

Abstract
Viruses can be classified into archaeoviruses, bacterioviruses, and eukaryoviruses according to the taxonomy of the infected host. The host-constrained perception of viruses implies preference of genetic exchange between viruses and cellular organisms of their host superkingdoms and viral origins from host cells either via escape or reduction. However, viruses frequently establish non-lytic interactions with organisms and endogenize into the genomes of bacterial endosymbionts that reside in eukaryotic cells. Such interactions create opportunities for genetic exchange between viruses and organisms of non-host superkingdoms. Here, we take an atypical approach to revisit virus-cell interactions by first identifying protein fold structures in the proteomes of archaeoviruses, bacterioviruses, and eukaryoviruses and second by tracing their spread in the proteomes of superkingdoms Archaea, Bacteria, and Eukarya. The exercise quantified protein structural homologies between viruses and organisms of their host and non-host superkingdoms and revealed likely candidates for virus-to-cell and cell-to-virus gene transfers. Unexpected lifestyle-driven genetic affiliations between bacterioviruses and Eukarya and eukaryoviruses and Bacteria were also predicted in addition to a large cohort of protein folds that were universally shared by viral and cellular proteomes and virus-specific protein folds not detected in cellular proteomes. These protein folds provide unique insights into viral origins and evolution that are generally difficult to recover with traditional sequence alignment-dependent evolutionary analyses owing to the fast mutation rates of viral gene sequences.
Keywords: virus host, protein structure, fold superfamily, comparative genomics, horizontal gene transfer, virus evolution
Introduction
Depending on the nature of the infected host, viruses can be broadly classified into three major groups, archaeoviruses, bacterioviruses (Krupovic et al., 2016), and eukaryoviruses, in addition to the lesser-known virophages that parasitize giant viruses (La Scola et al., 2003, 2008). While host jumps are common (Longdon et al., 2014; Geoghegan et al., 2017), such as HIV from chimps (Sharp and Hahn, 2010), SARS Coronavirus from bats (Li et al., 2005), H1N1 from birds (Webby and Webster, 2001), and arboviruses that replicate in mammalian cells and insect vectors, viruses are not known to infect cellular organisms separated by superkingdom (domain of life) boundaries (Nasir et al., 2014, 2017). This has been confirmed by recent studies revealing strong biases in the distribution of viral replicon types in superkingdoms such as the paucity of discovered RNA and retrotranscribing viruses in prokaryotes and their abundance and diversity in eukaryotic species such as mammals and vertebrates (Nasir et al., 2014; Koonin et al., 2015). The highly specific nature of virus-host interactions logically constrains genetic exchange to occur more frequently between the interacting partners. For example, bacterioviruses are known to capture bacterial genes involved in toxins and photosynthesis (Canchaya et al., 2004; Lindell et al., 2004). Similarly, eukaryoviruses often capture genes involved in antiviral immunity from eukaryotic cells (Elde and Malik, 2009; Rappoport and Linial, 2012). Thus, host-constrained evolution of viral lineages has led to favoring either the “escape” or “reduction” models for the origin of modern viruses, both attributing viral origins from modern or ancient host cells (reviewed in Hendrix et al., 2000; Forterre and Krupovic, 2012; Nasir et al., 2012b).
Virus-host affiliations however are largely established by observing the cytopathic effects of viral infection or by microscopy detection of virion particles. These properties relate to the lytic mode of viral reproduction that has historically remained on focus due to the noxious effects that lysis has on human health, livestock, and agriculture. However, viruses can also frequently endogenize by integration into cellular genomes (Feschotte and Gilbert, 2012), sometimes providing useful novel genes to make them evolutionarily competitive (Cornelis et al., 2012). Moreover, many viruses either infect bacterial symbionts of eukaryotic cells (e.g., the bacterial component of the human microbiota, Turnbaugh et al., 2007) or reside as prophages in the genomes of obligate intracellular bacteria that infect a wide range of eukaryotic hosts (Brüssow et al., 2004). These virus-cell interactions are largely non-lytic in nature and because they do not yield the classic phenotypic effects of viral infection, have likely remained underestimated through established methods of virus discovery (reviewed in Nasir et al., 2017). Importantly, such interactions blur the traditional concept of “virus host” and raise the possibility of viruses interacting (not necessarily in a lytic manner) and exchanging genetic material simultaneously with more than one superkingdom of life. Bordenstein and Bordenstein (2016) recently reported an example of a eukaryotic gene module in bacteriophage WO residing as prophage in the intracellular α-proteobacterium Wolbachia, which infects a large group of insects. In order to produce viral progeny, the bacteriophage WO must neutralize antiviral defense and enter/exit the membranes of both bacterial and eukaryal organisms (Bordenstein and Bordenstein, 2016). The study therefore offered unique insights into virus-cell interactions that extend beyond their known hosts and identified viruses of endosymbiotic bacteria as interesting examples of vectors with genetic material from non-host superkingdoms.
Here we take a comparative genomic approach to revisit virus-cell interactions by identifying the repertoires of protein structural domains (proteomes) in 3,440 viruses categorized into archaeoviruses, bacterioviruses, and eukaryoviruses and tracing their spread in the proteomes of 1,620 “hosts” from Archaea, Bacteria, and Eukarya. Protein domains were grouped into fold superfamilies (FSFs), as defined by the structural classification of proteins (SCOP) database (Andreeva et al., 2008; Fox et al., 2014) to include distantly related domains that show negligible sequence identity (can be < 15%) but recognizable common three-dimensional (3D) cores and biochemical functions that are likely indicative of shared ancestry. The evolutionary conservation of FSFs makes them useful molecular characters for inferring long-term viral evolutionary patterns, especially since fast mutational rates of viral gene sequences (Sanjuán et al., 2010) sometimes prohibit meaningful global evolutionary analyses (Abroi and Gough, 2011; Caetano-Anollés and Nasir, 2012; Nasir and Caetano-Anollés, 2015).
The comparative exercise of tracing the spread of each viral FSF in cellular proteomes was made explicit with an f-value representing the fraction of cellular proteomes encoding individual FSFs (see Methods). The f-values of viral FSFs in cellular proteomes and their reported biochemical functions were then used to postulate hypotheses regarding the direction of gene transfer, virus-to-cell or cell-to-virus (see Figure 1 for demonstration). For example, an FSF with a viral hallmark function (e.g., virion synthesis) that had negligible presence in proteomes of a cellular superkingdom (e.g., f < 1%) was considered a candidate for horizontal gene transfer (HGT) event from virus-to-cell rather than from cell-to-virus as the latter would require invoking multiple gene loss events in related cellular species. This approach of inferring the likely direction of gene transfer is thus similar to considering anomalous phylogenetic distributions of genes in closely related species as more likely a result of HGT rather than vertical inheritance and loss. This method reliably detects HGT events (Philippe and Douady, 2003), especially in viral genes where sequence identity with cellular counterparts may be too low to produce meaningful alignment-dependent phylogenetic trees (Nasir and Caetano-Anollés, 2015). The tracings yielded unique insights into genetic transfers between viruses and cells, highlighted the quantitatively greater cross-superkingdom genetic exchange occurring between bacterioviruses and eukaryotes and eukaryoviruses and bacteria, and supported models of viral origins from ancient cells (Nasir et al., 2012b). The genetic crosstalk between viral and cellular proteomes that we uncover with this comparative genomics approach presents a more global picture for evolutionary understanding of virus-cell interactions that goes beyond the perceived textbook definitions of virus hosts (Nasir et al., 2017).
Figure 1.
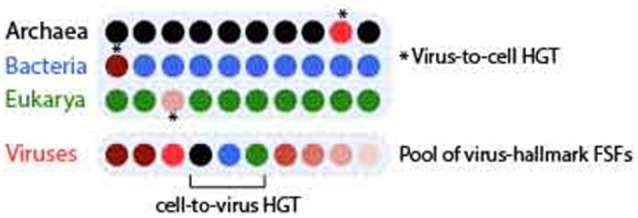
Demonstration of virus-to-cell and cell-to-virus HGT events. Ten genomes are displayed as colored closed disks each for Archaea (black), Bacteria (blue), Eukarya (green), and viruses. Seven out of 10 viral genomes encode different virus hallmark FSFs (with incidence represented by different shades of red) such as those involved in virion synthesis and capsid assembly. If any of these virus-hallmark FSFs is detected in no more than 1/10 cellular genomes (real f-values are even lower), the event is determined to be virus-to-cell HGT. In turn, any of the cellular FSFs that are widespread in cells (i.e., present in 9/10 cellular genomes) are detected in a viral genome, that event is determined to be cell-to-virus HGT.
Results
A large number of protein folds shared exclusively between viruses and their host genomes were likely transferred from viruses to cells
A total of 98, 441, and 489 FSFs were detected in the proteomes of 62 archaeoviruses, 1,223 bacterioviruses, and 2,155 eukaryoviruses (Table S1), respectively (Figure 2). Based on the presence/absence of these viral FSFs in 1,620 cellular proteomes from Archaea (122 in number), Bacteria (1,115), and Eukarya (383), seven mutually exclusive Venn groups could be defined each for archaeoviruses, bacterioviruses, and eukaryoviruses: A (viral FSFs shared only with archaeal proteomes), B (shared only with bacterial proteomes), E (shared only with eukaryotic proteomes), AB (shared only with prokaryotic proteomes), AE (shared only with archaeal and eukaryal proteomes), BE (shared only with bacterial and eukaryal proteomes), and ABE (shared with proteomes of all three superkingdoms), in addition to virus-specific (V) FSFs not detected in cellular proteomes (Figure 2).
Figure 2.

Sharing of protein structural domains between viral and cellular proteomes. The Venn diagrams illustrate the number of FSFs detected in the proteomes of archaeoviruses, bacterioviruses, and eukaryoviruses and their distributions in the proteomes of superkingdoms Archaea (A), Bacteria (B), and Eukarya (E). n = total number of viral proteomes, m = total number of FSFs detected in viral proteomes. V represents virus-specific FSFs (Table 2).
Under the expected host-constrained evolution model of viral lineages, viruses of any superkingdom are expected to share more FSFs with organisms of their host superkingdom rather than with organisms of other superkingdoms. Indeed, archaeoviruses shared a single FSF exclusively with Archaea (Venn group A; Hypothetical protein D-63) but none exclusively with Bacteria (group B) and Eukarya (group E) (Figure 2, Table 1). In turn, bacterioviruses shared 29 FSFs exclusively with Bacteria (group B) but also 2 and 6 FSFs with Archaea and Eukarya in groups A and E, respectively. Similarly, eukaryoviruses shared 37 FSFs exclusively with Eukarya (group E) but also 1 and 5 FSFs with Archaea and Bacteria in groups A and B, respectively (Figure 2, Table 1). At first glance, the data support the idea that viruses tend to share/exchange more genes with their host groups relative to organisms they do not infect or associate with.
