

Abstract
Biological rhythms lie at the center of regulatory schemes that control many aspects of living systems. At the cellular level, meaningful responses to external stimuli depend on propagation and quenching of a signal to maintain vigilance for subsequent stimulation or changes that serve to shape and modulate the response. The hypothalamus–pituitary–gonad endocrine axis that controls reproductive development and function relies on control through rhythmic stimulation. Central to this axis is the pulsatile stimulation of the gonadotropes by hypothalamic neurons through episodic release of the neuropeptide gonadotropin-releasing hormone. Alterations in pulsatile stimulation of the gonadotropes result in differential synthesis and secretion of the gonadotropins LH and FSH and changes in the expression of their respective hormone subunit genes. The requirement to amplify signals arising from activation of the gonadotropin-releasing hormone (GnRH) receptor and to rapidly quench the resultant signal to preserve an adaptive response suggests the need for rapid activation and feedback control operating at the level of intracellular signaling. Emerging data suggest that reactive oxygen species (ROS) can fulfill this role in the GnRH receptor signaling through activation of MAP kinase signaling cascades, control of negative feedback, and participation in the secretory process. Results obtained in gonadotrope cell lines or other cell models indicate that ROS can participate in each of these regulatory cascades. We discuss the potential advantage of reactive oxygen signaling for modulating the gonadotrope response to GnRH stimulation and the potential mechanisms for this action. These observations suggest further targets of study for regulation in the gonadotrope.
Keywords: reactive oxygen species, gonadotropin-releasing hormone, pulsatility, DUSP1, gonadotropins, mitogen-activated protein kinase, ERK, pituitary, metabolism
The Challenge of Pulsatile Gonadotropin-Releasing Hormone (GnRH) Signaling in Gonadotropes
The fundamental role of pulsatile stimulation of gonadotropes by the hypothalamic neuropeptide GnRH, or GnRH-I in maintaining function of the hypothalamic–pituitary–gonad (HPG) axis is one of the earliest principal findings after discovery of the hormone (1, 2). Studies in nonhuman primates demonstrated the requirement for pulsatile stimulation of the pituitary to maintain the reproductive axis (3). The identity of the signaling molecules and mechanisms that contribute to pulse interpretation has been the subject of extensive study since many models developed to explain signaling control of gene expression (4–13). But questions remain concerning the mechanism of pulse interpretation and the signaling factors responsible. The hypothalamic neuropeptide GnRH and its receptor GnRHR are the prototypic members of a superfamily that has evolutionary roots reaching to the emergence of the bilateria (14). In vertebrates, a feature of this pair is its central role in regulating the anterior pituitary gonadotropes (15, 16). The hypothalamic GnRH neurons release hormone into the adenohypophyseal portal circulation in an episodic manner that is central to the development and operation of the HPG axis and fertility. In this system, the GnRHR governs the release of the gonadotropins LH and FSH and regulates expression of their subunit genes. The mammalian GnRHR is unique in structure, lacking a cytoplasmic tail that is normally associated with β-arrestin-mediated downregulation of receptor signaling. Thus, GnRHR itself faces unique challenges in transmitting an episodic signal in which alterations in amplitude and frequency are meaningful, yet, receptor homologous desensitization is not an accessible regulatory scheme. It is likely that pulse interpretation is accomplished by the operation of the signaling cascades themselves rather than desensitization or receptor availability at the membrane.
In mouse LβT2 cells, the switch between LH and FSH preference occurs at the 60-min pulse interval (17). A general switching mechanism is achieved by the expression and decay of activating and repressing transcription factors that create high- or low-pass filters to govern gene expression. For Lhb, this is the pairing of the immediate-early Egr1 family of transcriptional activators with the Nab1/2 family of repressors. Transient frequency and amplitude-dependent stimulation of Egr1 expression is countered by pulse-insensitive expression of Nab1/2, establishing a high-pass filter that requires sustained stimulation to overcome suppression (17). Features of this model have been confirmed by in vivo studies and mixed primary pituitary culture in rats and in αT3-1 cells that do not express gonadotropin β-subunit genes (18). On the other hand, Fshb prefers low frequencies for promoter activator (c-Fos and c-Jun) upregulation, and high frequencies lead to upregulation of Fshb promoter inhibitors, such as Skil and Tgif1 (19). A feature of pulse decoding in the GnRH system is the occurrence of maximal responses at submaximal stimulation, creating a bell-curve frequency response that requires complex regulation but imparts true frequency decoding (20, 21). Components of the signaling network may exhibit digital tracking in which each response is resolved between pulses and acts dependently, or in the case of slower, incomplete resolution, exhibits integrative tracking in which the cumulative stimulation creates a maximal response (10). Transcriptional regulation of gonadotropin subunit genes is modest overall but exhibits integrative interpretation (22). GnRH also regulates protein synthesis and the distribution of mRNA in polyribosomes (23–26). Each of these may utilize different interpretative mechanisms.
MAP Kinase Signaling in Response to GnRH
GnRHR is a G protein-coupled receptor that signals primarily via the Gαq/11 G protein subfamily, although interaction with other G proteins is also documented in vivo (27, 28). Stimulation of gonadotropes or gonadotrope-derived cell lines causes activation of phospholipase C, resulting in inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) production. IP3 mobilizes Ca2+ from intracellular stores and influx via L-type voltage-gated Ca2+ channels. The mobilization of Ca2+ is associated with initiation of the secretory response and fusion of secretory granules with the extracellular membrane. In a related signaling branch, DAG along with Ca2+ activates multiple PKC isozymes, including the conventional isoforms PKCα, PKCβII, the novel isoforms PKCδ and PKCε, and the atypical PKCζ in αT3-1 and LβT2 cells (29, 30). These activated signals link to downstream induction of mitogen-activated protein kinases (MAPK) (18, 31–33). The role of MAPK1/3 (ERK1/2) is sexually dimorphic and essential in female reproduction (34). Phosphorylation of MAPK1/3 is highly stimulated within a few minutes and rapidly resolved such that MAPK1/3 activation is restored to prestimulation levels well within the 60-min interval switch point of differential gene expression (Figure 1) (35). The connection between PKC and MAPK1/3 activation is well appreciated, but the intervening sequence of Ras/Raf/MAPK kinase (MEKK) signaling is not well described (29, 30). MAPK1/3 activation can occur through the c-SRC-mediated RAS activation (30, 36) and, in other cells, RAS activation occurs through DAG-dependent GRP1/2. However, recent evidence has shown that GnRH-stimulated MAPK1/3 activation in gonadotropes depends on reactive oxygen species (ROS) production by the NADPH oxidases (37). This suggests that multiple pathways contribute to MAPK1/3 activation and examination may shed light on their contribution to pulse interpretation.
