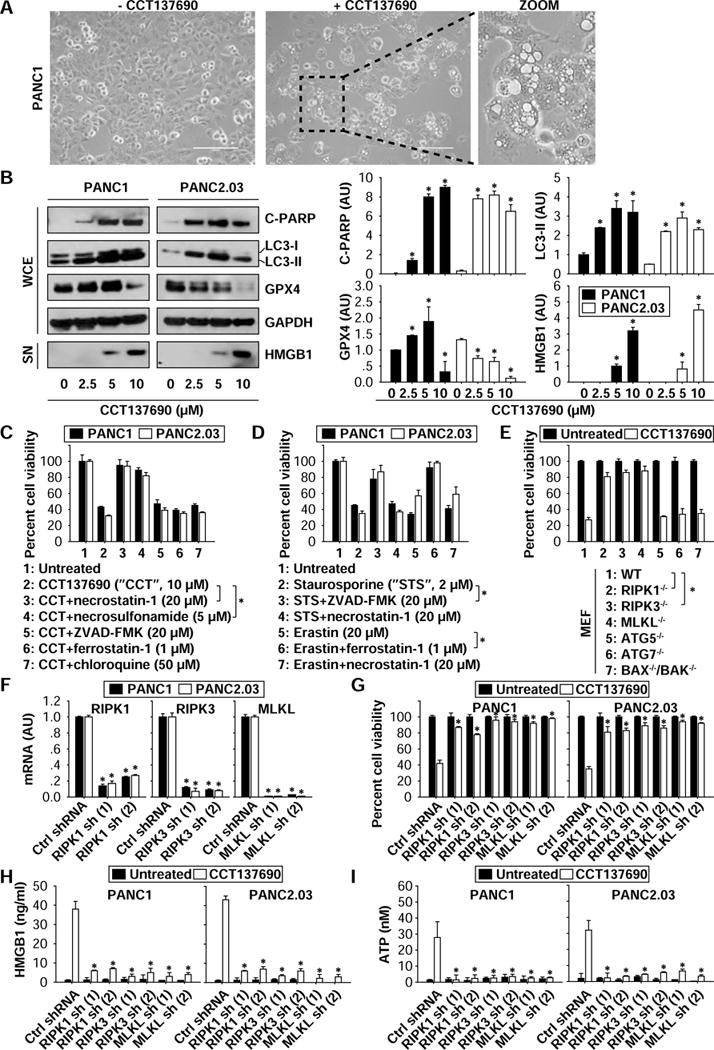
(A) Necrotic-like morphology was observed in PANC1 cells following treatment with CCT137690 (10 μM) for 24 hours. (B) Western blot analysis of indicated proteins in PANC1 and PANC2.03 cells following treatment with CCT137690 (2.5–10 μM) for 24 hours (n=3, *p < 0.05 versus untreated group). (C, D) PANC1 and PANC2.03 cells were treated with CCT137690 (C) or staurosporine (D) or erastin (D) in the absence or presence of indicated cell death inhibitors for 24 hours. Cell viability was assayed (n=3, *p < 0.05). (E) Indicated MEFs were treated with CCT137690 (10 μM) for 24 hours, and then cell viability was assayed (n=3, *p < 0.05). (F–I) Knockdown of RIPK1, RIPK3, and MLKL by shRNAs inhibited CCT137690 (10 μM, 24 hours)-induced cell death and HMGB1 and ATP release (n=3, *p < 0.05 versus control shRNA group).
We next compared CCT137690-induced cell death in wild type (WT) MEFs and necroptosis-deficient MEFs, including RIPK1−/−, RrPK3−/−, and MLKL−/− cells. Knockout of RIPK1, RIPK3, and MLKL restored cell viability following CCT137690 treatment (Fig. 2E). Knockdown of RIPK1, RIPK3, and MLKL by two different shRNAs (Fig. 2F and Fig. S1C) also restored cell viability (Fig. 2G) and reduced cell death (Fig. S1D) in multiple PDAC cells following CCT137690 treatment. In contrast, depletion of autophagic (e.g., ATG5−/− or ATG7−/−) or apoptotic (BAX−/− and BAK−/−) proteins had no significant influence on CCT137690-induced cell death in MEFs (Fig. 2E). Colony formation assays confirmed that the reproductive integrity of the PDAC cells after CCT137690 treatment was significantly restored in the inhibition of necroptosis by necrostatin-1 or knockdown of MLKL (Fig. S1D). These findings further demonstrate that induction of necroptosis contributes to the anticancer activity of CCT137690.
Immunogenic cell death (ICD) constitutes a prominent pathway for the activation of the immune system against cancer, which in turn determines the long-term success of anticancer therapies 14, 15. Necroptosis is a form of ICD associated with release of damage-associated molecular patterns (DAMPs) such as HMGB1 and ATP 16. We found that knockdown of RIPK1, RIPK3, and MLKL blocked CCT137690-induced release of HMGB1 (Fig. 2H) and ATP (Fig. 2I) in PANC1 and PANC2.03 cells, suggesting that the RIPK1-RIPK3-MLKL pathway contributes to CCT137690-induced ICD in human PDAC cells.
AURKA inhibits necroptosis in a cell cycle-independent manner
Human aurora kinases, including the three primary members AURKA, AURKB, and AURKC, are activated by one or more phosphorylations 17. CCT137690 inhibits phosphorylation of individual aurora kinase members, depending on the tumor type 18–20. The phosphorylation of AURKA (but not AURKB and AURKC) was suppressed by CCT137690 treatment in PANC1 and PANC2.03 cells (Fig. 3A). Moreover, phosphorylation of histone H3 (the principal substrate of AUKRA 21) was blocked following treatment with CCT137690 (Fig. 3A), further supporting that the AURKA pathway is inhibited by CCT137690. Stable knockdown of AURKA with two different shRNAs (Fig. 3B) also resulted in cell growth inhibition in PANC1 and PANC2.03 cells by colony formation assay (Fig. 3C and 3D). This process was reversed by inhibitors of necroptosis (e.g., necrostatin-1 and necrosulfonamide), but not by specific inhibitors of other cell death modalities (e.g., Z-VAD-FMK, chloroquine, and ferrostatin-1) (Fig. 3C and 3D). Moreover, the release of HMGB1 (Fig. 3E) and ATP (Fig. 3F) was increased in AURKA-knockdown cells; this process was also inhibited by necrostatin-1 and necrosulfonamide. Together, these findings suggest that AURKA acts as an endogenous repressor of necroptosis and ICD in PDAC cells.