Table 1.
Virus-Host FSF sharing.
| SCOP Id | SCOP ccs | FSF description | f-value (A) | f-value (B) | f-value (E) | f-value (AV) | f-value (BV) | f-value (EV) |
|---|---|---|---|---|---|---|---|---|
| FSFS ONLY IN ARCHAEOVIRUSES AND ARCHAEAL PROTEOMES (n = 1) | ||||||||
| 109801 | a.30.5 | Hypothetical protein D-63 | 0.0082 | 0.0000 | 0.0000 | 0.0968 | 0.0000 | 0.0000 |
| FSFS ONLY IN BACTERIOVIRUSES AND BACTERIAL PROTEOMES (n = 29) | ||||||||
| 160570 | d.368.1 | YonK-like | 0.0000 | 0.0018 | 0.0000 | 0.0000 | 0.0008 | 0.0000 |
| 64210 | d.186.1 | Head-to-tail joining protein W, gpW | 0.0000 | 0.0170 | 0.0000 | 0.0000 | 0.0123 | 0.0000 |
| 159865 | d.186.2 | XkdW-like | 0.0000 | 0.0054 | 0.0000 | 0.0000 | 0.0049 | 0.0000 |
| 54857 | d.57.1 | DNA damage-inducible protein DinI | 0.0000 | 0.0520 | 0.0000 | 0.0000 | 0.0090 | 0.0000 |
| 51327 | b.90.1 | Head-binding domain of phage P22 tailspike protein | 0.0000 | 0.0135 | 0.0000 | 0.0000 | 0.0074 | 0.0000 |
| 143749 | d.323.1 | Phage tail protein-like | 0.0000 | 0.0278 | 0.0000 | 0.0000 | 0.0098 | 0.0000 |
| 89064 | a.179.1 | Replisome organizer (g39p helicase loader/inhibitor protein) | 0.0000 | 0.0009 | 0.0000 | 0.0000 | 0.0016 | 0.0000 |
| 54328 | d.15.5 | Staphylokinase/streptokinase | 0.0000 | 0.0036 | 0.0000 | 0.0000 | 0.0041 | 0.0000 |
| 56826 | e.27.1 | Upper collar protein gp10 (connector protein) | 0.0000 | 0.0009 | 0.0000 | 0.0000 | 0.0147 | 0.0000 |
| 46575 | a.237.1 | DNA polymerase III theta subunit-like | 0.0000 | 0.0493 | 0.0000 | 0.0000 | 0.0025 | 0.0000 |
| 140919 | a.263.1 | DNA terminal protein | 0.0000 | 0.0009 | 0.0000 | 0.0000 | 0.0025 | 0.0000 |
| 159871 | d.230.6 | YdgH-like | 0.0000 | 0.0502 | 0.0000 | 0.0000 | 0.0016 | 0.0000 |
| 68918 | a.140.4 | Recombination endonuclease VII, C-terminal and dimerization domains | 0.0000 | 0.0009 | 0.0000 | 0.0000 | 0.0311 | 0.0000 |
| 160582 | d.100.2 | MbtH-like | 0.0000 | 0.1623 | 0.0000 | 0.0000 | 0.0008 | 0.0000 |
| 141658 | b.163.1 | Bacteriophage trimeric proteins domain | 0.0000 | 0.0027 | 0.0000 | 0.0000 | 0.0139 | 0.0000 |
| 51274 | b.85.2 | Head decoration protein D (gpD, major capsid protein D) | 0.0000 | 0.0072 | 0.0000 | 0.0000 | 0.0090 | 0.0000 |
| 58046 | h.1.17 | Fibritin | 0.0000 | 0.0009 | 0.0000 | 0.0000 | 0.0417 | 0.0000 |
| 58059 | h.2.1 | Tetramerization domain of the Mnt repressor | 0.0000 | 0.0027 | 0.0000 | 0.0000 | 0.0041 | 0.0000 |
| 50789 | b.57.1 | Herpes virus serine proteinase, assemblin | 0.0000 | 0.0682 | 0.0000 | 0.0000 | 0.0147 | 0.0255 |
| 50017 | b.32.1 | gp9 | 0.0000 | 0.0009 | 0.0000 | 0.0000 | 0.0581 | 0.0000 |
| 58091 | h.4.2 | Clostridium neurotoxins, “coiled-coil” domain | 0.0000 | 0.0018 | 0.0000 | 0.0000 | 0.0008 | 0.0000 |
| 57987 | h.1.4 | Inovirus (filamentous phage) major coat protein | 0.0000 | 0.0099 | 0.0000 | 0.0000 | 0.0074 | 0.0000 |
| 101059 | a.159.3 | B-form DNA mimic Ocr | 0.0000 | 0.0009 | 0.0000 | 0.0000 | 0.0123 | 0.0000 |
| 158668 | a.285.1 | MtlR-like | 0.0000 | 0.0753 | 0.0000 | 0.0000 | 0.0008 | 0.0000 |
| 103370 | d.262.1 | NinB | 0.0000 | 0.0386 | 0.0000 | 0.0000 | 0.0368 | 0.0000 |
| 118010 | d.64.2 | TM1457-like | 0.0000 | 0.2161 | 0.0000 | 0.0000 | 0.0057 | 0.0000 |
| 48657 | a.136.1 | FinO-like | 0.0000 | 0.1686 | 0.0000 | 0.0000 | 0.0033 | 0.0000 |
| 50610 | b.48.1 | mu transposase, C-terminal domain | 0.0000 | 0.0700 | 0.0000 | 0.0000 | 0.0139 | 0.0000 |
| 47681 | a.49.1 | C-terminal domain of B transposition protein | 0.0000 | 0.0135 | 0.0000 | 0.0000 | 0.0025 | 0.0000 |
| FSFS ONLY IN EUKARYOVIRUSES AND EUKARYAL PROTEOMES (n = 55) | ||||||||
| 58069 | h.3.2 | Virus ectodomain | 0.0000 | 0.0000 | 0.0757 | 0.0000 | 0.0000 | 0.0362 |
| 90229 | g.66.1 | CCCH zinc finger | 0.0000 | 0.0000 | 1.0000 | 0.0000 | 0.0000 | 0.0074 |
| 49749 | b.121.2 | Group II dsDNA viruses VP | 0.0000 | 0.0000 | 0.0131 | 0.0000 | 0.0008 | 0.0381 |
| 101912 | b.69.12 | Sema domain | 0.0000 | 0.0000 | 0.3211 | 0.0000 | 0.0000 | 0.0060 |
| 57567 | g.22.1 | Serine protease inhibitors | 0.0000 | 0.0000 | 0.3316 | 0.0000 | 0.0000 | 0.0023 |
| 54117 | d.9.1 | Interleukin 8-like chemokines | 0.0000 | 0.0000 | 0.1540 | 0.0000 | 0.0000 | 0.0084 |
| 47836 | a.61.1 | Retroviral matrix proteins | 0.0000 | 0.0000 | 0.0366 | 0.0000 | 0.0000 | 0.0186 |
| 50353 | b.42.1 | Cytokine | 0.0000 | 0.0000 | 0.3264 | 0.0000 | 0.0000 | 0.0292 |
| 52087 | c.13.1 | CRAL/TRIO domain | 0.0000 | 0.0000 | 0.9948 | 0.0000 | 0.0000 | 0.0005 |
| 103417 | e.48.1 | Major capsid protein VP5 | 0.0000 | 0.0000 | 0.0026 | 0.0000 | 0.0000 | 0.0241 |
| 56994 | g.1.1 | Insulin-like | 0.0000 | 0.0000 | 0.3055 | 0.0000 | 0.0000 | 0.0009 |
| 57535 | g.18.1 | Complement control module/SCR domain | 0.0000 | 0.0000 | 0.3760 | 0.0000 | 0.0000 | 0.0125 |
| 57180 | g.3.8 | Cellulose-binding domain | 0.0000 | 0.0000 | 0.2846 | 0.0000 | 0.0000 | 0.0005 |
| 161008 | e.76.1 | Viral glycoprotein ectodomain-like | 0.0000 | 0.0000 | 0.0131 | 0.0000 | 0.0000 | 0.0390 |
| 54277 | d.15.2 | CAD & PB1 domains | 0.0000 | 0.0000 | 0.9530 | 0.0000 | 0.0000 | 0.0023 |
| 47195 | a.24.5 | TMV-like viral coat proteins | 0.0000 | 0.0000 | 0.0418 | 0.0000 | 0.0000 | 0.0190 |
| 82856 | e.42.1 | L-A virus major coat protein | 0.0000 | 0.0000 | 0.0104 | 0.0000 | 0.0000 | 0.0019 |
| 158235 | a.271.1 | SOCS box-like | 0.0000 | 0.0000 | 0.3159 | 0.0000 | 0.0000 | 0.0005 |
| 47943 | a.73.1 | Retrovirus capsid protein, N-terminal core domain | 0.0000 | 0.0000 | 0.0522 | 0.0000 | 0.0000 | 0.0190 |
| 47353 | a.28.3 | Retrovirus capsid dimerization domain-like | 0.0000 | 0.0000 | 0.1723 | 0.0000 | 0.0000 | 0.0125 |
| 88645 | b.121.5 | ssDNA viruses | 0.0000 | 0.0000 | 0.0418 | 0.0000 | 0.0139 | 0.0320 |
| 101399 | a.206.1 | P40 nucleoprotein | 0.0000 | 0.0000 | 0.0104 | 0.0000 | 0.0000 | 0.0005 |
| 110132 | b.147.1 | BTV NS2-like ssRNA-binding domain | 0.0000 | 0.0000 | 0.0026 | 0.0000 | 0.0000 | 0.0046 |
| 49599 | b.8.1 | TRAF domain-like | 0.0000 | 0.0000 | 0.9974 | 0.0000 | 0.0000 | 0.0005 |
| 57302 | g.7.1 | Snake toxin-like | 0.0000 | 0.0000 | 0.3211 | 0.0000 | 0.0000 | 0.0005 |
| 50122 | b.34.7 | DNA-binding domain of retroviral integrase | 0.0000 | 0.0000 | 0.0235 | 0.0000 | 0.0000 | 0.0097 |
| 140809 | a.260.1 | Rhabdovirus nucleoprotein-like | 0.0000 | 0.0000 | 0.0183 | 0.0000 | 0.0000 | 0.0125 |
| 46919 | a.4.10 | N-terminal Zn binding domain of HIV integrase | 0.0000 | 0.0000 | 0.0261 | 0.0000 | 0.0000 | 0.0084 |
| 57924 | g.52.1 | Inhibitor of apoptosis (IAP) repeat | 0.0000 | 0.0000 | 0.7441 | 0.0000 | 0.0000 | 0.0376 |
| 57933 | g.53.1 | TAZ domain | 0.0000 | 0.0000 | 0.4700 | 0.0000 | 0.0000 | 0.0005 |
| 103575 | g.16.2 | Plexin repeat | 0.0000 | 0.0000 | 0.3316 | 0.0000 | 0.0000 | 0.0023 |
| 57059 | g.3.6 | omega toxin-like | 0.0000 | 0.0000 | 0.0444 | 0.0000 | 0.0000 | 0.0014 |
| 140586 | a.242.1 | Dcp2 domain-like | 0.0000 | 0.0000 | 0.9034 | 0.0000 | 0.0000 | 0.0005 |
| 57501 | g.17.1 | Cystine-knot cytokines | 0.0000 | 0.0000 | 0.3185 | 0.0000 | 0.0000 | 0.0046 |
| 69340 | b.80.5 | C-terminal domain of adenylylcyclase associated protein | 0.0000 | 0.0000 | 0.9765 | 0.0000 | 0.0000 | 0.0009 |
| 49830 | b.20.1 | ENV polyprotein, receptor-binding domain | 0.0000 | 0.0000 | 0.0313 | 0.0000 | 0.0000 | 0.0042 |
| 81382 | a.157.1 | Skp1 dimerisation domain-like | 0.0000 | 0.0000 | 0.9687 | 0.0000 | 0.0000 | 0.0032 |
FSFs shared exclusively between the proteomes of host superkingdoms, Archaea (A), Bacteria (B), and Eukarya (E), and the proteomes of their viruses, archaeoviruses (AV), bacterioviruses (BV), and eukaryoviruses (EV). FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). FSF distribution (f-values, number of proteomes in a superkingdom or virus group encoding an FSF/total number of proteomes in that superkingdom or virus group) are also listed. FSF b.57.1 was also detected in eukaryoviruses in addition to Bacteria and bacterioviruses and FSFs b.121.2 and b.121.5 were also detected in bacterioviruses in addition Eukarya and eukaryoviruses possibly indicating genetic crosstalk or ancient ancestry (read text). These FSFs are highlighted in bold.