Figure 1.

Reactive oxygen species (ROS) involvement in mitogen-activated protein kinases (MAPK) 1/3 activation by gonadotropin-releasing hormone (GnRH) and resolution in gonadotrope cells. Activation profiles of MAPK1/3 and DUSP1 as determined by phosphorylation in response to a single GnRH pulse [adapted from Ref. (35)]. GnRH receptor-signaling via Gaq/11 activates phospholipase C, leading to diacylglycerol (DAG) and IP3 production. The DAG and IP3-induced rise in intracellular Ca2+ activate both NOX and DUOX family members, resulting in increased ROS production. ROS stimulates MAPK1/3 activation by promoting Ras and Raf activation of the MEKn cascade ultimately targeting MAPK1/3. Oxidative activation of epidermal growth factor receptor (EGFR) contributes to MAPK1/3 activation through Raf. ROS may also transiently inactivate negative feedback through reversible oxidation of the DUSP active-site cysteine. ROS is normally reduced by peroxiredoxin (PRDX) by conversion of active reducing site cysteine thiol C–SH to sulfenic C–SOH. Sulfenic cysteine is recycled by thioredoxin (TRX) reduction. Excess ROS contributes to PRDX hyperoxidation that further oxidizes the sulfenic C–SOH to the sulfinic C–SOOH, which is reduced by the ATP-dependent reductase activity of sulfiredoxin 1 (SRXN1), preserving PRDX capacity but allowing transient DUSP inactivation. DUSP activity is resumed after ROS level declines, permitting feedback control of MAPK1/3. ROS activation of L-type VGCC promotes intracellular Ca2+ that supports exocytosis and activation of DUOX.
ROS Integration into Pathways Promoting MAP Kinase Signaling
Reactive oxygen species are partially reduced metabolites of oxygen produced through intracellular mechanisms or encountered in extracellular environments. Mitochondrial ROS are produced by aerobic respiration and incomplete oxidation of fatty acids and can indicate mitochondrial and endoplasmic reticulum stress. ROS are also employed as rapid signaling molecules through production by the NADPH/Dual Oxidase (NOX/DUOX) family, which are targets of activation by intracellular kinases or elevated intracellular Ca2+ (38). Exposure to ROS can cause oxidative damage to many biomolecules, resulting in nucleic acid damage or mutation, enzymatic dysfunction, or cell death. Therefore, management of ROS is a focus of cellular homeostasis. Reducing systems operating through peroxiredoxins (PRDX1-6), thioredoxin (TRX), and glutathione (GSH) exchange of free radical oxygen are present in all cells and PRDX isoforms partition into subcellular regions for specialized action. Each of the six mammalian PRDX isoforms is represented in to the top 5% of cellular protein content (39), collectively constituting a high proportion of cellular protein and a significant investment in localizing ROS action and limiting oxidative damage.
Elevated ROS is associated with MAPK activation in multiple cell types (40). Insulin-like growth factor I activation of MAPK1/3 increases ROS production and antioxidants inhibit activation of the MAPK1/3 pathway, showing dependence on ROS (38–41). Similarly, MAPK8/9 (JNK) and MAPK14 (p38 MAPK) phosphorylation is associated with ROS generation (42–44). The MAP3K-related kinase ASK1 associates with TRX and is released upon TRX oxidation, permitting activation of MAPK8/9/14 (44). In other professional secretory cells, ROS is central to secretion and activation of biosynthesis. In the endocrine pancreas, NOX enzymes are involved in stimulated insulin secretion and excess ROS production increases oxidative stress and loss of function (45–47). NOX/DUOX participate in the signaling response activating thyroid hormone biosynthesis (48–50). ROS mediates enhanced MAP kinase activation in activated eosinophils, contributing to IL-5-mediated cell death (51). In contrast, NOX is a target of MAP kinase activation in neutrophils and ROS signaling is utilized in formation of neutrophil extracellular traps (52). NOX proteins are, therefore, both upstream activators of MAP kinase signaling and targets of MAP kinase action, suggesting plasticity in how NOX/DUOX-derived ROS is deployed.
Gonadotropin-releasing hormone stimulation of mouse primary pituitary and LβT2 cells that endogenously express all gonadotropin subunit genes (53, 54), results in ROS production that is blocked by pharmacological inhibition of NOX/DUOX enzymes with diphenyleneiodonium (DPI). N-acetyl cysteine (NAC), which is general ROS scavenger, also attenuates MAPK1/3 and MAPK8/9 activation. Both DPI and NAC attenuate activation of Lhb and Fshb transcription (37). Further, GnRH-mediated activation of ROS depends on PKC and Ca2+ availability. This places ROS between PKC and MAPK1/3 and supports an intermediate role in activation similar to ROS-mediated activation of Ras through kinase and regulatory subunit regulation (55, 56). The rapid and transient activation of MAPK1/3 by GnRH is similar to that reported by direct activation by H2O2 via epidermal growth factor receptor (EGFR) (57). The cysteine-rich motifs of growth factor receptors including EGFR are proposed targets of activation by oxidation (42). But demonstration using suramin and its known broad actions that include inhibition of G-protein receptor signaling suggests that revisiting this may be warranted. GnRH signaling is also associated with EGFR activation possibly through matrix metalloprotease liberation of extracellular ligand (58–60). An alternative ligand-independent activation pathway through oxidative activation of EGFR could support the rapid elevation of MAPK 1/3 activation after GnRH stimulation (41).
In addition to activation of MAP kinase pathways through positive regulation of signaling cascades, ROS may also play a central role in promoting MAP kinase phospho-activation through inactivation of negative feedback. Dual-specificity protein phosphatases (DUSP’s, also MKP’s) serve a primary role as negative feedback regulators of MAP kinase signaling through dephosphorylation of activated MAP kinases (Figure 1) (61). DUSP’s and other protein tyrosine phosphatase superfamily members share a common catalytic site motif of [I/V]HCXXGXXR[S/T] in which the invariant cysteine residue serves as a catalytic nucleophile that is susceptible to reversible inactivating oxidation (62). Oxidative suppression of DUSP’s can support sustained activation of MAPK 8/9 and drive TNF-α-mediated cell death (63) and oxidative control of DUSP and MAPK signaling has been observed in pancreatic β-cells (46, 64), supporting this mechanism in professional secretory cells. Oxidation of DUSP’s also promotes their proteosomal degradation, limiting their availability to inhibit MAPK kinase (65). In LβT2 gonadotropes, high amplitude GnRH stimulation causes sustained activation of MAPK1/3 similar to that observed with ROS-mediated suppression of DUSP feedback (35). Chronic stimulation with GnRH also results in ROS production (37) but the status of DUSP after prolonged exposure to ROS or chronic GnRH stimulation has not been directly examined. The participation of ROS in rapid activation of MAP kinase signaling in gonadotropes through positive control of signaling cascades and negative control of feedback suggests that ROS contributes to the rapid activation of MAP kinases that is observed in response to GnRH stimulation. Involvement in both MAPK 1/3 and MAPK 8/9 activation suggests that both Fshb and Lhb transcription can be regulated through ROS.
