

Abstract
In recent years, a new neurodegenerative tauopathy labeled Chronic Traumatic Encephalopathy (CTE), has been identified that is believed to be primarily a sequela of repeated mild traumatic brain injury (TBI), often referred to as concussion, that occurs in athletes participating in contact sports (e.g. boxing, football, football, rugby, soccer, ice hockey) or in military combatants, especially after blast-induced injuries. Since the identification of CTE, and its neuropathological finding of deposits of hyperphosphorylated tau protein, mechanistic attention has been on lumping the disorder together with various other non-traumatic neurodegenerative tauopathies. Indeed, brains from suspected CTE cases that have come to autopsy have been confirmed to have deposits of hyperphosphorylated tau in locations that make its anatomical distribution distinct for other tauopathies. The fact that these individuals experienced repetitive TBI episodes during their athletic or military careers suggests that the secondary injury mechanisms that have been extensively characterized in acute TBI preclinical models, and in TBI patients, including glutamate excitotoxicity, intracellular calcium overload, mitochondrial dysfunction, free radical-induced oxidative damage and neuroinflammation, may contribute to the brain damage associated with CTE. Thus, the current review begins with an in depth analysis of what is known about the tau protein and its functions and dysfunctions followed by a discussion of the major TBI secondary injury mechanisms, and how the latter have been shown to contribute to tau pathology. The value of this review is that it might lead to improved neuroprotective strategies for either prophylactically attenuating the development of CTE or slowing its progression.
Keywords: traumatic brain injury, chronic traumatic encephalopathy, tauopathy, concussion, repetitive head injury
Graphical abstract
1. Introduction
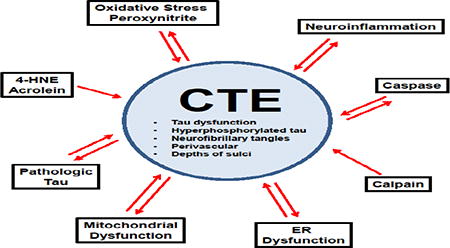
Chronic traumatic encephalopathy (CTE) is classified as a neurodegenerative tauophathy (McKee et al., 2016). In addition to CTE several other tauopathies exist including Alzheimer's disease (AD), frontotemporal dementia (FTD), progressive supra nuclear palsy (PSP), corticobasal degeneration, Pick's disease, and argyrophilic grain disease (Lee and Leugers, 2012). One defining characteristic of tauopathies is hyperphosphorylation of the protein tau. Once hyperphosphorylated, tau assumes a tightly folded confirmation, increasing susceptibility to aggregation, conformational change, filament assembly, polymerization into paired helical filaments, and bundling of paired helical filaments into neurofibrillary tangles and neuropil threads (Mietelska-Porowska et al., 2014).
Clinically, CTE is characterized by cognitive, behavioral, and/or mood dysfunction, with the possible accompaniment of motor symptoms (Montenigro et al., 2014). Unfortunately, the current incidence and prevalence of CTE is unknown (Kiernan et al., 2015) because although CTE can be suspected clinically, a definitive diagnosis can only be made post-mortem (McKee et al., 2016). Recently, a consensus meeting was held to define the neuropathological criteria of CTE, and concluded that a CTE diagnosis should be based upon identification of an irregular pattern of abnormally hyperphosphorylated tau accumulations in astrocytes and neurons located around small blood vessels and at the depths of cortical sulci (McKee et al., 2016). Additional non-specific, but supportive features of CTE, include hippocampal tangles, neurofibrillary tangles in the subcortical nuclei, and TAR DNA-binding protein 43 (TDP-43) reactivity in the temporal cortex, hippocampus and amygdala (McKee et al., 2016). However, in reaching the consensus, definition other neuropathologic characteristics of CTE were not addressed including gliosis, inflammation, hemosiderin deposition, and the presence of comorbid pathologies such as Aβ and α-synuclein deposition (McKee et al., 2016). Although the neuropathologic criteria for CTE have now been defined, identification of CTE as a unique entity remains controversial for several reasons including reliance on retrospective case reports subject to selection bias for identification of cases (Maroon et al., 2015), as well a lack of prospective and epidemiologic studies (McCrory et al., 2013). Indeed, the clinical diagnostic criteria of CTE vs. other neurodegenerative disorders (e.g. AD) for which the history of one or more traumatic brain injuries is an established risk factor is still being intensely debated.
Nevertheless, to date, CTE pathology has only been identified in individuals with a history of traumatic brain injury (TBI) (McKee et al., 2016). CTE pathology has been observed in the brains of military personnel who have sustained blast injuries, and in athletes who participate in sports where the risk of sustaining repetitive head injuries is high including boxing, football, soccer, hockey and rugby (Goldstein et al., 2012; Kiernan et al., 2015; McKee et al., 2009; McKee and Robinson, 2014; McKee et al., 2013; Omalu et al., 2011b; Omalu et al., 2006; Omalu et al., 2005). Although there are several tauopathies, the pathoanatomic location of tau deposition in CTE makes it distinct from the others (Kiernan et al., 2015; McKee et al., 2016). However, the neuronal tau found in CTE does share a similar profile in regard to isoform ratio and phosphorylation state as the tau in AD (Kiernan et al., 2015; McKee et al., 2014; Schmidt et al., 2001). CTE and AD share other similarities; brain injury also increases the risk of developing AD (Guo et al., 2000; Nemetz et al., 1999; Plassman et al., 2000). In fact both pathologic tau (Ikonomovic et al., 2004; Johnson et al., 2012; Uryu et al., 2007) and Aβ deposition (Johnson et al., 2012; Reynolds et al., 2005; Reynolds et al., 2006) are seen following a single severe TBI.
Although advancements have been made in defining the core neuropathological features of CTE, much about CTE is still unknown. Similar to brain injury itself, CTE is heterogeneous both in its supporting neuropathological features (McKee et al., 2016) and in its clinical presentation. Clinically, CTE is suspected of having at least two subtypes, a younger onset subtype predominated by behavior and mood symptoms but with minimal cognitive or motor impairment, and an older onset subtype characterized by cognitive impairment and motor dysfunction (Stern et al., 2013). However, up to four clinical subtypes have been proposed (Montenigro et al., 2014).
The pathophysiologic processes which lead to the development of CTE, the main characteristic of which is pathologic tau deposition, are not well understood. Although repetitive TBI has been singled out as a major etiologic factor in CTE and brain injury is considered necessary for its development (McKee et al., 2016), individuals who have experienced repetitive TBI do not always go on to develop CTE (Hazrati et al., 2013; McKee et al., 2013; Omalu et al., 2011a). Additional factors hypothesized to contribute to CTE include environment, genetics and injury characteristics, e.g. age at injury, type and severity of injury, number of hits, duration between injuries, etc. (Ojo et al., 2016) Although animal models are being utilized to further characterize CTE and repetitive TBI, many have failed to recapitulate the tau pathology seen in CTE or have required the use of transgenic mice already predisposed to develop tau pathology (Ojo et al., 2016). Therefore, there is much still to be elucidated regarding the progression of CTE following injury. However, despite the paucity of information regarding how CTE tau pathology progresses over time, the tau protein itself and processes that contribute to its dysfunction have been well characterized in the literature, thanks in large part to AD, FTD and TBI models in which tauopathy is a.commonly occurring pathology.
In this review, which focuses on CTE, we attempt to integrate what is known about canonical post-traumatic neurodegenerative processes and the tau protein in order to identify mechanisms which could possibly lead to, or contribute to, chronic tau dysfunction following TBI that might serve as pharmacological neuroprotective targets. We have chosen to direct our attention to CTE because, by definition it is associated with the previous occurrence of repetitive, and possibly singular TBI even though, a history of TBI is known to be a risk factor for other neurodegenerative disorders that are involve the same secondary post-TBI degenerative mechanisms. Thus, throughout the rest of this review, we consistently mention what is known about the interaction of post-TBI secondary injury processes such as mitochondrial dysfunction oxidative stress, intracellular calcium overload, etc., with the pathogenesis of other neurodegenerative disorders as AD, that involve tauopathies that appear to be somewhat pathologically distinct from CTE. While this review is mainly directed at CTE, our literature review has revealed a rich interaction between acute post-TBI secondary injury mechanisms and tauopathy development in general.