Remarkably, the 29 FSFs shared exclusively between bacterioviral and bacterial proteomes included several viral hallmark proteins involved in phage (virus) assembly such as the gp9 and gp10 proteins, head-binding, head-to-tail joining, head decoration, and tail proteins, along with the major coat proteins of ssDNA harboring bacterioviruses (Inoviridae) and the dimerization domain of bacteriophage T4 recombination endonuclease VII (Table 1). In addition, the coiled-coiled domain of bacterial neurotoxin involved in host virulence was also detected. Interestingly, the majority of FSFs in group B had f-values close to 0 indicating their rare presence in bacterial proteomes (Table 1). Collectively, therefore, the enrichment of the B Venn group in viral hallmark functions with negligible presence in bacterial proteomes suggests that these genes were likely acquired by bacterial cells from viruses via virus-to-cell HGT, a phenomenon that has been assumed to be relatively less frequent than cell-to-virus HGT (Moreira and Lopez-Garcia, 2009), though now increasingly being revisited (Forterre, 2016). Similarly, viral hallmark proteins such as the viral capsid and coat-related proteins (e.g., the “jelly-roll” and “double jelly-roll” folds) (Abrescia et al., 2012), viral glycoproteins and matrix proteins, the integrase proteins of retroviruses and HIV, and toxins were part of the 37 FSFs shared exclusively between eukaryoviruses and Eukarya (the E Venn group) with low f-values in eukaryal proteomes (Table 1). These viral hallmark proteins shared exclusively between eukaryoviruses and eukaryotes could therefore also represent episodes of virus-to-cell gene transfer. In turn, other E FSFs such as the CCCH zinc finger domains (involved in regulation and DNA binding), CAD and PB1 domains (cell cycle and apoptosis), CRAL/TRIO domains (likely functional components of the visual cycle), TRAF-domain like (involved in stress response, immunity, apoptosis, among other roles), and others were near ubiquitous in eukaryotic proteomes (i.e., f-value close to 1.0, Table 1). These “cell-like” proteins detected in eukaryoviruses could therefore suggest recent gene capture by viruses from cells (i.e., cell-to-virus HGT) as likely part of viral mimicry of cellular proteins to interfere with the antiviral response (Elde and Malik, 2009).
In summary, a large number of FSFs shared exclusively between viruses and their host genomes had rare presence in hosts and were involved in virus-hallmark functions suggesting these genes likely originated in viral lineages and were later transferred to their host cells.
Traces of genetic crosstalk between viruses and non-host superkingdoms could be recovered from the comparative genomic data
While the data of Figure 2 indicated significant levels of genetic exchange restricted between viruses and their known host superkingdoms, some bacterioviral and eukaryoviral FSFs were also shared with Eukarya and Bacteria, respectively (archaeoviruses shared no domains exclusively with either Bacteria or Eukarya) (Figure 2, Tables S2–S4). For example, bacterioviruses shared 2 FSFs exclusively with Archaea (group A) and 6 with Eukarya (group E) (Table S3). Interestingly, 4/6 E FSFs in bacterioviruses could be considered viral hallmark proteins such as FSFs b.121.2 (the “double jelly-roll” fold hallmark of capsid proteins of the PRD1/Adenovirus-like lineage) (Bamford, 2003; Abrescia et al., 2012), b.121.5 (the “jelly-roll” fold in ssDNA viruses members of the Picornavirus-like lineage), d.85.1 (capsid/coat related fold in RNA bacteriophages), and a.251.1 (the phage replication organizer domain) (Table S3). Viruses have been recently (re)-classified into structure-based lineages based on 3D structural similarities in capsid/coat architectures or common principles of functional virion construction (Bamford, 2003; Abrescia et al., 2012; Nasir and Caetano-Anollés, 2017). Some of the lineages such as the PRD1/Adenovirus-like lineage (characterized by the so-called “double jelly-roll” fold) include member viruses infecting the three cellular superkingdoms (Bamford, 2003; Abrescia et al., 2012). Thus, it is no surprise that bacterioviruses share capsid/coat related protein folds characteristic of eukaryoviruses. It is however indeed intriguing to note that these FSFs were present in eukaryotic proteomes, especially because the capsid is considered to be a virus hallmark (Benson et al., 2004; Abrescia et al., 2010). Thus, rare occurrences of capsid/coat related genes in cellular proteomes are more likely due to virus-to-cell HGT or their utilization in the assembly of capsid-like architectures in cells (e.g., carboxysomes and protein microcompartments in prokaryotes, Yeates et al., 2007, 2011) that are hitherto believed to be rare in cells (Cheng and Brooks, 2013; Nasir and Caetano-Anollés, 2017).
In turn, eukaryoviruses shared a single FSF exclusively with Archaea (group A; Chromosomal protein MC1) and 5 FSFs exclusively with Bacteria (group B) (Table S4). The MC1 protein is associated with thermophilic archaeal species (f-value = 0.26 in Archaea) and is involved in protecting DNA denaturation at high temperature (Chartier et al., 1989). Its presence in eukaryoviruses (but not in eukaryotic proteomes!) is therefore intriguing and could signal undiscovered viral-mediated interactions between eukaryotic and archaeal species. In turn, the 5 FSFs shared exclusively between eukaryoviruses and bacterial proteomes (the B Venn group) included capsid proteins (Outer capsid protein sigma 3) and other virus and cell-like proteins likely indicating a mixed ancestry (Table S4).
Finally, the BE Venn group for archaeoviruses, the AE group for bacterioviruses, and the AB group for eukaryoviruses may also represent genetic exchanges occurring between viruses and non-host superkingdoms. For archaeoviruses, the d.285.1 (DNA-binding domain of intron-encoded endonucleases), a.118.25 (TROVE domain-like), and b.22.1 (TNF-like) FSFs were detected in the BE group. In turn, only one bacterioviral FSF (d.282.1, SSo0622-like) was detected in the AE group and 2 eukaryoviral FSFs were detected in the AB group (a.18.1, T4 endonuclease V and g.90.1, E6 C-terminal domain-like) (highlighted in Tables S2–S4). These FSFs are likely candidates of genetic transfer occurring between viruses and non-host superkingdoms, more likely in the cell-to-virus direction because of the “cell-like” nature of these FSFs.
An unanticipated relatively greater genetic affiliation between Bacterioviruses and eukaryal proteomes and Eukaryoviruses and bacterial proteomes
Bacterioviruses shared 66 FSFs and eukaryoviruses shared 65 FSFs with both Bacteria and Eukarya (the BE groups), respectively, which constituted 15 and 13% of total bacterioviral and eukaryoviral FSFs, respectively (Figure 2). Only 20 BE FSFs overlapped and the remaining (46/66 in bacterioviruses and 45/65 in eukaryoviruses) were uniquely shared with bacterial and eukaryal proteomes (Figure 3A), thus extending the total number of BE FSFs to 111. The 46 BE FSFs unique to bacterioviruses (i.e., FSFs detected in bacterioviral, bacterial and eukaryal proteomes but not in eukaryoviral proteomes) were significantly more widespread in bacterial proteomes (Welch two-sample t-test, P = 0.01) while the 45 BE FSFs unique to eukaryoviruses (FSFs detected in eukaryoviral, bacterial and eukaryal proteomes but not in bacterioviral proteomes) were significantly more widespread in eukaryotic proteomes (P < 0.0001) with f-values approaching 1.0 in some cases (Figure 3B).
Figure 3.

The structural domain composition of the BE Venn group in bacterioviruses and eukaryoviruses. (A) The Venn diagram describes how the BE FSFs were shared between bacterioviruses and eukaryoviruses. (B) Boxplots display the distribution of f-values (number of proteomes in a superkingdom encoding an FSF divided by the total number of proteomes in that superkingdom) for BE FSFs unique to bacterioviruses, unique to eukaryoviruses, and common to both the bacterial and eukaryal proteomes (see also Table S5). P-values were calculated from two-sample Welch t-tests.
We hypothesize that bacterioviruses and eukaryoviruses acquired BE FSFs directly from their host cells (i.e., Bacteria and Eukarya, respectively) without the need to invoke genetic crosstalk between viruses and non-host superkingdoms and also between bacterioviruses and eukaryoviruses (since these FSFs were absent in one of the two viral groups except for the 20 common BE FSFs, Figure 3A). While, this may represent the generic trend, we noticed that 6 BE FSFs unique to bacterioviruses had an f-value that was 25% higher in eukaryotic proteomes than the corresponding f-value in bacterial proteomes (highlighted in Table S5). For example, FSF d.344.1 (PriA/YqbF domain) was present in roughly 95% of eukaryotic proteomes and in only 2% bacterial proteomes (i.e., f-value differential of 93%). For another 22 BE FSFs, the differential in f-values was either negligible or below the 25% cutoff making it difficult to establish the likely direction of origin (i.e., from Eukarya to bacterioviruses or from Bacteria to bacterioviruses). In fact, only 18 out of 46 BE FSFs in bacterioviruses had an f-value > 25% in bacterial proteomes than the corresponding f-value in eukaryotic proteomes (Table S5). Therefore, closer inspection of BE FSFs in bacterioviruses indicated that both sources of origin could be considered likely, especially when accounting for the relative preference of bacterial species to become endosymbionts of eukaryotes and considering mechanical similarities between bacterial and eukaryotic cells (read below). The same was also true for 20 BE FSFs common to bacterioviruses and eukaryoviruses where the f-value differential was under 25% for 11 out of 20 FSFs. However, 37 out of 45 BE FSFs in eukaryoviruses had an f-value of 25% or greater in eukaryotic proteomes than the corresponding f-value in bacterial proteomes (Table S5) indicating that eukaryoviruses perhaps did not engage in genetic exchange directly from Bacteria (or bacterioviruses).
