

Abstract
Rationale:
Methylmalonic acidemia (MMA) is a common organic acidemia, mainly due to methylmalonyl-CoA mutase (MCM) or its coenzyme cobalamin (VitB12) metabolic disorders. Cobalamin C (CblC) type is the most frequent inborn error of cobalamin metabolism; it can develop symptoms in childhood and often combine multisystem damage, which leads to methylmalonic acid, propionic acid, methyl citrate, and other metabolites abnormal accumulation, causing nerve, liver, kidney, bone marrow, and other organ damage.
Patient concerns:
A 4-year-old girl presented with paleness, fatigue, severe normochromic anemia, and acute kidney injury.
Diagnosis:
Based on severe normochromic anemia and acute kidney injury, renal biopsy showed membranous proliferative glomerular lesions and thrombotic microvascular disease, supporting the diagnosis of aHUS. Although the serum vitamin B12 was normal, further investigation found the concentration of urinary methylmalonic acid and serum homocysteine increased obviously, genetic analysis revealed a heterozygous MMACHC mutation (exonl: c. 80A >G, c. 609G >A). The final diagnosis was aHUS induced by inherited methylmalonic acidemia (MMACHC heterozygous mutation exonl: c. 80A >G, c. 609G >A).
Interventions:
The patient was treated with a 1mg vitamin B12 intramuscular injection daily for 4 days after which the dose was then adjusted to a 1mg intramuscular injection twice a week. At the same time, the girl was given levocarnitine, betaine, folic acid, along with supportive treatment.
Outcomes:
After treated by vitamin B12 for 10 days, the patient condition significantly improved, Follow-up results showed complete recovery of hemoglobin and renal function.
Lessons:
Although the majority of MMA onset from neurological damage, our case illustrates that partial CblC-type MMA can onset with severe metabolic aHUS. On the basis of chronic thrombotic microangiopathy (TMA)-induced renal damage, it can be complicated by acute hemolytic lesions. MMA should be considered in those patients with unclear microangiopathic hemolytic anemia accompany significant megaloblastic degeneration in bone marrow. We should pay attention to the causes and adopt a reasonable treatment strategy.
Keywords: atypical hemolytic uremic syndrome, CblC subtype, children, methylmalonic acidemia
1. Introduction
Methylmalonic acidemia (MMA) is a common organic acidemia, mainly due to methylmalonyl-CoA mutase (MCM) or its coenzyme cobalamin (VitB12) metabolic disorders. Cobalamin C (CblC) type is the most frequent inborn error of cobalamin metabolism; it can develop symptoms in childhood and often combine multisystem damage, which leads to methylmalonic acid, propionic acid, methyl citrate, and other metabolites abnormal accumulations, causing nerve, liver, kidney, bone marrow, and other organ damage.[1,2] The majority of MMA onset from neurological damage, but here we have another interesting case that rarely presenting predominantly with hemolytic uremic syndrome (HUS) as the first manifestation.
2. Case report
A 4-year-old girl presented to our hospital with a 20-day history of paleness and fatigue, with no other clinical manifestations such as hematemesis, melena, diarrhea, dermatorrhagia, gross hematuria, edema, joint swelling, and fever. She had been diagnosed with severe anemia and received erythrocyte suspension transfusion before hospital admission. When she was 1 year old, her Hb was found to be 90 g/L, for which she was given iron but she was not brought to the hospital for a follow-up. Her parents and her younger sister are all healthy.
On physical examination, she had an ear temperature of 37°C, a pulse of 90 beats per minute, and a blood pressure of 140/90 mm Hg. Except for moderate anemic appearance, no special abnormality was found. She was diagnosed with anemia and admitted to our hospital for further investigation and management.