2. The Tau Protein – Functions and Dysfunctions
2.1 Functions
There are several detailed reviews that cover structure, function, and post-translational modification of the tau protein (Fontaine et al., 2015; Lee and Leugers, 2012; Morris et al., 2011). Tau is encoded for by the MAPT gene found on chromosome 17, a gene that is mutated in several tauopathies including FTD (Ferrari et al., 2011) and PSP (Im et al., 2015). In humans, alternative splicing generates six isoforms of tau containing either three (3R) or four (4R) microtubule binding repeats (Goedert and Jakes, 1990), leading to formation of isoforms with differing characteristics (Lee and Leugers, 2012). Additional heterogeneity exists in the n-terminal region as well (Goedert and Jakes, 1990). As stated previously, the neuronal tau of CTE has a similar isoform ratio and phosphorylation state as the tau of AD (Kiernan et al., 2015; McKee et al., 2014; Schmidt et al., 2001). AD is reported to have a 2:1 ratio of 4R:3R tau (Chen et al., 2010b; Conrad et al., 2007; Ginsberg et al., 2006), a shift from the equal 4R:3R ratio found in healthy adult brains (Goedert and Jakes, 1990; Kosik et al., 1989).
The most well-known function of tau is its ability to bind microtubules, which promotes microtubule assembly and stabilization (Fontaine et al., 2015; Lee and Leugers, 2012; Morris et al., 2011). However, tau has several other functions, including axonal transport (Cuchillo-Ibanez et al., 2008; Lee and Leugers, 2012; Morris et al., 2011), regulation of actin and neurite outgrowth (Lee and Leugers, 2012; Morris et al., 2011), and regulation of signaling pathways (Morris et al., 2011). Tau can associate with heat shock proteins, chaperone proteins involved in the tau degradation pathway (Fontaine et al., 2015; Lee and Leugers, 2012), and although tau is primarily found associated with the cytoskeleton of axons, it can also localize to the nucleus, as well as interact with the plasma membrane and post-synaptic density (Fontaine et al., 2015; Lee and Leugers, 2012; Morris et al., 2011).
The binding of tau to microtubules can be regulated by the tau phosphorylation state, and phosphorylation of tau within the microtubule binding sites decreases the ability of tau to bind microtubules (Fontaine et al., 2015). Similarly, several of the tau mutations utilized in transgenic tau mice, such as P301L and P301S, function to decrease binding of tau to microtubules and can lead to decreases in microtubule assembly (Fontaine et al., 2015; Hasegawa et al., 1998; Iovino et al., 2014). Several protein kinases and phosphatases regulate tau phosphorylation, and thus its ability to interact with microtubules (Fontaine et al., 2015; Martin et al., 2013; Morris et al., 2011).
2.2 Dysfunctions
One characteristic feature of tauopathies, such as CTE and AD, is tau hyperphosphorylation, where phosphorylation of tau at specific residues occurs as an ordered process, leading to tau aggregation (Alonso et al., 2001; Fontaine et al., 2015; Jeganathan et al., 2008; Morris et al., 2011) and oligomer formation (Tepper et al., 2014). In addition to tauopathies, TBI is also capable of inducing tau hyperphosphorylation (Ikonomovic et al., 2004; Uryu et al., 2007; Yang et al., 2016).
In addition to phosphorylation other post-translational modifications to tau occur such as acetylation, glycosylation, sumolyation, ubiquitination, polyamination, oxidation, nitration, isomerization and truncation (Fontaine et al., 2015; Morris et al., 2011). Many of these post-translational modifications have been implicated in the formation of pathologic tau. For example, oxidation of tau induces cross-linkages and aggregation (Reynolds et al., 2005) and nitration of tau decreases microtubule binding and assembly (Reynolds et al., 2005). Isomerization of pThr231 tau to the cis isoform attenuates tau dephosphorylation, decreases microtubule binding, and promotes aggregation (Wang and Zhang, 2015), and proteolytic cleavage of tau by calpain or caspase (Liu et al., 2011) can result in formation of truncated tau species which are prone to aggregation (Fontaine et al., 2015).
The mechanisms regarding tau toxicity in neurodegenerative disease remain controversial (Morris et al., 2011), with both loss of function and gain of function theories being proposed (Trojanowski and Lee, 2005). However, although hyperphosphorylated tau results in decreased binding of tau to microtubules, several studies indicate that knock-down of tau results in normal behavioral phenotypes, synaptic transmission and microtubule stability, without compensatory increases in other microtubule-binding proteins, such as MAP1 or MAP2, suggesting that tau toxicity may not be a direct result of loss of function (Morris et al., 2011). Rather, the toxicity associated with pathologic tau may in part be due to tau mislocalization and/or dysfunctional axonal transport. The majority of tau is localized to axons (Morris et al., 2011); however, tau hyperphosphorylation results in redistribution of tau from the axon to the somatodendritic compartment, an effect which is sufficient to impair synaptic function (Hoover et al., 2010), and several studies have shown that pathologic tau isoforms impair fast axonal transport (Lee and Leugers, 2012).
Evidence has also accumulated suggesting that tau oligomers, i.e. pre-filamentous tau aggregates, are more toxic than filamentous or monomeric tau (Spires-Jones et al., 2011). For example, injection of human tau oligomers into mouse brains results in impaired memory consolidation, as well as synaptic and mitochondrial dysfunction, whereas administration of tau monomers or fibrils does not (Lasagna-Reeves et al., 2011), while repression of human tau in transgenic mice results in attenuation of neuronal loss and cognitive impairment despite continued increases in neurofibrillary tangle formation (Santacruz et al., 2005). Similarly, in a drosophila model of tauopathy, neurodegeneration can still be seen in the absence of neurofibrillary tangle formation (Wittmann et al., 2001). Interestingly, It has been hypothesized that one reason oligomeric tau is able to accumulate in tauopathies is due to the fact that both oligomeric tau and cleaved forms of tau, which have an increased propensity for aggregation compared to full-length tau, are preferentially degraded via autophagy (Chesser et al., 2013), a pathway reported to be defective in tauopathies (Piras et al., 2016) and TBI (Sarkar et al., 2014). Although proteasome dysfunction, the mechanism which preferentially degrades monomeric tau, has also been reported to occur following TBI due to mechanisms such as oxidative stress (Bader and Grune, 2006; Weih et al., 2001; Yao et al., 2008), monomeric full-length tau has a decreased propensity to aggregate compared to cleaved tau (Chesser et al., 2013) and is less toxic than oligomeric tau (Spires-Jones et al., 2011).
Tau is considered to have prion-like properties (Alonso et al., 2016; Medina and Avila, 2014), which likely contribute to intracellular, intraregional, and trans-synaptic spread of pathologic tau (Liu et al., 2012; Medina and Avila, 2014). Tau can be secreted from neurons in its naked form or within exosomes or membrane vesicles (Chai et al., 2012; Medina and Avila, 2014; Saman et al., 2012; Simon et al., 2012). In healthy neurons the release of tau is induced by neuronal activity, however, neuronal activity is altered following TBI (Carron et al., 2016) and in tauopathy brains (Pooler et al., 2013), likely resulting in abnormal tau release (Pooler et al., 2013). Additionally, both c-terminal proteolytic cleavage of tau and tau overexpression are capable of enhancing tau secretion (Medina and Avila, 2014; Plouffe et al., 2012; Simon et al., 2012). In-vitro, extracellular tau is known to be toxic (Gomez-Ramos et al., 2006; Medina and Avila, 2014), through a mechanism which may involve increases in intracellular calcium levels via tau stimulation of muscarinic receptors (Gomez-Ramos et al., 2008), and in further support of the toxic oligomeric theory, only extracellular tau aggregates, not tau monomers, are taken up by cells (Alonso et al., 2016; Frost et al., 2009; Wu et al., 2013). Once tau is secreted, the extracellular tau has the ability to enhance tau pathology. For example, transplanting TBI induced tau oligomers into naive brains of hTau transgenic mice results in oligomeric spread of tau and accelerated cognitive impairment (Gerson et al., 2016), secreted tau fibrils are capable of inducing transcellular misfolding and tau aggregation (Kfoury et al., 2012), and hyperphosphorylated tau is capable of forming filaments and tangles with non-phosphorylated tau (Alonso et al., 1996). Therefore, it is possible that in non-familial tauopathies, such as CTE, an initial event, such as repetitive TBI, triggers a misfolding cascade which can then be transmitted in a prion-like manner (Morales et al., 2015).
