

Abstract
Osteoarthritis (OA) is a chronic disease that degrades the joints and is often associated with increasing age and obesity. The two most common sites of OA in adults are the knee and hip joints. Increased mechanical stress on the joint from obesity can cause the articular cartilage to degrade and release damage associated molecular patterns (DAMPs). These DAMPs are involved in various molecular pathways that interact with nuclear factor-kappa B (NF-κB) and result in the transcription of inflammatory cytokines and activation of matrix metalloproteinases (MMP) that progressively destroy cartilage. This review focuses on the interactions and contribution to the pathogenesis and progression of OA through the DAMPs: high mobility group box 1 (HMGB-1), the receptor for advanced glycation end-products (RAGE), the alarmin proteins S100A8 and S100A9, and heparan sulfate. HMGB-1 is released from damaged or necrotic cells and interacts with toll-like receptors (TLRs) and RAGE to induce inflammatory signals, as well as behave as an inflammatory cytokine to activate innate immune cells. RAGE interacts with HMGB-1, advanced glycation end-products, and innate immune cells to increase local inflammation. The alarmin proteins are released following cell damage and interact through TLRs to increase local inflammation and cartilage degradation. Heparan sulfate has been shown to facilitate the binding of HMGB-1 to RAGE and could play a role in the progression of OA. Targeting these DAMPs may be potential therapeutic strategies for the treatment of OA.
Keywords: Osteoarthritis, Cartilage damage, Damage associated molecular patterns (DAMPs), Alarmins, Inflammation, Targeted therapy
1. Introduction
Osteoarthritis (OA) is a significant cause of joint pain and disability [1]. There are many risk factors for OA including age, joint trauma, biomechanics, and obesity [2]. OA causes loss of the joint space through degradation of articular cartilage. The unbalanced process of degradation and repair is the ultimate process of OA. Understanding how degradation occurs in the joint is paramount to understanding how the disease progresses, and how to best proceed with treatment. Studies have demonstrated the increased presence of damage associated molecular proteins (DAMPs) in joints affected by OA and their probable role in the pathogenesis of the disease [2–6]. However, other factors such as obesity and increased mechanical loads have also been associated with its pathogenesis [7].
The damage caused by mechanical stress on the joint results in the release of DAMPs that trigger an innate immune response [2]. One known cause of an increased mechanical stress on joints is obesity. Therefore, obesity may facilitate the release of DAMPs to potentiate OA. These DAMPs can originate from the extracellular matrix, intracellular alarmins, plasma proteins, and crystal deposits [2]. DAMPs interact with pattern recognition receptors (PRRs) to initiate inflammation. Overexpression of DAMPs in joints has been linked to the pathogenesis of OA [2]. The aim of this review is to provide a summary of five DAMPs associated with OA in the knee and hip joint, as well as summarize possible therapies to affect the role of those DAMPs in the disease (Fig. 1).
Fig. 1. The role of DAMPs in the pathogenesis of osteoarthritis.
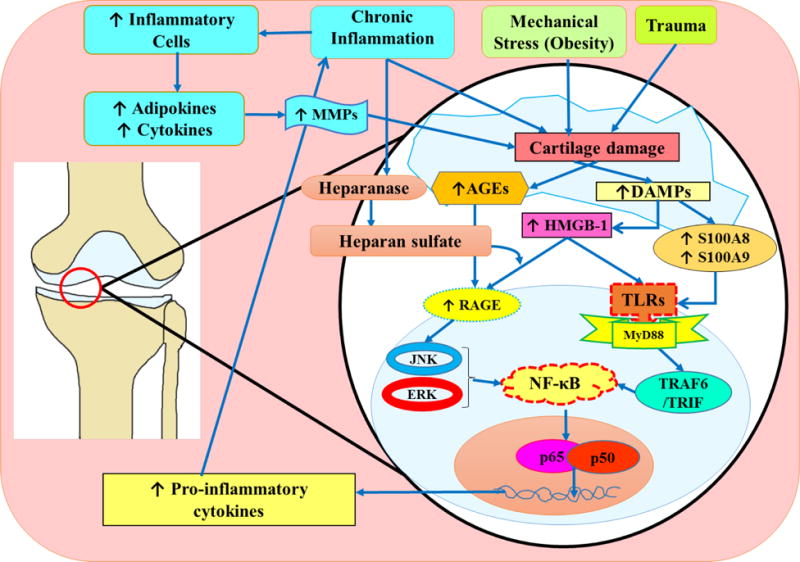
Cartilage damaged by trauma, mechanical stress such as obesity, and chronic inflammation initiates the release of DAMPs including HMGB-1, AGEs, and S100A8 and S100A9 calgranulin proteins. HMGB-1 released from the nucleus of the damaged cell into the extracellular environment binds to both TLRs and RAGE. The binding of HMGB-1 and RAGE can be facilitated by the proteoglycan and heparan sulfate. Heparan sulfate is activated via interactions with heparanase. Homodimers of S100A8 and S100A9 have been shown to have a proinflammatory effect, while the S100A8 and S100A9 heterodimer has not been shown to facilitate inflammation. Binding of HMGB-1with RAGE and TLRs, and calgranulin with TLRs initiates a signaling cascade involving JAK, ERK, MyD88 TRAF/TRIM pathway and acts through the transcription factor NF-κB. This results in secretion of pro-inflammatory cytokines. These cytokines are translated and released from the host cell to interact with immune cells such as macrophages leading to secretion of additional inflammatory cytokines to recruit more immune cells, as well as MMPs that break down collagen and degrade cartilage. The degradation of cartilage triggers the release of additional DAMPs and the process continues. DAMPs, damage associated molecular patterns; JNK, Jun amino-terminal kinases; HMGB-1, high mobility group box-1; NF-κB, nuclear factor-kappa beta; RAGE, receptor for advanced glycation end product; TLRs, toll like receptors;
2. Pathophysiology of Osteoarthritis
The two most common locations of OA are the knee and hip [8]. However, each joint experiences and handles increased mechanical loads differently. Unbalanced mechanical loading on the joint may lead to chondral damage in the knee [1]. It is not only the magnitude of the load but also the location of the load that influences damage, as more damage is observed on the medial surface of the tibia [9]. Knee adduction in the gait has been associated with femoral cartilage changes, and external knee flexion has been associated with more tibial cartilage changes [10]. Obesity has been associated with increased mechanical loading on the joint, as well as altering one’s gait. Like the knee joint, the hip is more likely to be affected by OA in obese patients, but the relationship between obesity and OA is not as strong in the hip as in the knee [11]. There is an elevated risk of hip OA in men involved in heavy manual work such as construction and farming, implying the hip is better at bearing increased load compared to the knee but is still vulnerable to increased weight [11]. Increased body weight in obese patients