Resolution of GnRH-Stimulated MAPK Signaling
Feedback control of MAPK activation by DUSP family members is central to the control of MAP kinase signaling networks. For cells to remain vigilant for change in GnRH pulses, sensitivity to a subsequent pulse is maintained and interpreted in context, which implies a capacity for hysteresis. In either digital or integrative pulse tracking, some balance between activation, negative feedback, and response decay must be achieved. Signaling networks may switch between modes by changing this relationship. Hysteresis in cell signaling was initially proposed and tested in the model of bistable MAPK1/3 activation by platelet-derived growth factor receptor, which showed that MAPK1/3 response amplitude is dictated by the degree of DUSP1 feedback activated by a previous signaling response (66). The role of DUSP’s in negative feedback control of MAPK1/3 activation has been examined extensively in the context of GnRHR signaling. In LβT2 cells, activation of MAPK1/3 by physiological levels of GnRH is resolved within 30 min (35). Overexpression or knockdown of nuclear-resident DUSP1 suppresses or increases activation of MAPK1/3 in response to GnRH, respectively (35). But studies in cells that do not natively express GnRHR or gonadotropin genes or using reporters of translocation have questioned this observation (67). LβT2 gonadotropes show elevated DUSP1 in unstimulated cells, suggesting that they are primed for suppression of MAP kinase signaling activation. This available phosphatase activity is subject to rapid inactivation by ROS but the reversibility of inactivation suggests some capacity is maintained or quickly recovered, contributing to the rapid resolution of MAPK1/3 activation. Another possibility is the involvement of cellular mechanisms limiting ROS through reduction by PRDX and TRX. These proteins are part of a larger network of factors controlling oxidative stress that includes GSH, catalase, superoxide dismutase, and the ATP-dependent redox factor sulfiredoxin 1 (SRXN1, also NPN3). These factors contribute to the maintenance of reductive capacity through resolution of oxidized or hyperoxidized PRDX, returning it to the pool of available reductase. Although the 2-cysteine PRDX1-4 family members are efficient ROS scavengers, they can be hyperoxidized by conversion of their nucleophilic thiol to sulfinic acid (Figure 1). Hyperoxidized PRDX is recycled through ATP-dependent reduction by SRXN1 (68, 69). In LβT2 cells, Srxn1 gene expression is proportionally induced by increasing pulsatile and tonic GnRH stimulation (17), implying a role in resolution of oxidative stress.
The restoration of feedback control can also be achieved through increased DUSP synthesis in response to GnRH stimulation. In LβT2 cells, GnRH stimulation causes transient activation of the unfolded protein response (UPR) (24). Translation is largely inhibited by the UPR but Dusp1 and Dusp8 mRNA escape translation inhibition and DUSP1 is increased during the time the UPR is active (23, 35). Translational control of Dusp1 mRNA is MAPK1/3 dependent and is attributed to the 3′UTR ELAVL1 binding site known to contribute to mRNA stability (23, 70). Pulsatile GnRH increases Dusp1, Dusp8, and Dusp16 expression, all of which target MAP kinases (17). Although DUSPs are subject to rapid inactivation by ROS, the reversibility of oxidative inhibition, the rapid translational response of the UPR, and the long-term transcriptional response to GnRH stimulation provide mechanisms for preserving feedback regulation while permitting short-term activation of MAP kinase signaling.
On a broad time scale, ROS may regulate gonadotropins through regulation of gene expression and sensitivity to GnRH through microRNA modulation of gonadotropin and Gαq/11 signaling component gene expression. MiR132/212 regulates Fshb mRNA expression and secretion via SIRT1 deacetylation in gonadotropes (71). MiR-7a2 or miR-200b and miR-429 participate in maintenance of gonadotropin gene expression (72, 73). Further, miR125b contributes to desensitization of sustained GnRH stimulation by targeting components of the Gαq/11 pathway (74). MicroRNA regulation occurs through oxidation-sensitive transcription factors such as CEBPB and ZEB1 and microRNA also directly affects MAPK signaling (75). The ROS-sensitive regulation of microRNA may further link GnRH signaling to ROS directly through GnRH receptor stimulation of ROS production or to ROS derived from other sources.
ROS in GnRH-Induced Gonadotropin Secretion
Hormone secretion by exocytosis in endocrine cells is triggered by Ca2+ released from intracellular stores. A rise of intracellular Ca2+ regulates several steps of exocytosis; including, vesicle priming and fusion to the plasma membrane (76). Localized Ca2+ increase with IP3 stimulation is necessary for gonadotropin exocytosis (77). Increased Ca2+ also induces ROS production by DUOX activation (37) and both voltage-gated and L-type calcium channels are activated by ROS (78, 79). This may tie localized Ca2+ to enzymatic ROS generation by DUOX. Thrombin promotes Ca2+ influx in smooth muscle cells by NOX-derived ROS activation of L-type calcium channels (80). Also, insulin-induced NOX increases IP3 receptor activity and Ca2+ release in skeletal muscle (81). In gonadotrope cells, DPI blocks GnRH-induced gonadotropin secretion, indicating dependence on ROS for secretion (37) and suggesting integration of Ca2+ and ROS signaling in exocytosis.
Interestingly, ROS in the form of nitric oxide may play an important role in maturation and regulation of the hypothalamus in concert with pituitary ROS. Nitric oxide production in the hypothalamus elicits GnRH secretion and expression of GnRH mRNA is modulated through miR-200 and miR-155 expression before puberty by controlling the nitric oxide-sensitive regulators CEBPB and ZEB1 (82, 83). These phenomena indicate that microRNA and ROS are tightly linked to rapid and long-term control of the HPG axis.