3. Traumatic Brain Injury - Pathophysiological Mechanisms
Although the specific mechanisms by which repetitive TBI can lead to CTE or by which a single severe TBI can lead to AD are still being elucidated, much of the pathology that occurs in tauopathies is paralleled in TBI. TBIs range from mild to severe, with at least 2.5 million TBIs occurring in the United States annually (Faul M, 2010), the majority of which are classified as mild (Holm et al., 2005). However, these numbers are underestimates because mild TBIs often go unreported, especially in sports and military communities (Jordan, 2013; Marion et al., 2011), populations which are at additional risk for sustaining repeat injuries. In fact, it is estimated that sports-related TBIs alone occur at a rate of 3.8 million annually (Langlois et al., 2006).
3.1 Primary Injury Mechanisms
TBI consists of a primary injury followed by a secondary injury cascade. The primary injury occurs immediately and is caused by external forces, such as a direct impact, rapid acceleration/deceleration, or blast a wave. Primary injury can result in contusion, hemorrhage, ischemia, shearing and straining of axons and blood vessels, and diffuse axonal injury from the mechanical insult (Maas et al., 2008; McAllister, 2011; Weber, 2012). Of important relevance to CTE, which consists of tau pathology located at the depths of cortical sulci and around blood vessels, are deceleration/acceleration and blast forces (McKee et al., 2016). Deceleration/acceleration forces are particularly damaging to long white matter tracts and the grey-white matter junctions of the cerebral cortex (McAllister, 2011), and blast injury has been reported to damage perivascular neural tissue due to transmission of pressure waves throughout the cerebral vasculature (McAllister, 2011).
3.2 Secondary Injury Mechanisms
Cellular strain and deformation caused by the primary injury results in membrane depolarization, mechanoporation of membranes, ionic imbalances, and neurotransmitter release, resulting in initiation of the secondary injury cascade, a process which occurs hours to weeks following injury (Maas et al., 2008; McAllister, 2011), and is heavily influenced by alterations in calcium homeostasis (Weber, 2012).
3.2.1 Intracellular Calcium Overload, Oxidative Damage and Mitochondrial Dysfunction
Activation of calcium channels by high levels of extracellular glutamate following injury, along with activation of voltage-gated calcium channels and membrane leakage lead to large increases in intracellular calcium (Weber, 2012). As essential regulators or calcium homeostasis (Rizzuto et al., 2000; Rizzuto et al., 1999) mitochondria buffer the increases in intracellular calcium following TBI (Lifshitz et al., 2003; Xiong et al., 1997). Increases in mitochondrial calcium lead to decreased respiration and increased generation of reactive oxygen and nitrogen species (ROS/RNS) (Fiskum, 2000; Sullivan et al., 2005). Following TBI, as the electron transport chain becomes impaired, single electrons leak from complex I, generating superoxide radicals (O2•-), which rapidly react the nitric oxide (NO•) generated by calcium activated mitochondrial nitric oxide synthase (mtNOS), forming peroxynitrite (PN) anion (ONOO-) (Bringold et al., 2000; Radi et al., 2002). Protonation of ONOO- results in peroxynitrious acid (ONOOH) which decomposes into nitrogen dioxide (NO•2) and hydroxyl (OH•) radicals. Alternatively, ONOO- can react with carbon dioxide to form nitrosoperoxocarbonate (ONOOCO2-) which decomposes into the radicals NO•2 and (CO•-3) (Bains and Hall, 2012; Hall et al., 2010). PN is demonstratively increased following TBI (Deng et al., 2007; Hall et al., 2004; Hall et al., 2012; Singh et al., 2007) and because of its unique diffusion radius, mitochondrial derived PN is capable of damaging multiple cellular structures (Hall et al., 2010).
The highly reactive PN-derived radicals, NO•2, OH•, CO•-3, initiate lipid peroxidation (LP) of polyunsaturated fatty acids, such as arachidonic acid, which are highly enriched in neurons and cellular and organelle membranes, forming lipid peroxyl radicals (Bains and Hall, 2012; Hall et al., 2010). LP propagates throughout the membrane as lipid peroxyl radicals react with adjacent polyunsaturated acids, and although lipid peroxidation is considered to be a self-propagating process, it is also catalyzed by the presence of iron, particularly in the acidic tissues of TBI (Hall et al., 2010). Following TBI, decreases in pH cause iron to be released from the iron storage proteins, ferritin and transferritin, and additional iron is released from the hemoglobin deposited during hemorrhages and microbleeds (Hall et al., 2010). Therefore, similar to tau deposition in CTE, iron-catalyzed LP may be enhanced near the vasculature following TBI. LP terminates with formation of neurotoxic aldehydes, such as 4-hydroxynonenal (4-HNE) and 2-propenal (acrolein). Both LP and its derivatives, 4-HNE and acrolein, are well known to be increased following TBI (Bayir et al., 2007; Hall et al., 2004; Mustafa et al., 2010; Mustafa et al., 2011; Singh et al., 2013) (Cebak et al., 2016; Hill et al., Submitted).
The neurotoxic aldehydes, 4-HNE and acrolein, covalently bind proteins via the amino acids lysine, histidine, or cysteine, resulting in enzyme inhibition and protein dysfunction (Hall et al., 2010; Petersen and Doorn, 2004; Stevens and Maier, 2008). In addition to protein dysfunction, neurotoxic aldehydes, ROS/RNS and lipid peroxyl radicals are capable of inducing DNA damage to both nuclear and mitochondrial DNA (Dalleau et al., 2013; Hall et al., 2010). As a major site of PN formation, mitochondria are particularly susceptible to attack by LP-derived neurotoxic aldehydes. Binding of 4-HNE and acrolein to mitochondria results in extensive mitochondrial dysfunction through impairment of mitochondrial respiration and enhanced generation of ROS/RNS (Singh et al., 2013; Vaishnav et al., 2010) (Cebak et al., 2016; Hill et al., Submitted; Miller et al., 2013; Picklo et al., 1999; Picklo and Montine, 2001). Following TBI, the mitochondrial dysfunction induced by LP-derived neurotoxic aldehydes and increased intra-mitochondrial calcium concentrations leads to formation of the mitochondrial permeability transition pore (mPTP) (Bringold et al., 2000; Hansson et al., 2008; Sullivan et al., 2005). Opening of the mPTP results in collapse of the mitochondrial membrane potential, loss of ATP production, mitochondrial swelling, rupture of the outer mitochondrial membrane and release of calcium and cytochrome c into the cytosol (Galluzzi et al., 2009; Sullivan et al., 2005).
3.2.2 Calcium-Mediated Proteolytic Degradation
Extrusion of calcium back into the cytosol leads to neurodegeneration, necrosis, and activation of the calcium-dependent cysteine protease, calpain, which is capable of breaking down a variety of cytoskeletal proteins including MAP2, spectrin and tau (Galluzzi et al., 2009; Kampfl et al., 1997; Sullivan et al., 2005; Wang, 2000). In fact, calpain-cleaved αII-spectrin breakdown products are frequently used to assess post-TBI axonal damage and neuroprotection following TBI (Bains et al., 2013; Deng-Bryant et al., 2008; Deng et al., 2007; Mbye et al., 2009; Miller et al., 2014; Mustafa et al., 2011; Saatman et al., 1996). Similarly, mitochondrial release of cytochrome c leads to activation of the protease caspase-3 and induction of apoptosis (Galluzzi et al., 2009; Sullivan et al., 2005; Wang, 2000).
One effect of cytoskeletal degradation following TBI is impairment of axonal transport. Following injury, axonal transport of amyloid precursor protein (APP), the precursor to Aβ, a pathologic protein found in the tauopathy, AD, and some cases of CTE, is impaired and results in accumulations of APP in axonal varicosities and bulbs of damaged neurons; as such, the accumulation of APP following TBI is often used as a marker for traumatic axonal injury (Chauhan, 2014). In addition to cytoskeletal degradation, protein accumulation following TBI can also occur due to proteasomal dysfunction (Yao et al., 2008). In particular, the proteasome is responsible for the degradation of oxidatively damaged proteins, however, it is also subject to oxidative stress-induced impairment itself (Bader and Grune, 2006; Weih et al., 2001; Yao et al., 2008). In addition to protein accumulation following injury, impairment of axonal transport also leads to somatodendritic accumulation of organelles, such as the mitochondria (Kilinc et al., 2008), although, impairment of mitochondrial dynamics following TBI is complex and includes additional processes such as alterations in fission and fusion (Fischer et al., 2016).
