

Abstract
Detailed characterization of chromosomal abnormalities, a common cause for congenital abnormalities and pregnancy loss, is critical for elucidating genes for human fetal development. Here, 2186 product of conception (POC) samples were tested for copy number variations (CNVs) at two clinical diagnostic centers using whole genome sequencing and high-resolution chromosomal microarray analysis. We developed a new gene discovery approach to predict potential developmental genes and identified 275 candidate genes from CNVs detected from both datasets. Based on Mouse Genome Informatics (MGI) and Zebrafish model organism database (ZFIN), 75% of identified genes could lead to developmental defects when mutated. Genes involved in embryonic development, gene transcription and regulation of biological processes were significantly enriched. Especially, transcription factors and gene families sharing specific protein domains predominated, which included known developmental genes such as HOX, NKX homeodomain genes and helix-loop-helix containing HAND2, NEUROG2 and NEUROD1 as well as potential novel developmental genes. We observed that developmental genes were denser in certain chromosomal regions, enabling identification of 31 potential genomic loci with clustered genes associated with development.
Keywords: Essential gene prediction, pregnancy loss, congenital malformations, CNVs, WGS, CMA
Introduction
Chromosomal abnormalities contribute significantly to various congenital anomalies and pregnancy loss in humans. Prior studies showed that about half of the spontaneous pregnancy losses had chromosomal abnormalities [Goddijn and Leschot, 2000] and approximately 5–17% of congenital anomalies were linked to genomic aberrations [Lu et al., 2008; Sagoo et al., 2009; Wellesley et al., 2012; Brady et al., 2014]. Among all chromosomal abnormalities associated with pregnancy losses, chromosomal aneuploidies that involve the copy number change of an entire chromosome, e.g., trisomy 21, trisomy 13, trisomy 18, and monosomy X, are the most frequently detected and their occurrence increases substantially with maternal age [Eiben et al., 1990; Wellesley et al., 2012]. Chromosomal structural abnormalities that occur due to a change in the structure or the parts of a chromosome, e.g., deletions and duplications, are less common than chromosomal aneuploidy, but their detection rate has been greatly improved with the application of microarray-based cytogenetic techniques [Breman et al., 2012; Wapner et al., 2012]. While critical disease genes have been successfully identified from small overlapped deletions or duplications detected in patients with similar clinical phenotypes [Weischenfeldt et al., 2013], it is a greater challenge to elucidate such genes associated with pregnancy loss or severe congenital malformations that display multiple phenotypes, since the structural abnormalities are often larger, affecting a greater number of genes.
In this study, we performed whole genome sequencing (WGS) and chromosomal microarray analysis (CMA) on clinical samples from spontaneous pregnancy losses and terminations of pregnancies for fetal anomaly (TOPFA) to detect copy-number variations (CNVs). Considering that the majority of our cases of pregnancy losses occurred during the first trimester, we rationalized that genes affected by these CNVs might be critical for early embryonic development. Because the detected CNVs are often very large and methodologies for identifying essential genes from those large CNVs are still limited, we therefore developed a gene discovery approach that integrated several important characteristics of essential genes and identified 275 potential developmental genes located in the CNVs detected in pregnancy tissues. Further functional annotation indeed revealed that a significant number of genes identified using our approach were functionally important for organ development or general survival, and potentially novel developmental genes were also identified. Moreover, we observed that some of these essential genes were physically clustered on the genome, forming critical regions associated with human early development.
Materials and Methods
Subjects
China
DNA samples of 1810 pregnancy tissues from spontaneous pregnancy loss or TOPFA were received by Jiahui Hospital in China. A total of 817 (45%) cases had copy number variations and were included in the current study. 776 cases were from natural conceptions and 41 cases were from assisted conception. Informed consents were received from all participating couples. The mean maternal age of subjects of the 817 cases is 31.05 years (SD = ±5.08). All pregnancy losses occurred before the 28th week of gestation with a mean gestational period of 10.48 week (SD = ±3.58). 811 cases had prior pregnancy records. 26.51% (215/811) of the cases were the first pregnancy loss, while 35.64% (289/811) and 37.85% (307/811) were from the second and third or more pregnancy losses, respectively.
USA
376 pregnancy loss cases were referred to Baylor Miraca Genetics Laboratory for CMA. 112 (30%) cases were diagnosed with chromosomal abnormalities. The mean maternal age is 33.86 years (SD = ±5.74) and the mean gestational week at the time of the pregnancy loss is 9.4 weeks (SD = ±3.44).
Whole genome sequencing of products of conception
Depending on gestational weeks, products of conception tissues, including those from fetus, chorionic villi, or umbilical cord, were collected according to standard clinical procedures. DNA was extracted from tissues using DNeasy Blood & Tissue Kit (Qiagen, #69506) and purified with Genomic DNA Clean & Concentrator Kit (Zymo, #D4011). The sequencing and calling of CNVs were performed as previously described [Liang et al., 2014; Wang et al., 2014; Liu et al., 2015]. Briefly, about 3 million sequencing reads of each sample were mapped to the reference genome using the Burrows-Wheeler Aligner (BWA) and allocated to 20 Kb sequencing bins with a 5 Kb sliding to achieve a higher resolution in identifying CNVs. CNV profiles of each chromosome was represented as log2 of mean sequencing reads of each sequencing bin along the chromosome.
Chromosomal microarray-based analysis of pregnancy tissues
DNA was extracted from products of conception using a modified Qiagen method [Breman et al., 2012]. After digestion and labeling, DNA was subjected to oligonucleotide-based chromosomal microarray analysis (CMA Version 7.6 OLIGO), which contains probes covering the whole genome with an average resolution of 30 Kb. CNV calling was performed using an in-house analysis package [Cheung et al., 2005; El-Hattab et al., 2009]. Clinical data was reviewed according to a protocol approved by the Baylor College of Medicine Institutional Review Board. Detected copy number gains or losses were systematically evaluated for clinical significance by examining their minimum and maximum size, genomic position, and the number of genes located in the region by overlapping their coordinates with the UCSC genome browser (http://www.genome.ucsc.edu/).