To test, we divided eukaryoviruses into five subgroups representing viruses of fungi, plants, metazoa, protozoa, and invertebrates-plants (viruses that can replicate in both plants and insect vectors), as defined by the NCBI Viral Genomes Resource (Figure 4). FSF distributions of the five subgroups of eukaryoviruses were mapped to the seven Venn groups already defined for eukaryoviruses (Figure 2). The majority of eukaryoviruses belonged to metazoa (n = 1,057) and plant hosts (963) revealing strong biases in the sequencing of human infection, livestock and agriculture related viruses. Interestingly, only 27 viruses were associated to protozoa. These viruses encoded a total of 291 FSFs (the second largest amongst the five eukaryoviral subgroups after 306 FSFs of metazoan viruses). This is expected since protozoa act as natural hosts of many “giant viruses” (e.g., Acanthamoeba polyphaga), which surpass parasitic cellular species both in particle and genome sizes and sometimes encode more than a thousand proteins (La Scola et al., 2003; Arslan et al., 2011; Philippe et al., 2013; Legendre et al., 2015). However, out of the total 65 BE FSFs detected in eukaryoviruses (Figure 2), 40 (62%) were detected in metazoan viruses and 32 (49%) in protozoan viruses (overlap of 14 common FSFs) (Figure 4). Animals are known hosts for symbiotic bacteria and also harbor large microbiota communities, especially in the gastrointestinal tract that is considered to be a “melting pot” for HGT (Shterzer and Mizrahi, 2015). Similarly, free-living amoeba (e.g., Acanthamoeba) are notorious reservoirs for both facultative and obligate intracellular bacteria and serve as “training grounds” to facilitate bacterial adaptation in eukaryotic cells (Barker and Brown, 1994; Molmeret et al., 2005). These two eukaryotic host subgroups therefore provide ample opportunities for eukaryoviruses to exchange genetic material either directly with bacterial proteomes or through prophages integrated in bacterial genomes.
Figure 4.

Breakdown of the 489 FSFs detected in eukaryoviruses. Eukaryoviruses were divided into viruses of plants (included all plants, blue-green algae, and diatoms), metazoa (vertebrates and invertebrates), protozoa (animal-like protists), fungi, and a group that includes invertebrates and plants (IP). For each subgroup, bars indicate the percentage of FSFs present in one of the seven Venn groups listed on the right (see also Figure 2) and the percentage of virus-specific FSFs. Numbers on bars indicate actual count. n = total number of viral proteomes, m = total number of FSFs detected in viral proteomes.
The ABE and virus-specific protein folds (V) provide unique insights into viral origins and evolution
The ABE group was the largest Venn group for viruses of the three superkingdoms (i.e., 93 ABE FSFs out of 98 total FSFs in archaeoviruses, 319 out of 441 in bacterioviruses, and 315 out of 449 in eukaryoviruses, Figure 2). The ABE domains are, by definition, detected in the proteomes of all three superkingdoms and are more likely to evolve vertically and hold a deep history (Nasir and Caetano-Anollés, 2013, 2015). Indeed, ABE domains were widespread in cellular species (median f-value > 0.6 for all, Figure 5) and were enriched in “cell-like” functions such as metabolism, information, DNA repair, among others, and occasionally viral proteins (Tables S2–S4). Therefore, the ABE group stands in contrast to the “viral-like” nature of the other Venn groups, especially, B and E FSFs that had limited spread in cellular proteomes (Table 1). The presence of a large number of universal “cell-like” proteins in viral proteomes is therefore intriguing and worthy of exploration. It suggests two possible scenarios. First, the detection of ABE FSFs in viral proteomes effectively transforms ABE into an ABEV group, which now represents a large core of (near)-universal FSF domains shared by both cells and viruses. The mere existence of this FSF core supports an early “cell-like” phase in the evolution of modern viruses, an idea that has recently become popular (Nasir et al., 2012a,b) following the discovery of several “giant viruses” that overlap parasitic cells in physical and genome size (La Scola et al., 2003; Philippe et al., 2013; Legendre et al., 2014, 2015). Under this proposal, viruses are secondarily acellular as they either “escaped” or “reduced” from primordial cells before these cells diversified into superkingdoms (Nasir and Caetano-Anollés, 2015, see Schulz et al., 2017 for an opposite view). In an alternative second scenario, the ABEV group points to recent HGTs occurring between viruses and cells in either direction, more likely from cell-to-virus considering the cell-like nature of ABE FSFs. It is important to note that a single HGT event is sufficient to invoke transformation from the ABE to the ABEV group. For example, ABE FSFs can be transferred directly from Archaea to archaeoviruses, from Bacteria to bacterioviruses, and Eukarya to eukaryoviruses, in addition to indirect cross-superkingdom genetic transfers. All of these transfers suffice for ABE to ABEV transformation.
Figure 5.
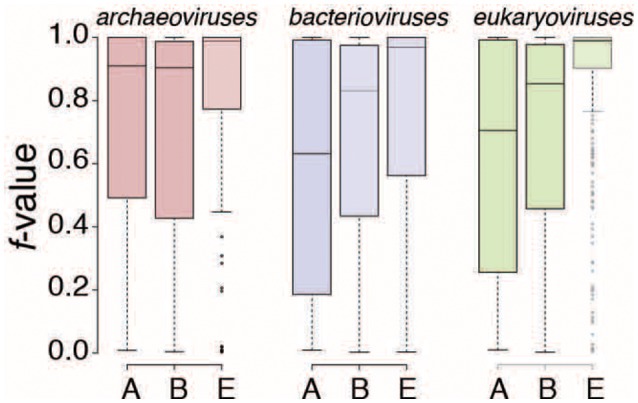
ABE FSFs are widespread in the proteomes of Archaea, Bacteria, and Eukarya. The f-value (number of proteomes in a superkingdom encoding an FSF / total number of proteomes in that superkingdom) distribution is plotted for the ABE Venn group of FSF domains for archaeoviruses, bacterioviruses, and eukaryoviruses. The three boxplots in each viral group describe FSF spread individually for Archaea (A), Bacteria (B), and Eukarya (E).
To evaluate these alternatives, we pooled the ABE FSFs of archaeoviruses (n = 93), bacterioviruses (319), and eukaryoviruses (315) into a non-redundant list of 442 FSFs (Table S6). Next, we dissected the 442 FSFs into seven new Venn groups: a (ABE FSFs detected only in the proteomes of archaeoviruses), b (ABE FSFs detected only in bacterioviruses), e (ABE FSFs detected only in eukaryoviruses), ab (ABE FSFs detected only in prokaryotic viruses), ae (ABE FSFs detected only in archaeoviruses and eukaryoviruses), be (ABE FSFs detected only in bacterioviruses and eukaryoviruses), and abe (ABE FSFs detected in archaeoviruses, bacterioviruses, and eukaryoviruses) (Figure 6). This classification enabled evaluation of virus-to-virus HGTs in contrast to either virus-to-cell or cell-to-virus candidate HGT events postulated above. The majority of the ABE FSFs were part of the be group (n = 130) once again suggesting relatively high activity of cross-superkingdom genetic exchange between bacterioviruses and eukaryoviruses (or their cellular proteomes) possibly driven by bacteria-eukarya lifestyle affiliations or, as an alternative, loss of these FSFs in archaeoviruses. The next larger groups included e (117) and b (102) (Figure 6). These could represent direct HGT events from bacterial proteomes to bacterioviruses and eukaryal proteomes to eukaryoviruses, respectively.
Figure 6.
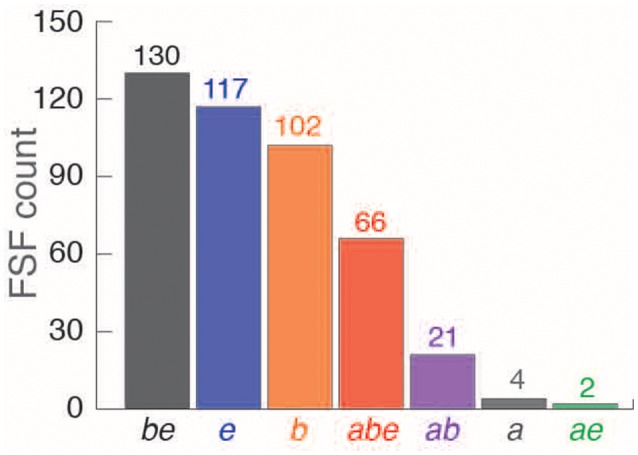
Breakdown of pooled non-redundant 442 viral ABE FSFs (see text) into seven possible Venn groups for archaeoviruses, bacterioviruses, and eukaryoviruses. Numbers on bars indicate actual count (see also Table S6).
A total of 66 ABE FSFs were detected in the proteomes of archaeoviruses, bacterioviruses, and eukaryoviruses (Venn group abe), again enriched in cell-like functions (highlighted in Table S6). The abe “universal” or “core” group of FSFs therefore included FSFs detected in the proteomes of all virus groups (archaeoviruses, bacterioviruses, and eukaryoviruses) and the three superkingdoms (Archaea, Bacteria, and Eukarya). While significant cross-superkingdom indirect genetic exchange cannot be ruled out, the possibility of the same HGT event occurring three times independently and in different ecological habitats should be considered unlikely. The origin of abe FSFs is therefore better and more parsimoniously reconciled with an origin of modern viral lineages in ancient cells that existed prior to the diversification of cellular life and experienced high levels of genome reduction (Nasir et al., 2012a; Claverie and Abergel, 2016). One interesting observation was the existence of only 2 FSFs belonging to the ae group. An origin of Eukarya from within Archaea has recently been postulated following the reconstruction of the genomes of the candidate archaeal phylum “Lokiarchaeota” and “Asgard” archaea (Zaremba-Niedzwiedzka et al., 2017), which harbor several eukaryote-specific proteins (Guy et al., 2014; Spang et al., 2015, see Nasir et al., 2016; Da Cunha et al., 2017 for opposite views). Under this scenario, one should expect stronger affiliation of eukaryoviruses with archaeoviruses, which however does not materialize in the FSF data.