ROS as a Metabolic Reporter in the Gonadotrope
Energy balance has a profound influence on reproductive fitness and operation of the HPG axis. The critical fat hypothesis suggests that an optimal level of body fat is permissive to menarche and may be necessary for optimal operation of the HPG axis (84, 85). Adipose-associated changes in gonadotropin levels imply the presence of a sensing mechanism that reports energy status to the reproductive endocrine axis (86–91). Reproductive disorders such as polycystic ovary syndrome and hypogonadotropic hypogonadism are associated with metabolic dysfunction and obesity. However, not all metabolic signals associated with obesity explain the inverse relationship between adiposity and gonadotropin levels observed in men and women (92–95). Adipose-derived endocrine signals such as leptin play a role in modulating hypothalamic or pituitary function (96). Another potential modulator of the HPG axis are free fatty acids (FFA). Data suggest that FFA have a direct impact on gonadotropes in ruminants, and FFA suppresses gonadotropin secretion in cultured primary pituitary (97, 98). Unsaturated FFA can induce mitochondrial ROS production and activation of the UPR (99). We examined the ability of the monounsaturated fatty acid oleate (OLA) to induce mitochondrial ROS in LβT2 cells (Figure 2). We found that moderate physiological OLA, 500 µM, can induce mitochondrial. Unlike OLA, GnRH does not impact mitochondrial ROS production as measured by conversion of the indicator dye MitoSOX nor does there appear to be any synergistic action, although GnRH induces ROS production in the same cells via NOX/DUOX (37). This supports the enzymatic ROS production with GnRH stimulation, and casts us the question that mitochondria produced ROS needs to be further examined in the aspect of both gonadotropin secretion and GnRH-induced gene expressions.
Figure 2.
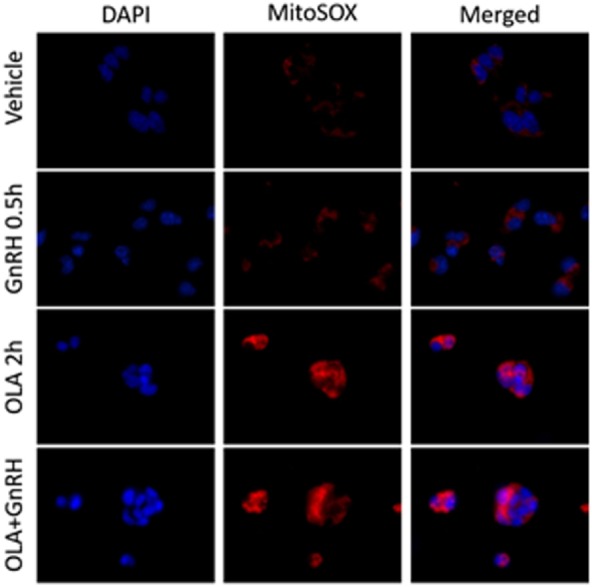
Oleate (OLA), but not gonadotropin-releasing hormone (GnRH), induces mitochondrial superoxide production. LβT2 cells (RRID:CVCL_0398) cultured on Poly-l-Lysine (Sigma-Aldrich Inc.) coated Nunc Lab-Tek II chamber slides (Thermo Fisher Scientific) for 24 h were serum starved overnight, then exposed to either vehicle or 500 µM oleate for 2 h, then with vehicle or 10 nM GnRH for 30 min. Cells were subsequently treated with 5 µM red-fluorescent MitoSOX probe (Thermo Fisher Scientific) for 5 min. Afterward, cells were directly fixed with 2% paraformaldehyde solution for 15 min, washed, and cover slipped with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs) to visualize nuclei in blue. Blue DAPI and Red MitoSOX fluorescence was captured by wide-field fluorescent microscopy using Nikon TE2000-U microscope (Nikon America Inc., Melville, NY, USA) equipped with an X-Cite 120PC collimated light source (Lumen Dynamics Group Inc.) and a DAPI-1160A or mCherry-C000 filter set (Semrock, Inc.) using a CoolSNAP DYNO CCD camera (Photometrics Inc.). In LβT2 cells, GnRH alone does not enhance mitochondrial ROS production as determined by changes in red fluorescence, whereas OLA-treated cells showed a highly elevated signal. Co-treatment with GnRH and Oleic acid did not appear to alter overall staining intensity.
Conclusion
The emerging evidence that ROS are integrated into multiple cell signaling cascades has led to appreciation as an important signaling molecule. In gonadotropes, it appears that enzymatic ROS plays a role in GnRH receptor signaling. The rapid and transient nature of ROS signaling is well-suited for the episodic pattern of GnRH stimulation and ROS signaling can contribute to both rapid activation and rapid resolution of activated signaling cascades. Further studies are needed to confirm or disprove the role of PRDX and SRXN1 in resolution of MAPK activation. It is also necessary to examine the impact of FFA-induced ROS on cell stress and to confirm the potential role of enzymatic ROS on L-type calcium stimulation in gonadotrope cells. The integration of ROS in GnRH receptor signaling provides an opportunity for other ROS sources to impact gonadotrope function at the cellular level. Incorporating ROS into our view of GnRH signaling is likely to yield useful insight into the mechanism of pulse interpretation and integration of stress signaling into the reproductive axis.
Author Contributions
TT authored the manuscript and prepared figures. MA produced new data presented in Figure 2 under direction and technical support of TT and DL. TK and SL developed the original concept, experimental approaches, and produced pilot data not reported here. ET participated in editing and writing the manuscript. ML edited the manuscript and secured support for all co-authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Terri Stoner for advice and technical assistance.
Footnotes
Funding. Supported by the NIH Eunice Kennedy Shriver National Institute of Child Health and Human Development grants R01 HD037568, R01 HD 082567, and P50 HD12303 to ML and training support for TK was provided by T32 HD007203. TT was supported by a fellowship from the Uehara Memorial Foundation.
Abbreviations
DAG, diacylglycerol; DPI, diphenyleneiodonium; DUOX, dual oxidase; DUSP, dual-specificity protein phosphatase; EGFR, epidermal growth factor receptor; FFA, free fatty acids; GnRH, gonadotropin-releasing hormone; GnRHR, gonadotropin-releasing hormone receptor; GSH, glutathione; HPG, hypothalamic–pituitary–gonad; IP3, inositol 1,4,5-trisphosphate; MAPK, mitogen-activated protein kinase; MEKK, MAPK kinase; NAC, N-acetyl cysteine; NOX, NADPH oxidase; OLA, oleate; PRDX, peroxiredoxin; ROS, reactive oxygen species; SRXN1, sulfiredoxin 1; TRX, thioredoxin; UPR, unfolded protein response.