Prediction of developmental genes from CNVs
The gene discovery approach integrated genomic location, evolution conservation, human fetal or placental expression, as well as the Haploinsufficient (HI) score and Residual Variance Intolerance Score (RVIS) percentiles of the genes. All analyses were performed based on the human genome assembly GRCh37/hg19. CNVs from healthy human controls were downloaded from the 1000 genomes project database (release v5) (www.1000genomes.org) and CNVs with all frequencies were included for following analysis. PhyloP scores were downloaded from the UCSC genome browser. The mean phyloP scores were calculated for each gene and a cut-off of 0.4 was used to define the conserved gene. The expression profiles of genes in human tissues were obtained from the GNF Atlas 2 (http://www.genome.ucsc.edu/). The mean expression value of all tissues was calculated for each gene. Genes with an expression value in any fetal of placental tissues one standard deviation higher than the mean expression value of all tissues were defined with a higher expression in human fetal or placental tissues. HI and RVIS scores were downloaded from the Decipher database (https://decipher.sanger.ac.uk/) and RVIS website (http://genic-intolerance.org/), respectively. Both RVIS_ExAC and RVIS original scores were used in the analysis with the same score percentile threshold. The gene ontology (GO) analysis was performed using the DAVID bioinformatics database (https://david.ncifcrf.gov/) [Huang da et al., 2009b, a]. Protein domain analysis was carried out using GENEMANIA (www.genemania.org). OMIM (Online Mendelian Inheritance in Man) genes were downloaded from OMIM website (www.omim.org). Plots were generated using R core functions, ggplot2 [Wichham, 2009], ggbio [Yin et al., 2012], and Cytoscape [Shannon et al., 2003].
Results
Copy number variants detected by WGS and CMA
Of the 2186 samples of product of conception due to spontaneous pregnancy loss or TOPFA, whole genome sequencing was performed on 1810 cases from Jiahui Hospital in China with CNVs detected in 817 (45%) cases, while 112 of the 376 (30%) cases tested by Baylor Miraca Genetics Laboratories using high-resolution CMA [El-Hattab et al., 2009; Bi et al., 2013] had CNVs, for a combined total of 929 (42.5%) CNV cases. Among these CNV cases, 439 (47.3%) cases were diagnosed with autosomal trisomy, followed by autosomal mosaicism (94/929, 10.1%), 45, XO (92/929, 9.9%) and polyploidy (83/929, 8.9%) (Fig. 1). Among cases with gain or loss of an entire chromosomal, or chromosomal aneuploidy, 132 (20.1%) cases occurred on chromosome 16, followed by 117 (17.8%) on chromosome X and 72 (11.0%) on chromosome 22. By contrast, no such cases were found on chromosome 1 (Fig. 2A). Moreover, 36 cases contained chromosomal aneuploidy on two chromosomes, among which trisomy 16-trisomy 21 was observed in 3 cases and trisomy 13-trisomy 18, trisomy 13-trisomy 22, trisomy 15-trisomy 16, trisomy 16–45, XO was found in 2 cases, respectively (Fig. 2B).
Figure 1. Chromosomal abnormalities detected in products of conception using whole genome sequencing and chromosomal microarray-based analysis.
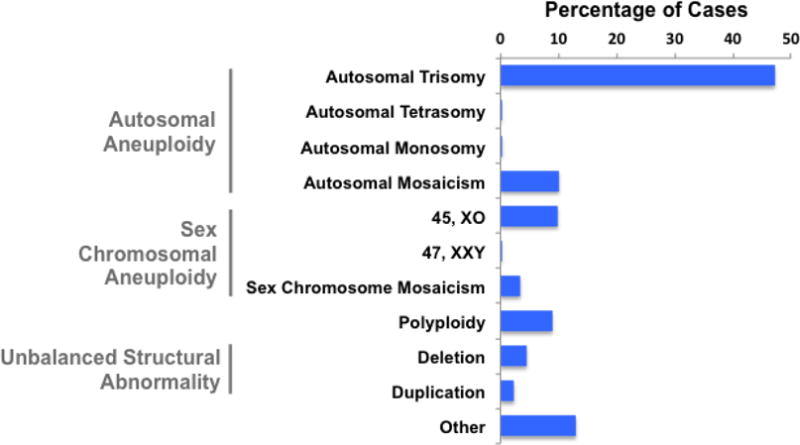
Figure 2. Chromosomal aneuploidies associated with pregnancy loss or TOPFA phenotypes.
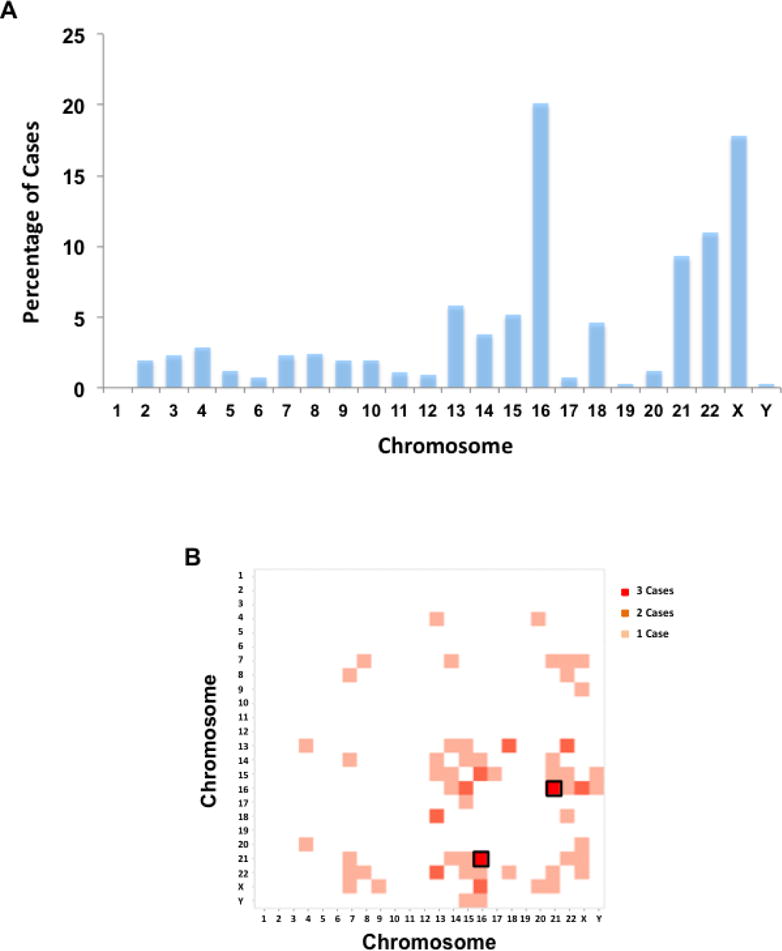
A) The distribution of chromosomal aneuploidy on each chromosome.