After admission, multiple blood routine tests showed normochromic anemia. The lowest Hb was 61 g/L. It was difficult to maintain her Hb to >90 g/L even after doing the erythrocyte suspension transfusion 3 times. The platelet count fluctuated between 102 and 276 × 109/L, the reticulocytes at 0.8% (0.5%–1.5%), vitamin B12 at 592 pg/mL (180–914 pg/mL), and folic acid at 9.64 ng/mL (3.7–19.8 ng/mL). Ferritin (248.7 ng/mL) and transferrin (119.0 mg/dL) were all within the normal range. Thalassemia gene screening was also negative. Fecal routine and occult blood tests were negative, and the coagulation functions were normal. Haptoglobin had decreased significantly and was only <5.83 mg/dL (30–200 mg/dL), whereas lactate dehydrogenase had significantly increased and was 629 U/L (109–245 U/L). Erythrocyte fragmentation was observed in peripheral blood erythrocyte morphology examination. Serum creatinine was 38.4 μmol/L at first, which then rapidly increased to 88.4 μmol/L within 4 days. Hematuria (35.38/HP) and proteinuria (3+) were present in the urine, and the urinary red blood cell morphology suggested it to be glomerular hematuria. Urinary albumin 2260 mg/L (0–30 mg/L) and urinary N-acetylglucosidase 32.9 U/L (0.3–12.0 U/L) were both high. The patient had no acidemia, and the pH (7.39), HCO3− (22.1 mmol/L), BE (−2.0 mmol/L), and the anion gap (15 mmol/L) were all within the normal range.
Direct and indirect antihuman ball tests were all negative. ASO <25 IU/mL (0–100 IU/mL), complement C3, C4, and immunoglobulin A, G, M were normal as well. Anti-double-stranded DNA antibodies were at 2.0% (<20%). ANA and ANCA series, lupus anticoagulant substances, and anticardiolipin antibodies were all negative. B ultrasound showed an enlargement of the kidneys (right kidney size 91 × 42 mm, left kidney size 90 × 40 mm), but no change in the echo.