is an increase in fat and muscle mass without a change in muscular strength. In order to compensate for the increased load, gait is adjusted and that adjustment results in the location of load changing within the joint, which adds to the degeneration [7]. The extra weight in obese patients may compress mechanoreceptors on chondrocytes and illicit a local proinflammatory response within the joint that further contributes to OA. Additionally, obesity leads to the release of local and systemic proinflammatory markers, and in fact, low-grade systemic inflammation is considered a hallmark of obesity [7]. Obesity contributes to OA through two pathways, a weight dependent pathway where the mechanical load is increased on the joint, and a weight-independent pathway that results in both systemic and local inflammation [12]. Heavy mechanical load remains the largest contributor to the development of OA in the knee and hip joint, along with the cycle of inflammation and repair.
3. Damage Associated Molecular Patterns
DAMPs play a role in OA by binding to various receptors such as: toll-like receptors (TLRs), advanced glycated end product receptors (RAGE), and the NLRP3 inflammasome on macrophages or chondrocytes, and activate signaling cascades such as: nuclear factor-kappa B (NF-κB) and mitogen -activated protein kinases (MAPK) pathways (Fig. 1). The NF-κB pathway is a regulator for inflammatory processes in chondrocytes, and allows those cells to express matrix metalloproteinase (MMPs), nitrous oxide synthase 2 (NOS2), cyclooxygenase-2 (COX2) and interleukin-1 (IL-1) which can cause apoptosis, shift in cell phenotype, and over-expression of proinflammatory genes that ultimately contribute to further destruction of cartilage [13]. Activation of the MAPK pathway via mechanical and inflammatory stimuli works through ERK, JNK and p38 kinase signaling cascade to induce gene expression of catabolic and inflammatory genes that participate in the induction of MMP-13, a collagenase with a higher selectivity toward collagen-II [13]. The majority of DAMPs implicated in OA are components of the extracellular matrix such as biglycan, fibronectin, heparin sulfate, tenascin-c, and hyaluronan, while others are cellular proteins such as high mobility group box-1 (HMGB-1), heat shock proteins (HSP), and S100A protein families, while others are plasma proteins like fibronectin, or crystals like basic calcium phosphate [2, 14]. Biglycan, fibrinogen, hyaluronan, fibrinogen, HMGB-1, HSPs, tenascin-c, and S100 family proteins bind to TLRs and RAGE, and basic calcium phosphate and other crystals interact with NLRP3 inflammasome [2, 13]. Since upregulation of RAGE has been associated with OA, this review will focus on the role of RAGE in OA, as well as its ligands HMGB-1, S100A protein family, and heparin sulfate [15].
3a. High Mobility Group Box-1
HMGB-1 is a non-histone nuclear protein that facilitates the binding of transcription factors and aids in the maintenance of nucleosomal structure [16]. HMGB-1 is present in all eukaryotic cells, and its release from the nucleus is associated with an inflammatory response, meaning the molecule has a dual role: as a nuclear protein, and as a cytokine [17]. HMGB-1 can be released following damage to cells, like necrosis or hypoxia, or following stimulation from cytokines like tumor necrosis factor (TNF)-α [18]. Since HMGB-1 is nearly ubiquitous, and its extracellular presence can trigger an inflammatory response, HMGB-1 is thought to be a large contributor to the pathogenesis of chronic inflammatory diseases like OA [3]. A study by Terada et al. [16] found that significantly increased ratios of HMGB-1 positive cells were present with higher grades of OA in knee cartilage, relative to cartilage normal joints, and in grade 1 OA joints. However, HMGB-1 is not just an expresser of inflammation but also acts as a trigger that stimulates many other pro-inflammatory cascades [16]. IL-1β and TNF-α have been shown to stimulate the release of HMGB-1 from chondrocytes, where HMGB-1 will then bind to TLRs and RAGE on macrophages and other neighboring cells [16]. HMGB-1 binding to TLR-4 will proceed down the MyD88 dependent pathway, and activate transcription factors NF-κB, AP (activator protein)-1 and MAPK [14] (Fig. 1). On macrophages, this pathway will induce the production of IL-1β and TNF-α, which will, in turn, increase inflammation and stimulate more release of HMGB-1 on chondrocytes [16]. Additionally, IL-1β and TNF-α will upregulate TLR-2, another receptor for HMGB-1, suppress extracellular matrix synthesis, and induce the expression of other cytokines such as IL-6 and IL-8 which promote cartilage catabolism by producing matrix-degrading enzymes [19]. HMGB-1 has been known to induce the release NO, MMP-3, MMP-13, and a disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS-5) via secretion by chondrocytes, and therefore exhibits the same proinflammatory responses observed by TNF-α and IL-1β [20]. HMGB-1 is involved in a cyclic proinflammatory process which the release of HMGB-1 can be triggered by chondrocyte damage or the release of cytokines such as IL-1β. The binding of released HMGB-1 with TLRs and RAGE will induce a signaling cascade largely through NF-κB pathway. This, in turn, triggers the expression of more proinflammatory cytokines, such as IL-1β which will continue the inflammatory cycle by releasing more HMGB-1. TLR2, TLR4 and their associated molecule MyD88 with HMGB-1 are central to chondrocyte matrix catabolism and chondrocyte hypertrophy, however HMGB-1 interactions with RAGE in addition to TLRs comprise the total chondrocyte catabolic processes triggered by HMGB-1 [20]. Since HMGB-1 is so involved in chondrocyte inflammation, especially in OA, HMGB-1 can be used as a marker to assess the severity of cartilage damage in OA [18]. RAGE, in addition to binding to HMGB-1, is upregulated in OA to a significant degree and contributes to the disease [16] (Fig. 1).