Following injury, the endoplasmic reticulum (ER) also functions to regulate calcium homeostasis (Weber, 2012). Increases in ER stress are reported to occur in both single (Begum et al., 2014; Krajewska et al., 2011; Larner et al., 2004; Logsdon et al., 2014) and repetitive TBI (Lucke-Wold et al., 2016). In fact, calcium release from the ER has been hypothesized to be toxic to neurons and white matter tracts (Weber, 2012). In addition to regulation of intracellular calcium stores, the ER is involved in protein folding and quality control of misfolded proteins and protein aggregates through a process termed, the unfolded protein response (UPR) (Hoozemans and Scheper, 2012). Therefore, in addition to calcium-induced pathology, ER stress can also lead to several additional aspects of cellular dysfunction, including inhibition of protein synthesis (Doutheil et al., 1997), apoptosis (Nakagawa et al., 2000), accumulation of protein aggregates (Hoozemans and Scheper, 2012), and activation of the tau kinase, GSK-3β (Song et al., 2002).
3.2.3 Neuroinflammation
Another important contributory factor to the pathology that occurs following TBI is inflammation. Inflammation has been observed acutely following TBI in both severe and mild injury, an effect which is amplified in mild TBI by repeated injury (Collins-Praino and Corrigan, 2016). Following injury, resident immune cells, such as astrocytes and microglia, are activated by damaged tissue and cellular debris (Collins-Praino and Corrigan, 2016; Karve et al., 2016), but can also be activated by other mechanisms such as ROS/RNS (Collins-Praino and Corrigan, 2016). Additionally, peripheral immune cells are recruited to the site of injury (Collins-Praino and Corrigan, 2016; Karve et al., 2016). Immune cells are capable of releasing both pro-inflammatory and pro-survival cytokines and chemokines; therefore, they are capable of serving both destructive and reparative roles following injury (Collins-Praino and Corrigan, 2016; Karve et al., 2016). For example, the microglial phenotype M1 is neurotoxic, while the microglial phenotype M2 is neuroprotective (Karve et al., 2016). Chronic microglial activation has been observed following both human and experimental TBI and has been linked to chronic neurodegeneration (Faden and Loane, 2015). In fact, the neurotoxic M1 phenotype has been shown to persist longer than the M2 phenotype following TBI (Collins-Praino and Corrigan, 2016; Kumar et al., 2016; Wang et al., 2013a) Furthermore, some studies have demonstrated that persistent neuroinflammation can occur decades following severe TBI or repetitive concussion (Collins-Praino and Corrigan, 2016; Coughlin et al., 2015; Ramlackhansingh et al., 2011). Physiologically, one explanation for the persistence of neuroinflammation following repetitive TBI focuses on microglial priming. Microglial priming is a process in which microglia develop exaggerated immune responses and decreased activation thresholds following an insult such as TBI; therefore, neuroinflammation can be chronically exacerbated upon additional TBIs or systemic inflammation (Collins-Praino and Corrigan, 2016).
3.2.4 Tau Phosphorylation
In direct relation to CTE, TBI also results in increased levels of total and phosphorylated tau. In human TBI, injury severity is correlated with increased tau phosphorylation, increased activity of the tau kinase GSK-3β, and decreased activity of the tau phosphatase, PP2A, (Yang et al., 2016). Tau pathology has been reported following cases of single severe TBI (Ikonomovic et al., 2004; Johnson et al., 2012; Uryu et al., 2007), in patients whom have died within six months of sustaining a concussion (McKee et al., 2014), and in young military veterans with a history of blast exposure (McKee and Robinson, 2014). Therefore, there is not only increasing interest in investigating the development of CTE-like tau pathology in animal models of single and repetitive TBI, but also in the development of tau as a biomarker for TBI (Section 5. Tau – A Biomarker for TBI). Figure 1 hypothesizes the possible contribution of single, or perhaps multiple, pathophysiological mechanisms to the development of CTE.
Figure 1. Interplay between TBI pathophysiology and mechanisms of pathologic tau formation demonstrating several cyclic relationships between the two.
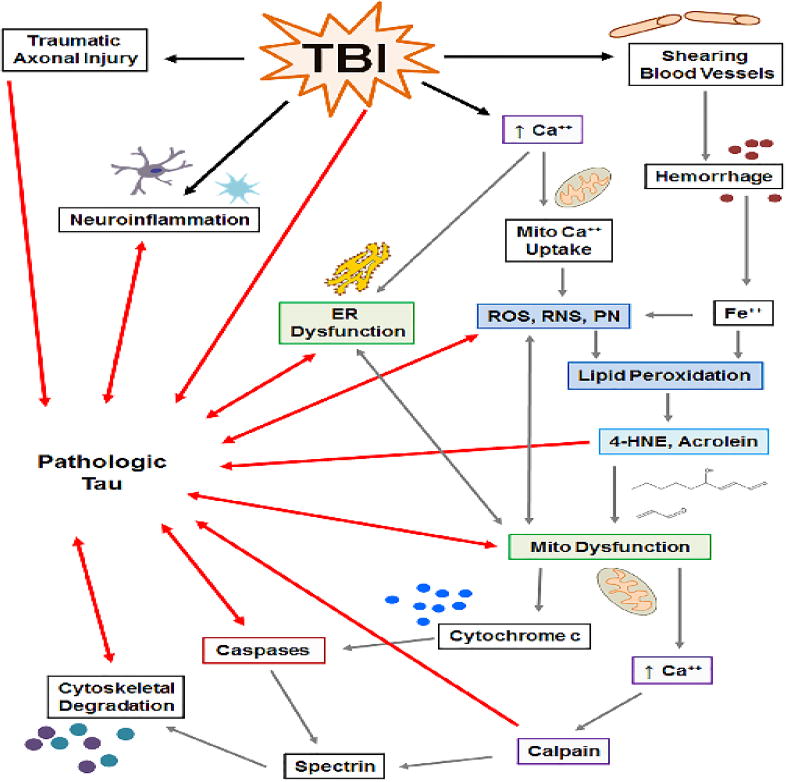
TBI induces traumatic axonal injury (TAI), neuroinflammation, increases in intracellular Ca++, shearing of blood vessels, and tau hyperphosphorylation. TAI causes microtubule detachment of tau and tau mislocalization. Ca++ uptake by the endoplasmic reticulum (ER) and mitochondria leads to organelle dysfunction, including increases in ROS, RNS, and peroxynitrite (PN), initiation of lipid peroxidation (LP), and 4-HNE and acrolein formation. Hemorrhage-derived Fe++ catalyzes formation of reactive species and LP. 4-HNE and acrolein bind mitochondrial proteins, exacerbating dysfunction and generation of reactive species. Dysfunctional mitochondria enhance ER dysfunction, release cytochrome c, which activates caspases, and extrude Ca++ into the cytosol, which activates calpain. Calpain cleaves cytoskeletal proteins (e.g. spectrin) resulting in cytoskeletal degradation. Neuroinflammation, ER dysfunction, ROS/RNS/PN, mitochondrial dysfunction, caspase/calpain activation, and cytoskeletal degradation contribute to formation of pathologic tau (e.g. post-translational modification, conformational change, aggregation, etc.). In return, pathologic tau contributes to neuroinflammation, ER dysfunction, generation of ROS/RNS/PN, mitochondrial dysfunction, caspase activation and cytoskeletal degeneration.
4. The Role of Tau in Cellular Dysfunction
The process in which acute TBI pathology develops into the chronic tau pathology seen in the tauopathies CTE and AD remains to be established, however, the mechanisms that can initiate formation of pathologic tau as well as the mechanisms by which tau can induce cellular dysfunction parallel many of the secondary injury mechanisms of TBI, including damage by ROS/RNS and LP-derived neurotoxic aldehydes, mitochondrial dysfunction, ER stress, calpain and caspase activation, and inflammation.
4.1 Oxidative Stress
Oxidative stress is elevated in several tauopathies (Alavi Naini and Soussi-Yanicostas, 2015; Castellani et al., 1995; Litvan, 2004; Martinez et al., 2008a). It is well known that in the tauopathy AD, Aβ is capable of inducing oxidative stress, a process which is hypothesized to be upstream of the formation of pathologic tau (Giraldo et al., 2014). Several studies confirm both the ability of oxidative stress to induce tau pathology and the ability of pathologic tau to induce oxidative stress, suggesting the possibility that in tauopathies such as CTE a self-propagating cycle of pathologic tau formation and oxidative stress occurs and leads to cellular dysfunction and neurodegeneration.