After admission, she was given supportive treatment including erythrocyte suspension transfusion and oxygen. Her blood pressure was successively lowered by nifedipine, amlodipine metoprolol, losartan potassium, and furosemide. The condition of patient still had not improved. Her blood pressure fluctuated between 103 and 173 mm Hg/66 and 107 mm Hg, and she still had severe anemia, with Hb at 64 g/L even after blood transfusions. Renal function was also abnormal, with the lowest eGFR at 56.5 mL/min/1.73 m2 (>90 mL/min/1.73 m2).
We further performed conventional bone marrow examination. The smear showed a proliferation of nucleated cell with a G/E ratio of 1.33:1 (2.76:1). Granulocyte and erythroid hyperplasia were obvious and showed significant megaloblastic degeneration.
Renal biopsy showed that 5 of the 10 glomeruli were sclerotic spheroids, 4 of which were concentrated, and suggestive of membranous proliferative glomerular lesions. Thrombotic microvascular disease is highly likely (Figs. 1 and 2).
Figure 1.
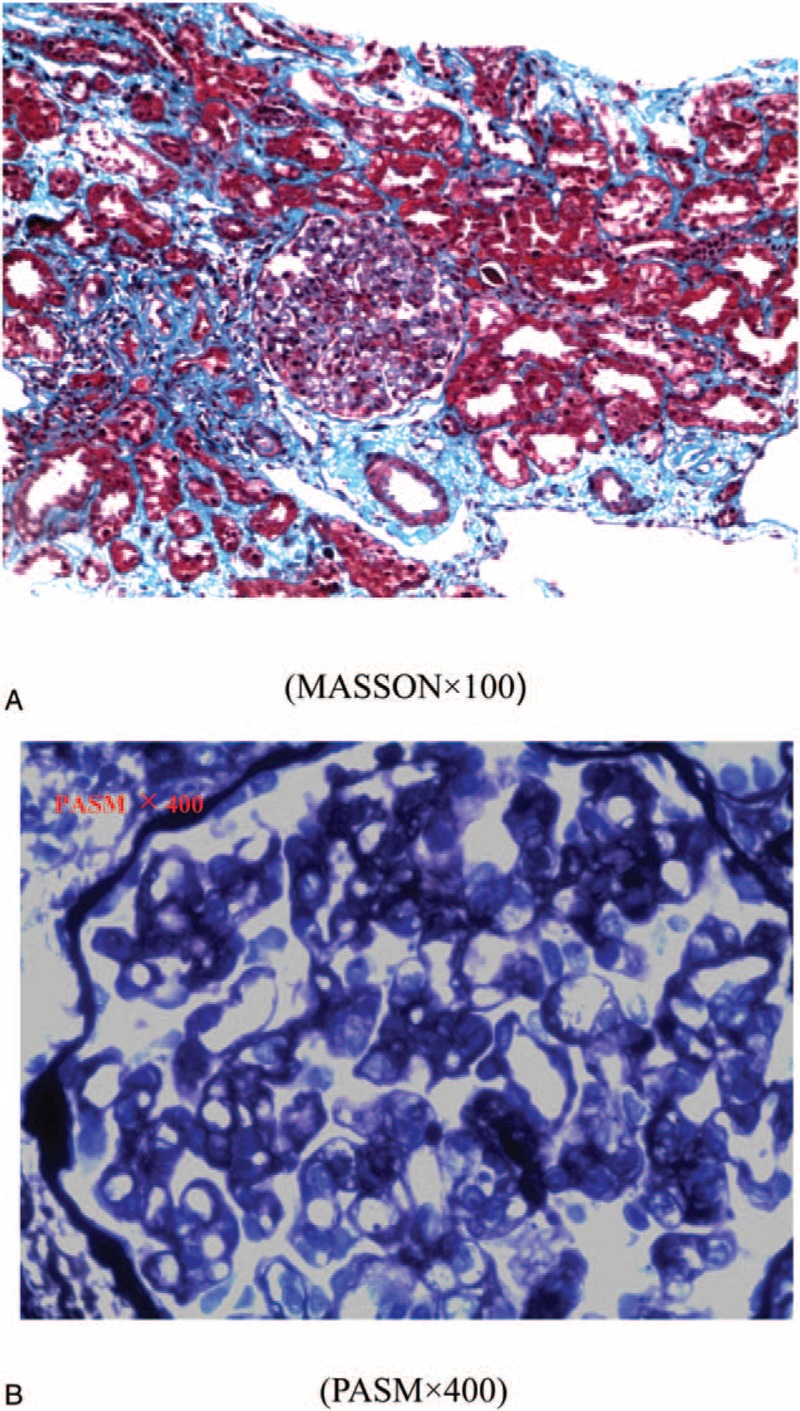
LM: (A) renal pathology suggests that there is severe extensive glomerulosclerosis, showing glomerular segmental basement membrane thickening, capillary endothelial cell swelling, and mild proliferation of segmental mesangial cells. Mesangial cells showed segmental insert phenomenon. Red blood cell debris was visible in part of the capillary blood vessels and polymorphonuclear leukocyte infiltration exists in part of the glomerulus. There is also an expansion in the glomerular Bowman's capsule cyst and a capillary loop ischemic shrinkage. There are moderate tubulointerstitial lesions, multifocal interstitial infiltration of renal interstitial cells with edema and fibrosis, multifocal lamellar tubular atrophy, part of the renal tubular epithelial cells swelling, vacuolar degeneration, and partial tubular expansion. Part of the tubule has tubular protein present and there is focal hyalinosis in the endomembrane of renal arterioles. (B) Light microscopy shows glomerular segmental basement membrane thickening and “double-track sign” formation.
Figure 2.