3b. Receptor of Advanced Glycation End-product
RAGE is a member of immunoglobulin family, and acts a receptor of advanced glycation end-products, and is expressed in a diverse array of cell types to include macrophages and endothelial cells [21]. However, an increase in RAGE expression is noted in OA and its ability to stimulate NF-κB and MAPK pathways leading to MMP-13 stimulation suggest that RAGE and its ligand play a direct role in the degradation of the joint [21]. Advanced glycated end-products (AGEs), a ligand for RAGE, are formed by glycation of proteins with sugars; their accumulation is related to protein turnover. The long life of collagen contributes to an increase in AGEs, especially in cartilage, and may explain an age-related predisposition to OA [4]. Increasing AGE would increase signaling cascades initiated by RAGE and lead to the production of MMPs and other proinflammatory cytokines, possibly contributing to OA (Fig. 1). In addition to AGEs, RAGE also binds HMGB-1, S100 alarmins, which induced oxidative stress and stimulates MMP-13 production to degrade collagen II in cartilage [21]. HMGB-1 interacts with RAGE and initiates signaling pathways through NF-κB that propagate further expression of RAGE through an amplification loop in addition to inducing inflammatory cytokines [22]. MMP-3 expression in chondrocytes has been shown to proceed through HMGB-1 and RAGE signaling; binding of RAGE with HMGB-1 can stimulate c-Src/Akt/NF-κB cascade leading to MMP-3 production [22]. RAGE has been shown to play a role in OA, with noted increase where its ligand is more prevalent; in aging populations where AGEs are increased, as well as in patients where HMGB-1 and S100 alarmins are increased [4] (Fig. 1). Decreased RAGE density is noted in healthy joints [21]. NF-κB can bind to the RAGE promoter; any stimuli that increase NF-κB activity can activate RAGE transcription [21]. Ligands such as HMGB-1 and S100 that can interact through TLRs and the NF-κB pathway can induce RAGE production, as well as cytokines such as IL-1β [21]. Furthermore, inhibition of RAGE with sRAGE, a truncated form of RAGE, suppressed the development of OA in mice models [21]. This suggests that RAGE plays a vital role in the pathogenesis and development of OA, and can help explain part of the reason aging populations are at higher risk for OA via the increase of AGEs. It has been revealed that chondrocytes express several types of RAGE that have undergone post-translational modifications, with the 55-kd form more prevalent in OA [21] (Fig. 1).
RAGE is expressed in a variety of cell types to include macrophages and chondrocytes. Inflammatory cytokines such as TNFα and IL-1β and DAMPs such as HMGB-1 and S100 proteins can induce RAGE expression through NF-κB/ERK pathways [21]. Binding of RAGE will further activate NF-κB pathways that induce oxidative stress and MMPs, to include MMP-13 in chondrocytes, as well as cytokines TNFα and IL-1β. Those molecules contribute to further degradation of cartilage as well as increase general inflammation and participate in an amplification loop that upregulates RAGE. Age and obesity are considered two of the greatest risk factors for OA, and since an increase in AGEs are correlated in aging populations, and obesity is correlated with increased release of DAMPs such as HMGB-1, both factors contribute to upregulation of RAGE [4]. RAGE upregulation may be vital to the pathogenesis of OA, and suppression of RAGE could lead to therapeutic outcomes in patients with OA.
3c. S100A8 and S100A9
S100A8 and S100A9 are calcium binding proteins that are involved in pathways regulating the cytoskeleton, movement, migration, and adhesion, as well as help in regulating the enzyme activities in a calcium-dependent manner [6, 23]. However, when released from the cell they act as proinflammatory cytokines known as alarmins [6]. S100A8 and S100A9 have been found in granulocytes, monocytes, and macrophages, and the increase of these proteins has been found in patients with inflammatory arthritis [6]. The alarmins can be secreted as homodimers and heterodimers and the heterodimer has been known to enhance leukocyte recruitment to the local environment. S100A8/9 complex can bind to endothelial surface receptors and interact with surface receptors such as heparin sulfate, and RAGE but the major receptor is TLR 4 [23] (Fig. 1). Chondrocytes have also been shown to express the S100A8 and S100A9 alarmins following stimulation via IL-1 and their increase is believed to play a role in cartilage degradation [6].