B) Cases with chromosomal aneuploidies on two different chromosomes. Trisomy 16 – Trisomy 21 is observed in 3 cases and highlighted with a black box.
A total of 130 deletions or duplications were detected from the WGS dataset and 24 from the CMA dataset (Supp. Table S1). These CNVs are mostly de novo and heterozygous. Deletions and duplications occurred most frequently at chromosomes 22 and 18, followed by chromosomes X and 8 (Fig. 3A). The size of the deletions identified in all cases was substantially smaller than those of duplications with a median size of 7.64 Mb (95%CI: 10.72–18.58 Mb) for deletions and 22.67 Mb (95%CI: 23.41–41.49 Mb) for duplications. Both deletions and duplications detected in products of conception were much larger than those detected in healthy humans, which were mostly less than 1 Mb [Sudmant et al., 2015] (P <= 2.523E-9) (Fig. 3B).
Figure 3. Deletions and duplications detected in products of conception (POC) by whole genome sequencing and chromosomal microarray-based analysis.
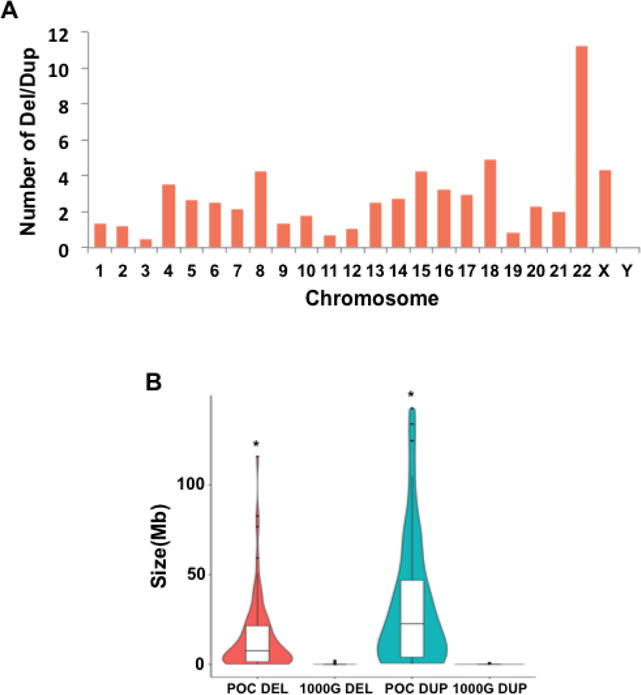
A) The number of deletions or duplications normalized by chromosome length on each chromosome.
B) Violin plot showing the size distribution of deletions and duplications detected from POC and 1000 Genomes project (1000G). The boxplot shows the median, 1st and 3rd quartile (*: P <= 2.523E-9).
Since there are 811 cases with prior pregnancy information, we also studied whether there was any correlation between number of prior miscarriages and chromosomal abnormalities. We found that the incidence of sex chromosome aneuploidy decreased, while that of polyploidy increased with the number of prior miscarriage (Supp. Figure S1A). In addition, we found that the frequency of deletions and duplications was not correlated with number of miscarriages. Though larger deletions were detected in mothers with multiple miscarriages, the difference was not statistically significant (Supp. Figure S1B).
Identification of developmental genes from CNV regions detected in products of conception
To deduce critical genes and genomic loci for early embryonic development from CNVs detected in products of conception, we developed an integrative gene discovery approach based on the genomic location, evolutionary conservation, human fetal or placental expression profile, RVIS and HI scores of genes (Fig. 4). RVIS and HI are two gene-level scoring systems commonly used to assess the susceptibility of genes to genetic variations [Huang et al., 2010; Petrovski et al., 2013]. RVIS score shows the evolutionary constraints on individual genes, as represented by the deviation of the observed functional variants from the expected number of common variants predicted based on total amount of variants on a gene, while HI score integrates genomic, functional and network properties to predict the possibility of a given gene affected by a deletion being haploinsufficient in terms of biological functions. Both methods have been used to prioritize potential disease causing genes and variants [Stittrich et al., 2014; Sanders et al., 2015]. Among 154 deletions and duplications identified using both WGS and CMA, 36.2% (47/130) of the deletions or duplications detected by WGS overlapped with those detected by CMA, which contained a total of 10,572 genes. We first applied the gene discovery analytical framework to this set of genes and deduced those that were critical for human early development. When comparing to the 1000 Genomes (1000G) datasets, any gene with at least 1 bp overlap with CNVs detected in 1000G was regarded as the controls. A total of 4463 genes were thus identified as control genes, whereas the remaining 6109 genes (57.8%) were not included in the deletions or duplications identified in the 1000G subjects and were thus treated as potential candidate genes associated with pregnancy loss or TOPFA phenotype. Compared with the control genes, we found that 22% more genes located outside of 1000G CNVs had a higher phyloP conservation score (P < 2.2E-16) (Fig. 5). An earlier study identified 2741 genes essential for the proliferation and survival of cell lines using the CRISPR/Cas9 system [Wang et al., 2015]. Considering the average phyloP conservation score of these identified essential genes is 0.36, we applied a similar threshold of 0.4 for the phyloP score in our approach. 2028 genes met this criterion, of which 1635 (80.6%) genes were not present in the CNVs from the 1000G.