Figure 3. AURKA inhibits necroptosis in a cell cycle-independent manner.
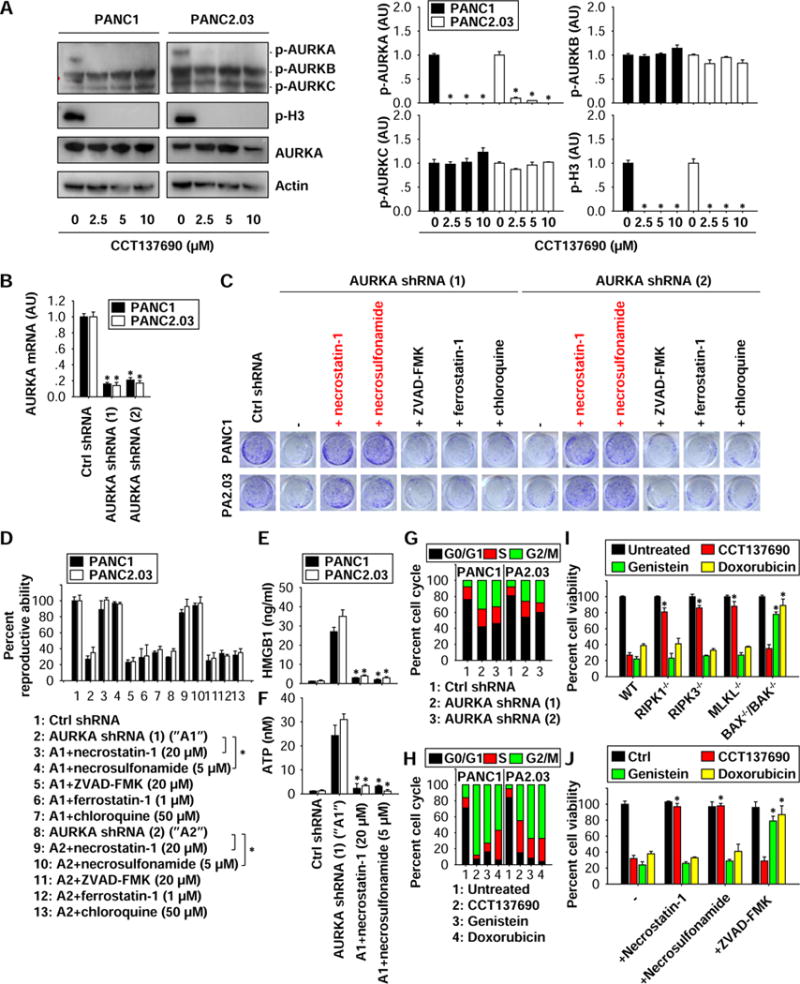
(A) Western blot analysis of indicated proteins in PDAC cells following treatment with CCT137690 (2.5–10 μM) for 24 hours (n=3, *p < 0.05 versus untreated group). AU=arbitrary unit. (B) Q-PCR analysis AURKA mRNA in stable AURKA-knockdown cells (n=3, *p < 0.05 versus control shRNA group). (C, D) Clonogenic cell survival assay determines the reproductive ability of AURKA-knockdown cells in the absence or presence of indicated cell death inhibitors (n=3, *p < 0.05). (E, F) ELISA analysis of HWGB1 and ATP levels in culture supernatants (n=3, *p < 0.05 versus AURKA shRNA group). (G) Cell cycle analysis in AURKA-knockdown cells. (H) Cell cycle analysis in PANC1 and PANC2.03 cells following CCT137690 (10 μM), genistein (50 μM), and doxorubicin (0.2 μg/ml) for 24 hours. (I) Indicated MEFs were treated with CCT137690 (10 μM), genistein (50 μM), and doxorubicin (0.2 μg/ml) for 24 hours, and cell viability was assayed (n=3, *p < 0.05 versus WT group). (J) PANC1 cells were treated with CCT137690 (10 μM), genistein (50 μM), and doxorubicin (0.2 μg/l) in the absence or presence of indicated cell death inhibitors for 24 hours, and then cell viability was assayed (n=3, *p < 0.05 versus control group).
Given that AURKA inhibits mitotic catastrophe through regulation of cell cycle 17,22, we next addressed whether impaired cell cycle regulation is required for AURKA-mediated necroptosis inhibition. Consistent with results from previous studies on laryngeal and lung cancer cells23,24, knockdown of AURKA expression by shRNA (Fig. 3G) or inhibition of AURKA activity by CCT137690 (Fig. 3H) arrested PANC1 and PANC2.03 cell growth within the G2/M phase of the cell cycle. We next tested the capacity of other cell cycle inhibitors other than CCT137690 to induce necroptosis. Cytotoxic drugs such as doxorubicin 25 and genistein 26 can induce cancer cell death, as they favor the accumulation of cells in the G2/M phase (Fig. 3H). However, genetic inhibition (by knockout of RIPK1, RIPK3, or MLKL) (Fig. 3I) or pharmacological inhibition (with necrostatin-1 or necrosulfonamide) (Fig. 3J) of necroptosis had no effects on cell death induced by doxorubicin or genistein. In contrast, measures that blocked apoptosis either genetically (by combined knockout of BAX−/− and BAK−/−) (Fig. 3I) or pharmacologically (by application of ZVAD-FMK) (Fig. 3J) prevented cell death induced by doxorubicin or genistein. These findings indicate that cell cycle arrest and mitotic catastrophe may not be essential for necroptosis induction in PDAC cells following AURKA inhibition.
AURKA inhibits necroptosis by binding the RIPK1/RIPK3 necrosome
AURKA inhibition by CCT137690 (Fig. 4A) or MLN8237 (Fig. S2A) or knockdown (Fig. 4B) significantly induced the phosphorylation of MKLK at Ser358 in PANC1 and PANC2.03 cells, suggesting that AURKA inhibits p-MLKL to suppress necroptosis. MLKL phosphorylation is controlled by its physical interaction with other core components of the necrosome, including RIPK1 and RIPK3 3, 4 Immunoprecipitation analysis showed that CCT137690 (Fig. 4C) or MLN8237 (Fig. S2B) or knockdown of AURKA (Fig. 4D) enhanced the formation of RIPK1-RIPK3 and RIPK3-MLKL complexes, suggesting that AURKA could be a negative regulator of necrosome formation and activation.