Cartilage is not the only tissue affected by OA. OA is driven by pathologic changes in the synovium and subchondral bone as well, with 50% of OA patients showing synovial inflammation in the early and late stages of the disease [24]. Studies by Van Lent et al. [24] showed that macrophage activation was crucial in regulating synovial inflammation and subsequent cartilage degradation in osteoarthritis [24]. While OA is typically thought of as a disease of cartilage degradation, more evidence is suggesting that synovial inflammation and synovial macrophage in synovitis play a role in cartilage degeneration [5, 24]. The synovial macrophages play a significant role in cartilage degradation, as the removal of these macrophages prior to the introduction of experimental OA resulted in almost complete halting of MMP induced destruction and osteophyte formation [24]. Extracellular S100A8 has been shown to stimulate the expression of various MMPs and is thought to be a major mediator of cartilage degradation [23]. Therefore, the current evidence suggests that extracellular alarmins released by chondrocytes and contributes to cartilage degradation and advancement of OA, however, their role in the onset and progression of OA remain unknown [6]. Their net catabolic effect is achieved through TLR-4 signaling and is significantly higher in OA chondrocytes than normal chondrocytes [5] (Fig. 1).
A study by Zreiqat et al. [6] found that the regulation of S100A8 and S100A9 secretion from chondrocytes could play a role in the early stages of OA and highlight differences between OA and other inflammatory joint diseases. S100A8 and S100A9 as homodimers promote increased catabolism, while the heterodimer does not tend to affect metabolism, so a dysregulation of the two proteins has potential pathogenic effects [6]. Furthermore, immunostaining in surgically-induced mouse OA of the alarmins was restricted to load-bearing cartilage which suggests that mechanical factors, rather than systemic factors such as cytokines, play a more significant role in the metabolism of the alarmins in OA [6]. The increase of the extracellular alarmins results in an increase in MMPs as well as stimulates the release of IL-6, IL-8, and IL-1β, which may attract granulocytes to the cartilage and IL-8 could initiate a positive feedback loop by increasing the number of inflammatory cells in the joint which release more S100A8/9 proteins [5] (Fig. 1). Patients with increased S100A8 and S100A9 levels were shown to have a highly increased risk of developing severe OA, and since S100A8/9 remains at high levels for a prolonged period of time, they may be effective biomarkers for assessing and predicting the cartilage destruction in OA [24].
S100A8/9 proteins can be released from synovial macrophages after macrophage activation from other cytokines, and play role in synovitis and cartilage degradation. Chondrocytes also release the alarmins, but evidence suggests chondrocytes depend more on mechanical stressors to initiate the release of alarmins than cytokines. Since the alarmins have increased levels in OA, and their levels remain high for prolonged periods of time, their potential as a biomarker that judges and predicts the severity of OA could have therapeutic value.
3d. Heparan Sulfate
Heparan sulfate proteoglycan surface receptors (HSGP) are among the most abundant receptors and serve to modulate the binding of extracellular proteins to other receptors on the cell [25]. Since HSGPs serve as modulators, they can affect ligand/receptor encounters by altering ligand concentrations or receptor oligomerization [25]. Heparan sulfate is non-selective molecule and can also facilitate the binding of cellular proteins to matrix components, allowing synovial macrophages to bind to cartilage in arthritis [26]. HMGB-1 and RAGE have also been shown to bind heparan sulfate [27]. HMGB-1 is heparan sulfate-dependent signaling factor but is controlled at the RAGE level opposed to HMGB-1 [27]. Evidence suggests that RAGE and heparan sulfate are preformed complexes at the endothelial surface that help facilitate the binding of RAGE and HMGB-1 [27]. Since HSGPs are non-selective and act on a variety of DAMPs to include RAGE, HMGB-1, and heat shock proteins, HSGPs may serve as vital coreceptor during inflammatory processes in damaged tissue [27]. HSGPs may also serve to localize that inflammation; HMGB-1 may bind heparan sulfate in the extracellular matrix thereby preventing greater diffusion of the DAMP (Fig. 1).
Heparan sulfate is a protein that exists in the ECM and serves to modulate binding between ligands and receptors. HSGP is a non-selective molecule that may play a role in local inflammation of damage tissue by binding HMGB-1 and RAGE. Regulation appears to occur at the level of RAGE, where preformed complexes between heparan sulfate and RAGE seem to facilitate binding of RAGE ligands such as HMGB-1. Additionally, heparan sulfate may aid in localization of inflammation by binding HMGB-1 in the ECM to restrict further diffusion of the molecule.
4. Therapeutic Aspects of DAMPs in OA
There are many therapies and proposed algorithms to treat damaged articular cartilage, but current treatments can often result in unsuccessful clinical outcomes [28]. More effective restorative therapies are needed to more effectively treat OA. One such therapy to treat cartilage loss can be the construction of scaffolds to create a substitute for native cartilage [28]. Marrow stimulation can sometimes be ineffective because the mature structure of cartilage does not form correctly. Alternatives to traditional marrow stimulation techniques include stem cells introduced to polymer scaffolds that are biocompatible, biodegradable and that allow for cell proliferation and adhesion present a viable option for interventionist therapy [28]. Chondrocytes should not be the cell of choice to populate the scaffold because the expansion of chondrocytes has been shown to lead to dedifferentiation [28]. Instead, mesenchymal stem cells present the most viable option to populate polymer scaffolds to create substitute articular cartilage [28]. Other potential therapies include weight management and exercise, controlling inflammation, drug therapy, mitigating DAMPs and signaling cascades, and addressing occupational risk factors that are associated with OA. The goals for these therapies are to replace arthritic cartilage in an attempt to reverse the damage caused by OA and reduce the effects of DAMPs in the joint through targeted molecular therapy and the removal risk factors that increase the activity of DAMPs such as obesity.