Figure 4. The outline of gene discovery analytic framework.
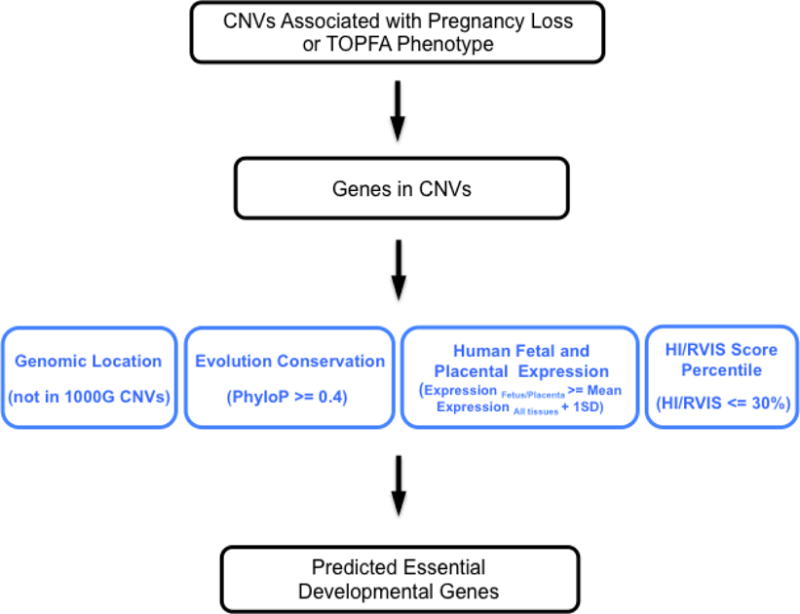
Figure 5. A higher percentage of candidate genes are conserved.
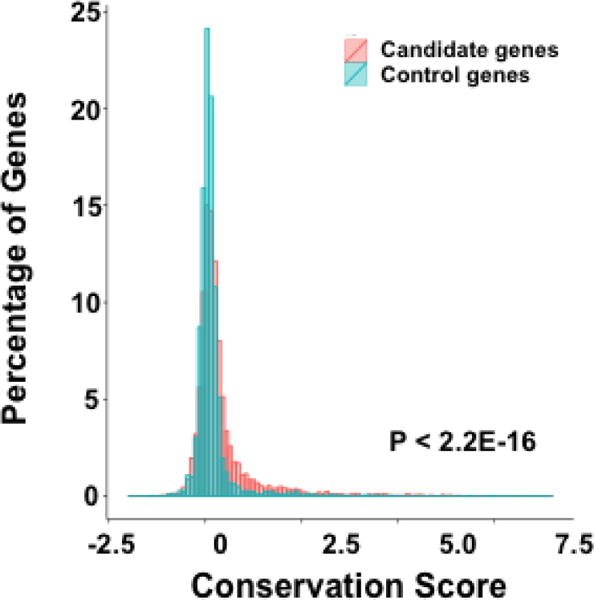
Compared with control genes, a higher percentage of pregnancy loss and TOPFA candidate genes have a higher conservation score.
We further examined the expression profiles of the 1635 genes using the GNF gene expression Atlas 2 [Su et al., 2004] and identified 360 genes with a higher expression in human fetal or placental tissues (see details in Methods). Based on the HI and RVIS score profiles of reported essential and loss-of-function tolerant genes [MacArthur et al., 2012; Sudmant et al., 2015; Wang et al., 2015; Zarrei et al., 2015], we used 30% as the threshold of both HI and RVIS score percentiles for our approach, which eventually led to the identification of 244 genes that are putatively critical for human early embryonic development (Supp. Table S2). We used same approach for the genes mapping within the CNVs detected in the CMA dataset and identified 97 potential developmental genes. 66 (68%) of genes are also within the CNV regions detected by WGS, which leaves 31 unique important developmental genes identified from the CMA dataset (Supp. Table S3). Therefore, a total of 275 potential developmental genes were identified from both datasets.
Transcription factors and gene families sharing specific protein domains were significantly enriched
Based on the Mouse Genome Informatics (MGI), Zebrafish model organism database (ZFIN) and previous publications, mutant alleles in 206 out of 275 genes (74.9%) could cause developmental defects in the model organisms. The deficiency of 84 of these 206 genes (40.8%) could lead to prenatal lethality in mammalian models. The GO analysis showed that genes involved in three major functional categories: embryonic development, gene transcription and regulation of biological process were significantly enriched among the 275 predicted developmental genes (P<0.05). In particular, genes involved in neuronal development and differentiation were overrepresented (Fig. 6A). 31.6% (87/275) of the predicted gene products had DNA-binding property, among which transcription factors, including both activators and repressors, were significantly enriched (P<1.6E-6) (Fig. 6B). In addition, we found that 61.8% (170/275) of predicted developmental genes shared the same protein domains with at least one additional gene and thus might belong to same gene families. Ten groups containing at least four predicted developmental genes sharing same functional domains were identified, including genes encoding proteins with the homeodomain, protein kinase domain, and basic helix-loop-helix (HLH) domains (Fig. 6C; Supp. Figure S2). Some of these genes are known developmental genes. For example, HOX homeodomain genes form a large conserved gene family that controls the anterior-posterior polarity of embryos [McGinnis and Krumlauf, 1992; Lawrence and Morata, 1994]. NKX-class homeodomain proteins, such as NKX2.1 and NKX6.1, and basic HLH proteins, NEUROD1 and NEUROG2, play an essential role not only in brain development but also in the organogenesis of lung, pancreas, thyroid and retina [Kimura et al., 1996; Fode et al., 1998; Miyata et al., 1999; Sander et al., 2000a; Sander et al., 2000b; Akagi et al., 2004]. HAND2 and SOX4 knockout mouse embryos died from heart malformations [Schilham et al., 1996; Srivastava et al., 1997], while the leucine-rich repeats containing SLITRK genes regulate the nervous system development and have high expression in human fetal brain [Proenca et al., 2011].