Figure 4. AURKA inhibits necroptosis by binding the RIPK1/RIPK3 necrosome.
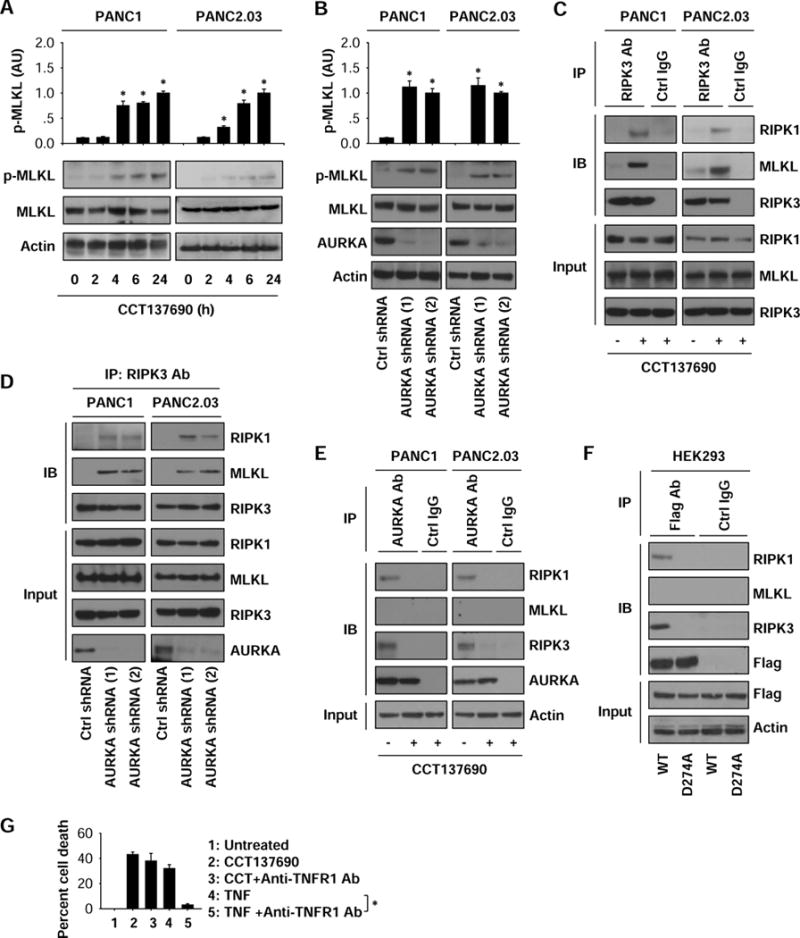
(A) Western blot analysis of indicated proteins in PDAC cells following treatment with CCT137690 (10 μM) for three to 24 hours (n=3, *p < 0.05 versus untreated group). (B) Western blot analysis of indicated proteins in control and stable AURKA-knockdown PDAC cells (n=3, *p < 0.05 versus control shRNA group). (C) Immunoprecipitation (IP) analysis of the levels of RIPK3 binding to RIPK1 and MLKL in PANC1 and PANC2.03 cells following treatment with CCT137690 (10 μM) for 24 hours. (D) IP analysis of the levels of RIPK3 binding to RIPK1 and MLKL in control and stable AURKA-knockdown PANC1 and PANC2.03 cells. (E) IP analysis of the levels of AURKA binding to RIPK1, RIPK3, and MLKL in PANC1 and PANC2.03 cells following treatment with CCT137690 (10 μM) for 24 hours. (F) IP analysis of the levels of AURKA binding to RIPK1, RIPK3, and MLKL in HEK293 cells after expressed AURKA wild type and D274A mutant. (G) PANC1 cells were treated with CCT137690 (10 μM) or TNF (50 ng/ml) in the absence or presence of anti-TNFR1 antibody (1 mg/ml) for 24 hours and cell death was analyzed (n=3, *p < 0.05).
To define the mechanisms of action of AURKA in the necrosome, we next evaluated whether AURKA binds to RIPK1, RIPK3, or MLKL in PANC1 and PANC2.03 cells. We found that AURKA directly formed a complex with RIPK1 or RIPK3 (but not MLKL) under normal conditions (Fig. 4E). CCT137690 blocked the formation of AURKA-RIPK1 and AURKA-RIPK3 complexes in human PDAC cells during necroptosis (Fig. 4E). These findings indicate that AURKA may be a previously unidentified component of the RIPK1-RIPK3 necrosome that acts as a local inhibitor.
The catalytic loop of AURKA spanning residues 274 to 299 is required for AURKA kinase activity 27. We hypothesized that the anti-necroptosis activity of AURKA is related to its kinase activity; therefore, we sought to determine whether depletion of kinase activity would facilitate necrosome formation. Towards this goal, HEK293 cells were transfected with WT AURKA or a kinase dead AURKA mutant (D274A) bearing FLAGs. While WT AURKA formed complexes with RIPK1 and RIPK3, D274 AURKA failed to do so (Fig. 4F). We conclude that the kinase activity of AURKA is required for its binding to the RIPK1/RIPK3 necrosome in necroptosis.
TNF receptor 1 (TNFR1), a death receptor, is involved in the regulation of necroptosis. To determine whether TNFR1 is required for AURKA inhibition-induced necroptosis, we treated PANC1 cells with anti-TNFR1 antibody. Anti-TNFR1 antibody inhibited TNF- (but not CCT137690-) induced death in PANC1 cells (Fig. 4G), indicating that TNFR1 is not required for necrosome formation under AURKA inhibition conditions.
GSK3ß contributes to AURKA-mediated necroptosis inhibition
Glycogen synthase kinase 3β (GSK3β) participates in multiple cellular processes and is a proposed target of AURKA in gastric cancer cells 28. Genetic inhibition (with shRNAs) (Fig. 5A) or pharmacological inhibition (with CCT137690 or MLN8237) (Fig. 5B and S2A) of AURKA significantly inhibited the phosphorylation levels of GSK3β at Ser 9 (p-GSK3β) in PANC1 and PANC2.03 cells, suggesting that GSK3β might indeed act as a downstream target of AURKA in necroptosis.