Finally, 10 and 55 virus-specific FSFs (V) were detected in the proteomes of bacterioviruses and eukaryoviruses, respectively (none in archaeoviruses, Figure 2, Table 2). These protein domains represent crucial viral functions involved in viral pathogenicity and virion synthesis and could therefore become hot targets for designing novel therapeutics against contemporary viruses. Their origin however cannot be attributed to cell-to-virus HGT, as these FSFs are completely absent in cellular proteomes. They could originate either directly in viral lineages during replication inside host cells (refer to the “virocell” concept, Forterre, 2011) or represent ancient proteins relics of an early origin of viruses. Testing either of these two scenarios via data-driven approaches remains an open challenge though insights are starting to emerge (Nasir and Caetano-Anollés, 2015).
Table 2.
Virus specific FSFs (VSFs).
| SCOP ID | SCOP ccs | FSF description | Virus classification | Member families/order |
|---|---|---|---|---|
| 158974 | b.170.1 | WSSV envelope protein-like | eukaryoviruses | Nimaviridae |
| 88648 | b.121.6 | Group I dsDNA viruses | eukaryoviruses | Polyomaviridae, Papillomaviridae |
| 101089 | a.8.5 | Phosphoprotein XD domain | eukaryoviruses | Mononegavirales |
| 69070 | a.150.1 | Anti-sigma factor AsiA | bacterioviruses | Caudovirales |
| 89433 | b.127.1 | Baseplate structural protein gp8 | bacterioviruses | Caudovirales, Unclassified |
| 160099 | d.346.1 | SARS Nsp1-like | eukaryoviruses | Nidovirales |
| 89428 | b.126.1 | Adsorption protein p2 | bacterioviruses | Tectiviridae |
| 143076 | d.302.1 | Coronavirus NSP8-like | eukaryoviruses | Nidovirales |
| 56502 | d.172.1 | gp120 core | eukaryoviruses | Retroviridae |
| 55671 | d.102.1 | Regulatory factor Nef | eukaryoviruses | Retroviridae |
| 56983 | f.10.1 | Viral glycoprotein, central and dimerisation domains | eukaryoviruses | Flaviviridae, Togaviridae |
| 50012 | b.31.1 | EV matrix protein | eukaryoviruses | Mononegavirales |
| 118208 | e.58.1 | Viral ssDNA binding protein | eukaryoviruses | Herpesvirales |
| 54957 | d.58.8 | Viral DNA-binding domain | eukaryoviruses | Papillomaviridae, Herpesvirales |
| 48493 | a.120.1 | gene 59 helicase assembly protein | bacterioviruses | Caudovirales, Unclassified |
| 101816 | b.140.1 | Replicase NSP9 | eukaryoviruses | Nidovirales |
| 48145 | a.95.1 | Influenza virus matrix protein M1 | eukaryoviruses | Orthomyxoviridae |
| 140506 | a.30.8 | FHV B2 protein-like | eukaryoviruses | Nodaviridae |
| 161240 | g.92.1 | T-antigen specific domain-like | eukaryoviruses | Polyomaviridae |
| 69922 | f.12.1 | Head and neck region of the ectodomain of NDV fusion glycoprotein | eukaryoviruses | Mononegavirales |
| 101156 | a.30.3 | Nonstructural protein ns2, Nep, M1-binding domain | eukaryoviruses | Orthomyxoviridae |
| 143021 | d.299.1 | Ns1 effector domain-like | eukaryoviruses | Orthomyxoviridae |
| 49818 | b.19.1 | Viral protein domain | eukaryoviruses | Nidovirales, Orthomyxoviridae, Reoviridae |
| 75347 | d.13.2 | Rotavirus NSP2 fragment, C-terminal domain | eukaryoviruses | Reoviridae |
| 48345 | a.115.1 | A virus capsid protein alpha-helical domain | eukaryoviruses | Reoviridae |
| 141666 | b.164.1 | 'SARS ORF9b-like | eukaryoviruses | Nidovirales |
| 82046 | b.116.1 | Viral chemokine binding protein m3 | eukaryoviruses | Herpesvirales |
| 56558 | d.182.1 | Baseplate structural protein gp11 | bacterioviruses | Caudovirales |
| 103145 | d.255.1 | Tombusvirus P19 core protein, VP19 | eukaryoviruses | Tombusviridae |
| 160892 | d.378.1 | Phosphoprotein oligomerization domain-like | eukaryoviruses | Mononegavirales |
| 103068 | d.254.1 | Nucleocapsid protein dimerization domain | eukaryoviruses | Nidovirales |
| 51289 | b.85.5 | Tlp20, baculovirus telokin-like protein | eukaryoviruses | Baculoviridae |
| 75574 | d.216.1 | Rotavirus NSP2 fragment, N-terminal domain | eukaryoviruses | Reoviridae |
| 49894 | b.28.1 | Baculovirus p35 protein | eukaryoviruses | Baculoviridae, Poxviridae, |
| 161003 | e.75.1 | flu NP-like | eukaryoviruses | Orthomyxoviridae |
| 110304 | b.148.1 | Coronavirus RNA-binding domain | eukaryoviruses | Nidovirales |
| 48045 | a.84.1 | Scaffolding protein gpD of bacteriophage procapsid | bacterioviruses | Microviridae |
| 58030 | h.1.13 | Rotavirus nonstructural proteins | eukaryoviruses | Reoviridae |
| 69652 | d.199.1 | DNA-binding C-terminal domain of the transcription factor MotA | bacterioviruses | Caudovirales |
| 58034 | h.1.14 | Multimerization domain of the phosphoprotein from sendai virus | eukaryoviruses | Mononegavirales |
| 55064 | d.58.27 | Translational regulator protein regA | bacterioviruses | Caudovirales, Unclassified |
| 50176 | b.37.1 | N-terminal domains of the minor coat protein g3p | bacterioviruses | Inoviridae |
| 118173 | d.293.1 | Phosphoprotein M1, C-terminal domain | eukaryoviruses | Mononegavirales |
| 47724 | a.54.1 | Domain of early E2A DNA-binding protein, ADDBP | eukaryoviruses | Adenoviridae |
| 57917 | g.51.1 | Zn-binding domains of ADDBP | eukaryoviruses | Adenoviridae |
| 143587 | d.318.1 | SARS receptor-binding domain-like | eukaryoviruses | Nidovirales |
| 75404 | d.213.1 | VSV matrix protein | eukaryoviruses | Mononegavirales |
| 160957 | e.69.1 | Poly(A) polymerase catalytic subunit-like | eukaryoviruses | Poxviridae |
| 140367 | a.8.9 | Coronavirus NSP7-like | eukaryoviruses | Nidovirales |
| 160453 | d.361.1 | PB2 C-terminal domain-like | eukaryoviruses | Orthomyxoviridae |
| 56548 | d.180.1 | Conserved core of transcriptional regulatory protein vp16 | eukaryoviruses | Herpesvirales |
| 49889 | b.27.1 | Soluble secreted chemokine inhibitor, VCCI | eukaryoviruses | Poxviridae |
| 144251 | g.87.1 | Viral leader polypeptide zinc finger | eukaryoviruses | Picornavirales |
| 89043 | a.178.1 | Soluble domain of poliovirus core protein 3a | eukaryoviruses | Picornavirales, Theilovirus |
| 144246 | g.86.1 | Coronavirus NSP10-like | eukaryoviruses | Nidovirales |
| 47852 | a.62.1 | Hepatitis B viral capsid (hbcag) | eukaryoviruses | Hepadnaviridae |
| 69903 | e.34.1 | NSP3 homodimer | eukaryoviruses | Reoviridae |
| 159936 | d.15.14 | NSP3A-like | eukaryoviruses | Nidovirales |
| 69908 | e.35.1 | Membrane penetration protein mu1 | eukaryoviruses | Reoviridae |
| 101257 | a.190.1 | Flavivirus capsid protein C | eukaryoviruses | Flaviviridae |
| 111379 | f.47.1 | VP4 membrane interaction domain | eukaryoviruses | Reoviridae |
| 90246 | h.1.24 | Head morphogenesis protein gp7 | bacterioviruses | Caudovirales |
| 57647 | g.34.1 | HIV-1 VPU cytoplasmic domain | eukaryoviruses | Retroviridae |
| 117066 | b.1.24 | Accessory protein X4 (ORF8, ORF7a) | eukaryoviruses | Nidovirales |
| 51332 | b.91.1 | E2 regulatory, transactivation domain | eukaryoviruses | Papillomaviridae |
FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). See Nasir and Caetano-Anollés (2015) for an expanded list.
Discussion
A simple comparative genomic analysis calculating the spread of viral protein domain structure FSFs in reported host and non-host cellular proteomes revealed that proteomes of virus hosts harbored several viral hallmark proteins necessary for virion assembly and successful viral infection cycles (Table 1). These viral hallmark proteins however were absent from the majority of closely-related organisms within the same superkingdom indicating that their rare presence in some host cellular proteomes could be an outcome of virus-to-cell gene transfer. In turn, proteomes not presumed to serve as natural hosts for viruses also shared homologous FSFs with viral proteomes. These FSFs included both viral- and cell-like proteins. This was especially obvious for FSFs shared between bacterioviruses and eukaryotic proteomes indicating either direct or indirect cross-superkingdom genetic exchange. This sharing could have been driven by the endosymbiotic and pathogenic lifestyle of bacteria that sometimes associate with eukaryotic cells. Interestingly, despite sharing the same ecosystem with Bacteria (e.g., the human gastrointestinal tract, Lurie-Weinberger and Gophna, 2015), our results suggested that little or no FSF sharing (or genetic exchange) occurred between archaeoviruses and the proteomes of Bacteria and Eukarya (e.g., AB was 1 and AE 0 in archaeoviruses, Figure 2). Bacteria are established pathogens and (endo)-symbionts of eukaryotes but Archaea are not known to infect eukaryotic organisms (Aminov, 2013). The membranes of Archaea also differ in lipid composition with the membranes of Bacteria and Eukarya (ether-linked vs. ester-linked, Jain et al., 2014), along with other differences (Gill and Brinkman, 2011). These differences could therefore pose a barrier for archaeoviruses to cross/traverse bacterial and eukaryal membranes and participate in horizontal genetic exchange. In contrast and thanks to the relatively similar lipid organization of Bacteria and Eukarya, bacterioviruses may either directly traverse eukaryotic membranes or alternatively transduce benign bacterial species into human pathogens by transferring virulence factors (Brüssow et al., 2004), which in turn infect Eukarya. As stated by Gill and Brinkman (2011), “eukaryotic viruses infect eukaryotes, and bacteriophages transduce Bacteria, which allows them to infect Eukarya”. Moreover, there are many more known examples of obligate and facultative intracellular bacteria (e.g., Chlamydia, Rickettsia, Mycoplasma) in eukaryotes. Therefore, viral infection of bacterial endosymbionts or prophage integration into their genomes will create more opportunities for genetic interactions with eukaryoviruses and eukaryotic proteomes explaining more “crosstalk” between bacterioviruses and Eukarya (and perhaps eukaryoviruses and Bacteria) than between archaeoviruses and Bacteria/Eukarya. However, it must be noted that both archaeoviruses and archaeal species are relatively underrepresented in sequence databases. Thus, a global picture of the true contribution of archaeoviruses and archaeal proteomes to protein structure space remains elusive despite increased metagenome sequencing efforts. Indeed, Archaea constitute an important part of the animal microbiota (Hoffmann et al., 2013; Lurie-Weinberger and Gophna, 2015), an ecosystem that is considered a “hot spot” for genetic exchange (Shterzer and Mizrahi, 2015).