The greatest stressors leading to the development of OA are biomechanical and are largely associated with increasing BMI and body weight. In fact, for every 5 kg of weight gained, there is an increased 36% chance of developing OA [7]. The most therapeutic treatment for OA in patients with high body mass index (BMI) is to lose weight. Losing weight can lead to a decrease in pain and less fear of movement and a loss of 10% body weight have been associated with moderate to large clinical improvements in joint pain [7]. The most successful programs have been a combination of aerobic exercise to lose body weight and resistance exercises to increase muscle strength around the joint to ameliorate arthritis associated pain [7]. Increased mechanical load along with increased inflammation within joints are two of effects from increased body fat. A loss of weight can decrease mechanical load and possibly fix altered gait, both of which are implicated in cartilage degradation. Decreased load and stresses on the chondrocytes translate to a lower expression of DAMPs than those with OA [2] (Fig. 2).
Fig. 2. Potential therapeutic targets in OA.
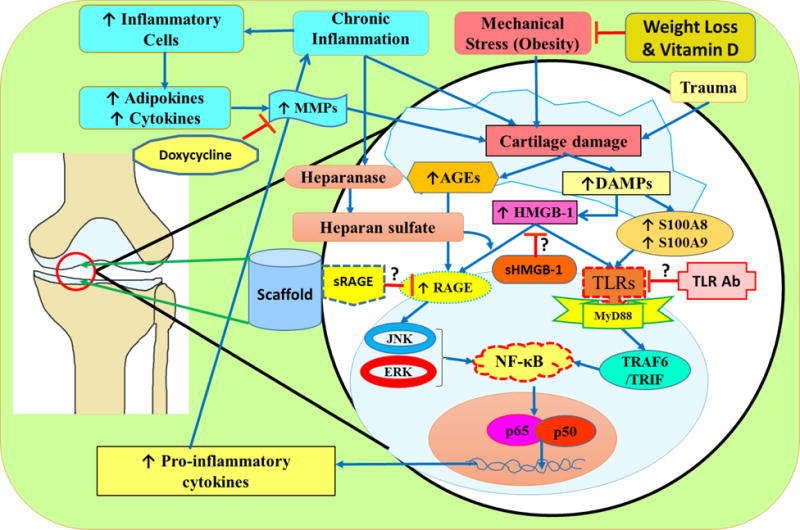
Possible targets for therapeutic effects in OA include risk factors for the disease, DAMPs such as HMGB-1 and RAGE, TLRs, MMPs, and the construction of a protein scaffold to substitute damaged cartilage. Weight loss or vitamin D supplementation has been shown to mitigate mechanical stress on the joint, and therefore cause less DAMPs and inflammatory signals to be released; decreasing cartilage degradation and progression of OA. Glycyrrhizin has been shown to be an extracellular inhibitor of HMGB-1, and inhibits binding of HMGB-1 to RAGE and TLRs. This inhibition mitigates the transcription of genes that encode proinflammatory cytokines and mitigate cartilage degradation. Similarly, sRAGE and TLR antibodies inhibit RAGE and TLRs respectively from triggering signaling cascades that contribute to the progression of OA. Drugs such as doxycycline and TIMPs inhibit the action of MMPs, and therefore, halt the effects of collagenase and mitigate cartilage degradation. A protein scaffold implanted with mesenchymal stem cells could act as a cartilage substitute and enhance proliferation of chondrocyte to replace damaged cartilage in the joint.
Controlling inflammation and muscle weakness can be another viable option to treat OA, and some relief can be achieved by removing synovial macrophages or treatment with vitamin D [24, 29]. Vitamin D can have therapeutic effects in patients with OA because vitamin D modulates mechanical issues by addressing quadriceps weakness [29]. Furthermore, vitamin D deficiency has been associated with OA, with vitamin D-deficient men twice as likely to have hip OA compared to those with normal levels of vitamin D [30]. Modulating strength of the quadriceps is important because stronger quadriceps translates to a stronger knee joint, and those with knee OA were found to have significantly weaker quadriceps, hamstrings, and hip muscles compared with age-matched controls [31]. Quadriceps weakness is considered an early symptom of knee OA because weakness in the muscle increases load-bearing on the knee joint, and this increased load contributes to cartilage damage and loss [29]. It was believed that vitamin D deficiency also associated with an increase in inflammatory cytokines, but data by Barker et al. [29] suggests that a decrease in vitamin D with an increase in γ-tocopheral leads to increased inflammatory cytokines; while purely vitamin D deficiency associated with quadriceps dysfunction and decreasing γ-tocopheral in vitamin D sufficient patients could have therapeutic inflammatory effects [29]. Since vitamin D deficiency is associated with incidence and progression of OA, increasing vitamin D levels can have therapeutic effects by increasing quadriceps strength and reducing inflammation in the joint [32]. While not a true interventionist therapy that actively fights against cartilage damage, vitamin D can reduce risk factors and slow the progression of the disease. Weight loss and vitamin D can relieve compressive forces on load bearing joints, but other methods should be used to mitigate the effects of DAMPs in the progression of OA (Fig. 2).