Figure 5. GSK3β contributes to AURKA-mediated necroptosis inhibition.
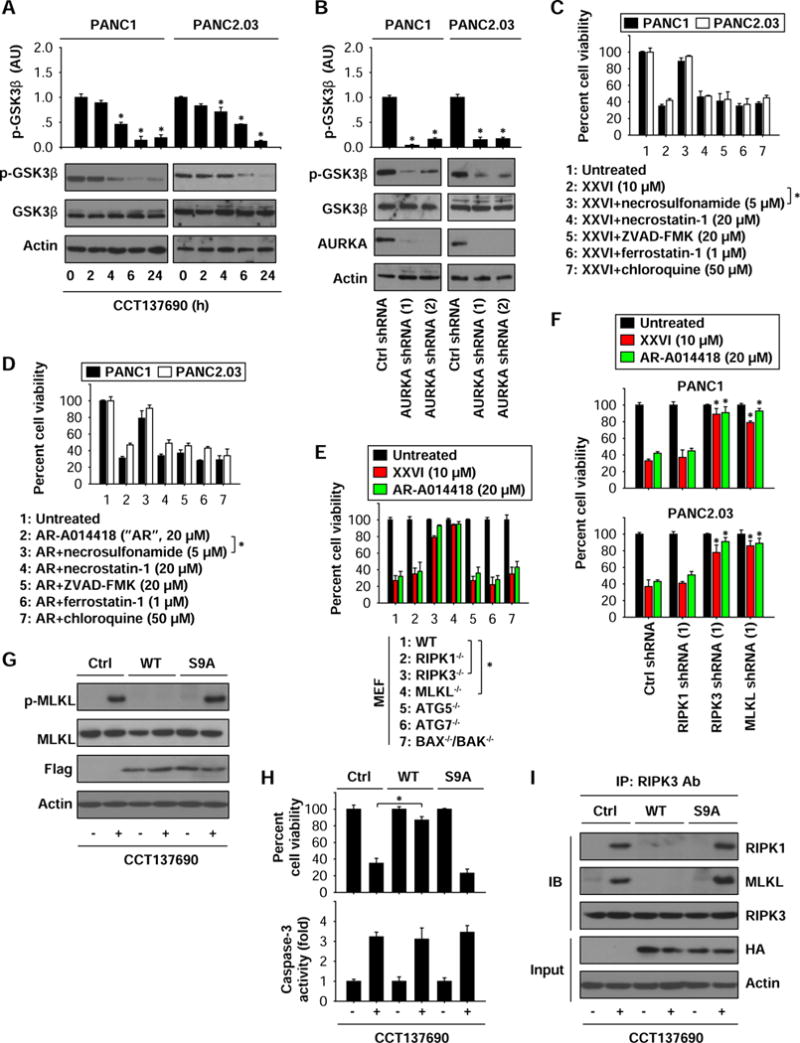
(A) Western blot analysis of indicated proteins in PDAC cells following treatment with CCT137690 (10 μM) for three to 24 hours (n=3, *p < 0.05 versus untreated group). (B) Western blot analysis of indicated proteins in control and stable AURKA-knockdown PANC1 and PANC2.03 cells (n=3, *p < 0.05 versus control shRNA group). (C, D) PANC1 and PANC2.03 cells were treated with XXVI (C) or AR-A014418 (D) in the absence or presence of indicated cell death inhibitors for 24 hours. Cell viability was assayed (n=3, *p < 0.05). (E) Indicated MEFs were treated with XXVI or AR-A014418 for 24 hours, and then cell viability was assayed (n=3, *p < 0.05). (F) Knockdown of RIPK3 and MLKL, but not RIPK1, inhibited XXVI- or AR-A014418-induced cell death (n=3, *p < 0.05 versus control shRNA group). (G–I) HEK293 cells were expressed with GSK3β WT and S9A mutant and then treated with CCT137690 (10 μM) for 24 hours. Protein level (G), cell viability (H), caspase-3 activity (H), and complex formation (I) were assayed.
Next, we tested whether inhibition of GSK3β causes necroptosis in PDAC cells, although impaired GSK3β activity reportedly triggers apoptosis 29. Treatment with GSK3β inhibitors (XXVI or AR-A014418) resulted in the death of PANC1 and PANC2.03 cells (Fig. 5C and 5D). This process was prevented by the MLKL inhibitor necrosulfonamide. However, a RIPK1 inhibitor (necrostatin-1) and other cell death inhibitors (Z-VAD-FMK, chloroquine, and ferrostatin-1) failed to block XXVI- or AR-A014418-induced cell death (Fig. 5C and 5D), suggesting that GSK3β acts downstream of RIPK1 in the induction of necroptosis (but not apoptosis, autophagy, and ferroptosis). This notion was further confirmed using genetic approaches. Knockout or knockdown of RIPK3 and MLKL (but not RIPK1) blocked XXVI- or AR-A014418-induced cell death in MEFs (Fig. 5E), PANC1 (Fig. 5F), and PANC2.03 cells (Fig. 5F). In contrast, ablation of genes involved in autophagy (ATG5−/− or ATG7−/−) or apoptosis (BAX−/− and BAK−/−) failed to interfere with the cytotoxic activity of GSK3β inhibitors (Fig. 5E).
We next investigated whether phosphorylation of GSK3β at Ser9 is required for AURKA-mediated necroptosis inhibition. Overexpression of unmutated WT GSK3β, but not that of the S9A mutant, prevented CCT137690-induced MLKL phosphorylation (Fig. 5G) and cell growth inhibition (Fig. 5H) in HEK293 cells. CCT137690-induced caspase-3 activity was not affected by WT and S9A mutant (Fig. 5H), indicating that GSK3β is not involved in the regulation of CCT137690-induced apoptosis. Furthermore, overexpression of WT GSK3β, but not S9A mutant, inhibited CCT137690-induced formation of the RIPK3-MLKL complex (Fig. 5I). In contrast, the RIPK1-RIPK3 complex was not affected by the overexpression of GSK3β WT or S9A mutant (Fig. 5I). These findings indicate that phosphorylation of GSK3β at Ser9 suppresses necroptosis through interfering with formation of the RIPK3-MLKL complex.