References
- 1.Amoss M, Burgus R, Blackwell R, Vale W, Fellows R, Guillemin R. Purification, amino acid composition and N-terminus of the hypothalamic luteinizing hormone releasing factor (LRF) of ovine origin. Biochem Biophys Res Commun (1971) 44:205–10. 10.1016/S0006-291X(71)80179-1 [DOI] [PubMed] [Google Scholar]
- 2.Schally AV, Arimura A, Baba Y, Nair RM, Matsuo H, Redding TW, et al. Isolation and properties of the FSH and LH-releasing hormone. Biochem Biophys Res Commun (1971) 43:393–9. 10.1016/0006-291X(71)90766-2 [DOI] [PubMed] [Google Scholar]
- 3.Wildt L, HÄUsler A, Marshall G, Hutchison JS, Plant TM, Belchetz PE, et al. Frequency and amplitude of gonadotropin-releasing hormone stimulation and gonadotropin secretion in the rhesus monkey. Endocrinology (1981) 109:376–85. 10.1210/endo-109-2-376 [DOI] [PubMed] [Google Scholar]
- 4.Wurmbach E, Yuen T, Ebersole BJ, Sealfon SC. Gonadotropin-releasing hormone receptor-coupled gene network organization. J Biol Chem (2001) 276:47195–201. 10.1074/jbc.M108716200 [DOI] [PubMed] [Google Scholar]
- 5.Ruf F, Hayot F, Park MJ, Ge Y, Lin G, Roysam B, et al. Noise propagation and scaling in regulation of gonadotrope biosynthesis. Biophys J (2007) 93:4474–80. 10.1529/biophysj.107.115170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruf F, Park MJ, Hayot F, Lin G, Roysam B, Ge Y, et al. Mixed analog/digital gonadotrope biosynthetic response to gonadotropin-releasing hormone. J Biol Chem (2006) 281:30967–78. 10.1074/jbc.M606486200 [DOI] [PubMed] [Google Scholar]
- 7.Stern E, Ruf-Zamojski F, Zalepa-King L, Pincas H, Choi SG, Peskin CS, et al. Modeling and high-throughput experimental data uncover the mechanisms underlying Fshb gene sensitivity to gonadotropin-releasing hormone pulse frequency. J Biol Chem (2017) 292:9815–29. 10.1074/jbc.M117.783886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuen T, Wurmbach E, Ebersole BJ, Ruf F, Pfeffer RL, Sealfon SC. Coupling of GnRH concentration and the GnRH receptor-activated gene program. Mol Endocrinol (2002) 16:1145–53. 10.1210/mend.16.6.0853 [DOI] [PubMed] [Google Scholar]
- 9.Lim S, Pnueli L, Tan JH, Naor Z, Rajagopal G, Melamed P. Negative feedback governs gonadotrope frequency-decoding of gonadotropin releasing hormone pulse-frequency. PLoS One (2009) 4:e7244. 10.1371/journal.pone.0007244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong SP, Caunt CJ, Fowkes RC, Tsaneva-Atanasova K, McArdle CA. Pulsatile and sustained gonadotropin-releasing hormone (GnRH) receptor signaling: does the ERK signaling pathway decode GnRH pulse frequency? J Biol Chem (2010) 285:24360–71. 10.1074/jbc.M110.115964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garner KL, Perrett RM, Voliotis M, Bowsher C, Pope GR, Pham T, et al. Information transfer in gonadotropin-releasing hormone (GnRH) signaling: extracellular signal-regulated kinase (ERK)-mediated feedback loops control hormone sensing. J Biol Chem (2016) 291:2246–59. 10.1074/jbc.M115.686964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pratap A, Garner KL, Voliotis M, Tsaneva-Atanasova K, McArdle CA. Mathematical modeling of gonadotropin-releasing hormone signaling. Mol Cell Endocrinol (2017) 449:42–55. 10.1016/j.mce.2016.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsaneva-Atanasova K, Caunt CJ, Armstrong SP, Perrett RM, McArdle CA. Decoding neurohormone pulse frequency by convergent signalling modules. Biochem Soc Trans (2012) 40:273–8. 10.1042/BST20110645 [DOI] [PubMed] [Google Scholar]
- 14.Roch GJ, Busby ER, Sherwood NM. Evolution of GnRH: diving deeper. Gen Comp Endocrinol (2011) 171:1–16. 10.1016/j.ygcen.2010.12.014 [DOI] [PubMed] [Google Scholar]
- 15.Schlosser G. Evolutionary origins of vertebrate placodes: insights from developmental studies and from comparisons with other deuterostomes. J Exp Zool B Mol Dev Evol (2005) 304:347–99. 10.1002/jez.b.21055 [DOI] [PubMed] [Google Scholar]
- 16.Schlosser G. Induction and specification of cranial placodes. Dev Biol (2006) 294:303–51. 10.1016/j.ydbio.2006.03.009 [DOI] [PubMed] [Google Scholar]
- 17.Lawson MA, Tsutsumi R, Zhang H, Talukdar I, Butler BK, Santos SJ, et al. Pulse sensitivity of the luteinizing hormone beta promoter is determined by a negative feedback loop Involving early growth response-1 and Ngfi-A binding protein 1 and 2. Mol Endocrinol (2007) 21:1175–91. 10.1210/me.2006-0392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burger LL, Haisenleder DJ, Aylor KW, Marshall JC. Regulation of Lhb and Egr1 gene expression by GNRH pulses in rat pituitaries is both c-Jun N-terminal kinase (JNK)- and extracellular signal-regulated kinase (ERK)-dependent. Biol Reprod (2009) 81:1206–15. 10.1095/biolreprod.109.079426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mistry DS, Tsutsumi R, Fernandez M, Sharma S, Cardenas SA, Lawson MA, et al. Gonadotropin-releasing hormone pulse sensitivity of follicle-stimulating hormone-beta gene is mediated by differential expression of positive regulatory activator protein 1 factors and corepressors SKIL and TGIF1. Mol Endocrinol (2011) 25:1387–403. 10.1210/me.2011-0032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krakauer DC, Page KM, Sealfon S. Module dynamics of the GnRH signal transduction network. J Theor Biol (2002) 218:457–70. 10.1016/S0022-5193(02)93092-4 [DOI] [PubMed] [Google Scholar]
- 21.Perrett RM, McArdle CA. Molecular mechanisms of gonadotropin-releasing hormone signaling: integrating cyclic nucleotides into the network. Front Endocrinol (2013) 4:180. 10.3389/fendo.2013.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burger LL, Dalkin AC, Aylor KW, Haisenleder DJ, Marshall JC. GnRH pulse frequency modulation of gonadotropin subunit gene transcription in normal gonadotropes-assessment by primary transcript assay provides evidence for roles of GnRH and follistatin. Endocrinology (2002) 143:3243–9. 10.1210/en.2002-220216 [DOI] [PubMed] [Google Scholar]