In-vitro, oxidative stress can induce both increases in tau aggregation and phosphorylation. Exposing tau isolated from bovine brain to iron-catalyzed oxidation results in dimerization, polymerization and formation of tau filaments (Troncoso et al., 1993), and in primary rat cortical neurons, oxidative stress (Fe2+/H2O2) increases tau phosphorylation by increasing activity of the tau kinase, GSK-3β, an effect which is attenuated by the GSK-3β inhibitor, lithium (Lovell et al., 2004). Interestingly, lithium also has the ability to attenuate tau pathology in-vivo by decreasing GSK-3β dependent tau phosphorylation and aggregation in transgenic tau mice (Perez et al., 2003). Additional in-vitro studies indicate that tau phosphorylation is also induced by chronic oxidative stress. Inhibiting glutathione synthase in neuroblastoma cells induces a mild, chronic oxidative stress, which increases the activity of the tau kinase, JNK, decreases the activity of the tau phosphatase, PP2A, and results in increases in tau phosphorylation and tau aggregation (Su et al., 2010). Although these studies indicate oxidative stress has the ability to induce tau phosphorylation in-vitro, oxidative stress induced tau dephosphorylation has also been reported (Davis et al., 1997; Galas et al., 2006; Olivieri et al., 2001; Zambrano et al., 2004). However, dephosphorylation of tau following oxidative stress exposure seems to be limited to acute administration of H2O2.
In-vivo experiments also link tau phosphorylation to oxidative stress exposure. Transgenic mice deficient for the mitochondrial antioxidant enzyme, SOD2, have increases in mitochondrial-induced oxidative stress, which result in increased tau phosphorylation, an effect which is attenuated by antioxidant administration (Melov et al., 2007). Additionally, transgenic mice overexpressing the mutated form of human tau, P301S, show markers of oxidative stress such as, increased mitochondrial protein carbonyls and decreased SOD2, months prior to the appearance of tau hyperphosphorylation and tangle formation (Dumont et al., 2011).
While oxidative stress has a demonstrated ability to induce tau pathology, several studies show that tau itself has the ability to induce oxidative stress. In-vitro, overexpressing tau impairs trafficking of peroxisomes, organelles responsible for H2O2 detoxification and results in increased susceptibility to H2O2-induced oxidative stress (Alavi Naini and Soussi-Yanicostas, 2015; Stamer et al., 2002). Tauopathy animal models also suggest the ability of tau to induce oxidative stress. Cultured neurons obtained from transgenic rats expressing a truncated form of human tau analogous to that found in AD have increased levels of ROS and are more susceptible to oxidative stress, effects which are likely the result of decreased numbers of mitochondria and abnormal mitochondrial distribution (Alavi Naini and Soussi-Yanicostas, 2015; Cente et al., 2006). Furthermore, transgenic mice overexpressing mutant human tau, P301L, also show increased generation of ROS, another consequence of mitochondrial dysfunction (Alavi Naini and Soussi-Yanicostas, 2015; David et al., 2005).
In summary, oxidative stress is well-established to occur in neurodegenerative tauopathies, and there are several mechanisms by which oxidative stress can induce tau phosphorylation and aggregation and by which pathologic tau can induce oxidative stress (Figure 2). Therefore, a primary event capable of inducing either oxidative stress or tau dysfunction, such as TBI, could set off a chronic and self-propagating cyclical cascade of oxidative stress and pathologic tau formation.
Figure 2. Select pathways demonstrating the role of oxidative stress in formation of tau pathology.
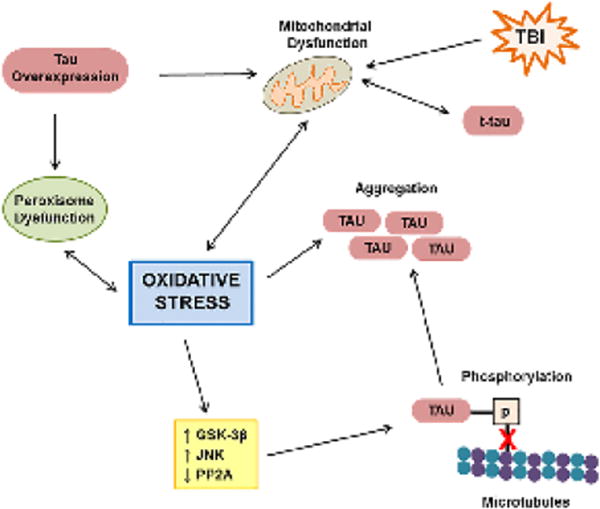
TBI induces mitochondrial dysfunction, resulting in increased oxidative stress, which in turn, exacerbates mitochondrial dysfunction. Proteases downstream of mitochondrial dysfunction cleave tau, forming truncated tau (t-tau), which in turn, exacerbates mitochondrial dysfunction. Experimental tau overexpression induces mitochondrial and peroxisome dysfunction. Peroxisome dysfunction exacerbates oxidative stress and is enhanced by oxidative stress. Oxidative stress increases tau kinases activity (GSK-3β, JNK) and decreases tau phosphatase activity (PP2A), resulting in hyperphosphorylation of tau, which decreases tau binding of microtubules (red X), leading to tau aggregation, which is enhanced by oxidative stress itself.
4.2 Peroxynitrite
Oxidative stress is often used as a broad term which encompasses a multitude of reactive species, one specific reactive species being PN. The PN derived radical, NO•2, is capable of nitrating tyrosine at the 3 position, thus forming 3-NT, a specific marker of PN-induced cellular damage (Hall et al., 2010). 3-NT modified tau has been identified in a variety of tauopathies, including AD, frontotemporal dementia, and Pick's disease (Horiguchi et al., 2003; Smith et al., 1997).
Peroxynitrite is capable of inducing tau dysfunction through both oxidation and nitration mechanisms (Reynolds et al., 2005; Reynolds et al., 2006). In-vitro, cysteine oxidation of tau by PN results in formation of disulfide crosslinks, tau oligomerization, and decreased microtubule assembly (Landino et al., 2004). PN can also promote tau oligomerization through addition of tyrosyl radicals which results in formation of 3,3′-dityrosine irreversible crosslinks and stabilization of insoluble tau filament aggregates characteristic of late stage paired helical filaments (PHF) (Reynolds et al., 2005; Reynolds et al., 2006). Furthermore, although PN-derived NO•2 nitration of tau has been shown to inhibit tau polymerization (Reynolds et al., 2005), 3-NT modification of tau does result in disrupted binding of tau to microtubules and inhibition of tubulin assembly (Reynolds et al., 2006; Zhang et al., 2005).
Peroxynitrite has also been shown to modify tau in-vivo. Injection of the PN donor, SIN-1, into rat hippocampus results in both nitration and hyperphosphorylation of tau, and increased activation of the tau kinases, GSK-3β and p38 MAPK, processes are attenuated by pre-administration of the PN scavenger, uric acid (Zhang et al., 2006). SIN-1 administration increased levels of nitrated p85, a regulator of GSK-3β, suggesting that PN is capable of inducing tau hyperphosphorylation by nitration of upstream proteins in tau phosphorylation cascade (Zhang et al., 2006). Furthermore, SIN-1 also led to a decrease in proteasome activity, which combined with the fact that the nitrated form of tau is less susceptible to proteasomal degradation, resulted in an increase in tau aggregation (Zhang et al., 2006).
In summary, there are several mechanisms by which the reactive species, PN, can induce tau pathology, including oxidation, nitration, and induction of tau hyperphosphorylation, processes which lead to accumulation of toxic tau aggregates and/or destabilization of microtubules. PN is generated following TBI; therefore it is possible that the PN formed acutely following TBI initiates tau dysfunction, while other mechanisms of PN generation, such as continued mitochondrial dysfunction, contribute to chronic PN-induced tau dysfunction as suggested in Figure 3.
Figure 3. Mechanisms by which peroxynitrite leads to formation of tau pathology.