Anticancer activity of CCT137690 in vivo
To determine whether induction of necroptosis by CCT137690 suppresses tumor growth in vivo, human PANC1 cells were implanted into the subcutaneous space of the right flank of immunodeficient nu/nu mice. Beginning on day seven post-tumor implantation, mice were administered CCT137690 orally at 80 mg/kg. Compared to the vehicle control group, administration of CCT137690 effectively reduced tumor growth (Fig. 6A and 6B) as it increased serum HMGB1 levels (Fig. 6C) and stimulated MLKL phosphorylation in the tumor (Fig. 6D). In contrast, CCT137690 reduced the local phosphorylation of AURKA and GSK3β (Fig. 6D). CCT137690 failed to induce signs of toxicity such as weight loss (Fig. 6E) and increased serum levels of tissue-dysfunction enzymes (ALT and BUN) (Fig. 6F and 6G). Oral administration of CCT137690 also inhibited tumor growth in immunocompetent B6 mice after subcutaneous implantation of mouse PANC02 (Fig. 6H) or KPC cells (Fig. 6I). In addition to the increased serum levels of HMGB1 (Fig. 6J) and ATP (Fig. 6K), the frequency of IFN-γ-producing tumor-infiltrating CD8+ T cells (Fig. 6L) was increased following CCT137690 treatment in B6 mice after subcutaneous implantation of mouse PANC02 or KPC cells, suggesting that CCT137690 induces so-called ICD in vivo. Furthermore, synergistic effects with combinations of gemcitabine (20 mg/kg) and CCT137690 (40 mg/kg) were observed in established PANC1 subcutaneous tumor models in vivo (Fig. 6M).
Figure 6. Anticancer activity of CCT137690 in subcutaneous tumor models.
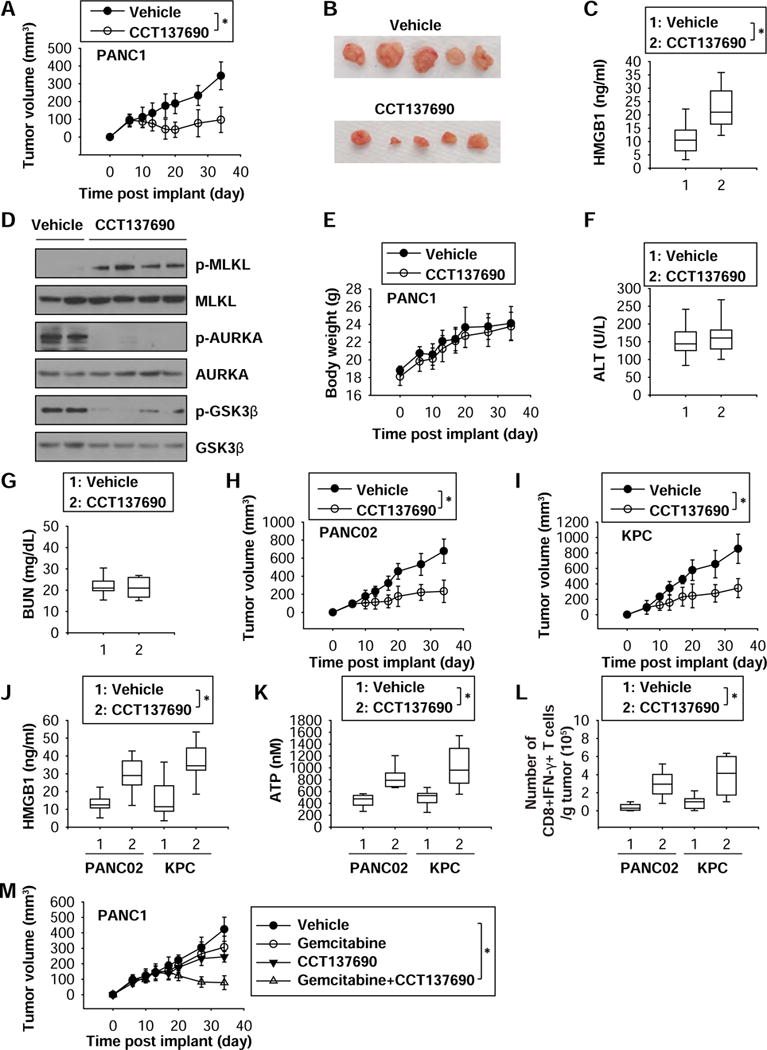
(A) Athymic nude mice were injected subcutaneously with PANC1 cells (5 × 106 cells/mouse) and treated with CCT137690 (80 mg/kg/p.o., twice every other day) at day seven for two weeks. Tumor volume was calculated weekly (n=10 mice/group, * p < 0.05). (B) Representative photographs of isolated tumors at day 34 after treatment. (C) ELISA analysis of serum HMGB1 levels in mice at day 34 after treatment (n=10 mice/group, * p < 0.05). (D) Western blot analysis of indicated protein expressions in isolated tumor at day 34 after treatment. (E–G) Average body weight (E) and serum ALT (F) and BUN (G) levels in mice at day 34 after treatment (n=10 mice/group, * p < 0.05). (H–L) B6 mice were injected subcutaneously with PANC02 cells (1 × 106 cells/mouse) or KPC cells (1 × 106 cells/mouse) and treated with CCT137690 (80 mg/kg/p.o., twice every other day) at day seven for two weeks. Tumor volume was calculated weekly (H, I). The serum HMGB1 (J) and ATP (K) levels and frequency of IFN-γ-producing tumor-infiltrating CD8+ T cells (L) were assayed in isolated tumor at day 34 after treatment (n=10 mice/group, * p < 0.05). (M) Athymic nude mice were injected subcutaneously with PANC1 cells (5 × 106 cells/mouse) and treated with CCT137690 (40 mg/kg/p.o., twice every other day) and/or gemcitabine (20 mg/kg/i.p., once every other day) at day 14 for two weeks. Tumor volume was calculated weekly (n=8 mice/group, * p < 0.05).