Figure 6. Functional characterization of predicted developmental genes.
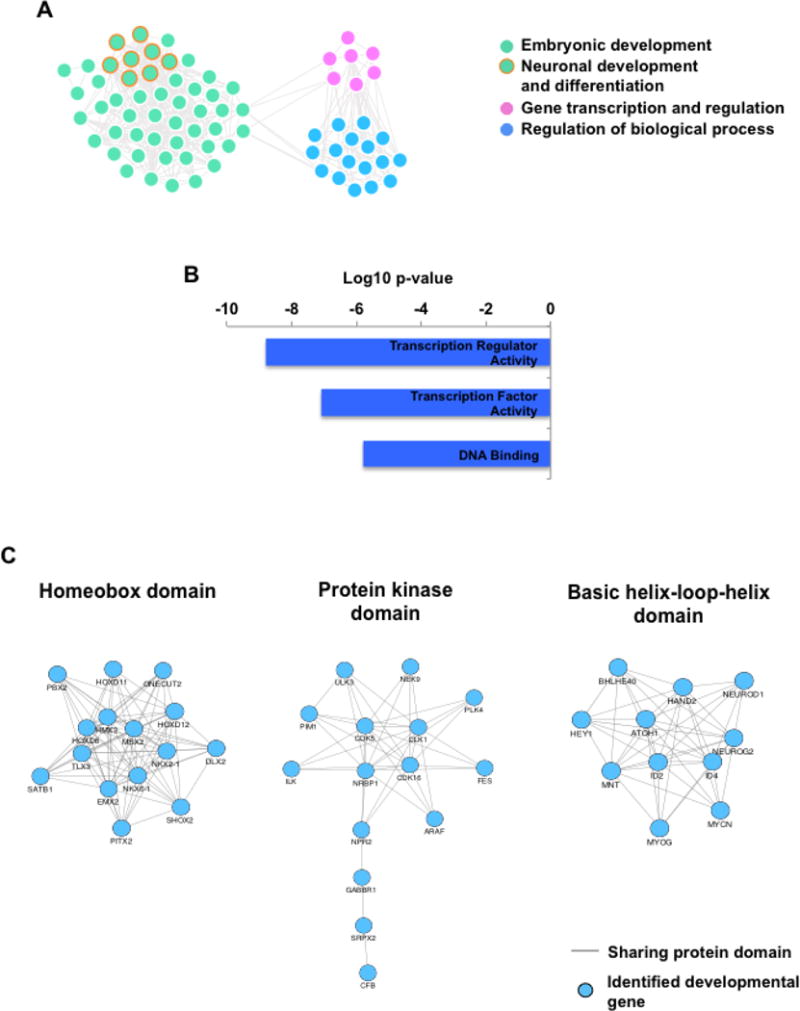
A) Gene ontology analysis showed that genes involved in embryonic development, gene transcription and regulation of biological processes are highly enriched among 275 predicted developmental genes. Each dot represents one GO term. GO terms related to neuronal development and differentiation are highlighted in orange.
B) Transcription factors and regulators are significantly enriched among predicted developmental genes.
C) Top three gene families enriched among 275 predicted developmental genes based on functional protein domains.
93.1% (256/275) identified genes are OMIM genes (Supp. Table S4). However, we found that 69 genes whose functions during development have not been well characterized and could be potential novel developmental genes (Supp. Table S5). For instance, ULK3 is a ULK family of serine/threonine kinase. Prior studies showed that ULK3 was involved in regulating the activity of Sonic hedgehog signaling pathways [Maloverjan et al., 2010], but its function during development has not been well characterized. We identified ULK3 as a potential developmental gene using our integrative gene discovery approach, which is further supported by a recent study showing that ULK3 is a critical factor of abscission checkpoint pathway, ensuring the proper segregation of chromosomes into two daughter cells during cell cycle and preventing the formation of aneuploidy cells [Caballe et al., 2015]. ZNF711 is another potential development gene identified by our analysis, which belongs to the zinc finger gene family. Although its function during development requires further characterization, ZNF711 has been identified as a disease gene for X-linked mental retardation 97 [Tarpey et al., 2009], suggesting its potential function during development. These results demonstrate that our approach provides one effective way to predict genes important for human early development. These identified genes are indeed involved in essential processes during embryonic development, among which transcription factors as well as genes families with specific functional domains are predominated.
Identification of genomic loci important for development
We noticed that some of the identified developmental genes were clustered in the genome. These clusters may contain genes of the same gene family, such as the HOX gene cluster, or genes with different functions and properties. To investigate such clusters of developmental genes in the human genome, we used the same analytic framework to search for potential developmental genes in the whole genome and identified 712 candidate genes, including 244 and 97 genes potential developmental genes identified from WGS and CMA datasets. Indeed, we found that these genes were clustered at certain genomic regions (Fig. 7). We therefore scanned the entire genome using a sliding-window analysis (1 Mb window) and identified 31 genomic loci that contained at least four predicted developmental genes each (Fig. 7, Supp. Table S6). Our findings showed that in addition to gene clusters of the same gene families, human genome also contained functionally clustered genes that defined genomic loci essential for human early development.
Figure 7. The map of genomic loci with clustered developmental genes.