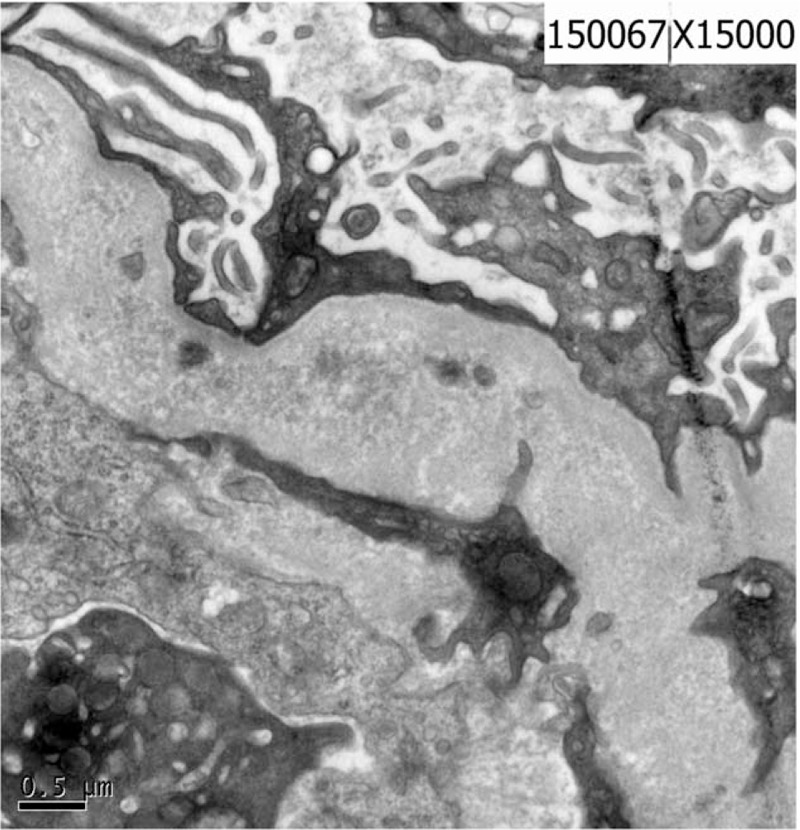
EM: the glomerular mesangial matrix is grossly increased and there is a thickening and shrinkage of the GBM. There is a segmental inner tectorium widening, extensive foot process fusion and the capillary is obstructed. There are no electron dense deposits.
Eye examination, adrenal and renal B-ultrasound, and echocardiography all showed no obvious abnormalities. Head MRI showed widened bilateral frontal temporal exterior brain space and a slightly enlarged bilateral lateral ventricle.
3. Results
By screening for blood and urinary metabolic diseases, compared with a normal child, we found that the concentration of urinary methylmalonic acid was 0.2017 (base line value is 0.001) in this patient, which was increased 201.7 times. Her urinary excretion of methyl citrate was 0.7501 (base line value is 0.017), which was also increased 44.12 times. At the same time, her serum homocysteine was 39.935 (5–20), increased 1.997 times; propionyl carnitine was 6.545 (0–5.14), increased 1.27 times; C3/C0 was 0.425(0.01–0.23), increased 1.846 times; C3/C2 was 0.674 (0.02–0.29), increased 2.325 times; and C3/C16 was 14.213 (0.00–6.6), increased 2.153 times. Further genetic analysis revealed a heterozygous MMACHC mutation (exonl: c. 80A >G, c. 609G>A) (Fig. 3). So the diagnosis of inherited methylmalonic acidemia (MMACHC heterozygous mutation exonl: c. 80A >G, c. 609G >A) had been established.
Figure 3.
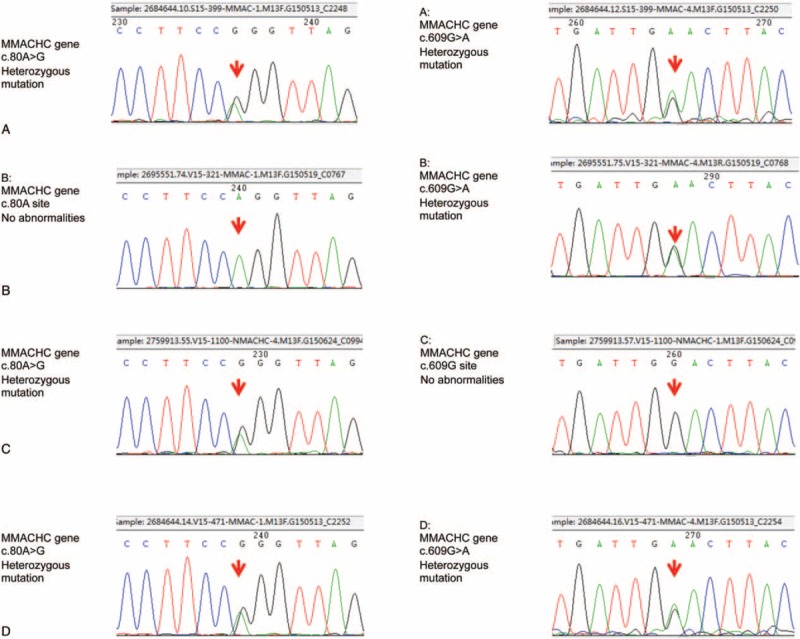
The MMACHC gene c. 80A >G and the MMACHC gene c. 609G >A. Heterozygous mutations were present in both, the patient (A) and her younger sister (D). The c. 80A site was not abnormal, but the c. 609G >A of MMACHC gene presented a heterozygous mutation in her mother (B). The MMACHC gene c. 609G site was normal, but c. 80A >G presented a heterozygous mutation in her father (C).
