

 This work is licensed under a
This work is licensed under a Abstract
Parathyroid carcinoma is an extremely rare endocrine malignancy that accounts for less than 1% of cases of primary hyperparathyroidism. We report a 44-year-old woman who presented with fatigue and diffuse bone pain. Laboratory findings revealed highly elevated serum calcium and parathyroid hormone (PTH) levels and a 4.5 × 3 × 2.5 cm cystic lesion in the lower pole of the right thyroid lobe that was shown histologically to be a parathyroid carcinoma. Ten years later, the patient developed brain and pulmonary metastases and recurrence of PTH-related hypercalcemia. Treatment of hypercalcemia along with localized radiotherapy and various chemotherapy regimens failed to induce a biochemical or radiological response. In conclusion, parathyroid carcinoma is a rare neoplasia that may develop metastases even after prolonged follow-up, for which there is no evidence-based treatment besides surgery. Different chemotherapeutic schemes did not prove to be of any benefit in our case highlighting the need for registering such patients to better understand tumor biology and develop specific treatment.
Learning points:
Metastases can develop many years after parathyroid cancer diagnosis.
Surgery is the only curative treatment for parathyroid carcinoma.
Chemotherapy and radiotherapy prove to be ineffective in parathyroid cancer treatment.
Patient registering is required in order to delineate underlining pathology and offer specific treatment.
Patient demographics: Adult, Female, White, Greece
Clinical overview: Parathyroid, Endocrine-related cancer, Parathyroid hormone, Parathyroid carcinoma, Metastatic carcinoma
Diagnosis & treatment: Bone pain, Fatigue, Polydipsia, Polyuria, Myasthaenia, Hypercalcaemia, Hypophosphataemia, MRI, PTH, Histopathology, CT scan, Sestamibi scan, Radionuclide imaging, Ultrasound scan, Calcium (serum), Phosphate (serum), Alkaline phosphatase, Fluid repletion, Parathyroidectomy, Thyroidectomy, Lymphadenectomy, Radiotherapy, Temozolomide, Furosemide, Pamidronate, Bisphosphonates, Calcium carbonate, Zoledronic acid, Cinacalcet, Fluorouracil, Capecitabine
Related disciplines: Oncology
Publication details: Unique/unexpected symptoms or presentations of a disease, October, 2017
Background
Parathyroid cancer is a rare endocrine disorder constituting less than 1% of parathyroid neoplasms. It is mainly a sporadic disease with a peak incidence in the fourth or fifth decade of life. Parathyroid carcinoma commonly presents with significantly elevated calcium levels due to high parathyroid hormone (PTH) levels and symptoms are related to severity of hypercalcemia. Distant metastases are developed in about 25% of patients, most commonly to lymph nodes, bone, lung and liver. It is usually difficult to diagnose malignant tumors pre-operatively or on histological examination and definitive diagnosis is made upon local recurrence or metastases development (1, 2). We report a female patient with parathyroid carcinoma who developed brain and pulmonary metastases ten years after the initial diagnosis. Different chemotherapeutic regimens and radiotherapy did not prove to be of any benefit.
Case presentation
A 44-year-old woman presented with one-month history of weakness, polydipsia, polyuria and diffuse bone pains. She had a past history of ureteral colic but no relevant familial history.
Initial laboratory investigations revealed hypercalcemia (19.3 mg/dL; normal range 8.4–10.1 mg/dL), hypophosphatemia (2 mg/dL; 2.7–4.5 mg/dL) and significantly increased PTH (1084 pg/mL; 15–65 pg/mL) and alkaline phosphatase (ALP) (308 U/L; 48–141) levels. Creatinine, BUN and thyroid function were normal. The patient was treated with intravenous fluids, furosemide and pamidronate that led to substantial reduction of calcium levels and resolution of her symptoms. A neck ultrasound and computerized tomography (CT) scanning revealed a 4.5 × 3 × 2.5 cm cystic lesion adjacent to the lower pole of the right thyroid lobe that was shown to represent a single hyperfunctioning parathyroid lesion on sestamibi (MIBI) parathyroid scintigraphy. No enlarged or suspicious lymph nodes were noted. In addition, chest, abdominal and brain CT revealed no pathological findings.