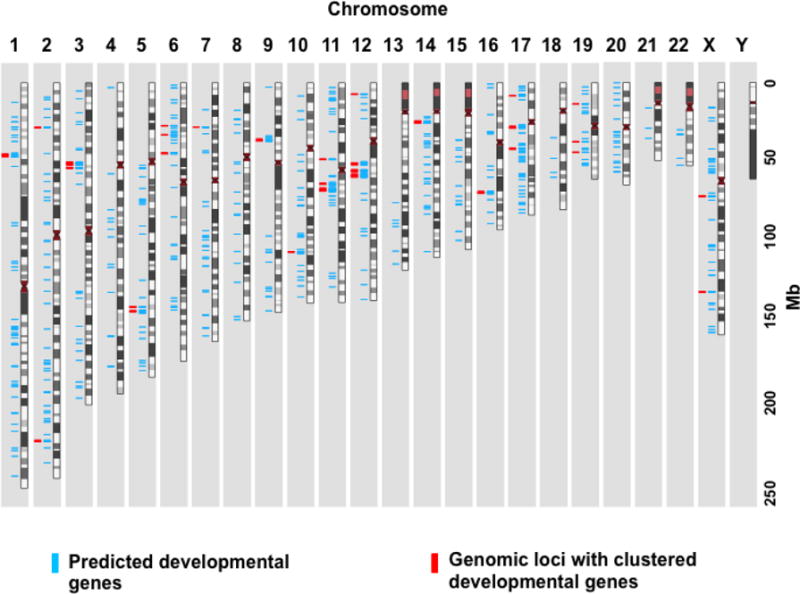
712 predicted developmental genes (blue) were plotted along the chromosomes and the genomic regions containing at least four predicted developmental gene are highlighted in red.
Discussion
Numerical and structural chromosomal abnormalities are major causes of pregnancy loss and congenital genetic disorders [Menasha et al., 2005; Breman et al., 2012]. The early and precise diagnosis of such anomalies during pregnancy has been the main goal of prenatal diagnostic attempts. With an increasingly lower cost, copy-number analyses based on next-generation sequencing has rapidly become an alternative technique for karyotyping and array-based molecular cytogenetic techniques [Talkowski et al., 2012; Shashi et al., 2014]. In this study, we applied whole genome sequencing to detect chromosomal abnormalities associated with pregnancy loss and TOPFA. We showed that low coverage sequencing was able to identify chromosomal copy-number variations of different extents, ranging from entire chromosome aneuploidy to unbalanced structural rearrangements as small as 0.12 Mb. The prevalence of major chromosomal abnormalities in this study was comparable to those analyzed using conventional techniques [Menasha et al., 2005] and 36% of the deletions and duplications detected by WGS overlapped with those identified from a smaller independent dataset using high-resolution CMA.
Identifying genes within CNV regions associated with pregnancy loss and congenital abnormalities is challenging because CNVs can be very large and usually have extensive gene content. The bioinformatic tools for identifying causal variants based on single nucleotide variations (SNVs) have been well established [Pabinger et al., 2014], which allows successful identification of genes associated with a lethal fetal phenotype or congenital malformations based on predicted deleterious variants [Putoux et al., 2011; Marshall et al., 2015; Shamseldin et al., 2015]. These studies are often performed on consanguineous families or subjects with well-defined clinical indications. Therefore, it is challenging when applying the same analytic strategy on pregnancy loss or TOPFA cases that often present with phenotypes resulting from a deficiency of different combinations of genes in an outbred population. An earlier study combined copy number variation and truncating SNV data to identify genes associated with developmental delay [Coe et al., 2014], but an effective approach for performing gene-based analysis within CNVs associated with pregnancy loss and TOPFA are still limited. In this study, we integrated five gene properties including genomic locations, evolution conservation, expression profiles, HI and RVIS scores to prioritize putatively developmental genes within the CNV loci detected in our cases with pregnancy loss or congenital malformations. 275 genes were identified as potential developmental genes from 154 CNVs detected in products of conception by WGS or CMA. Three fourths of the predicted genes led to developmental defects and approximately 40% of them caused embryonic lethality in animal models carrying mutant alleles. Correspondingly, genes involved in embryonic development, especially genes that regulate neuronal development and differentiation are significant enriched among predicted developmental genes. These results support that our approach is effective in predicting genes important for development.
Functional analysis revealed that predicted developmental genes were enriched for transcription factors and several specific gene families with particular protein domains, which includes genes known to be essential for development, such as HOX and NKX-class homeodomain genes, HMG-box containing SOX genes and SLITRK genes sharing the leucine-rich repeats. In addition, we observed clustering of predicted developmental genes in the genome. For example, HOX genes often form a cluster at the chromosome in the same order as these genes are expressed in the embryos [Pearson et al., 2005]. Although the gain or loss of function alleles of a single HOX gene could produce a phenotype, there were also cases when mutations in two HOX genes might lead to more severe embryonic defects, indicating an interaction between different members in the same gene family [Condie and Capecchi, 1994; Davenne et al., 1999]. In addition to HOX genes, we found that developmental genes were also clustered in the genome regardless of gene family, which led to the identification of 31 genomic loci containing at least four predicted developmental genes each. Our finding is consistent with a recent study showing the clustering of functionally related genes in the genome [Andrews et al., 2015]. Since CNVs detected in products of conception could affect multiple clusters of developmental genes, which eventually led to the pregnancy loss or severe congenital malformations, we further examined the clinical phenotypes caused by CNVs affecting each genomic loci with clustered developmental genes. According to DECIPHER database [Firth et al., 2009], 150 patients have CNVs covering one cluster of developmental genes. Although the difference is not statistically significant, these patients indeed displayed a higher percentage of intellectual disability, delayed speech and language development as well as Autism compared with all patients with clinical phenotypes collected in DECIPHER database (Supp. Figure S3). Moreover, our findings also suggest that genetic variations affecting genes within same cluster could be taken into consideration when identifying causal genes especially for cases with phenotypic variations. The extent by which such genomic loci are affected by CNVs can explain the phenotypic variations associated with prenatal manifestations or congenital disorders. Meanwhile, it can also serve as an indicator of the clinical severity of CNVs. Furthermore, these functionally clustered regions can be used to prioritize the disease-causing genetic variants, such as SNVs and small indels, in patients.