The patient was treated with a 1 mg vitamin B12 intramuscular injection daily for 4 days after which the dose was then adjusted to a 1 mg intramuscular injection twice a week. At the same time, the girl was given levocarnitine, betaine, folic acid, and nifedipine for her blood pressure, along with supportive treatment.
After being treated for 10 days, the patient's condition had significantly improved and she was then discharged from the hospital. She continued to receive 1 mg vitamin B12 intramuscular injections twice a week along with l-carnitine solution. Her follow-up results showed that the urinary methylmalonic acid concentration and the level of blood homocysteine had significantly declined. The proteinuria was also negative, and the hematuria had obviously reduced as well. The blood routine test and renal function changes were satisfactory (Figs. 4 and 5)
Figure 4.
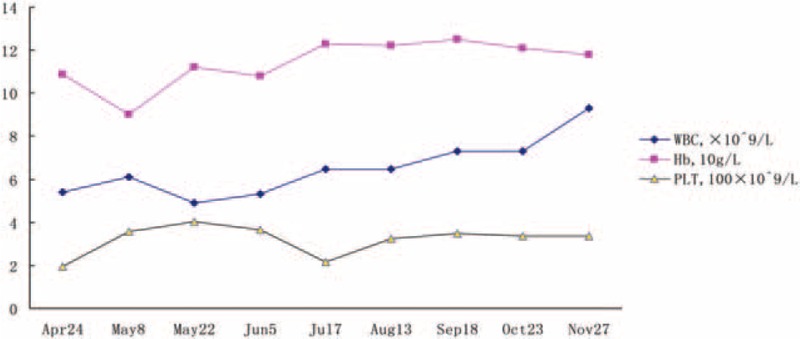
Follow-up of blood routine test.
Figure 5.
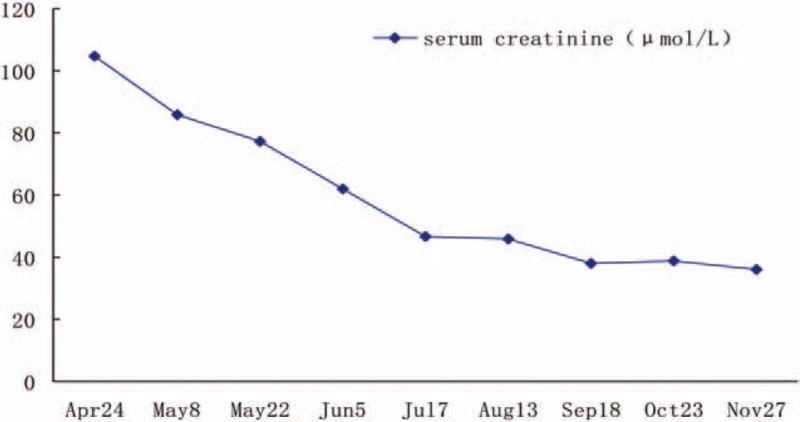
Follow-up of serum creatinin.
4. Discussion
HUS is a thrombotic microangiopathy, characterized by microangiopathic hemolytic anemia, renal impairment, and thrombocytopenia.[3] In this case, the outstanding performances were progressive hemolytic anemia, which was difficult to be corrected, and renal dysfunction, so we suspected that the diagnosis was HUS. The number of platelets was never <10 × 109/L during the whole course in this case, which was not a coincidence with HUS. However, sometimes patients can be diagnosed with HUS even without the presence of hemolytic anemia,[4] based on nonimmune hemolytic anemia combined with renal biopsy. A proven thrombotic microangiopathy, peripheral blood broken red blood cells, a significant increase in LDH, and obviously a decrease in haptoglobin all support the diagnosis of HUS.