MMPs degrade proteins in the extracellular matrix, and MMP-13 has been shown to be selective for collagen II, an important molecule in articular cartilage [13]. MMP-13 can be expressed through signal cascades initiated by binding of HMGB-1, RAGE, S100A8, and S100A9 to their appropriate ligands. Targeting MMPs does not affect DAMPs in OA, but does serve to mitigate the deleterious effects of DAMPs. Promising results have been achieved through MMP inhibitor doxycycline [2] (Fig. 2). While MMP inhibitors do not directly target DAMPs, MMP inhibitors can attenuate the effects of MMPs produced as a result of DAMPs activated downstream signaling pathways (Fig.1). MMP collagenases can be inhibited by tissue inhibitors of metalloproteinases (TIMPs) namely TIMP-1, TIMP-2, TIMP-3 and TIMP-4 [33]. Interestingly, drugs that target the inflammatory cytokines TNF-α and IL-1β were not shown to be effective in OA [2]. Instead, therapies that target the cytokine effects of HMGB-1, RAGE and TLRs that participate in the MyD88 dependent pathway are all viable targets for therapy.
HMGB-1 A box peptide has been found to mitigate the symptoms of OA by inhibiting the cytokine activity of HMGB-1 [34, 35]. Additionally, cytokine-release inhibitory drugs (CRIDs) have been shown to interfere with HMGB-1 release from the nucleus into the extracellular space, effectively preventing the inflammatory role of the molecule [35] (Fig. 2). Other steroid and nonsteroidal anti-inflammatory drugs (NSAID) failed to prevent HMGB-1 release and the subsequent signaling cascades [35]. Glycyrrhizin, a chelator of HMGB-1 derived from licorice plant, binds and stops HMGB-1 activities [36] (Fig. 2). Fortunately, the DNA binding capability of HMGB-1 is mildly blocked while glycyrrhizin more significantly blocks HMGB-1 cytokine activity [35]. Glycyrrhizin may show the greatest therapeutic potential by blocking directly interfering with HMGB-1 extracellular cytokine activities, while CRIDs and HMGB-1 A box peptides can only block the release of HMGB-1 from activated macrophages, but fail to act on HMBG-1 that is passively released by necrotic tissue, or upon extracellular HMGB-1 [35]. RAGE, a receptor for HMGB-1 and integrin binding site for inflammatory cells can be inhibited by using soluble RAGE or sRAGE [37] (Fig. 2).
sRAGE acts as a competitive inhibitor to RAGE and can completely inhibit downstream signaling and integrin binding [37]. Animal studies have shown promise using sRAGE to inhibit RAGE activity, but the long term effects of a RAGE blockade and the benefits and costs of blocking RAGE in humans are currently unknown [37] (Fig. 2). TLRs and the MyD88 pathway can be targeted for therapy via antibodies or the deubiquitinating enzyme A20 [38]. A20 limits the signaling cascade associated with TLRs and MyD88 and reduces inflammatory responses and the release of other cell signals such as HMGB-1 [38] (Fig. 2).
Osteoarthritis of the knee and hip have slightly different risk factors, with OA of the hip being more associated with heavy work or manual labor opposed to increased weight on the joint [1]. However, those laborers who sat greater than 2 hours a day had a decreased risk for developing OA in the hip joint [1]. The greatest complaint of the patient with OA is pain; identifying osteoarthritic pain earlier in the progression of the disease can have therapeutic effects. Weight-bearing-bending activities were hypothesized to cause pain in patients with OA of the knee, and activities such as climbing stairs are thought to be among the first to become painful [39]. Progression of knee OA is associated with pain while walking, then standing, then sitting, and finally pain while lying down [39]. Further studies to determine if pain associated with climbing stairs can be used as an effective screening tool for the development of OA.
Obesity has shown to be a significant risk factor in the development of OA due to increased load on joints and decreased musculature with fatty infiltration that leads to degradation of cartilage and the release of DAMPs. The most effective therapeutic strategies have been weight-loss and MMP inhibitors [2, 33]. Weight loss both relieves pressure on joints and decreases overall inflammation, while MMP inhibitors mitigate the degradative effects of DAMPs in the joint. Therapeutic options for treatment of OA include weight loss, drug therapy, scaffold construction, and mitigating occupational hazards and stressors. Weight loss and exercise appear to be the most effective therapies for OA because they are the most effective at reducing mechanical loads on joints and mitigating the progress of OA [7, 40]. Doxycycline and TIMPs to inhibit MMPs and stop cartilage degradation along with vitamin D to help strengthen quadriceps muscles may be effective drug therapy along with antibodies and molecules to target specific cells or cytokines [2, 24, 29, 33]. Glycyrrhizin to inhibit HMGB-1[35, 36], sRAGE to inhibit RAGE [37], and specific antibodies along with the enzyme A20 to remove or inhibit synovial macrophages and attenuate TLR and MyD88 signaling within the joint may help in decreasing the cartilage loss and OA progression [38]. Construction of a protein scaffold with mesenchymal stem cells to create substitute cartilage [28, 41] may be used as a better solution to osteoarthritis along with decreasing DAMP activity, because it could replace arthritic cartilage and return some function while alleviating pain opposed to focusing on preventing or slowing the disease like other therapies (Fig. 2).
5. Conclusion
OA is a significant cause of joint pain and disability that degrades articular cartilage. DAMPs such as HMGB-1, RAGE, S100A8, S100A9, and heparan sulfate have been associated with the progression of OA by activating and recruiting cells of the innate immune system and inducing the expression of MMPs. The activated macrophages will secrete cytokines that damage chondrocytes as well as clear away extracellular matrix within the joint. MMPs work in a similar manner by degrading collagen within the articular cartilage. The damage caused to chondrocytes by the activated macrophages will lead to the release of more DAMPs, creating a positive feedback loop. This loop results in a continued destruction of cartilage. This review examined the role of five DAMPs associated with OA in the knee and hip joints, as well as reviewed possible therapies to target or mitigates the effects of those DAMPs in an attempt to treat OA.