Orthotopic tumor and genetically engineered mouse models are considered more clinically relevant PDAC models. Oral administration of CCT137690 prolonged the survival of mice bearing orthotopic KPC PDACs (Fig. 7A), as it reduced tumor growth (Fig. 7B and 7C). This effect was associated with decreased pancreatic ductal/stromal response (Fig. 7D and 7E) and reduced liver invasion (Fig. 7F and 7G). Compared to treatment with vehicle only, CCT137690 administration increased serum HMGB1 (Fig. 7H) and ATP (Fig. 7I) levels, the frequency of IFN-γ-producing tumor-infiltrating CD8+ T cells (Fig. 7J), as well as intratumoral phosphorylation of MLKL, and dephosphorylaton of AURKA or GSK3β (Fig. 7K). Mutant K-Ras drives PDAC in mice with a Cre-inducible conditional allele (Pdx1-Cre;K-RasG12D/+, termed KC mice) targeting the endogenous K-Ras locus 30. Furthermore, oral administration of CCT137690 reduced pancreatic weight (Fig. 7L) and the ductal/stromal response (Fig. 7M and 7N) with increased serum HMGB1 (Fig. 7O), serum ATP (Fig. 7P), IFN-γ-producing tumor-infiltrating CD8+ T cells (Fig. 7Q), and intratumoral phosphorylation of MLKL (Fig. 7R) in old KC mice at the age of 10 months. These results further support the idea that AURKA inhibition by CCT137690 effectively reduces PDAC growth by induction of necroptosis and ICD.
Figure 7. Anticancer activity of CCT137690 in orthotopic tumor and genetically engineered mouse models.
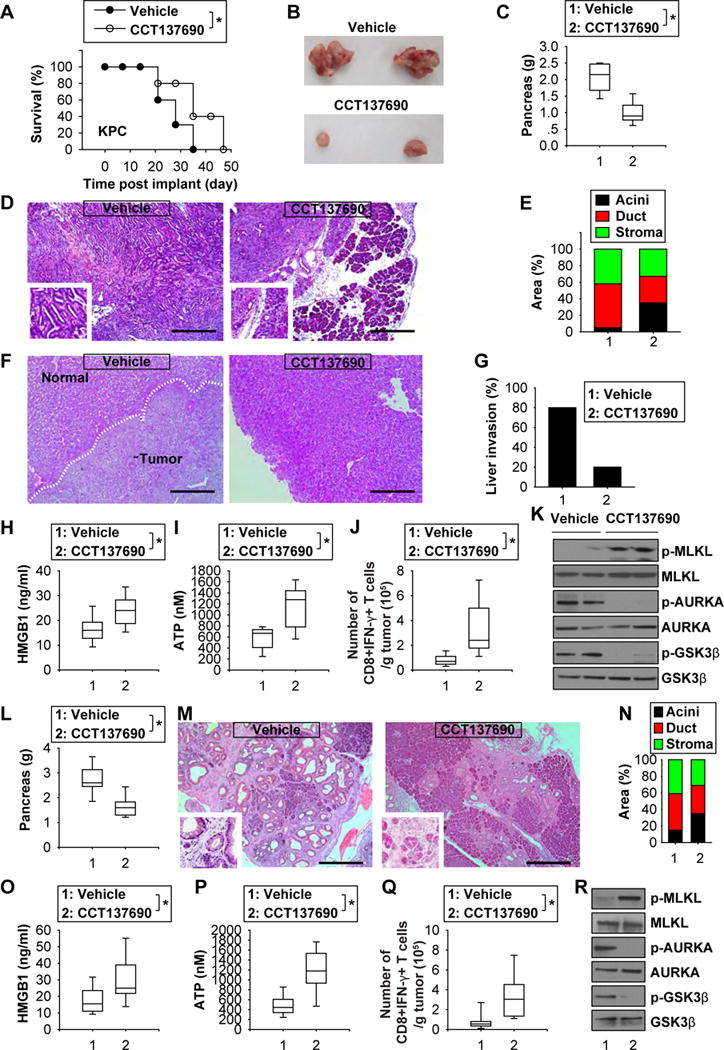
(A) Injection of CCT137690 (80 mg/kg/p.o., twice every other day, at day seven for two weeks) prolonged animal survival of orthotopic KPC (1 × 106 cells/mouse) tumor models (n=10 mice/group, * p < 0.05). (B) Representative photomicrographs of isolated tumors at day 21 after treatment. (C) Average pancreas weight at day 21 following treatment (n=10 mice/group, * p < 0.05). (D, E) Histological analysis of pancreas at day 21 after treatment (bar=400 03BCm, n=5 mice/group). (F, G) Histological analysis of liver at day 21 after treatment (bar=400 03BCm, n=5 mice/group). (H–J) Analysis of serum HMGB1 (H), serum ATP (I), and frequency of IFN-γ-producing tumor-infiltrating CD8+ T cells (H) in mice at day 21 after treatment (n=10 mice/group, * p < 0.05). (K) Western blot analysis of indicated protein expression in isolated tumor at day 21. (L–N) Injection of CCT137690 (80 mg/kg/p.o., twice every other day, for one month) reduced pancreatic weight and ductal/stromal response in KC mice at the age of 10 months (bar=400 03BCm). (O–Q) Analysis of serum HMGB1 (O), serum ATP (P), and frequency of IFN-γ-producing tumor-infiltrating CD8+ T cells (Q) in mice at day 30 after treatment (n=10 mice/group, * p < 0.05). (R) Western blot analysis of indicated protein expression in isolated tumor at day 30 after treatment.
AURKA and GSK3β are associated with poor prognosis in human pancreatic cancer
We next investigated the association between necroptosis-associated gene expression and survival in human pancreatic cancer (n=130) cohorts from the online R2 genomics analysis and visualization platform. High expression levels of the mRNA of AURKA (Fig. S3A) and GSK3β (Fig. S3B) were associated with decreased survival in pancreatic cancer patients. In contrast, there was no significant relationship between survival and the expression of mRNAs of RIPK1 (Fig. S3C), RIPK3 (Fig. S3D), and MLKL (Fig. S3E) in pancreatic cancer patients. These results suggest that negative regulators (e.g., AURKA and GSK3β) of necroptosis, but not positive regulators (e.g., RIPK1, RIPK3, and MLKL) of necroptosis may indeed be prognostic markers for patients with pancreatic cancer.