An onset of complement deficiency, pneumococcal infection, abnormal metabolism of cobalamin, and so on can lead to atypical hemolytic uremic syndrome (aHUS), which is related to genetic mutations and has a distinct pathophysiology.[3,6,7] So far, only a few cases of HUS have been triggered by a metabolic disease, such as diabetes mellitus[8,9] and metabolic disorder of cobalamin.[10,11] Recently, Adrovic et al described a case of a 6-year-old girl with CblC disorder, who was found to have a homozygous mutation in exon 4 of MMACHC, c. 484G >T.[11] Although the serum level of VitB12 was normal in the patient, the bone marrow showed an obvious degeneration, so we highly suspected that it was caused by metabolic abnormalities of cobalamin.
MMA is a common organic acidemia, which belongs to the autosomal recessive diseases, mainly due to MCM or its coenzyme cobalamin metabolic disorders, which leads to abnormal accumulations of methylmalonic acid, propionic acid, methyl citrate, and other metabolites, causing nerve, liver, kidney, bone marrow, and other organ damage.[2] The clinical diagnosis of MMA can be easily and rapidly diagnosed by gas chromatography–mass spectrometry (GC–MS) for the organic acid in urine, blood, and cerebrospinal fluid, or tandem mass spectrometry (MS/MS) for the determination of propanoylcarnitine (C3).[12] In this case, the methylmalonic acid and methyl citrate were significantly increased in the urine, and the blood C3, C3/C0 (free carnitine), and C3/C2 (acetyl carnitine) were also increased, so the clinical diagnosis was established.
So far, we have a very limited understanding of the genetics and epidemiology of aHUS.[5] As CblC type is the most frequent inborn error of cobalamin metabolism, it can develop symptoms in childhood and often combine multisystem damage.[1,13] In this case there was simultaneous methylmalonic aciduria and elevated serum homocysteine, therefore we presumed that this case probably belongs to CblC type.
Mutation analysis is the most reliable evidence for MMA classification. The CblC encoding gene named MMACHC, located in 1p34.1. Through the analysis of the gene mutation of this patient's family, the results were different from homozygous mutation of Adrovic's findings,[11] the MMACHC gene c. 80 A >G and c. 609G >A. Heterozygous mutations were all found in the patient and her sister. The c. 80A site was not abnormal, but there was a heterozygous mutation in the patient's mother in the c. 609G >A of MMACHC, and the MMACHC gene c. 609G site was normal, but c.80A >G heterozygous mutation in her father. The result confirmed this patient and her sister presented with ectopic heterozygous mutations, namely, MMACHC heterozygous mutations (exonl: c. 80A >G, c. 609G >A). It is particularly necessary to notice that normal C3, C4, CFH, and CFI plasma levels do not exclude the diagnosis of complement-dependent HUS.[14] Since the lack of complement analysis, in this patient we cannot completely exclude the coexisting of complement-dependent HUS, which is a limitation of our case.
The majority of MMA onset from different neurological damage,[15–17] but some of them may first onset with unusual presentations, such as mimicking diabetic ketoacidosis,[18] late-onset diffuse lung disease,[19] and Juvenile gout.[20] For this girl, although the brain CT and MRI scan were suggesting the existence of brain atrophy, there were no clinical manifestations such as vomiting, lethargy, convulsions, movement disorders, and mental retardation. Children with CblC type mainly present with megaloblastic anemia, growth disorders, and neurological symptoms. Clinical data also showed that renal damage exists in a large population of children with MMA,[2,21] but rarely onset from HUS.
Renal pathological changes of the girl were very interesting, which showed coexistence of acute and chronic pathological changes. Basement membrane ischemia shrinkage and a negative fluorescence suggested TMA, consistent with Beck et al, who demonstrated renal TMA in patients with CblC defect.[22] Combined with glomerular sclerosis hinted the possibility of chronic TMA. Although there was swelling of the endothelial cells, the treatment results were satisfactory. Follow-up results showed normal renal function, clinically difficult to explain. Based on the renal biopsy, glomerular segmental basement membrane thickening and more “double-track sign” formation prompted MPGN. Based on the basic disease, it should be MMA-related MPGN.