A large cohort of universal protein domains shared between archaeoviruses, bacterioviruses, eukaryoviruses, Archaea, Bacteria, and Eukarya, was also detected that provides support to an ancient co-existence of viral and cellular ancestors before the rise of a diversified cellular world (abe FSFs, highlighted in Table S6), a scenario supported by a recent large-scale phylogenomic study (Nasir and Caetano-Anollés, 2015) and a number of philosophical arguments (Claverie and Abergel, 2016). Cross-superkingdom genetic transfers were also likely after the rise of diversified cellular lineages. In turn, the list of virus-specific protein domains (Table 2) provides a useful set of molecular targets for antiviral research.
The comparative genomic approach presented here is useful to postulate data-driven hypotheses regarding viral evolution, especially because large-scale sequence-based phylogenetic analysis on viral genes and genomes is sometimes prohibitive due to high nucleotide and amino acid sequence variability within and between viral genome groups. The comparative genomic approach however does suffer from some limitations. First, in the absence of phylogenetic reconstruction, structural similarities are considered homologies. Protein domain sharing could be a result of convergent evolution, HGT and vertical evolution. However, protein domains grouped into FSFs are believed to have evolved from a common evolutionary ancestor and thus cannot (by SCOP definitions) be subject to convergent evolution. Specifically, the interlocking of amino acid side-chains in the buried cores of protein domain structures represents a distinctive “fingerprint,” which is recognizable among member domains of any particular superfamily. Amino acid substitutions that occur over evolutionary timespans do not distort the 3D fingerprint characteristic of each superfamily without risking loss of the protein fold, and ultimately its biochemical function (e.g., bacterial MreB and FtsZ proteins that are prokaryotic homologs of eukaryotic actin and tubulin, respectively). That is the reason why despite low sequence identities, member protein domains of SCOP FSFs share recognizable structural and biochemical similarities, which are taken as evidence for common origin. Empirically, the odds of originating the same fingerprint (a product of multiple interactions occurring between many amino acid side chains) independently are considered to be extremely low (e.g., between 3 and 5% in Gough, 2005). In other words, each known fold or FSF is a unique discovery in evolution. Given the small number of expected folds that exist in nature (~1,500), convergence becomes an unlikely scenario.
Second, HGT can transfer protein domains and thus increase their representation in modern proteomes. We used the f-value as a proxy to evaluate the relative evolutionary spread of each FSF in cellular proteomes. When linked with the biochemical function of the protein fold (i.e., viral-like or cellular-like), the analysis indicated a likely direction of gene transfer (i.e., virus-to-cell or cell-to-virus). For example, FSFs involved in a viral hallmark function such as virion synthesis and/or capsid assembly that had negligible presence in either host or non-host superkingdoms (e.g., f < 1%) were treated as candidate virus-to-cell gene transfers. In turn, FSFs involved in cellular functions such as metabolism that were widespread among cellular proteomes (e.g., f > 60%) were treated as cell-to-virus candidate HGTs, except when these FSFs were also detected in the three virus groups (i.e., the abe group of 66 “universal” FSFs). This approach of inferring a qualitative likelihood of HGT is thus similar to the method of detecting anomalous phylogenetic distributions of genes where rare presence of a gene in closely related members is more likely a result of HGT rather than vertical evolution, especially because the latter would require invoking multiple events of gene loss that are less parsimonious than considering fewer HGT events (Philippe and Douady, 2003). The f-value approach is especially useful for viral genes that exhibit fast mutation rates and prohibit utilizing genome-scale alignment-dependent phylogenetic analysis (Abroi and Gough, 2011; Nasir and Caetano-Anollés, 2015).
Third, it can be argued that the f-value may not reflect phylogenetic diversity. For example, an f-value of 0.05 indicates rare presence but the FSF could be specific to a particular phylum or group of organisms (e.g., Firmicutes). However, and to emphasize, the f-value was coupled with known biochemical functions of the protein fold, i.e., viral-like (e.g., virion assembly) or cell-like (e.g., metabolism) functions, which was then used as composite variable to postulate the direction of candidate HGT event (see Nasir and Caetano-Anollés, 2013 for previous applications of the approach). When the molecular function of an FSF is well-known (i.e., cell-like or virus-like), it becomes easier to postulate a direction of gene transfer and to also exclude convergence as an alternative scenario. Moreover, FSFs that are specific to only one phylum (or a group of organisms) are likely not to be inherited vertically but after the divergence from the common ancestor of that group, a time period that follows virus-cell divergence.
Fourth, we raise the issue of coverage of viral proteomes, where coverage is defined by the number of viral genes (proteins) with significant homologs in either sequence or structure databases. We have previously shown that roughly >60% of viral proteins did not match to known FSFs (Figure 2B in Nasir and Caetano-Anollés, 2015). It is already well-known that the majority of viral genes lack sequence homologies, putatively termed ORFans (Ogata and Claverie, 2007; Yin and Fischer, 2008; Cortez et al., 2009). These viral genes either evolved fast and hence are no longer recognizable at either sequence or structure levels, or represent genes that originated directly in viruses (Forterre, 2011) (e.g., VSFs in Table 2). Determining the origin of viral ORFans remains an open and important question in virology research.
Finally, we only considered f-values of virus-encoded FSFs in cellular proteomes and not in viral proteomes. Viruses are notorious for encoding small-sized genomes that are likely a result of extreme genome reduction (Nasir et al., 2012a; Claverie and Abergel, 2013). In a recent analysis, we showed that only three viral FSFs had an f-value of over 0.3 (Nasir and Caetano-Anollés, 2015). This result is unsurprising considering that the 3,440 viruses of this study belong to seven different replication strategies, infect the many diverse groups of cellular organisms (see Nasir et al., 2014 for a mapping of virus replicons to their hosts), and in general harbor genomes and particle sizes that are minimalistic. The tendency of viruses to reduce genome size over long evolutionary timespans has effectively led to loss of information when extant virus genomes are comparatively analyzed with cellular genomes. Indeed, no single FSF could be detected in all seven viral replicon types (Nasir and Caetano-Anollés, 2015). Therefore, we caution the readers that the strategy reported in this study takes a modern-day snapshot of the proteomes of both viruses and cellular organisms and does not benefit from phylogenomic reconstruction. It is also dependent on the size of available genomic databases that are severely under-represented, especially, in archaeal and viral genome sequences. However, we do not expect that sequencing and discovery of novel viral and cellular lineages will drastically compromise our conclusions since we used a very strict threshold (e.g., f < 1%) in classifying an FSF to be acquired horizontally from viruses along with investigation of its biochemical function (e.g., virion synthesis). That is, future discovery of a virus-hallmark FSF in hundreds of newly sequenced genomes of a superkingdom that would significantly increase the f-values should be considered a highly unlikely event. However, discovery of novel viruses/cellular lineages can definitely add more virus-derived genes in cellular organisms thus adding to the lists of virus-acquired genes in cells or virus-specific genes (Table 2). Moreover, we restricted our analysis to the reference genomes of viruses and corresponding host organisms and to coding DNA. The next logical step is to perform a similar exercise on viruses recovered from metagenomic samples that are increasingly populating bioinformatics databases due to the continuous decline in sequencing cost and availability of fast and reliable high-throughput sequencing platforms. However, it can be sometimes challenging to establish host tropism in metagenome samples. Furthermore, there is no single universal gene (i.e., ribosomal RNA gene in cellular organisms) that can taxonomically classify short sequencing reads of viral metagenomes (Rohwer and Edwards, 2002). That is why we restricted our analysis to only well-curated reference genomes with virus host information available from experimental studies. Similarly, many viral genetic elements are permanently integrated into cellular genomes (Katzourakis and Gifford, 2010). This DNA also originated in viruses and thus should be considered the horizontal transfer of non-coding virus-to-cell transfer. While the virology community remains divided whether or not to include viruses in the realm of life (Claverie and Ogata, 2009; Moreira and Lopez-Garcia, 2009; Forterre, 2016), there have been recent important phylogenomic data-driven breakthroughs unfolding viral origins (Nasir and Caetano-Anollés, 2015). Past events such as FSFs lost via reductive evolution or species extinction leading to loss of ancestral FSFs cannot be accounted for in this analysis without phylogenomic reconstruction. Moreover, inferences drawn in this study are best parsimonious explanations consistent with reported data and lead to testable hypotheses. It is expected that a global dissection of viral and host proteomes will inspire debates and improve studies to better understand viral-host co-evolution.
Methods
Data retrieval and manipulation
Proteome data used in this study was taken from Nasir and Caetano-Anollés (2015). In brief, a total of 190,610 protein sequences corresponding to 3,966 completely-sequenced reference genomes of viruses that were available on the NCBI Viral Genomes Resource in June 2014 (Brister et al., 2015) were downloaded. Reference virus genomes are well-curated first genomes submitted for any virus species. Subsequent submissions of new genomes for that virus species are termed “genome neighbors.” In our study, we only kept reference viral genomes, corresponding to any of the seven known viral replicon types (i.e., dsDNA, ssDNA, plus-ssRNA, minus-ssRNA, dsRNA, and retrotranscribing viruses) and excluding genomic neighbors, viruses that were listed as either “unclassified” or “unassigned” and deltaviruses.
Protein structure assignment
Viral proteins were scanned against a library of hidden Markov models (HMMs) (Gough et al., 2001; Gough and Chothia, 2002) maintained by the SUPERFAMILY database (ver. 1.75) (de Lima Morais et al., 2011) to detect SCOP FSFs using stringent E-value cutoff of 10−4. Viral genomes with no hits were discarded from the analysis. This reduced the viral dataset to include a total of 3,460 viruses including 1,649 dsDNA, 534 ssDNA, 166 dsRNA, 991 ssRNA, and 120 retrotranscribing viruses. Virus host information was available for 3,440/3,460 viruses (Bao et al., 2004) and was used to identify 62 archaeoviruses, 1,223 bacterioviruses, and 2,155 eukaryoviruses (Table S1). In parallel, pre-calculated FSF assignments for ~11 million proteins encoded by the completely-sequenced genomes of a total of 1,620 cellular organisms including 122 Archaea, 1,115 Bacteria, and 383 Eukarya were retrieved directly from the local installation of SUPERFAMILY MySQL database (July 2014). HMM assignments for viral proteomes can be downloaded from https://figshare.com/articles/Nasir_and_Caetano-Anolles_2015_zip/4833641.