The patient underwent a parathyroidectomy along with en bloc right thyroid lobectomy. Histological examination revealed anaplastic regions and giant cells with abundant cytoplasm and polymorphous nuclei consistent with parathyroid carcinoma. In addition, capsular and vascular invasion were observed, whereas the Ki-67 labeling index (LI) was approximately 10%. No invasion in the thyroid has been observed while resection margins were free. Postoperatively, calcium and PTH levels were substantially reduced, and replacement therapy with calcidiol and calcium carbonate was initiated (Fig. 1). Post-surgery ultrasound, magnetic resonance imaging (MRI) of the neck, chest and abdominal CT and whole-body MIBI scintigraphy did not reveal any evidence of residual or metastatic disease.
Figure 1.
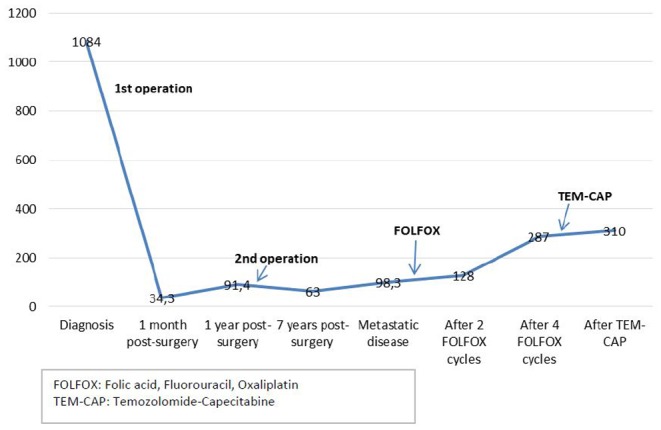
PTH levels (pg/mL) at initial diagnosis and during different treatments.
One year later, laboratory investigations showed mildly raised PTH levels (91.4 pg/mL). Mediastinal MRI, abdominal CT and whole-body MIBI scintigraphy showed no evidence of distant metastatic disease, whereas a neck MRI revealed a 7–8 mm lesion behind the left thyroid lobe that was excised with concomitant right neck lymph node dissection. Histology revealed chief cell hyperplasia in the resected lesion, whereas all 15 lymph nodes excised were free of neoplastic disease. Postoperative calcium and PTH levels were normal. As the patient had no relevant family history neither fulfilled any criteria of a familial syndrome, she only underwent genetic analysis for CDC73 (cell division cycle 73) gene somatic and germline mutations that was negative.
The patient remained normocalcemic with normal neck ultrasonography and MRI during the following six years. Subsequently, a progressive increase of calcium levels was observed and a neck MRI revealed three 4–5 mm structures, one on the right and two on the left, in the anatomic region of parathyroid glands. During the following four years, serum calcium and PTH levels remained marginally raised while the abovementioned neck structures displayed no significant change (Fig. 1).
Ten years after initial diagnosis, the patient developed numbness and right upper limb weakness. Physical examination revealed diminished right upper limb muscle strength and increased deep tendon reflex.
Investigation
A brain MRI showed a 37 mm left parietal lobe lesion that was entirely removed followed by radiotherapy (Fig. 2). Histopathology demonstrated positive immunostaining for PTH while the Ki-67 LI was 15%, confirming a parathyroid carcinoma metastasis (Fig. 3). A subsequent chest CT revealed a 7.5 cm solid lesion in the lower right lung lobe with satellite lesions and a further 8 mm lesion in the upper left lobe that all exhibited increased uptake to whole-body MIBI scintigraphy. There were no other MIBI avid lesions or any uptake on tectrotyd somatostatin receptor and bone scintigraphy. Biopsy of the lung lesion was suggestive of metastatic parathyroid carcinoma with a Ki-67 LI of 40% (Fig. 3).