Acknowledgments
Funding
This work was supported by research grants R01 HL112597, R01 HL116042, and R01 HL120659 to DK Agrawal from the National Heart, Lung and Blood Institute, National Institutes of Health, USA. The content of this review article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest
As the corresponding author, I declare that this manuscript is original; that the article does not infringe upon any copyright or other proprietary right of any third party; that neither the text nor the data have been reported or published previously. All the authors have no conflict of interest and have read the journal’s authorship statement.
References
- 1.Yucesoy B, Charles LE, Baker B, Burchfiel CM. Occupational and genetic risk factors for osteoarthritis: A review. Work. 2015;50(2):261–273. doi: 10.3233/WOR-131739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. 2013;5(2):77–94. doi: 10.1177/1759720X12467868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ulloa L, Batliwalla FM, Andersson U, Gregersen PK, Tracey KJ. High mobility group box chromosomal protein 1 as a nuclear protein, cytokine, and potential therapeutic target in arthritis. Arthritis Rheum. 2003;48(4):876–881. doi: 10.1002/art.10854. [DOI] [PubMed] [Google Scholar]
- 4.Chayanupatkul M, Honsawek S. Soluble receptor for advanced glycation end products (sRAGE) in plasma and synovial fluid is inversely associated with disease severity of knee osteoarthritis. Clin Biochem. 2010;43(13–14):1133–1137. doi: 10.1016/j.clinbiochem.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Schelbergen RF, Blom AB, van den Bosch MH, Slöetjes A, Abdollahi-Roodsaz S, Schreurs BW, et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on toll-like receptor 4. Arthritis Rheum. 2012;64(5):1477–1487. doi: 10.1002/art.33495. [DOI] [PubMed] [Google Scholar]
- 6.Zreiqat H, Belluoccio D, Smith MM, Wilson R, Rowley LA, Jones K, et al. S100A8 and S100A9 in experimental osteoarthritis. Arthritis Res Ther. 2010;12(1):R16. doi: 10.1186/ar2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vincent HK, Heywood K, Connelley J, Hurley RW. Weight Loss and Obesity in the Treatment and Prevention of Osteoarthritis. Am Acad Phys Med Rehabil. 2012;4(50):S59–S67. doi: 10.1016/j.pmrj.2012.01.005.Weight. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turkiewicz A, Petersson IF, Björk J, Hawker G, Dahlberg LE, Lohmander LS, Englund M. Current and future impact of osteoarthritis on health care: A population-based study with projections to year 2032. Osteoarthr Cartil. 2014;22(11):1826–1832. doi: 10.1016/j.joca.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 9.Favre J, Erhart-Hledik JC, Chehab EF, Andriacchi TP. Baseline ambulatory knee kinematics are associated with changes in cartilage thickness in osteoarthritic patients over 5 years. J Biomech. 2016:1–6. doi: 10.1016/j.jbiomech.2016.04.029. [DOI] [PubMed] [Google Scholar]
- 10.Varady NH, Grodzinsky AJ. Osteoarthritis year in review 2015: Mechanics. Osteoarthr Cartil. 2016;24(1):27–35. doi: 10.1016/j.joca.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris EC, Coggon D. Hip Osteoarthritis and Work. 2016;29(3):462–482. doi: 10.1016/j.berh.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wen L, Kang JH, Yim YR, Kim JE, Lee JW, Lee KE, et al. Associations between body composition measurements of obesity and radiographic osteoarthritis in older adults: Data from the Dong-gu Study. BMC Musculoskelet Disord. 2016;17(1):192. doi: 10.1186/s12891-016-1040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldring MB. Articular Cartilage Degradation in Osteoarthritis. HSS J. 2012;8(1):7–9. doi: 10.1007/s11420-011-9250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010;14(11):2592–2603. doi: 10.1111/j.1582-4934.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larkin DJ, Kartchner JZ, Doxey AS, Hollis WR, Rees JL, Wilhelm SK, et al. Inflammatory markers associated with osteoarthritis after destabilization surgery in young mice with and without Receptor for Advanced Glycation End-products (RAGE) Front Physiol. 2013;4:121. doi: 10.3389/fphys.2013.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terada C1, Yoshida A, Nasu Y, Mori S, Tomono Y, Tanaka M, et al. Gene expression and localization of high-mobility group box chromosomal protein-1 (HMGB-1) in human osteoarthritic cartilage. Acta Med Okayama. 2011;65(6):369–77. doi: 10.18926/AMO/47262. [DOI] [PubMed] [Google Scholar]
- 17.Hamada T, Torikai M, Kuwazuru A, Tanaka M, Horai N, Fukuda T, et al. Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthritis Rheum. 2008;58(9):2675–2685. doi: 10.1002/art.23729. [DOI] [PubMed] [Google Scholar]
- 18.Heinola T, Kouri VP, Clarijs P, Ciferska H, Sukura A, Salo J, Konttinen YT. High mobility group box-1 (HMGB-1) in Osteoarthritic cartilage. Clin Exp Rheumatol. 2010;28(4):511–518. doi:297 [pii] [PubMed] [Google Scholar]