Discussion
In this study, we report that the AURKA kinase inhibitor CCT137690 significantly suppressed PDAC growth in vitro and in vivo through induction of necroptosis, a form of regulated necrosis. Necroptosis signaling requires the RIPK3-catalyzed phosphorylation of the executor protein MLKL, although the detailed regulatory mechanism remains to be fully defined. As shown here, AURKA acts as an endogenous repressor of necroptosis by inhibiting MLKL phosphorylation, presumably by impeding necrosome formation and GSK3β phosphorylation (Fig. S3F). These findings not only provide new insights into the molecular function of AURKA in necroptosis 27, but also reinforce the notion that induction of regulated non-apoptotic forms of cell death may be critical for anti-cancer therapies 31.
Aurora kinases contribute tumorigenesis because of their profound effects on mitosis and multiple signaling pathways 32–34. Although there are more than 30 aurora kinase inhibitors in different stages of pre-clinical and clinical development, the mechanism responsible for the anticancer activity of these inhibitors might differ with respect of the modality of cell death they trigger 7. Thus, aurora kinase inhibitors have been reported to trigger mitotic catastrophe 22, apoptosis 35, and autophagy 36. Here, we demonstrate that CCT137690 is capable of inducing PDAC cell death with phenotypic features of apoptosis, autophagy, necroptosis, mitotic catastrophe, and ferroptosis. However, only necroptotic signaling played a major role in mediating the anticancer activity of CCT137690 in PDAC cells. Notably, pharmacologic or genetic inhibition of necroptosis (but not that of other types of regulated cell death) significantly blocked CCT137690-induced PDAC cell death.
Necroptosis depends on activation of the necrosome, which is a protein complex including three core components: RIPK1, RIPK3, and MLKL 3, 4 Our results indicate that AURKA is a component and regulator of necrosome activation. Indeed, it appears that a full-length AURKA protein containing the catalytic domain limits necrosome activation through binding to RIPK1 and RIPK3. In contrast, expression of an AURKA kinase dead mutant D274A lacks the ability to inhibit necroptosis by binding to RIPK1 and RIPK3. Pharmacological or genetic inhibition of AURKA increased the interaction between RIPK1 and RIPK3, correlating with enhanced RIPK3-mediated phosphorylation of MLKL. Phosphorylated MLKL forms pores in the plasma membrane to precipitate the final, lethal step of necroptosis 37, 38. Beyond direct inhibition of RIPK1-RIPK3 complex activation by AURKA, it appears that AURKA-mediated phosphorylation of GSK3β at Ser9 prevented RIPK3-MLKL complex activation in necroptosis. Previous studies indicate that phosphorylation of GSK3β at Ser9 limits GSK3β activity, which in turn promotes Wnt/β-catenin signaling activation 39. Interestingly, lymphoid enhancer-binding factor 1 (LEF1), a downstream effector of the Wnt/β-catenin pathway, is a negative regulator of necroptosis in chronic lymphocytic leukemia (CLL) 40. Suppression of LEF1 expression increased necroptosis in CLL cells following TNFα/ZVAD-FMK treatment40. Unlike most kinases, GSK3β is constitutively active by dephosphorylation and high levels of GSK3β activity promote cell death 39. Our findings indicate a potential link between AURKA, GSK3β, and Wnt/β-catenin pathways in the regulation of necroptosis 28, 41, 42.
Preclinical experiments in rodent models of PDAC revealed that the induction of necroptosis by CCT137690 warrants their further clinical exploration as a therapeutic opportunity. Of note, cell death may play a different role in tumorigenesis and tumor therapy. Induction of cell death by chemotherapy, radiation, and immunotherapy is a major treatment for established cancer, whereas excess death may promote the release of DAMPs, which contribute to protumerigenic inflammation and immunosuppression. A recent preclinical study revealed that pancreatic knockout of RIPK1 and RIPK3 limited K-Ras-driven spontaneous PDAC formation due to activation of anti-tumor immunity by suppression of DAMP (e.g., CXCL1) release 43. Our current findings demonstrate that induction of necroptosis by CCT137690 promotes the release of ICD mediators (e.g., HMGB1 and ATP), which increases antitumor T cell immunity in established K-Ras-driven PDAC. We also found that high expression of AURKA and GSK3β (but not RIPK1, RIPK3, or MLKL) are biomarkers for poor prognosis in human pancreatic cancer. These findings suggest that necroptosis-relevant genes and proteins play rather distinct roles in the initial stages of oncogenesis (which are targeted for cancer prevention) and later tumor progression (which are targeted for treatments).
Supplementary Material
Acknowledgments
We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript.
Grant Support: This work was supported by grants from the US National Institutes of Health (R01GM115366 and R01CA160417), the Natural Science Foundation of Guangdong Province (2016A030308011), the American Cancer Society (Research Scholar Grant RSG-16-014-01-CDD), and the National Natural Science Foundation of China (31671435 and 81570154). This project partly utilized University of Pittsburgh Cancer Institute shared resources supported by award P30CA047904. G.K. is supported by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR)-Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); 1 Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Institut Universitaire de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière, the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Author Contributions: D.T. conceived and designed the experiments. Y.X., S.Z., M.Z., Y.Y., X.S., J.L., R.K., and D.T conducted the experiments. Y.X., S.Z., M.Z., G.K., M.T.L, H.J.Z, R.K., and D.T analyzed the data. Y.X. and D.T. wrote the paper. G.K., M.T.L., and H.J.Z. edited the manuscript.
Author names in bold designate shared co-first authorship.