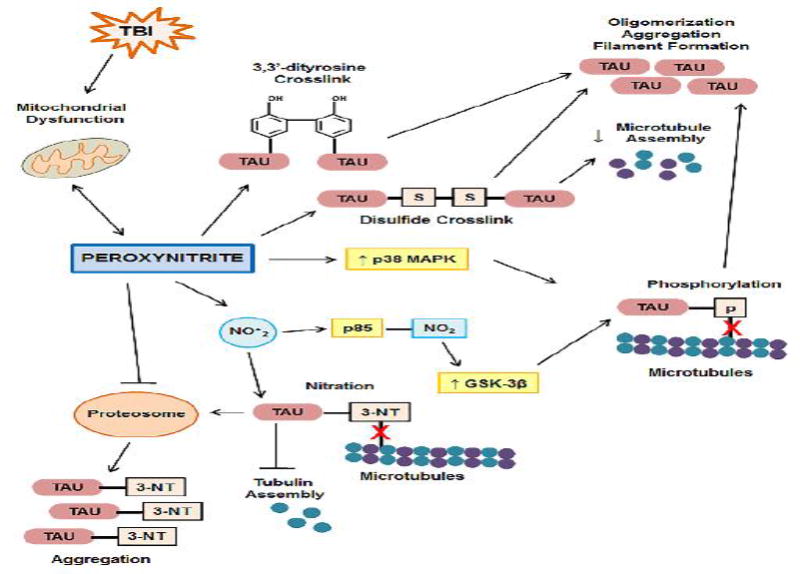
TBI-induced mitochondrial dysfunction generates peroxynitrite (PN), which in turn, exacerbates mitochondrial dysfunction. PN nitrates or oxidizes tau forming 3,3′-dityrosine or disulfide crosslinks, respectively. Crosslinked tau induces tau oligomerization, aggregation and filament formation. Disulfide crosslinked tau also decreases microtubule assembly. PN increases the tau kinase, p38 MAPK. PN-derived NO•2 increases the tau kinase, GSK-3β, through nitration of the GSK-3β upstream regulator, p85. Increased tau kinase activity results tau hyperphosphorylation which decreases binding of tau to microtubules (red X). NO•2 directly nitrates tau at the 3 position of tyrosine, generating tau-3NT. Tau-NT has decreased microtubule binding properties (red X) and inhibits tubulin assembly. Tau-3NT is preferentially degraded by the proteasome, an organelle that's function is inhibited by PN, resulting in build-up and aggregation of tau-NT.
4.3 Lipid Peroxidation-Derived Neurotoxic Aldehydes
Lipid peroxidation-derived aldehydes have been found to be elevated in the brains of several tauopathies (Butterfield et al., 2010; Lovell et al., 2001; Markesbery and Lovell, 1998; Martinez et al., 2008a; Martinez et al., 2008b; Montine et al., 1997; Muntane et al., 2006; Odetti et al., 2000; Sayre et al., 1997). In fact, in PSP, a tauopathy with paired helical filament morphology distinct from AD, significant increases in 4-HNE are correlated with tau aggregation, suggesting that accumulation of 4-HNE may contribute to impairment of tau degradation (Odetti et al., 2000).
The LP-derived aldehydes 4-HNE and acrolein are both capable of inducing tau pathology. In-vitro, administration of 4-HNE to cultured rat hippocampal neurons results in direct binding of 4-HNE to tau, preventing dephosphorylation of tau by alkaline phosphatases (Mattson et al., 1997). Acrolein, a more potent neurotoxic aldehyde than HNE (Vaishnav et al., 2010), also induces hyperphosphorylation of tau, both in neuroblastoma cells and in cultured cortical mouse neurons, an effect which can be attenuated by inhibiting the tau kinases, GSK-3β and p38 MAPK (Gomez-Ramos et al., 2003).
In addition to inducing tau hyperphosphorylation, 4-HNE and acrolein are also capable of promoting tau aggregation. In P19 neuroglial cultures, 4-HNE addition results in cytotoxicity and formation of high molecular weight tau species characteristic of neurofibrillary tangles (Montine et al., 1996). However, the effect of 4-HNE on tau aggregation is greatest for tau which has already been hyperphosphorylated. In-vitro, administration of 4-HNE promotes assembly of phosphorylated tau, but not native tau, into neurofibrillary tangle-like fibrillary polymers (Perez et al., 2000). Phosphorylation of tau is also required in order for 4-HNE to induce conformational changes to tau, such as the Alz50 epitope conformational change characteristic of early AD (Liu et al., 2005; Takeda et al., 2000). Therefore, 4-HNE may play a role in NFT formation by stabilizing pathologic tau confirmations (Liu et al., 2005). However, 4-HNE does not further enhance aldehydic modification of paired helical filaments, suggesting that paired helical filaments are either already extensively modified by aldehydes or that they are in a confirmation which prevents further aldehyde modifications (Liu et al., 2005). Acrolein also promotes tau aggregation in a concentration-dependent manner, with the fastest aggregation rates occurring in pseudophosphorylated tau, further suggesting that hyperphosphorylation of tau increases its susceptibility to aldehyde-induced aggregation (Kuhla et al., 2007).
While LP-derived aldehydes have a demonstrated ability to induce tau pathology, tau itself can enhance formation of LP-derived aldehydes. For example, in brain homogenates derived from transgenic mice overexpressing mutant human tau, P301L, ferric iron-induced LP results in increased formation of the LP-derived aldehyde, malondialdehyde, compared to wild-type mice (David et al., 2005).
In summary, LP-derived aldehydes are known to be present in human tauopathies, and LP-derived aldehydes, such as 4-HNE and acrolein, are capable of inducing tau phosphorylation and promoting and stabilizing tau aggregation, in particular the aggregation of hyperphosphorylated tau. Therefore, it is possible that following TBI, increases in 4-HNE and acrolein and increases in hyperphosphorylated tau work synergistically to promote tau aggregation, while other mechanisms of 4-HNE and acrolein generation, such as continued mitochondrial dysfunction and tau-induced LP, contribute to a chronic, self-promoting and cyclic cascade of LP and pathologic tau formation (Figure 4).
Figure 4. The role of 4-hydroxynonenal (4-HNE) and acrolein (ACR) in formation of tau pathology.
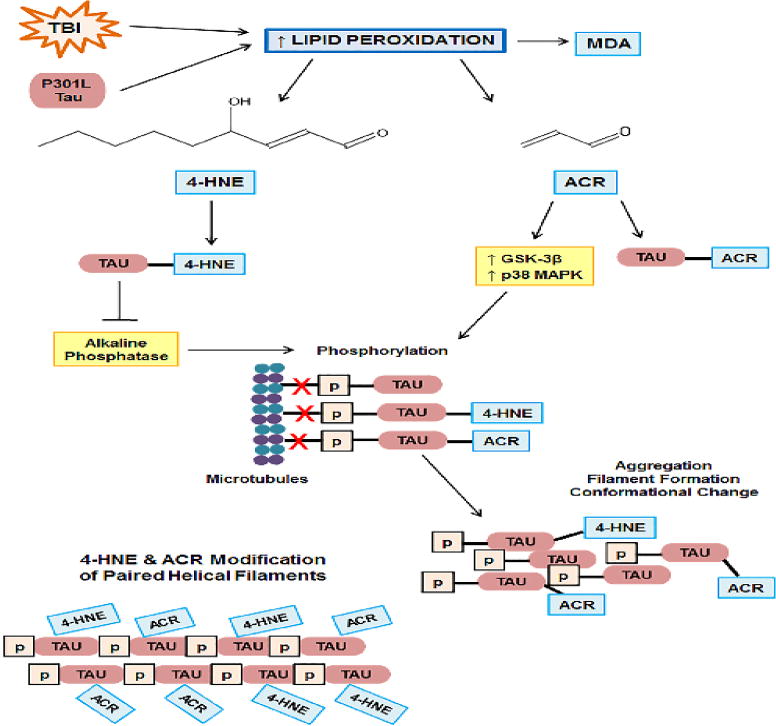
TBI induces lipid peroxidation (LP). Overexpression of P301L mutant also increases LP as measured by the LP-derived, malondialdeyde (MDA). LP results in the formation of the neurotoxic aldehydes, 4-HNE and ACR, which covalently bind proteins, including tau, causing protein dysfunction. ACR increases activity of the tau kinases, GSK-3β and p38 MAPK, and 4-HNE-modified tau inhibits alkaline phosphatase, resulting in hyperphosphorylation of tau, 4-HNE modified-tau, and ACR modified-tau, which decreases binding of tau to the microtubules (red X), leading to conformational changes, aggregation, and filament formation. Paired helical tau filaments are extensively modified by 4-HNE and ACR.
4.4 Mitochondrial Dysfunction
Alterations in mitochondrial function are known to occur in the tauopathies, AD and FTD (Baloyannis, 2006; David et al., 2005), and in normal aging (Swerdlow, 2011). Several studies indicate that mitochondrial dysfunction is capable of inducing tau dysfunction, and that tau itself is capable of inducing mitochondrial dysfunction.