The treatment principles of MMA are to reduce the generation of metabolic poisons and/or to accelerate their clearance.[23–25] Intramuscular cobalamin is more effective in lowering homocysteine and methylmalonic acid levels than oral administration, so we chose intramuscular cobalamin.[26] In addition, the methylmalonic acid and homocysteine heighten at the same time. As l-carnitine promotes excretion of methylmalonic acid and acyl carnitine and increases the body's tolerance to natural protein and betaine can promote the reduction of blood homocysteine levels, therefore these 2 drugs were used in this patient. In this case, patient's hemolytic anemia had rapidly improved and renal function quickly returned to normal. Repeat tests in children with increased methylmalonic acid and homocysteine levels were significantly reduced after intramuscular injections of cobalamin, confirming a good therapeutic effect.
The prognosis of children with MMA depends mainly on the type of disease, the onset, and clinical compliance. The type of vitamin B12 effective showed a better prognosis, delayed type of more stable clinical progression to a lesser extent. The remission of the clinical symptoms rapidly after treatment is suggestive of a good prognosis in the near future but long-term effects should still be further observed.
5. Conclusions
This case suggests that partial CblC-type MMA can onset with severe metabolic aHUS. On the basis of chronic TMA-induced renal damage, it can be complicated by acute hemolytic lesions. MMA should be considered in those patients with unclear microangiopathic hemolytic anemia accompanied by significant megaloblastic degeneration in the bone marrow. We should pay attention to the causes and adapt a reasonable treatment strategy.
Acknowledgments
The authors thank the combination and support of whole family of the patient in this study, and the Yuying Children's Hospital of Wenzhou Medical University, and also thank Amber Kaur for revising the manuscript.
Footnotes
Abbreviations: aHUS = atypical hemolytic uremic syndrome, CblC = cobalamin C, MMA = methylmalonic academia, TMA = thrombotic microangiopathy.
Availability of data and materials: All data and material were presented in this manuscript.
Consent for publication: Written informed consent was obtained from the patient for publication of this case report and images in it. A copy of the written consent is available for review by the Editor of this journal.
Authors’ contributions: QY, MC, and JZ wrote the manuscript and were treating physicians for the patient. JY and DW performed pathological analysis and interpretation and contributed to writing the manuscript. All authors read and approved the final manuscript.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Morel CF, Lerner-Ellis JP, Rosenblatt DS. Combined methylmalonic aciduria and homocystinuria (cblC): phenotype-genotype correlations and ethnic-specific observations. Mol Genet Metab 2006;88:315–21. [DOI] [PubMed] [Google Scholar]
- [2].Hörster F, Hoffmann GF. Pathophysiology, diagnosis, and treatment of methylmalonic aciduria-recent advances and new challenges. Pediatr Nephrol 2004;19:1071–4. [DOI] [PubMed] [Google Scholar]
- [3].Talarico V, Aloe M, Monzani A, et al. Hemolytic uremic syndrome in children. Minerva Pediatr 2016;68:441–55. [PubMed] [Google Scholar]
- [4].Akashi Y, Yoshizawa N, Oshima S, et al. Hemolytic uremic syndrome without hemolytic anemia: a case report. Clin Nephrol 1994;42:90–4. [PubMed] [Google Scholar]
- [5].Rafiq A, Tariq H, Abbas N, et al. Atypical hemolytic-uremic syndrome: a case report and literature review. Am J Case Rep 2015;16:109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ariceta G, Besbas N, Johnson S, et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol 2009;24:687–96. [DOI] [PubMed] [Google Scholar]
- [7].Weisser C, Feber J, Tsampalieros A, et al. An atypical case of acute kidney injury: hemolytic uremic syndrome (HUS). Pediatr Nephrol 2016;31: 917, 919–921. [DOI] [PubMed] [Google Scholar]