- 23.Do MH, Kim T, He F, Dave H, Intriago RE, Astorga UA, et al. Polyribosome and ribonucleoprotein complex redistribution of mRNA induced by GnRH involves both EIF2AK3 and MAPK signaling. Mol Cell Endocrinol (2014) 382:346–57. 10.1016/j.mce.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Do MH, Santos SJ, Lawson MA. GNRH induces the unfolded protein response in the LbetaT2 pituitary gonadotrope cell line. Mol Endocrinol (2009) 23:100–12. 10.1210/me.2008-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen KA, Santos SJ, Kreidel MK, Diaz AL, Rey R, Lawson MA. Acute regulation of translation initiation by gonadotropin-releasing hormone in the gonadotrope cell line LbetaT2. Mol Endocrinol (2004) 18:1301–12. 10.1210/me.2003-0478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sosnowski R, Mellon PL, Lawson MA. Activation of translation in pituitary gonadotrope cells by gonadotropin-releasing hormone. Mol Endocrinol (2000) 14:1811–9. 10.1210/mend.14.11.0550 [DOI] [PubMed] [Google Scholar]
- 27.Kanasaki H, Oride A, Hara T, Mijiddorj T, Sukhbaatar U, Kyo S. Interactions between two different G protein-coupled receptors in reproductive hormone-producing cells: the role of PACAP and its receptor PAC1R. Int J Mol Sci (2016) 17:E1635. 10.3390/ijms17101635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roch GJ, Busby ER, Sherwood NM. GnRH receptors and peptides: skating backward. Gen Comp Endocrinol (2014) 209:118–34. 10.1016/j.ygcen.2014.07.025 [DOI] [PubMed] [Google Scholar]
- 29.Mugami S, Dobkin-Bekman M, Rahamim-Ben Navi L, Naor Z. Differential roles of PKC isoforms (PKCs) in GnRH stimulation of MAPK phosphorylation in gonadotrope derived cells. Mol Cell Endocrinol (2017). 10.1016/j.mce.2017.04.004 [DOI] [PubMed] [Google Scholar]
- 30.Naor Z. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol (2009) 30:10–29. 10.1016/j.yfrne.2008.07.001 [DOI] [PubMed] [Google Scholar]
- 31.Roberson MS, Misra-Press A, Laurance ME, Stork PJ, Maurer RA. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone alpha-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol (1995) 15:3531–9. 10.1128/MCB.15.7.3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roberson MS, Zhang T, Li HL, Mulvaney JM. Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology (1999) 140:1310–8. 10.1210/endo.140.3.6579 [DOI] [PubMed] [Google Scholar]
- 33.Zhang T, Roberson MS. Role of MAP kinase phosphatases in GnRH-dependent activation of MAP kinases. J Mol Endocrinol (2006) 36:41–50. 10.1677/jme.1.01881 [DOI] [PubMed] [Google Scholar]
- 34.Bliss SP, Miller A, Navratil AM, Xie J, McDonough SP, Fisher PJ, et al. ERK signaling in the pituitary is required for female but not male fertility. Mol Endocrinol (2009) 23:1092–101. 10.1210/me.2009-0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen KA, Intriago RE, Upadhyay HC, Santos SJ, Webster NJ, Lawson MA. Modulation of gonadotropin-releasing hormone-induced extracellular signal-regulated kinase activation by dual-specificity protein phosphatase 1 in LbetaT2 gonadotropes. Endocrinology (2010) 151:4882–93. 10.1210/en.2009-1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobkin-Bekman M, Naidich M, Rahamim L, Przedecki F, Almog T, Lim S, et al. A preformed signaling complex mediates GnRH-activated ERK phosphorylation of paxillin and FAK at focal adhesions in L beta T2 gonadotrope cells. Mol Endocrinol (2009) 23:1850–64. 10.1210/me.2008-0260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim T, Lawson MA. GnRH regulates gonadotropin gene expression through NADPH/dual oxidase-derived reactive oxygen species. Endocrinology (2015) 156:2185–99. 10.1210/en.2014-1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katsuyama M, Matsuno K, Yabe-Nishimura C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J Clin Biochem Nutr (2012) 50:9–22. 10.3164/jcbn.11-06SR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, et al. PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics (2012) 11:492–500. 10.1074/mcp.O111.014704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors (2003) 17:287–96. 10.1002/biof.5520170128 [DOI] [PubMed] [Google Scholar]
- 41.Meng D, Shi X, Jiang BH, Fang J. Insulin-like growth factor-I (IGF-I) induces epidermal growth factor receptor transactivation and cell proliferation through reactive oxygen species. Free Radic Biol Med (2007) 42:1651–60. 10.1016/j.freeradbiomed.2007.01.037 [DOI] [PubMed] [Google Scholar]
- 42.Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct (2011) 2011:792639. 10.1155/2011/792639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J (1995) 9:726–35. [PubMed] [Google Scholar]
- 44.Sarker KP, Biswas KK, Yamakuchi M, Lee KY, Hahiguchi T, Kracht M, et al. ASK1-p38 MAPK/JNK signaling cascade mediates anandamide-induced PC12 cell death. J Neurochem (2003) 85:50–61. 10.1046/j.1471-4159.2003.01663.x [DOI] [PubMed] [Google Scholar]
- 45.Jiao J, Dou L, Li M, Lu Y, Guo HB, Man Y, et al. NADPH oxidase 2 plays a critical role in dysfunction and apoptosis of pancreatic beta-cells induced by very low-density lipoprotein. Mol Cell Biochem (2012) 370:103–13. 10.1007/s11010-012-1402-z [DOI] [PubMed] [Google Scholar]
- 46.Munhoz AC, Riva P, Simoes D, Curi R, Carpinelli AR. Control of insulin secretion by production of reactive oxygen species: study performed in pancreatic islets from fed and 48-hour fasted Wistar rats. PLoS One (2016) 11:e0158166. 10.1371/journal.pone.0158166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weaver JR, Grzesik W, Taylor-Fishwick DA. Inhibition of NADPH oxidase-1 preserves beta cell function. Diabetologia (2015) 58:113–21. 10.1007/s00125-014-3398-2 [DOI] [PubMed] [Google Scholar]
- 48.Milenkovic M, De Deken X, Jin L, De Felice M, Di Lauro R, Dumont JE, et al. Duox expression and related H2O2 measurement in mouse thyroid: onset in embryonic development and regulation by TSH in adult. J Endocrinol (2007) 192:615–26. 10.1677/JOE-06-0003 [DOI] [PubMed] [Google Scholar]