Calculation of FSF spread in proteomes
The spread of each viral FSF in proteomes was calculated by an f-value, which represents the number of proteomes in a superkingdom (or virus group) encoding an FSF divided by the total number of proteomes in that superkingdom (or group). The resulting statistic is given on a scale from 0 to 1 indicating range from either complete absence (i.e., f-value = 0) to ubiquitous presence (f-value = 1). The index does not evaluate how heterogeneous is that distribution.
Determination of virus-to-cell and cell-to-virus HGT events
Two factors were considered when postulating the direction of gene transfer (Figure 1): (i) the reported biochemical function of an FSF (e.g., virion synthesis or ATP synthesis), and (ii) spread of that FSF in the proteomes of cellular superkingdom(s). For example, if an FSF involved in capsid assembly (a virus hallmark function) was detected in only few cellular proteomes (e.g., <1%), then this FSF was determined to have transferred horizontally from virus-to-cell. In turn, if an FSF involved in cellular hallmark function (e.g., metabolism) and widespread in cellular proteomes (e.g., >60% presence) was detected in some viral proteomes then this was FSF was determined to have transferred horizontally from cell to viruses. The exception would be the presence of “cell-like” FSFs in the proteomes of all three virus groups, i.e., archaeoviruses, bacterioviruses, and eukaryoviruses, suggesting a cellular co-existence between viral and cellular ancestors prior to diversification of modern life (Nasir et al., 2012a,b). Thus, both the FSF spread and its biochemical function were considered when postulating the direction of gene transfer.
Author contributions
AN conceived the study. AN, KMK, and GCA designed the experiments. SSM and SAZ performed the preliminary experiments and wrote the first draft. AN, KMK, and GCA edited and improved the manuscript. All authors approved the study, final manuscript, and conclusions.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AN would like to thank members of the Computational Biology and Bioinformatics Group at COMSATS, Islamabad, for their support.
Footnotes
Funding. This work was supported by grants from the Higher Education Commission, Start-up Research Grant Program (Project no. 21-519/SRGP/R&D/HEC/2014), Pakistan to AN, from the National Science Foundation (OISE-1132791) and the National Institute of Food and Agriculture (ILLU-802-909 and ILLU-483-625) to GCA, and from the Collaborative Genome Program (20140428) funded by the Ministry of Oceans and Fisheries, Korea to KMK.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02110/full#supplementary-material
List of viruses analyzed in this study. Also listed are virus replicon type, virus host, and known family/order.
FSFs detected in archaeoviruses. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are also listed for Archaea, Bacteria, and Eukarya. FSFs shared exclusively with non-host superkingdoms are highlighted.
FSFs detected in bacterioviruses. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are also listed for Archaea, Bacteria, and Eukarya. FSFs shared exclusively with non-host superkingdoms are highlighted.
FSFs detected in eukaryoviruses. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are also listed for Archaea, Bacteria, and Eukarya. FSFs shared exclusively with non-host superkingdoms are highlighted.
The composition of BE Venn group in bacterioviruses and eukaryoviruses. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are listed for Bacteria, and Eukarya. The difference in f-values was calculated by subtracting f-value of an FSF in Bacteria from the corresponding f-value for that FSF in Eukarya. FSFs with an f-value difference of over 25% are highlighted (yellow when f-value in Eukarya exceeds that of in Bacteria and blue for vice versa). Data sorted in an increasing manner for FSFs common to both bacterioviruses and eukaryoviruses, unique to bacterioviruses, and unique to eukaryoviruses.
A non-redundant list of 442 ABE viral FSFs detected in all three superkingdoms. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are also listed for Archaea, Bacteria, and Eukarya, and archaeoviruses, bacterioviruses, and eukaryoviruses. abe FSFs are highlighted.
References
- Abrescia N. G. A., Grimes J. M., Fry E. E., Ravantti J. J., Bamford D. H., Stuart D. I. (2010). What does it take to make a virus: the concept of the viral ‘self,’ in Emerging Topics in Physical Virology, eds Stockley P. G., Twarock R. (London, UK: Imperial College Press; ), 35–58. [Google Scholar]
- Abrescia N. G., Bamford D. H., Grimes J. M., Stuart D. I. (2012). Structure unifies the viral universe. Annu. Rev. Biochem. 81, 795–822. 10.1146/annurev-biochem-060910-095130 [DOI] [PubMed] [Google Scholar]
- Abroi A., Gough J. (2011). Are viruses a source of new protein folds for organisms? - Virosphere structure space and evolution. Bioessays 33, 626–635. 10.1002/bies.201000126 [DOI] [PubMed] [Google Scholar]
- Aminov R. I. (2013). Role of archaea in human disease. Front. Cell. Infect. Microbiol. 3:42. 10.3389/fcimb.2013.00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva A., Howorth D., Chandonia J. M., Brenner S. E., Hubbard T. J., Chothia C., et al. (2008). Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 36, D419–D425. 10.1093/nar/gkm993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arslan D., Legendre M., Seltzer V., Abergel C., Claverie J. M. (2011). Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. U.S.A. 108, 17486–17491. 10.1073/pnas.1110889108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford D. H. (2003). Do viruses form lineages across different domains of life? Res. Microbiol. 154, 231–236. 10.1016/S0923-2508(03)00065-2 [DOI] [PubMed] [Google Scholar]
- Bao Y., Federhen S., Leipe D., Pham V., Resenchuk S., Rozanov M., et al. (2004). National center for biotechnology information viral genomes project. J. Virol. 78, 7291–7298. 10.1128/JVI.78.14.7291-7298.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker J., Brown M. R. W. (1994). Trojan horses of the microbial world: protozoa and the survival of bacterial pathogens in the environment. Microbiology 140, 1253–1259. 10.1099/00221287-140-6-1253 [DOI] [PubMed] [Google Scholar]
- Benson S. D., Bamford J. K. H., Bamford D. H., Burnett R. M. (2004). Does common architecture reveal a viral lineage spanning all three domains of life? Mol. Cell 16, 673–685. 10.1016/j.molcel.2004.11.016 [DOI] [PubMed] [Google Scholar]
- Bordenstein S. R., Bordenstein S. R. (2016). Eukaryotic association module in phage WO genomes from Wolbachia. Nat. Commun. 7:13155. 10.1038/ncomms13155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister J. R., Ako-Adjei D., Bao Y., Blinkova O. (2015). NCBI viral genomes resource. Nucleic Acids Res. 43, D571–D577. 10.1093/nar/gku1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüssow H., Canchaya C., Hardt W.-D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602. 10.1128/MMBR.68.3.560-602.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano-Anollés G., Nasir A. (2012). Benefits of using molecular structure and abundance in phylogenomic analysis. Front. Genet. 3:172. 10.3389/fgene.2012.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canchaya C., Fournous G., Brüssow H. (2004). The impact of prophages on bacterial chromosomes. Mol. Microbiol. 53, 9–18. 10.1111/j.1365-2958.2004.04113.x [DOI] [PubMed] [Google Scholar]
- Chartier F., Laine B., Belaïche D., Touzel J. P., Sautière P. (1989). Primary structure of the chromosomal protein MC1 from the archaebacterium Methanosarcina sp. CHTI 55. Biochim. Biophys. Acta 1008, 309–314. 10.1016/0167-4781(89)90021-3 [DOI] [PubMed] [Google Scholar]
- Cheng S., Brooks C. L. (2013). Viral capsid proteins are segregated in structural fold space. PLoS Comput. Biol. 9:e1002905. 10.1371/journal.pcbi.1002905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claverie J. M., Abergel C. (2013). Open questions about giant viruses. Adv. Virus Res. 85, 25–56. 10.1016/B978-0-12-408116-1.00002-1 [DOI] [PubMed] [Google Scholar]
- Claverie J. M., Ogata H. (2009). Ten good reasons not to exclude giruses from the evolutionary picture. Nat. Rev. 7:615 10.1038/nrmicro2108-c3 [DOI] [PubMed] [Google Scholar]
- Claverie J.-M., Abergel C. (2016). Giant viruses: the difficult breaking of multiple epistemological barriers. Stud. Hist. Philos. Biol. Biomed. Sci. 59, 89–99. 10.1016/j.shpsc.2016.02.015 [DOI] [PubMed] [Google Scholar]
- Cornelis G., Heidmann O., Bernard-Stoecklin S., Reynaud K., Veron G., Mulot B., et al. (2012). Ancestral capture of syncytin-Car1, a fusogenic endogenous retroviral envelope gene involved in placentation and conserved in Carnivora. Proc. Natl. Acad. Sci. U.S.A. 109, E432–E441. 10.1073/pnas.1115346109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D., Forterre P., Gribaldo S. (2009). A hidden reservoir of integrative elements is the major source of recently acquired foreign genes and ORFans in archaeal and bacterial genomes. Genome Biol. 10:r65. 10.1186/gb-2009-10-6-r65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cunha V., Gaia M., Gadelle D., Nasir A., Forterre P. (2017). Lokiarchaea are close relatives of Euryarchaeota, not bridging the gap between prokaryotes and eukaryotes. PLoS Genet. 13:e1006810 10.1371/journal.pgen.1006810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima Morais D. A., Fang H., Rackham O. J., Wilson D., Pethica R., Chothia C., et al. (2011). SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 39, D427–D434. 10.1093/nar/gkq1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde N. C., Malik H. S. (2009). The evolutionary conundrum of pathogen mimicry. Nat. Rev. Microbiol. 7, 787–797. 10.1038/nrmicro2222 [DOI] [PubMed] [Google Scholar]
- Feschotte C., Gilbert C. (2012). Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 13, 283–296. 10.1038/nrg3199 [DOI] [PubMed] [Google Scholar]
- Forterre P. (2011). Manipulation of cellular syntheses and the nature of viruses: the virocell concept. Comptes Rendus Chim. 14, 392–399. 10.1016/j.crci.2010.06.007 [DOI] [Google Scholar]
- Forterre P. (2016). To be or not to be alive: how recent discoveries challenge the traditional definitions of viruses and life. Stud. Hist. Philos. Biol. Biomed. Sci. 59, 100–108. 10.1016/j.shpsc.2016.02.013 [DOI] [PubMed] [Google Scholar]
- Forterre P., Krupovic M. (2012). The origin of virions and virocells: the escape hypothesis revisited, in Viruses: Essential Agents of Life, ed Witzany G. (Dordrecht: Springer; ), 43–60. [Google Scholar]
- Fox N. K., Brenner S. E., Chandonia J. M. (2014). SCOPe: Structural Classification of Proteins–extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 42, D304–D309. 10.1093/nar/gkt1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. L., Duchêne S., Holmes E. C. (2017). Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLoS Pathog. 13:e1006215. 10.1371/journal.ppat.1006215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill E. E., Brinkman F. S. L. (2011). The proportional lack of archaeal pathogens: do viruses/phages hold the key? Bioessays 33, 248–254. 10.1002/bies.201000091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough J. (2005). Convergent evolution of domain architectures (is rare). Bioinformatics 21, 1464–1471. 10.1093/bioinformatics/bti204 [DOI] [PubMed] [Google Scholar]