Impairment of oxidative phosphorylation can lead to tau dysfunction and pathologic tau formation both in-vitro and in-vivo. For example, in primary rat striatal rat neurons, inhibition of complex-I, inhibition of complex-II, or uncoupling of oxidative phosphorylation leads to decreased ATP production, cellular death and retrograde transport of tau and mitochondria from the axons to the soma (Escobar-Khondiker et al., 2007). In fact, complex-I inhibited neurons contain somal mitochondria which associate with tau at the outer mitochondrial membrane (Escobar-Khondiker et al., 2007). In-vivo, inhibition of complex-I can also induce tau pathology. Chronically infusing the complex-I inhibitor, rotenone, into rats results in cell death, motor dysfunction, and increases in cytosolic tau, phosphorylated tau, and neuronal, glial, and oligodendrocytic tau fibrils (Hoglinger et al., 2005). Furthermore, increased levels of the PN marker, 3-NT, and ubiquitin, a marker for defective protein degradation, are found within cells containing pathologic tau (Hoglinger et al., 2005). Mitochondrial dysfunction is known to lead to PN formation (Bringold et al., 2000; Radi et al., 2002) and can indirectly reduce proteosomal activity (Hoglinger et al., 2005). Therefore, it is possible that complex-I induced mitochondrial dysfunction can lead to formation of pathologic tau through a combination of mechanisms including mitochondrial and tau redistribution, generation of reactive nitrogen species, and impairment of proteosomal degradation pathways.
Pathologic tau has also been observed in transgenic mice lacking the mitochondrial antioxidant enzyme SOD2. Mitochondrial SOD2 catalyzes the dismutation of superoxide (O2•-), formed when single electrons leak from complex I of the electron transport chain (Brand et al., 2004), to the less reactive oxygen species, H2O2 (Flynn and Melov, 2013). Thus, SOD2 is an essential enzyme without which the undismutated O2•- wil react with mitochondrial NO• to form the reactive nitrogen species PN. . However, pharmacological administration of the catalytic antioxidant EUK-189 is able to attenuate tau phosphorylation in SOD2 deficient mice (Melov et al., 2007), confirming that mitochondrial generated O2•- or downstream PN (formed upon reaction of mitochondrial O2•- and NO•) is capable of driving tau phosphorylation (Melov et al., 2007). Additional evidence supports the theory that mitochondrial dysfunction can induce tau phosphorylation. In transgenic mice overexpressing the human tau mutation, P301S, mitochondrial abnormalities, including carbonyl modification of mitochondrial proteins and decreases in mitochondrial enzyme activities, precede tau hyperphosphorylation and tangle formation by at least three months (Dumont et al., 2011). However, while it is likely that the mitochondrial abnormalities contributed to pathologic tau development, it is also likely that the overexpression of mutated human tau itself induces mitochondrial abnormalities.
Multiple studies provide evidence that tau can induce mitochondrial dysfunction. Mitochondria are synthesized in neuronal cell bodies and then transported throughout the neuron along microtubules by the anterograde motor protein, kinesin (Chang et al., 2006; Morris and Hollenbeck, 1995), while damaged mitochondria are transported toward the soma by the retrograde motor protein, dynein (Miller and Sheetz, 2004). Therefore, neuronal health relies extensively on proper axonal transport of mitochondria. In CTE, fast axonal transport along microtubules is impaired, with decreases being seen both in kinesin and dynein (Kokjohn et al., 2013). Tau is primarily found in axons, functioning to stabilize microtubules (Maccioni and Cambiazo, 1995); however, tau also serves to regulate the attachment and detachment of motor proteins (Trinczek et al., 1999). Therefore, it has been hypothesized that overexpression of tau or accumulation of pathologic tau can impair axonal transport of mitochondria to the synapse, resulting in decreased synaptic ATP, calcium dysregulation and neurodegeneration (Sheng and Cai, 2012). However, it should be noted that in addition to mitochondria, tau-induced impairment of axonal transport affects additional organelles, vesicles, and proteins, and that each likely contributes to their own downstream pathologies. For example, tau overexpression can inhibit trafficking of peroxisomes, neurofilaments, Golgi-derived vesicles, the ER, and amyloid precursor protein (APP) (Darios et al., 2005; Ebneth et al., 1998; Stamer et al., 2002), the precursor to Aβ, a pathologic species found in the tauopathy AD and in a subset CTE cases.
In regard specifically to axonal transport of mitochondria, several mechanisms by which tau-induced impairment of axonal transport have been reported. In-vitro, overexpression of tau in neuroblastoma cells leads to preferential impairment of anterograde mitochondrial transport, resulting in mitochondria that are clustered near nuclei but absent in neurites (Ebneth et al., 1998). The degree to which tau overexpression interferes with mitochondrial transport is influenced by the number of microtubule binding domain repeats. For instance, overexpression of either 3R or 4R tau leads to redistribution of mitochondria from axons to the soma (Stoothoff et al., 2009); however, 4R tau, the isoform expressed to a greater degree in AD and CTE (McKee et al., 2014; Schmidt et al., 2001) has the greatest effect on redistribution (Stoothoff et al., 2009). The N-terminus of tau has also been shown to be sufficient for inhibiting anterograde transport (LaPointe et al., 2009). In fact, filamentous tau, which contains an exposed N-terminus, inhibits kinesin-dependent fast axonal transport, whereas monomeric tau confirmations with unexposed N-terminal regions do not (LaPointe et al., 2009). Hyperphosphorylation of tau at AD-specific sites also results in decreased mitochondrial transport due to extension of the N-terminal projection domain and expansion of inter-microtubule distances (Shahpasand et al., 2012). Additional studies confirm the ability of phosphorylated tau impair anterograde transport. Inhibition of anterograde transport by filamentous tau is dependent upon on activation of the tau kinase, GSK-3β (LaPointe et al., 2009). In PC12 cells, ceramide-induced CDK5-dependent tau phosphorylation results in dissociation of phosphorylated tau from the microtubules, clustering of mitochondria and ER near the centrosome, and cell death, effects which are attenuated by preventing tau phosphorylation (Darios et al., 2005).
Tau-induced impairment of mitochondrial transport likely has pathologic implications beyond decreased delivery of mitochondria to the synapse. For example, the clustering of mitochondria and ER near the centrosome in PC12 cells results in an increase in mitochondria-ER associations, neurotoxic transfer of Ca2+ from the ER to the mitochondria, and induction of apoptosis (Darios et al., 2005). Pathologic mitochondria-ER associations have also been observed in the soma of spinal motor neurons in transgenic mice overexpressing the human tau mutation, P301L (Perreault et al., 2009), and correlate with accumulation of hyperphosphorylated tau at the surface of the rough ER (rER) (Perreault et al., 2009). In fact, hyperphosphorylated tau is found on the surface of rER in the tauopathy, AD (Perreault et al., 2009). Because mitochondria-ER associations require the ER to be absent ribosomes, it is possible that tau is capable of inducing mitochondria-ER associations by displacing ribosomes from the ER surface (Perreault et al., 2009).
In addition to impairment of mitochondrial transport and alterations in mitochondrial-ER dynamics, other tau-induced mitochondrial impairments have been reported such as abnormal morphology. For example, neurons from transgenic mice overexpressing the human tau mutation, P301L, have decreased numbers of mitochondria and mitochondria which have abnormally swollen morphology (Yoshiyama et al., 2007).
Tau-induced decreases in oxidative phosphorylation capacity have also been reported. The mitochondria from P301L mice have reductions in complex I (David et al., 2005; Rhein et al., 2009) and complex V (ATP synthase) protein and activity levels (David et al., 2005). Additionally, aged P301L mice have decreases in ATP production and increases in ROS due to decreases in complex-I driven respiration (David et al., 2005). Importantly, reductions in complex V (ATP synthase) have also been seen in the human tauopathy, FTD (David et al., 2005).
Although many studies that investigate the effect of tau on mitochondrial function rely on mutated forms of human tau, the mutant tau constructs, such as P301, do induce tau hyperphosphorylation. Therefore, the mutant tau constructs are representative of non-genetic, hyperphosphorylated tau disease states. However, the possibility exists that mutated tau isoforms cause greater mitochondrial dysfunction than wild type tau. For example, stable overexpression of human mutant tau, P301L, into SY5Y cells results in complex I impairment, decreased ATP production, increased susceptibility to oxidative stress, abnormal morphology, and fission-fusion dysfunction (Schulz et al., 2012); however, overexpression of wild-type 4R human tau results in increases in complex I activity, normal morphology and normal fission-fusion dynamics (Schulz et al., 2012).