- 49.Massart C, Hoste C, Virion A, Ruf J, Dumont JE, Van Sande J. Cell biology of H2O2 generation in the thyroid: investigation of the control of dual oxidases (DUOX) activity in intact ex vivo thyroid tissue and cell lines. Mol Cell Endocrinol (2011) 343:32–44. 10.1016/j.mce.2011.05.047 [DOI] [PubMed] [Google Scholar]
- 50.Lacroix L, Nocera M, Mian C, Caillou B, Virion A, Dupuy C, et al. Expression of nicotinamide adenine dinucleotide phosphate oxidase flavoprotein DUOX genes and proteins in human papillary and follicular thyroid carcinomas. Thyroid (2001) 11:1017–23. 10.1089/105072501753271699 [DOI] [PubMed] [Google Scholar]
- 51.Kano G, Almanan M, Bochner BS, Zimmermann N. Mechanism of Siglec-8-mediated cell death in IL-5-activated eosinophils: role for reactive oxygen species-enhanced MEK/ERK activation. J Allergy Clin Immunol (2013) 132:437–45. 10.1016/j.jaci.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol (2011) 7:75–7. 10.1038/nchembio.496 [DOI] [PubMed] [Google Scholar]
- 53.Alarid ET, Windle JJ, Whyte DB, Mellon PL. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development (1996) 122:3319–29. [DOI] [PubMed] [Google Scholar]
- 54.Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang HJ, Miller WL, et al. Cell-specific transcriptional regulation of follicle-stimulating hormone-beta by activin and gonadotropin-releasing hormone in the LbetaT2 pituitary gonadotrope cell model. Endocrinology (2001) 142:2284–95. 10.1210/endo.142.6.8185 [DOI] [PubMed] [Google Scholar]
- 55.Abe J, Berk BC. Fyn and JAK2 mediate Ras activation by reactive oxygen species. J Biol Chem (1999) 274:21003–10. 10.1074/jbc.274.30.21003 [DOI] [PubMed] [Google Scholar]
- 56.Accorsi K, Giglione C, Vanoni M, Parmeggiani A. The Ras GDP/GTP cycle is regulated by oxidizing agents at the level of Ras regulators and effectors. FEBS Lett (2001) 492:139–45. 10.1016/S0014-5793(01)02251-7 [DOI] [PubMed] [Google Scholar]
- 57.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem (1996) 271:4138–42. 10.1074/jbc.271.8.4138 [DOI] [PubMed] [Google Scholar]
- 58.Childs GV, Armstrong J. Sites of epidermal growth factor synthesis and action in the pituitary: paracrine and autocrine interactions. Clin Exp Pharmacol Physiol (2001) 28:249–52. 10.1046/j.1440-1681.2001.03423.x [DOI] [PubMed] [Google Scholar]
- 59.Mouihate A, Verrier D, Lestage J. EGF release by rat gonadotroph cells: characteristics and effects of LHRH. Life Sci (1996) 58:107–14. 10.1016/0024-3205(95)02263-5 [DOI] [PubMed] [Google Scholar]
- 60.Roelle S, Grosse R, Aigner A, Krell HW, Czubayko F, Gudermann T. Matrix metalloproteinases 2 and 9 mediate epidermal growth factor receptor transactivation by gonadotropin-releasing hormone. J Biol Chem (2003) 278:47307–18. 10.1074/jbc.M304377200 [DOI] [PubMed] [Google Scholar]
- 61.Patterson KI, Brummer T, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J (2009) 418:475–89. 10.1042/BJ20082234 [DOI] [PubMed] [Google Scholar]
- 62.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell (2005) 121:667–70. 10.1016/j.cell.2005.05.016 [DOI] [PubMed] [Google Scholar]
- 63.Kamata H, Honda S-I, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell (2005) 120:649–61. 10.1016/j.cell.2004.12.041 [DOI] [PubMed] [Google Scholar]
- 64.Hou N, Torii S, Saito N, Hosaka M, Takeuchi T. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology (2008) 149:1654–65. 10.1210/en.2007-0988 [DOI] [PubMed] [Google Scholar]
- 65.Choi B-H, Hur E-M, Lee J-H, Jun D-J, Kim K-T. Protein kinase Cδ-mediated proteasomal degradation of MAP kinase phosphatase-1 contributes to glutamate-induced neuronal cell death. J Cell Sci (2006) 119:1329–40. 10.1242/jcs.02837 [DOI] [PubMed] [Google Scholar]
- 66.Bhalla US, Ram PT, Iyengar R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science (2002) 297:1018–23. 10.1126/science.1068873 [DOI] [PubMed] [Google Scholar]
- 67.Perrett RM, Voliotis M, Armstrong SP, Fowkes RC, Pope GR, Tsaneva-Atanasova K, et al. Pulsatile hormonal signaling to extracellular signal-regulated kinase: exploring system sensitivity to gonadotropin-releasing hormone pulse frequency and width. J Biol Chem (2014) 289:7873–83. 10.1074/jbc.M113.532473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neumann CA, Cao J, Manevich Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle (2009) 8:4072–8. 10.4161/cc.8.24.10242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature (2003) 425:980–4. 10.1038/nature02075 [DOI] [PubMed] [Google Scholar]
- 70.Kuwano Y, Kim HH, Abdelmohsen K, Pullmann R, Jr, Martindale JL, Yang X, et al. MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Mol Cell Biol (2008) 28:4562–75. 10.1128/MCB.00165-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lannes J, L’Hote D, Garrel G, Laverriere JN, Cohen-Tannoudji J, Querat B. Rapid communication: a microRNA-132/212 pathway mediates GnRH activation of FSH expression. Mol Endocrinol (2015) 29:364–72. 10.1210/me.2014-1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hasuwa H, Ueda J, Ikawa M, Okabe M. miR-200b and miR-429 function in mouse ovulation and are essential for female fertility. Science (2013) 341:71–3. 10.1126/science.1237999 [DOI] [PubMed] [Google Scholar]
- 73.Crowley WF, Balasubramanian R. MicroRNA-7a2 suppression causes hypogonadotropism and uncovers signaling pathways in gonadotropes. J Clin Invest (2017) 127:796–7. 10.1172/JCI92846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lannes J, L’Hote D, Fernandez-Vega A, Garrel G, Laverriere JN, Joelle Cohen T, et al. A regulatory loop between miR-132 and miR-125b involved in gonadotrope cells desensitization to GnRH. Sci Rep (2016) 6:31563. 10.1038/srep31563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pan Q, Liao X, Liu H, Wang Y, Chen Y, Zhao B, et al. MicroRNA-125a-5p alleviates the deleterious effects of ox-LDL on multiple functions of human brain microvessel endothelial cells. Am J Physiol Cell Physiol (2017) 312:C119–30. 10.1152/ajpcell.00296.2016 [DOI] [PubMed] [Google Scholar]