- Gough J., Chothia C. (2002). SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 30, 268–272. 10.1093/nar/30.1.268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough J., Karplus K., Hughey R., Chothia C. (2001). Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 313, 903–919. 10.1006/jmbi.2001.5080 [DOI] [PubMed] [Google Scholar]
- Guy L., Saw J. H., Ettema T. J. G. (2014). The archaeal legacy of eukaryotes: a phylogenomic perspective. Cold Spring Harb. Perspect. Biol. 6:a016022. 10.1101/cshperspect.a016022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix R. W., Lawrence J. G., Hatfull G. F., Casjens S. (2000). The origins and ongoing evolution of viruses. Trends Microbiol. 8, 504–508. 10.1016/S0966-842X(00)01863-1 [DOI] [PubMed] [Google Scholar]
- Hoffmann C., Dollive S., Grunberg S., Chen J., Li H., Wu G. D., et al. (2013). Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS ONE 8:e66019. 10.1371/journal.pone.0066019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Caforio A., Driessen A. J. M. (2014). Biosynthesis of archaeal membrane ether lipids. Front. Microbiol. 5:641. 10.3389/fmicb.2014.00641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzourakis A., Gifford R. J. (2010). Endogenous viral elements in animal genomes. PLoS Genet. 6:e1001191. 10.1371/journal.pgen.1001191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V., Dolja V. V., Krupovic M. (2015). Origins and evolution of viruses of eukaryotes: the ultimate modularity. Virology 479, 2–25. 10.1016/j.virol.2015.02.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M., Dutilh B. E., Adriaenssens E. M., Wittmann J., Vogensen F. K., Sullivan M. B., et al. (2016). Taxonomy of prokaryotic viruses: update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 161, 1095–1099. 10.1007/s00705-015-2728-0 [DOI] [PubMed] [Google Scholar]
- La Scola B., Audic S., Robert C., Jungang L., de Lamballerie X., Drancourt M., et al. (2003). A giant virus in amoebae. Science 299:2033. 10.1126/science.1081867 [DOI] [PubMed] [Google Scholar]
- La Scola B., Desnues C., Pagnier I., Robert C., Barrassi L., Fournous G., et al. (2008). The virophage as a unique parasite of the giant mimivirus. Nature 455, 100–104. 10.1038/nature07218 [DOI] [PubMed] [Google Scholar]
- Legendre M., Bartoli J., Shmakova L., Jeudy S., Labadie K., Adrait A., et al. (2014). Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. U.S.A. 111, 4274–4279. 10.1073/pnas.1320670111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M., Lartigue A., Bertaux L., Jeudy S., Bartoli J., Lescot M., et al. (2015). In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. U.S.A. 112, E5327–E5335. 10.1073/pnas.1510795112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J. H., et al. (2005). Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679. 10.1126/science.1118391 [DOI] [PubMed] [Google Scholar]
- Lindell D., Sullivan M. B., Johnson Z. I., Tolonen A. C., Rohwer F., Chisholm S. W. (2004). Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc. Natl. Acad. Sci. U.S.A. 101, 11013–11018. 10.1073/pnas.0401526101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longdon B., Brockhurst M. A., Russell C. A., Welch J. J., Jiggins F. M. (2014). The evolution and genetics of virus host shifts. PLoS Pathog. 10, e1004395. 10.1371/journal.ppat.1004395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurie-Weinberger M. N., Gophna U. (2015). Archaea in and on the human body: health implications and future directions. PLoS Pathog. 11:e1004833. 10.1371/journal.ppat.1004833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molmeret M., Horn M., Wagner M., Santic M., Abu Kwaik Y. (2005). Amoebae as training grounds for intracellular bacterial pathogens. Appl. Environ. Microbiol. 71, 20–28. 10.1128/AEM.71.1.20-28.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira D., Lopez-Garcia P. (2009). Ten reasons to exclude viruses from the tree of life. Nat. Rev. 7, 306–311. 10.1038/nrmicro2108 [DOI] [PubMed] [Google Scholar]
- Nasir A., Caetano-Anollés G. (2013). Comparative analysis of proteomes and functionomes provides insights into origins of cellular diversification. Archaea 2013:648746. 10.1155/2013/648746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A., Caetano-Anollés G. (2015). A phylogenomic data-driven exploration of viral origins and evolution. Sci. Adv. 1:e1500527. 10.1126/sciadv.1500527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A., Caetano-Anollés G. (2017). Identification of capsid/coat related protein folds and their utility for virus classification. Front. Microbiol. 8:380. 10.3389/fmicb.2017.00380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A., Forterre P., Kim K. M., Caetano-Anollés G. (2014). The distribution and impact of viral lineages in domains of life. Front. Microbiol. 5:194. 10.3389/fmicb.2014.00194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A., Kim K. M. K. M., Da Cunha V., Caetano-Anollés G. (2016). Arguments reinforcing the three-domain view of diversified cellular life. Archaea 2016, 1–11. 10.1155/2016/1851865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A., Kim K. M., Caetano-Anolles G. (2012a). Giant viruses coexisted with the cellular ancestors and represent a distinct supergroup along with superkingdoms Archaea, Bacteria and Eukarya. BMC Evol. Biol. 12:156. 10.1186/1471-2148-12-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A., Kim K. M., Caetano-Anollés G. (2012b). Viral evolution Primordial cellular origins and late adaptation to parasitism. Mob. Genet. Elements 2, 247–252. 10.4161/mge.22797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A., Kim K. M., Caetano-Anollés G. (2017). Long-term evolution of viruses: a Janus-faced balance. Bioessays 39:e201700026. 10.1002/bies.201700026 [DOI] [PubMed] [Google Scholar]
- Ogata H., Claverie J.-M. (2007). Unique genes in giant viruses: regular substitution pattern and anomalously short size. Genome Res. 17, 1353–1361. 10.1101/gr.6358607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H., Douady C. J. (2003). Horizontal gene transfer and phylogenetics. Curr. Opin. Microbiol. 6, 498–505. 10.1016/j.mib.2003.09.008 [DOI] [PubMed] [Google Scholar]
- Philippe N., Legendre M., Doutre G., Couté Y., Poirot O., Lescot M., et al. (2013). Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 341, 281–286. 10.1126/science.1239181 [DOI] [PubMed] [Google Scholar]
- Rappoport N., Linial M. (2012). Viral proteins acquired from a host converge to simplified domain architectures. PLoS Comput. Biol. 8:e1002364. 10.1371/journal.pcbi.1002364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer F., Edwards R. (2002). The phage proteomic tree: a genome based taxonomy for phage. J. Bacteriol. 184, 4529–4535 10.1128/JB.184.16.4529-4535.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., Nebot M. R., Chirico N., Mansky L. M., Belshaw R. (2010). Viral mutation rates. J. Virol. 84, 9733–9748. 10.1128/JVI.00694-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz F., Yutin N., Ivanova N. N., Ortega D. R., Lee T. K., Vierheilig J., et al. (2017). Giant viruses with an expanded complement of translation system components. Science 356, 82–85. 10.1126/science.aal4657 [DOI] [PubMed] [Google Scholar]
- Sharp P. M., Hahn B. H. (2010). The evolution of HIV-1 and the origin of AIDS. Philos. Trans. R. Soc. B Biol. Sci. 365, 2487–2494. 10.1098/rstb.2010.0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shterzer N., Mizrahi I. (2015). The animal gut as a melting pot for horizontal gene transfer. Can. J. Microbiol. 61, 603–605. 10.1139/cjm-2015-0049 [DOI] [PubMed] [Google Scholar]
- Spang A., Saw J. H., Jørgensen S. L., Zaremba-Niedzwiedzka K., Martijn J., Lind A. E., et al. (2015). Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179. 10.1038/nature14447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P. J., Ley R. E., Hamady M., Fraser-Liggett C. M., Knight R., Gordon J. I. (2007). The human microbiome project. Nature 449, 804–810. 10.1038/nature06244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webby R. J., Webster R. G. (2001). Emergence of influenza A viruses. Philos. Trans. R. Soc. B Biol. Sci. 356, 1817–1828. 10.1098/rstb.2001.0997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeates T. O., Thompson M. C., Bobik T. A. (2011). The protein shells of bacterial microcompartment organelles. Curr. Opin. Struct. Biol. 21, 223–231. 10.1016/j.sbi.2011.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeates T. O., Tsai Y., Tanaka S., Sawaya M. R., Kerfeld C. A. (2007). Self-assembly in the carboxysome: a viral capsid-like protein shell in bacterial cells. Biochem. Soc. Trans. 35, 508–511. 10.1042/BST0350508 [DOI] [PubMed] [Google Scholar]
- Yin Y., Fischer D. (2008). Identification and investigation of ORFans in the viral world. BMC Genomics 9:24. 10.1186/1471-2164-9-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba-Niedzwiedzka K., Caceres E. F., Saw J. H., Bäckström D., Juzokaite L., Vancaester E., et al. (2017). Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541, 353–358. 10.1038/nature21031 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of viruses analyzed in this study. Also listed are virus replicon type, virus host, and known family/order.
FSFs detected in archaeoviruses. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are also listed for Archaea, Bacteria, and Eukarya. FSFs shared exclusively with non-host superkingdoms are highlighted.
FSFs detected in bacterioviruses. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are also listed for Archaea, Bacteria, and Eukarya. FSFs shared exclusively with non-host superkingdoms are highlighted.
FSFs detected in eukaryoviruses. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are also listed for Archaea, Bacteria, and Eukarya. FSFs shared exclusively with non-host superkingdoms are highlighted.
The composition of BE Venn group in bacterioviruses and eukaryoviruses. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are listed for Bacteria, and Eukarya. The difference in f-values was calculated by subtracting f-value of an FSF in Bacteria from the corresponding f-value for that FSF in Eukarya. FSFs with an f-value difference of over 25% are highlighted (yellow when f-value in Eukarya exceeds that of in Bacteria and blue for vice versa). Data sorted in an increasing manner for FSFs common to both bacterioviruses and eukaryoviruses, unique to bacterioviruses, and unique to eukaryoviruses.
A non-redundant list of 442 ABE viral FSFs detected in all three superkingdoms. FSFs are identified both by SCOP numeric IDs and alpha-numeric concise classification strings (ccs). The f-values are also listed for Archaea, Bacteria, and Eukarya, and archaeoviruses, bacterioviruses, and eukaryoviruses. abe FSFs are highlighted.