- 19.Liu-Bryan R. Synovium and the innate inflammatory network in osteoarthritis progression topical collection on osteoarthritis. Curr Rheumatol Rep. 2013;15(5) doi: 10.1007/s11926-013-0323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu-Bryan R, Terkeltaub R. Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous toll-like receptor 2/toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein. Arthritis Rheum. 2010;62(7):2004–2012. doi: 10.1002/art.27475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loeser RF, Yammani RR, Carlson CS, Chen H, Cole A, Im HJ, Bursch LS, Yan SD. Articular chondrocytes express the receptor for advanced glycation end products: Potential role in osteoarthritis. Arthritis Rheum. 2005;52(8):2376–2385. doi: 10.1002/art.21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou CH, Fong YC, Tang CH. HMGB-1 induces IL-6 production in human synovial fibroblasts through c-Src, Akt and NF-κB pathways. J Cell Physiol. 2011;226(8):2006–2015. doi: 10.1002/jcp.22541. [DOI] [PubMed] [Google Scholar]
- 23.Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H, et al. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and 38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther. 2006;8(3):R69. doi: 10.1186/ar1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Lent PL, Blom AB, Schelbergen RF, Slöetjes A, Lafeber FP, Lems WF, et al. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 2012;64(5):1466–1476. doi: 10.1002/art.34315. [DOI] [PubMed] [Google Scholar]
- 25.Park PW, Reizes O, Bernfield M. Cell surface heparan sulfate proteoglycans: Selective regulators of ligand-receptor encounters. J Biol Chem. 2000;275(39):29923–29926. doi: 10.1074/jbc.R000008200. [DOI] [PubMed] [Google Scholar]
- 26.Korb-Pap A, Stratis A, Mühlenberg K, Niederreiter B, Hayer S, Echtermeyer F, et al. Early structural changes in cartilage and bone are required for the attachment and invasion of inflamed synovial tissue during destructive inflammatory arthritis. Ann Rheum Dis. 2012;71:1004–1011. doi: 10.1136/annrheumdis-2011-200386. [DOI] [PubMed] [Google Scholar]
- 27.Xu D, Young J, Song D, Esko JD. Heparan sulfate is essential for high mobility group protein 1 (HMGB1) signaling by the receptor for advanced glycation end products (RAGE) J Biol Chem. 2011;286(48):41736–41744. doi: 10.1074/jbc.M111.299685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cucchiarini M, Henrionnet C, Mainard D, Pinzano A, Madry H. New trends in articular cartilage repair. J Exp Orthop. 2015;2(1):8. doi: 10.1186/s40634-015-0026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barker T, Henriksen VT, Rogers VE, Aguirre D, Trawick RH, Lynn Rasmussen G, Momberger NG. Vitamin D deficiency associates with ??-tocopherol and quadriceps weakness but not inflammatory cytokines in subjects with knee osteoarthritis. Redox Biol. 2014;2(1):466–474. doi: 10.1016/j.redox.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heidari B, Heidari P, Hajian-Tilaki K. Association between serum vitamin D deficiency and knee osteoarthritis. Int Orthop. 2011;35(11):1627–1631. doi: 10.1007/s00264-010-1186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heidari B, Javadian Y, Babaei M, Yousef-Ghahari B. Restorative Effect of Vitamin D Deficiency on Knee Pain and Quadriceps Muscle Strength in Knee Osteoarthritis. Acta Med Iran. 2015;53(8):466–70. http://www.ncbi.nlm.nih.gov/pubmed/26545990. [PubMed] [Google Scholar]
- 32.Rai V, Dietz NE, Dilisio MF, Radwan MM, Agrawal DK. Vitamin D attenuates inflammation, fatty infiltration, and cartilage loss in the knee of hyperlipidemic microswine. Arthritis Res Ther. 2016:1–17. doi: 10.1186/s13075-016-1099-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee DE, Trowbridge RM, Ayoub NT, Agrawal DK. High-mobility Group Box Protein-1, Matrix Metalloproteinases, and Vitamin D in Keloids and Hypertrophic Scars. Plast Reconstr surgery Glob open. 2015;3(6):e425. doi: 10.1097/GOX.0000000000000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oh B, Lee M. Combined delivery of HMGB-1 box A peptide and S1PLyase siRNA in animal models of acute lung injury. J Control Release. 2014;175:25–35. doi: 10.1016/j.jconrel.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Girard JP. A Direct Inhibitor of HMGB1 Cytokine. Chem Biol. 2007;14(4):345–347. doi: 10.1016/j.chembiol.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Seol D, McCabe DJ, Choe H, Zheng H, Yu Y, Jang K, et al. Chondrogenic progenitor cells respond to cartilage injury. Arthritis Rheum. 2012;64(11):3626–3637. doi: 10.1002/art.34613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim W, Hudson BI, Moser B, Guo J, Rong LL, Lu Y, et al. Receptor for Advanced Glycation End Products and Its Ligands. Ann N Y Acad Sci. 2005;561:553–561. doi: 10.1196/annals.1338.063. [DOI] [PubMed] [Google Scholar]
- 38.Shembade N, Ma A, Harhaj EW. Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327(5969):1135–9. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hensor EMA, Dube B, Kingsbury SR, Tennant A, Conaghan PG. Toward a clinical definition of early osteoarthritis: Onset of patient-reported knee pain begins on stairs. Data from the osteoarthritis initiative. Arthritis Care Res. 2015;67(1):40–47. doi: 10.1002/acr.22418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vincent HK, Heywood K, Connelly J, Hurley RW. Obesity and weight loss in the treatment and prevention of osteoarthritis. PMR. 2012;4(5 Suppl):S59–67. doi: 10.1016/j.pmrj.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rai V, Dilisio MF, Dietz NE, Agrawal DK. Recent Strategies in Cartilage Repair: A Systemic Review of the Scaffold Development and Tissue Engineering. J Biomed Mater Res A. 2017 doi: 10.1002/jbm.a.36087. [DOI] [PubMed] [Google Scholar]