- [8].Iddings AC, Shenoi AN, Morales Pozzo A, et al. Hemolytic uremic syndrome complicated by Clostridium septicum bacteremia and new-onset type 1 diabetes mellitus. Report of a case? Clin Nephrol 2017;87:207–11. [DOI] [PubMed] [Google Scholar]
- [9].Zhu Z, Chen H, Gill R, et al. Diabetic ketoacidosis presenting with atypical hemolytic uremic syndrome associated with a variant of complement factor B in an adult: a case report. J Med Case Rep 2016;10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Medhioub Kaaniche F, Chaari A, Bacouch N, et al. Hemolytic uremic syndrome in young adult with metabolic disorder of cobalamin: a case report. Presse Med 2016;45:148–50. [DOI] [PubMed] [Google Scholar]
- [11].Adrovic A, Canpolat N, Caliskan S, et al. Cobalamin C defect-hemolytic uremic syndrome caused by new mutation in MMACHC. Pediatr Int 2016;58:763–5. [DOI] [PubMed] [Google Scholar]
- [12].Han LS, Ye J, Qiu WJ, et al. Selective screening for inborn errors of metabolism on clinical patients using tandem mass spectrometry in China: a four-year report. J Inherit Metab Dis 2007;30:507–14. [DOI] [PubMed] [Google Scholar]
- [13].Lerner-Ellis JP, Tirone JC, Pawelek PD, et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet 2006;38:93–100. [DOI] [PubMed] [Google Scholar]
- [14].Loirat C, Fakhouri F, Ariceta G, et al. HUS International. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 2016;31:15–39. [DOI] [PubMed] [Google Scholar]
- [15].Sheikhmoonesi F, Shafaat A, Moarefian S, et al. Affective disorder as the first manifestation of methylmalonic acidemia: a case report. Iran J Pediatr 2013;23:245–6. [PMC free article] [PubMed] [Google Scholar]
- [16].Prada CE, Villamizar-Schiller IT. Globus pallidus involvement as initial presentation of methylmalonic acidemia. Mov Disord 2014;29:870. [DOI] [PubMed] [Google Scholar]
- [17].Ito H, Mori K, Ito M, et al. Case of methylmalonic acidemia presenting clinically Leigh encephalopathy. No To Hattatsu 2004;36:324–9. [PubMed] [Google Scholar]
- [18].Dejkhamron P, Wejapikul K, Unachak K, et al. Isolated methylmalonic acidemia with unusual presentation mimicking diabetic ketoacidosis. J Pediatr Endocrinol Metab 2016;29:373–8. [DOI] [PubMed] [Google Scholar]
- [19].Liu J, Peng Y, Zhou N, et al. Combined methylmalonic acidemia and homocysteinemia presenting predominantly with late-onset diffuse lung disease: a case series of four patients. Orphanet J Rare Dis 2017;12:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Charuvanij S, Pattaragarn A, Wisuthsarewong W, et al. Juvenile gout in methylmalonic acidemia. Pediatr Int 2016;58:501–3. [DOI] [PubMed] [Google Scholar]
- [21].Morath MA, Okun JG, Müller IB, et al. Neurodegeneration and chronic renal failure in methylmalonic aciduria—a pathophysiological approach. J Inherit Metab Dis 2008;31:35–43. [DOI] [PubMed] [Google Scholar]
- [22].Beck BB, van Spronsen F, Diepstra A, et al. Renal thrombotic microangiopathy in patients with cblC defect: review of an under-recognized entity. Pediatr Nephrol 2017;32:733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Deodato F, Boenzi S, Santorelli FM, et al. Methylmalonic and propionic aciduria. Am J Med Genet C Semin Med Genet 2006;142C:104–12. [DOI] [PubMed] [Google Scholar]
- [24].Yannicelli S, Acosta PB, Velazquez A, et al. Improved growth and nutrition status in children with methylmalonic or propionic acidemia fed an elemental medical food. Mol Genet Metab 2003;80:181–8. [DOI] [PubMed] [Google Scholar]
- [25].Carrillo-Carrasco N, Chandler RJ, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type I. Clinical presentations, diagnosis and management. J Inherit Metab Dis 2012;35:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Solomon LR. Oral pharmacologic doses of cobalamin may not be as effective as parenteral cobalamin therapy in reversing hyperhomocystinemia and methylmalonic acidemia in apparently normal subjects. Clin Lab Haematol 2006;28:275–8. [DOI] [PubMed] [Google Scholar]