- 76.Martin TF. Tuning exocytosis for speed: fast and slow modes. Biochim Biophys Acta (2003) 1641:157–65. 10.1016/S0167-4889(03)00093-4 [DOI] [PubMed] [Google Scholar]
- 77.Tse FW, Tse A, Hille B, Horstmann H, Almers W. Local Ca2+ release from internal stores controls exocytosis in pituitary gonadotrophs. Neuron (1997) 18:121–32. 10.1016/S0896-6273(01)80051-9 [DOI] [PubMed] [Google Scholar]
- 78.Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol (2015) 6:260–71. 10.1016/j.redox.2015.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tabet F, Savoia C, Schiffrin EL, Touyz RM. Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol (2004) 44:200–8. 10.1097/00005344-200408000-00009 [DOI] [PubMed] [Google Scholar]
- 80.Zimmerman MC, Takapoo M, Jagadeesha DK, Stanic B, Banfi B, Bhalla RC, et al. Activation of NADPH oxidase 1 increases intracellular calcium and migration of smooth muscle cells. Hypertension (2011) 58:446–53. 10.1161/HYPERTENSIONAHA.111.177006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Espinosa A, Garcia A, Hartel S, Hidalgo C, Jaimovich E. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. J Biol Chem (2009) 284:2568–75. 10.1074/jbc.M804249200 [DOI] [PubMed] [Google Scholar]
- 82.Messina A, Langlet F, Chachlaki K, Roa J, Rasika S, Jouy N, et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat Neurosci (2016) 19:835–44. 10.1038/nn.4298 [DOI] [PubMed] [Google Scholar]
- 83.Chachlaki K, Garthwaite J, Prevot V. The gentle art of saying NO: how nitric oxide gets things done in the hypothalamus. Nat Rev Endocrinol (2017) 13(9):521–35. 10.1038/nrendo.2017.69 [DOI] [PubMed] [Google Scholar]
- 84.Frisch RE. The right weight: body fat, menarche and fertility. Proc Nutr Soc (1994) 53:113–29. 10.1079/PNS19940015 [DOI] [PubMed] [Google Scholar]
- 85.Frisch RE, Revelle R. Height and weight at menarche and a hypothesis of critical body weights and adolescent events. Science (1970) 169:397–9. 10.1126/science.169.3943.397 [DOI] [PubMed] [Google Scholar]
- 86.Fu JF, Liang JF, Zhou XL, Prasad HC, Jin JH, Dong GP, et al. Impact of BMI on gonadorelin-stimulated LH peak in premenarcheal girls with idiopathic central precocious puberty. Obesity (Silver Spring) (2015) 23:637–43. 10.1002/oby.21010 [DOI] [PubMed] [Google Scholar]
- 87.Holte J, Bergh T, Berne C, Lithell H. Serum lipoprotein lipid profile in women with the polycystic ovary syndrome: relation to anthropometric, endocrine and metabolic variables. Clin Endocrinol (Oxf) (1994) 41:463–71. 10.1111/j.1365-2265.1994.tb02577.x [DOI] [PubMed] [Google Scholar]
- 88.Jain A, Polotsky AJ, Rochester D, Berga SL, Loucks T, Zeitlian G, et al. Pulsatile luteinizing hormone amplitude and progesterone metabolite excretion are reduced in obese women. J Clin Endocrinol Metab (2007) 92:2468–73. 10.1210/jc.2006-2274 [DOI] [PubMed] [Google Scholar]
- 89.Lim SS, Norman RJ, Davies MJ, Moran LJ. The effect of obesity on polycystic ovary syndrome: a systematic review and meta-analysis. Obes Rev (2013) 14:95–109. 10.1111/j.1467-789X.2012.01053.x [DOI] [PubMed] [Google Scholar]
- 90.Pirwany IR, Fleming R, Greer IA, Packard CJ, Sattar N. Lipids and lipoprotein subfractions in women with PCOS: relationship to metabolic and endocrine parameters. Clin Endocrinol (2001) 54:447–53. 10.1046/j.1365-2265.2001.01228.x [DOI] [PubMed] [Google Scholar]
- 91.Veldhuis JD, Liu PY, Keenan DM, Takahashi PY. Older men exhibit reduced efficacy of and heightened potency downregulation by intravenous pulses of recombinant human LH: a study in 92 healthy men. Am J Physiol Endocrinol Metab (2012) 302:E117–22. 10.1152/ajpendo.00450.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arroyo A, Laughlin GA, Morales AJ, Yen SS. Inappropriate gonadotropin secretion in polycystic ovary syndrome: influence of adiposity. J Clin Endocrinol Metab (1997) 82:3728–33. 10.1210/jc.82.11.3728 [DOI] [PubMed] [Google Scholar]
- 93.Lawson MA, Jain S, Sun S, Patel K, Malcolm PJ, Chang RJ. Evidence for insulin suppression of baseline luteinizing hormone in women with polycystic ovarian syndrome and normal women. J Clin Endocrinol Metab (2008) 93:2089–96. 10.1210/jc.2007-2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pagan YL, Srouji SS, Jimenez Y, Emerson A, Gill S, Hall JE. Inverse relationship between luteinizing hormone and body mass index in polycystic ovarian syndrome: investigation of hypothalamic and pituitary contributions. J Clin Endocrinol Metab (2006) 91:1309–16. 10.1210/jc.2005-2099 [DOI] [PubMed] [Google Scholar]
- 95.Srouji SS, Pagan YL, D’Amato F, Dabela A, Jimenez Y, Supko JG, et al. Pharmacokinetic factors contribute to the inverse relationship between luteinizing hormone and body mass index in polycystic ovarian syndrome. J Clin Endocrinol Metab (2007) 92:1347–52. 10.1210/jc.2006-2716 [DOI] [PubMed] [Google Scholar]
- 96.Akhter N, CarlLee T, Syed MM, Odle AK, Cozart MA, Haney AC, et al. Selective deletion of leptin receptors in gonadotropes reveals activin and GnRH-binding sites as leptin targets in support of fertility. Endocrinology (2014) 155:4027–42. 10.1210/en.2014-1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mattos R, Staples CR, Thatcher WW. Effects of dietary fatty acids on reproduction in ruminants. Rev Reprod (2000) 5:38–45. 10.1530/ror.0.0050038 [DOI] [PubMed] [Google Scholar]
- 98.Barb CR, Kraeling RR, Rampacek GB. Glucose and free fatty acid modulation of growth hormone and luteinizing hormone secretion by cultured porcine pituitary cells. J Anim Sci (1995) 73:1416–23. 10.2527/1995.7351416x [DOI] [PubMed] [Google Scholar]
- 99.Ly LD, Xu S, Choi SK, Ha CM, Thoudam T, Cha SK, et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med (2017) 49:e291. 10.1038/emm.2016.157 [DOI] [PMC free article] [PubMed] [Google Scholar]