In this study, we developed a gene discovery approach that could be used to effectively identify developmental genes affected by CNVs associated with pregnancy loss or congenital abnormality phenotypes. Based on CNVs detected in our cases, we identified 275 potential developmental genes that were enriched with genes involved in embryonic development, especially neuronal development and differentiation. The majority of these genes could cause developmental defects in animal models carrying the mutant alleles and deficiency of 40% of them resulted in prenatal lethality in model organisms. Potential novel developmental genes were also identified, some of which have been shown to play a role during development based on in vitro experiments but their function in vivo requires further characterization. Furthermore, we identified 31 genomic loci in the genome with clustered developmental genes, which could be used to assess the severity of CNVs or prioritize causal SNVs for later studies.
Supplementary Material
Acknowledgments
We thank Dr. Christian Patrick Schaaf for scientific advice and manuscript review. This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. The funding for DECIPHER project was provided by the Wellcome Trust.
Grant Sponsor: This study was supported by the startup fund from Baylor College of Medicine and a T32 training grant (GM08307-23).
Footnotes
Conflict of Interest Disclosure
The authors declare that they have no competing interests.
References
- Akagi T, Inoue T, Miyoshi G, Bessho Y, Takahashi M, Lee JE, Guillemot F, Kageyama R. Requirement of multiple basic helix-loop-helix genes for retinal neuronal subtype specification. The Journal of biological chemistry. 2004;279:28492–28498. doi: 10.1074/jbc.M400871200. [DOI] [PubMed] [Google Scholar]
- Andrews T, Honti F, Pfundt R, de Leeuw N, Hehir-Kwa J, Vulto-van Silfhout A, de Vries B, Webber C. The clustering of functionally related genes contributes to CNV-mediated disease. Genome research. 2015;25:802–813. doi: 10.1101/gr.184325.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Borgan C, Pursley AN, Hixson P, Shaw CA, Bacino CA, Lalani SR, Patel A, Stankiewicz P, Lupski JR, et al. Comparison of chromosome analysis and chromosomal microarray analysis: what is the value of chromosome analysis in today’s genomic array era? Genetics in medicine : official journal of the American College of Medical Genetics. 2013;15:450–457. doi: 10.1038/gim.2012.152. [DOI] [PubMed] [Google Scholar]
- Brady PD, Delle Chiaie B, Christenhusz G, Dierickx K, Van Den Bogaert K, Menten B, Janssens S, Defoort P, Roets E, Sleurs E, et al. A prospective study of the clinical utility of prenatal chromosomal microarray analysis in fetuses with ultrasound abnormalities and an exploration of a framework for reporting unclassified variants and risk factors. Genetics in medicine : official journal of the American College of Medical Genetics. 2014;16:469–476. doi: 10.1038/gim.2013.168. [DOI] [PubMed] [Google Scholar]
- Breman A, Pursley AN, Hixson P, Bi W, Ward P, Bacino CA, Shaw C, Lupski JR, Beaudet A, Patel A, et al. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenatal diagnosis. 2012;32:351–361. doi: 10.1002/pd.3861. [DOI] [PubMed] [Google Scholar]
- Caballe A, Wenzel DM, Agromayor M, Alam SL, Skalicky JJ, Kloc M, Carlton JG, Labrador L, Sundquist WI, Martin-Serrano J. ULK3 regulates cytokinetic abscission by phosphorylating ESCRT-III proteins. eLife. 2015;4:e06547. doi: 10.7554/eLife.06547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genetics in medicine : official journal of the American College of Medical Genetics. 2005;7:422–432. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nature genetics. 2014;46:1063–1071. doi: 10.1038/ng.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condie BG, Capecchi MR. Mice with targeted disruptions in the paralogous genes hoxa-3 and hoxd-3 reveal synergistic interactions. Nature. 1994;370:304–307. doi: 10.1038/370304a0. [DOI] [PubMed] [Google Scholar]
- Davenne M, Maconochie MK, Neun R, Pattyn A, Chambon P, Krumlauf R, Rijli FM. Hoxa2 and Hoxb2 control dorsoventral patterns of neuronal development in the rostral hindbrain. Neuron. 1999;22:677–691. doi: 10.1016/s0896-6273(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Eiben B, Bartels I, Bahr-Porsch S, Borgmann S, Gatz G, Gellert G, Goebel R, Hammans W, Hentemann M, Osmers R, et al. Cytogenetic analysis of 750 spontaneous abortions with the direct-preparation method of chorionic villi and its implications for studying genetic causes of pregnancy wastage. American journal of human genetics. 1990;47:656–663. [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Smolarek TA, Walker ME, Schorry EK, Immken LL, Patel G, Abbott MA, Lanpher BC, Ou Z, Kang SH, et al. Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping. Human genetics. 2009;126:589–602. doi: 10.1007/s00439-009-0706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. American journal of human genetics. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fode C, Gradwohl G, Morin X, Dierich A, LeMeur M, Goridis C, Guillemot F. The bHLH protein NEUROGENIN 2 is a determination factor for epibranchial placode-derived sensory neurons. Neuron. 1998;20:483–494. doi: 10.1016/s0896-6273(00)80989-7. [DOI] [PubMed] [Google Scholar]
- Goddijn M, Leschot NJ. Genetic aspects of miscarriage. Bailliere’s best practice & research Clinical obstetrics & gynaecology. 2000;14:855–865. doi: 10.1053/beog.2000.0124. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PLoS genetics. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes & development. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- Lawrence PA, Morata G. Homeobox genes: their function in Drosophila segmentation and pattern formation. Cell. 1994;78:181–189. doi: 10.1016/0092-8674(94)90289-5. [DOI] [PubMed] [Google Scholar]
- Liang D, Peng Y, Lv W, Deng L, Zhang Y, Li H, Yang P, Zhang J, Song Z, Xu G, et al. Copy number variation sequencing for comprehensive diagnosis of chromosome disease syndromes. The Journal of molecular diagnostics: JMD. 2014;16:519–526. doi: 10.1016/j.jmoldx.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Liu S, Song L, Cram DS, Xiong L, Wang K, Wu R, Liu J, Deng K, Jia B, Zhong M, et al. Traditional karyotyping vs copy number variation sequencing for detection of chromosomal abnormalities associated with spontaneous miscarriage. Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and Gynecology. 2015;46:472–477. doi: 10.1002/uog.14849. [DOI] [PubMed] [Google Scholar]