Figure 2.
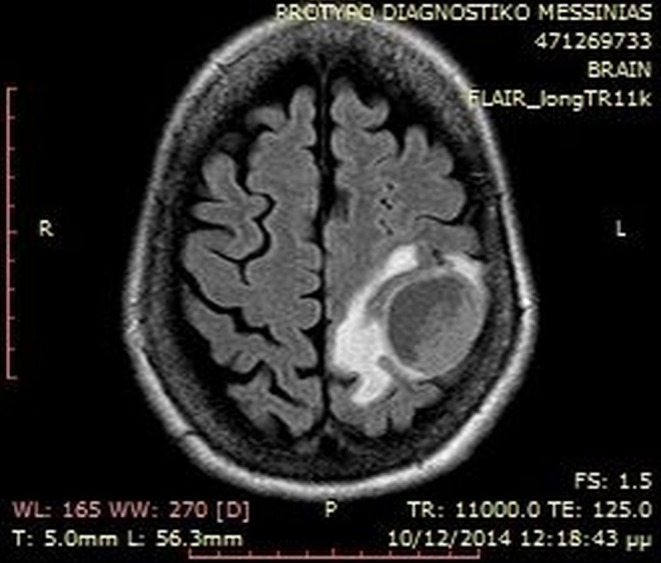
Brain MRI showing left parietal lobe metastatic lesion.
Figure 3.
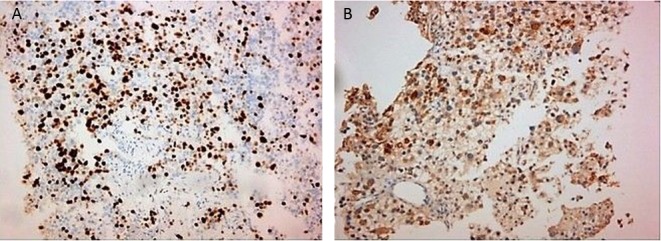
(A) Ki-67 labeling index of lung metastatic lesion. (B) PTH immunostaining of brain lesion.
Treatment
As the lung lesions were inoperable, and there is no specific therapeutic regimen for parathyroid carcinomas FOLFOX (folic acid, Fluorouracil, Oxaliplatin) chemotherapy was initiated. After three cycles, the patient was found to have stable disease that was regarded as a response, and further two FOLFOX cycles were administered. However, a subsequent chest CT revealed further increase of the right lower lung lesion that was extending into the right pulmonary artery also infiltrating the right lower pulmonary vein and left atrium. A heart MRI confirmed these findings.
Although radiation treatment to pulmonary metastases followed by two cycles of chemotherapy with temozolomide and capecitabine (capecitabine: 1500 mg/m2: d1–d14, temozolomide: 200 mg/m2: d10–d14 every 28 days) was administered, there was further disease progression.
Outcome and follow-up
Despite treatment with different chemotherapeutic regimens and radiotherapy, the disease displayed continuous progression. Persistent hypercalcemia was treated with intravenous fluids, furosemide, zoledronic acid and cinacalcet, but the patient died twelve years after initial diagnosis.
Discussion
Parathyroid carcinoma is an extremely rare, slowly evolving malignancy that accounts for less of 1% of cases of primary hyperparathyroidism. It occurs equally in males and females with a peak incidence in the fourth or fifth decade of life. Most cases are sporadic but may also occur as part of hyperparathyroidism-jaw tumor syndrome (HJTS) or rarely multiple endocrine neoplasia (MEN) type 1 or 2A syndromes (1). Our case demonstrates the slow evolution and increasing malignant potential of an initially localized parathyroid carcinoma that developed brain and lung metastases ten years after initial diagnosis and proved to be resistant to any conventional therapy.