References
- 1.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 3.Weinlich R, Oberst A, Beere HM, et al. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2016 doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Kepp O, Chan FK, et al. Necroptosis: Mechanisms and Relevance to Disease. Annu Rev Pathol. 2016 doi: 10.1146/annurev-pathol-052016-100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 6.Seifert L, Miller G. Molecular Pathways: The Necrosome – A Target for Cancer Therapy. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bavetsias V, Linardopoulos S. Aurora Kinase Inhibitors: Current Status and Outlook. Front Oncol. 2015;5:278. doi: 10.3389/fonc.2015.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torres MP, Rachagani S, Souchek JJ, et al. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: applications in diagnosis and therapy. PLoS One. 2013;8:e80580. doi: 10.1371/journal.pone.0080580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang R, Xie Y, Zhang Q, et al. Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer. Cell Res. 2017 doi: 10.1038/cr.2017.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glienke W, Maute L, Wicht J, et al. The dual PI3K/mTOR inhibitor NVP-BGT226 induces cell cycle arrest and regulates Survivin gene expression in human pancreatic cancer cell lines. Tumour Biol. 2012;33:757–65. doi: 10.1007/s13277-011-0290-2. [DOI] [PubMed] [Google Scholar]
- 11.Ochiai T, Saito Y, Saitoh T, et al. Inhibition of IkappaB kinase beta restrains oncogenic proliferation of pancreatic cancer cells. J Med Dent Sci. 2008;55:49–59. [PubMed] [Google Scholar]
- 12.Jani JP, Arcari J, Bernardo V, et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol Cancer Ther. 2010;9:883–94. doi: 10.1158/1535-7163.MCT-09-0915. [DOI] [PubMed] [Google Scholar]
- 13.Arbitrario JP, Belmont BJ, Evanchik MJ, et al. SNS-314, a pan-Aurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 2010;65:707–17. doi: 10.1007/s00280-009-1076-8. [DOI] [PubMed] [Google Scholar]
- 14.Galluzzi L, Buque A, Kepp O, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 15.Tang D, Kang R, Coyne CB, et al. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H, Ma Y, Chen G, et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology. 2016;5:e1149673. doi: 10.1080/2162402X.2016.1149673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu J, Bian M, Jiang Q, et al. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- 18.Faisal A, Vaughan L, Bavetsias V, et al. The aurora kinase inhibitor CCT137690 downregulates MYCN and sensitizes MYCN-amplified neuroblastoma in vivo. Mol Cancer Ther. 2011;10:2115–23. doi: 10.1158/1535-7163.MCT-11-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bavetsias V, Large JM, Sun C, et al. Imidazo[4,5-b]pyridine derivatives as inhibitors of Aurora kinases: lead optimization studies toward the identification of an orally bioavailable preclinical development candidate. J Med Chem. 2010;53:5213–28. doi: 10.1021/jm100262j. [DOI] [PubMed] [Google Scholar]
- 20.Wu X, Liu W, Cao Q, et al. Inhibition of Aurora B by CCT137690 sensitizes colorectal cells to radiotherapy. J Exp Clin Cancer Res. 2014;33:13. doi: 10.1186/1756-9966-33-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crosio C, Fimia GM, Loury R, et al. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–85. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheong JW, Jung HI, Eom JI, et al. Aurora-A kinase inhibition enhances the cytosine arabinoside-induced cell death in leukemia cells through apoptosis and mitotic catastrophe. Cancer Lett. 2010;297:171–81. doi: 10.1016/j.canlet.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Lu Y, Liu Y, Jiang J, et al. Knocking down the expression of Aurora-A gene inhibits cell proliferation and induces G2/M phase arrest in human small cell lung cancer cells. Oncol Rep. 2014;32:243–9. doi: 10.3892/or.2014.3194. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Chen X, Liu B, et al. Effects of stable knockdown of Aurora kinase A on proliferation, migration, chromosomal instability, and expression of focal adhesion kinase and matrix metalloproteinase-2 in HEp-2 cells. Mol Cell Biochem. 2011;357:95–106. doi: 10.1007/s11010-011-0879-1. [DOI] [PubMed] [Google Scholar]
- 25.Ling YH, el-Naggar AK, Priebe W, et al. Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34cdc2/cyclin B1 activity induced by doxorubicin in synchronized P388 cells. Mol Pharmacol. 1996;49:832–41. [PubMed] [Google Scholar]
- 26.Li Z, Liu W, Mo B, et al. Caffeine overcomes genistein-induced G2/M cell cycle arrest in breast cancer cells. Nutr Cancer. 2008;60:382–8. doi: 10.1080/01635580701861785. [DOI] [PubMed] [Google Scholar]
- 27.Nikonova AS, Astsaturov I, Serebriiskii IG, et al. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70:661–87. doi: 10.1007/s00018-012-1073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dar AA, Belkhiri A, E1-Rifai W. The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene. 2009;28:866–75. doi: 10.1038/onc.2008.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshino Y, Ishioka C. Inhibition of glycogen synthase kinase-3 beta induces apoptosis and mitotic catastrophe by disrupting centrosome regulation in cancer cells. Sci Rep. 2015;5:13249. doi: 10.1038/srep13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 31.Su Z, Yang Z, Xie L, et al. Cancer therapy in the necroptosis era. Cell Death Differ. 2016;23:748–56. doi: 10.1038/cdd.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly KR, Ecsedy J, Mahalingam D, et al. Targeting aurora kinases in cancer treatment. Curr Drug Targets. 2011;12:2067–78. doi: 10.2174/138945011798829410. [DOI] [PubMed] [Google Scholar]
- 33.Carvajal RD, Tse A, Schwartz GK. Aurora kinases: new targets for cancer therapy. Clin Cancer Res. 2006;12:6869–75. doi: 10.1158/1078-0432.CCR-06-1405. [DOI] [PubMed] [Google Scholar]
- 34.Li D, Zhu J, Firozi PF, et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res. 2003;9:991–7. [PubMed] [Google Scholar]
- 35.Katayama H, Wang J, Treekitkarnmongkol W, et al. Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73. Cancer Cell. 2012;21:196–211. doi: 10.1016/j.ccr.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zou Z, Yuan Z, Zhang Q, et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy. 2012;8:1798–810. doi: 10.4161/auto.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–46. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 38.Dondelinger Y, Declercq W, Montessuit S, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–81. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 39.Jacobs KM, Bhave SR, Ferraro DJ, et al. GSK-3beta: A Bifunctional Role in Cell Death Pathways. Int J Cell Biol. 2012;2012:930710. doi: 10.1155/2012/930710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu P, Xu B, Shen W, et al. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2012;26:1293–300. doi: 10.1038/leu.2011.357. [DOI] [PubMed] [Google Scholar]
- 41.Xia Z, Wei P, Zhang H, et al. AURKA governs self-renewal capacity in glioma-initiating cells via stabilization/activation of beta-catenin/Wnt signaling. Mol Cancer Res. 2013;11:1101–11. doi: 10.1158/1541-7786.MCR-13-0044. [DOI] [PubMed] [Google Scholar]
- 42.Grassilli E, Narloch R, Federzoni E, et al. Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin Cancer Res. 2013;19:3820–31. doi: 10.1158/1078-0432.CCR-12-3289. [DOI] [PubMed] [Google Scholar]
- 43.Seifert L, Werba G, Tiwari S, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532:245–9. doi: 10.1038/nature17403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.