- Lu XY, Phung MT, Shaw CA, Pham K, Neil SE, Patel A, Sahoo T, Bacino CA, Stankiewicz P, Kang SH, et al. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–1318. doi: 10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloverjan A, Piirsoo M, Kasak L, Peil L, Osterlund T, Kogerman P. Dual function of UNC-51-like kinase 3 (Ulk3) in the Sonic hedgehog signaling pathway. The Journal of biological chemistry. 2010;285:30079–30090. doi: 10.1074/jbc.M110.133991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Farrell SA, Cushing D, Paton T, Stockley TL, Stavropoulos DJ, Ray PN, Szego M, Lau L, Pereira SL, et al. Whole-exome analysis of foetal autopsy tissue reveals a frameshift mutation in OBSL1, consistent with a diagnosis of 3-M Syndrome. BMC genomics. 2015;16(Suppl 1):S12. doi: 10.1186/1471-2164-16-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis W, Krumlauf R. Homeobox genes and axial patterning. Cell. 1992;68:283–302. doi: 10.1016/0092-8674(92)90471-n. [DOI] [PubMed] [Google Scholar]
- Menasha J, Levy B, Hirschhorn K, Kardon NB. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: new insights from a 12-year study. Genetics in medicine : official journal of the American College of Medical Genetics. 2005;7:251–263. doi: 10.1097/01.gim.0000160075.96707.04. [DOI] [PubMed] [Google Scholar]
- Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes & development. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, Krabichler B, Speicher MR, Zschocke J, Trajanoski Z. A survey of tools for variant analysis of next-generation genome sequencing data. Briefings in bioinformatics. 2014;15:256–278. doi: 10.1093/bib/bbs086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JC, Lemons D, McGinnis W. Modulating Hox gene functions during animal body patterning. Nature reviews Genetics. 2005;6:893–904. doi: 10.1038/nrg1726. [DOI] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS genetics. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proenca CC, Gao KP, Shmelkov SV, Rafii S, Lee FS. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends in neurosciences. 2011;34:143–153. doi: 10.1016/j.tins.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G, Uz E, Buzas D, Gomes C, Patrier S, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nature genetics. 2011;43:601–606. doi: 10.1038/ng.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagoo GS, Butterworth AS, Sanderson S, Shaw-Smith C, Higgins JP, Burton H. Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects. Genetics in medicine : official journal of the American College of Medical Genetics. 2009;11:139–146. doi: 10.1097/GIM.0b013e318194ee8f. [DOI] [PubMed] [Google Scholar]
- Sander M, Paydar S, Ericson J, Briscoe J, Berber E, German M, Jessell TM, Rubenstein JL. Ventral neural patterning by Nkx homeobox genes: Nkx6.1 controls somatic motor neuron and ventral interneuron fates. Genes & development. 2000a;14:2134–2139. doi: 10.1101/gad.820400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A, German M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development. 2000b;127:5533–5540. doi: 10.1242/dev.127.24.5533. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilham MW, Oosterwegel MA, Moerer P, Ya J, de Boer PA, van de Wetering M, Verbeek S, Lamers WH, Kruisbeek AM, Cumano A, et al. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature. 1996;380:711–714. doi: 10.1038/380711a0. [DOI] [PubMed] [Google Scholar]
- Shamseldin HE, Tulbah M, Kurdi W, Nemer M, Alsahan N, Al Mardawi E, Khalifa O, Hashem A, Kurdi A, Babay Z, et al. Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome biology. 2015;16:116. doi: 10.1186/s13059-015-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashi V, McConkie-Rosell A, Rosell B, Schoch K, Vellore K, McDonald M, Jiang YH, Xie P, Need A, Goldstein DB. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genetics in medicine : official journal of the American College of Medical Genetics. 2014;16:176–182. doi: 10.1038/gim.2013.99. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Thomas T, Lin Q, Kirby ML, Brown D, Olson EN. Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nature genetics. 1997;16:154–160. doi: 10.1038/ng0697-154. [DOI] [PubMed] [Google Scholar]
- Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Lam P, Khromykh A, Iyer RK, Vockley JG, et al. Mutations in NOTCH1 cause Adams-Oliver syndrome. American journal of human genetics. 2014;95:275–284. doi: 10.1016/j.ajhg.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Ordulu Z, Pillalamarri V, Benson CB, Blumenthal I, Connolly S, Hanscom C, Hussain N, Pereira S, Picker J, et al. Clinical diagnosis by whole-genome sequencing of a prenatal sample. The New England journal of medicine. 2012;367:2226–2232. doi: 10.1056/NEJMoa1208594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O’Meara S, Latimer C, Dicks E, Menzies A, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nature genetics. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen Y, Tian F, Zhang J, Song Z, Wu Y, Han X, Hu W, Ma D, Cram D, et al. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clinical chemistry. 2014;60:251–259. doi: 10.1373/clinchem.2013.215145. [DOI] [PubMed] [Google Scholar]
- Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, Savage M, Platt LD, Saltzman D, Grobman WA, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. The New England journal of medicine. 2012;367:2175–2184. doi: 10.1056/NEJMoa1203382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Symmons O, Spitz F, Korbel JO. Phenotypic impact of genomic structural variation: insights from and for human disease. Nature reviews Genetics. 2013;14:125–138. doi: 10.1038/nrg3373. [DOI] [PubMed] [Google Scholar]
- Wellesley D, Dolk H, Boyd PA, Greenlees R, Haeusler M, Nelen V, Garne E, Khoshnood B, Doray B, Rissmann A, et al. Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. European journal of human genetics : EJHG. 2012;20:521–526. doi: 10.1038/ejhg.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: 2009. [Google Scholar]
- Yin T, Cook D, Lawrence M. ggbio: an R package for extending the grammar of graphics for genomic data. Genome biology. 2012;13:R77. doi: 10.1186/gb-2012-13-8-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nature reviews Genetics. 2015;16:172–183. doi: 10.1038/nrg3871. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.