More than 90% of parathyroid carcinomas are functional and symptoms are related to the severity of hypercalcemia similar to our case. Physical examination commonly reveals a palpable neck mass in 40–70% of patients (1, 2). It is often difficult to distinguish between benign and malignant tumors pre-operatively or at histological examination, and definite diagnosis is made in the presence of local recurrences or development of metastases. However, the presence of some predictive markers could raise the suspicion of malignancy pre-operatively.
Biochemically, parathyroid carcinoma presents with more severe hypercalcemia compared to benign disease, due to higher PTH levels. Alkaline phosphatase and α- and β-subunits of human chorionic gonadotropin (hCG) are particularly elevated in carcinomas (1, 2). Our patient was found to have significantly elevated serum alkaline phosphatase levels.
Histological criteria still remain the most established predictive method of malignancy irrespective of whether old or revised criteria are used (3). There are two prognostic classification systems derived from a retrospective literature review of 330 patients (4). Stage III disease in Schulte’s staging system (lymph node metastases) and a high-risk score in the Schulte Risk Classification (vascular and lymph node invasion, vital organ or distant metastases) are associated with parathyroid carcinoma recurrence (5).
Inactivating mutations of the CDC73 gene have been identified in up to 70% of apparently sporadic parathyroid carcinomas and approximately one-third of which are germline mutations. Loss of parafibromin protein immunostaining has also been found in most parathyroid carcinomas. The combination of CDC73mutations and loss of parafibromin protein expression predicts a worse clinical outcome and a lower overall 5- and 10-year survival (3). In contrast, the increased expression of the proliferative marker Ki-67 has produced contradictory results (6).
Genetic analyses should always be considered in parathyroid carcinomas. Except for CDC73gene, various genes have been found to be linked to parathyroid carcinomas including BRCA, p53, Rb and cyclin D1. However, no single molecular marker has proved to be reliable in distinguishing malignant from benign tumors (1, 2, 7). Our patient was not found to have a CDC73mutation, besides having an additional hyperplastic gland, neither had any clinical and/or biochemical stigmata of a familial syndrome.
Neck ultrasound is the most commonly employed radiological modality for parathyroid disease. Large tumors, displaying hypoechogenicity, inhomogeneity, irregular borders and calcifications are more likely to be malignant. However, only invasion into adjacent tissues or presence of metastases allows the pre-operative diagnosis of parathyroid cancer. MIBI scintigraphy remains a valuable tool to assess the presence and localization of hyperfunctioning parathyroid tissue (1, 2, 8).
Surgery is the only curative treatment for parathyroid carcinoma. Tumor resection en bloc with ipsilateral thyroidectomy is the procedure of choice. Surgery is also the treatment of choice in the presence of local recurrence or metastatic disease. However, neck reoperation is associated with a 3–5 times higher complication rate than the initial operation (2, 9, 10). Parathyroid carcinoma is considered resistant to radiotherapy, but recent case series suggest that adjuvant radiation therapy can be effective in preventing recurrence and extending disease-free survival (2).
Therapeutic options for unresectable parathyroid carcinoma are limited. Chemotherapy is considered to be ineffective in parathyroid carcinomas as shown in our case. A few case reports describing monotherapy with dacarbazine and several regimens (nitrogen mustard, vincristine, cyclophosphamide and actinomycin D, adriamycin, cyclophosphamide and 5-fluorouracil and adriamycin alone) showed that all have been relatively ineffective (1, 2).
In case of metastatic carcinoma, the primary aim is to control hypercalcemia. Mitramycin, plicamycin, gallium nitrate, bisphosphonates, calcitonin and glucocorticoids have been used, but newer drugs such as the calcimimetic cinacalcet are probably more effective (2). Anti-parathyroid hormone immunotherapy has been experimentally used with promising results but is not yet available, while ethanol ablation, radiofrequency ablation and trans-arterial embolization present palliative treatment methods (2).
Recurrence is very common in parathyroid carcinoma usually observed between 2.5 and 4.8 years after initial surgery but can occur even after 20 years (2). In our case, metastases developed 10 years after initial diagnosis. About 50% of patients develop local recurrence and 25% distant metastases, the most common being locoregional lymph nodes, lung, liver and bones (2). Five- and ten-year survival rates range from 76–85% to 49–77% respectively (1, 2). Local excision instead of en bloc resection at initial operation, high calcium levels and early recurrence are associated with 7-fold greater risk of mortality (5).
In conclusion, parathyroid carcinoma is an extremely rare endocrine malignancy that may develop metastases after prolonged follow-up. En bloc surgical resection with microscopically negative margins is the recommended treatment for cure, as these lesions are resistant to conventional chemotherapy and radiotherapy. International registries and inclusion of patients in experimental trials are required in order to develop tumor-oriented and specific therapies.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author contribution statement
Prof. Kaltsas, Dr Angelousi and Dr Tsoli were responsible for patient’s care. Histopathological examination was performed by Dr Rontogianni and genetic analysis by Dr Stratakis. Dr Tsoli and Dr Angelousi contributed equally to the manuscript.
References
- 1.McClenaghan F, Qureshi YA. 2015. Parathyroid cancer. Gland Surgery 4 329–338. ( 10.3978/j.issn.2227-684X.2015.05.09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei CH, Harari A. 2012. Parathyroid carcinoma: update and guidelines for management. Current Treatment Options in Oncology 13 11–23. ( 10.1007/s11864-011-0171-3) [DOI] [PubMed] [Google Scholar]
- 3.Cetani F, Pardi E, Marcocci C. 2016. Update on parathyroid carcinoma. Journal of Endocrinological Investigation 39 595–606. ( 10.1007/s40618-016-0447-3) [DOI] [PubMed] [Google Scholar]
- 4.Talat N, Schulte KM. 2010. Clinical presentation, staging and long-term evolution of parathyroid cancer. Annals of Surgical Oncology 17 2156–2174. ( 10.1245/s10434-010-1003-6) [DOI] [PubMed] [Google Scholar]
- 5.Xue S, Chen H, Lv C, Shen X, Ding J, Liu J, Chen X. 2016. Preoperative diagnosis and prognosis in 40 parathyroid carcinoma patients. Clinical Endocrinology 85 29–36. ( 10.1111/cen.13055) [DOI] [PubMed] [Google Scholar]
- 6.Farnebo F, Auer G, Farnebo LO, Tech BT, Twigg S, Aspenblad U, Thompson NW, Grimelius L, Larsson C, Sandelin K. 1999. Evaluation of retinoblastoma and Ki-67 immunostaining as diagnostic markers of benign and malignant parathyroid disease. World Journal of Surgery 23 68–74. ( 10.1007/s002689900567) [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Ranvier GG, Khanafshar E, Tacha D, Wong M, Kebebew E, Duh QY, Clark OH. 2009. Defining a molecular phenotype for benign and malignant parathyroid tumors. Cancer 115 334–344. ( 10.1002/cncr.24037) [DOI] [PubMed] [Google Scholar]
- 8.Sidhu PS, Talat N, Patel P, Mulholland NJ, Schulte KM. 2011. Ultrasound features of malignancy in the preoperative diagnosis of parathyroid cancer: a retrospective analysis of parathyroid tumours larger than 15 mm. European Radiology 21 1865–1873. ( 10.1007/s00330-011-2141-3) [DOI] [PubMed] [Google Scholar]
- 9.Harari A, Waring A, Fernandez-Ranvier G, Hwang J, Suh I, Mitmaker E, Shen W, Gosnell J, Duh QY, Clark O. 2011. Parathyroid carcinoma: a 43-year outcome and survival analysis. Journal of Clinical Endocrinology and Metabolism 96 3679–3686. ( 10.1210/jc.2011-1571) [DOI] [PubMed] [Google Scholar]
- 10.Kebebew E. 2001. Parathyroid carcinoma. Current Treatment Options in Oncology 2 347–354. ( 10.1007/s11864-001-0028-2) [DOI] [PubMed] [Google Scholar]