Tau oligomers, which have been implicated as the species responsible for tau-induced neurotoxicity (Spires-Jones et al., 2011), are also capable of inducing mitochondrial dysfunction. Injections of full-length human tau oligomers, but not tau fibrils or monomers, into mouse hippocampi induce memory impairment, neurodegeneration, loss of synaptic proteins, and mitochondrial dysfunction (Lasagna-Reeves et al., 2011). Specifically, the tau oligomers co-localized with mitochondria, decreasing complex I protein levels, and activating the intrinsic apoptotic protease, caspase-9 (Lasagna-Reeves et al., 2011). However, complex V protein levels were not altered, suggesting that complex I dysfunction occurs prior to tau-induced complex V dysfunction and ATP depletion (Lasagna-Reeves et al., 2011).
Truncated tau fragments are also capable of inducing mitochondrial dysfunction. In immortalized cortical neurons, Asp421 tau, a mutant mimicking C-terminus caspase-3 cleavage of tau at Asp421, induces mitochondrial fragmentation, decreases mitochondrial calcium buffering capacity, and increases production of mitochondrial ROS (Quintanilla et al., 2009), effects which are attenuated by administration of the calcineurin inhibitors cyclosporine A (CsA) or FK506, suggesting that the calcium-dependent phosphatase, calcineurin, plays a role in caspase-cleaved tau induction of mitochondrial dysfunction (Quintanilla et al., 2009). Tau is a known calcineurin substrate; therefore, it is possible that caspase-cleaved tau interacts differentially with calcineurin, increasing its activity level toward substrates which effect mitochondrial fragmentation (Quintanilla et al., 2009). Interestingly, both CsA and FK506 are neuroprotective in experimental TBI (Kilbaugh et al., 2011; Kulbe et al., 2016; Marmarou and Povlishock, 2006; Mbye et al., 2009; Mbye et al., 2008; Reeves et al., 2007; Sullivan et al., 2011), and CsA has been shown to attenuate levels of cleaved tau in the hippocampus of rats following severe controlled cortical impact injury (CCI) (Gabbita et al., 2005).
In addition to C-terminal cleavage, truncated tau fragments can also be generated by N-terminal processing. Although this process is not well understood, caspases, such as caspase-6 have the ability to cleave tau at the N-terminus (Horowitz et al., 2004), and N-terminal tau fragments have been reported to induce mitochondrial dysfunction. For example, addition of the NH2-26-44 tau fragment to cerebellar granule neuron homogenates results in decreases in cytochrome oxidase (COX) activity, adenine nucleotide translocator (ANT) activity, and ANT-dependent impairment of oxidative phosphorylation (Atlante et al., 2008). Importantly, N-terminal tau fragments have been identified in AD and other tauopathies (Amadoro et al., 2010). In fact, in AD, a 20-22kD NH2-tau fragment is highly enriched in synaptosomes, localizing with synaptic mitochondria, and correlating with synaptic and mitochondrial dysfunction (Amadoro et al., 2010). Synapses have high metabolic demands, met by enrichment of synaptic mitochondria (Khatri and Man, 2013; MacAskill et al., 2010). Not only are synaptic mitochondria essential for proper neurotransmission (Sheng and Cai, 2012) and synaptic plasticity (Cheng et al., 2010; MacAskill et al., 2010; Sheng and Cai, 2012), but their dysfunction is implicated in neurodegeneration (Cheng et al., 2010; Sheng and Cai, 2012). Therefore, localization of the 20-22kD NH2-tau with synaptic mitochondria has important implications for synaptic health, especially considering the fact that 20-22kD NH2-tau is capable of inducing mitochondrial dysfunction. In vitro, the 20-22kD NH2-tau fragment induces abnormal mitochondrial morphology, increases mitochondrial fragmentation, decreases mitochondrial fusion, enhances redistribution of mitochondria to the soma, decreases mitochondrial protein and mtDNA copy number, and enhances mitophagy, effects which correlate with in-vitro synaptic pathology and oxidative stress (Amadoro et al., 2014).
In addition to truncated tau species, phosphorylated tau has also been implicated in alteration of mitochondrial fission-fusion dynamics. Specifically, in the tauopathy AD, phosphorylated tau interacts with Drp1, a mitochondrial fission GTPase, and is associated with increases in GTPase activity indicative of mitochondrial fission, suggesting that phosphorylated tau may enhance mitochondrial fragmentation through interaction with Drp1 (Manczak and Reddy, 2012).
In summary, mitochondrial dysfunction is known to occur in human tauopathies, and there are several mechanisms by which mitochondrial dysfunction can induce tau pathology and by which pathologic tau species can induce mitochondrial dysfunction. Therefore, a primary event capable of inducing mitochondrial dysfunction or formation of pathologic tau species, such as TBI, could set off a self-propagating, cyclical cascade of mitochondrial dysfunction and pathologic tau formation, which work synergistically to induce cellular damage. In fact, in transgenic mice expressing mutations which induce both mitochondrial dysfunction and tau pathology, neurodegeneration and oxidative stress are increased over either mutation alone (Kulic et al., 2011). Furthermore, in addition to a self-propagating, cyclical cascade of mitochondrial-tau dysfunction, because mitochondrial impairment results in production of species capable of inducing damage to mitochondrial DNA (mtDNA), which encodes for electron transport chain proteins (Dalleau et al., 2013; Hall et al., 2010), mitochondrial dysfunction is capable of enhancing and perpetuating its own chronic damage. In fact, accumulations in mtDNA damage have been hypothesized to contribute to both aging and neurodegenerative disease (Keogh and Chinnery, 2015).
4.5 Endoplasmic Reticulum Stress
ER stress has been documented in several tauopathies, including AD, FTD, and CTE, and correlates with tau hyperphosphorylation (Hoozemans and Scheper, 2012; Hoozemans et al., 2009; Lucke-Wold et al., 2016; Nijholt et al., 2012). ER dysfunction has been reported to occur both upstream and downstream of pathologic tau formation (Ho et al., 2012). Upstream of tau, pharmacological induction of ER stress increases tau phosphorylation both in-vitro and in-vivo (Fu et al., 2010; Ho et al., 2012; Lin et al., 2014). One mechanism by which ER stress can increase tau phosphorylation is through activation of the tau kinase, GSK-3β (Fu et al., 2010; Lin et al., 2014). However, pathologic tau species can also induce ER dysfunction. The ER is responsible for protein folding and degradation; therefore, ER stress results in the build-up of misfolded and unfolded proteins, inhibition of ER-associated degradation (ERAD), initiation of the Unfolded Protein Response (UPR), and decreased protein synthesis (Abisambra et al., 2013; Radford et al., 2015).
Tau is capable of activating the UPR in-vitro. Pharmacologic induction of tau phosphorylation in primary rat cortical neurons results in UPR activation (Ho et al., 2012). Similarly, stable expression of tau in HEK cells activates the UPR and is accompanied by increased ubiquitin levels suggestive of ERAD impairment (Abisambra et al., 2013). In-vivo, the UPR is known to be increased in both transgenic tau animal models and human tauopathies (Abisambra et al., 2013; Hoozemans and Scheper, 2012; Hoozemans et al., 2009; Nijholt et al., 2012; Radford et al., 2015). It has been hypothesized that the increased UPR seen in tauopathies is the result of tau-induced ERAD impairment (Abisambra et al., 2013). For example, transgenic mice overexpressing human mutant tau, P301L, show ERAD impairment concomitant with associations between tau and the ERAD hetereocomplex (Abisambra et al., 2013). However, activation of UPR in tauopathies may also be due to the buildup of misfolded tau proteins (Radford et al., 2015). UPR activation has also been suggested to play a direct role in neurotoxicity. In fact, pharmacologic inhibition of the UPR in P301L mice decreases tau phosphorylation, neurodegeneration, and behavioral impairment (Radford et al., 2015).
An additional mechanism by which tau induced ER dysfunction can contribute to cellular dysfunction is inhibition of protein synthesis. Microsomes are vesicle-like fragments which contain ER and ribsosomes. In brain microsomes isolated from the tauopathy, AD, oligomeric tau associates more strongly with ribosomes than in control brains, suggesting that pathologic tau may have an effect on RNA translation (Meier et al., 2016). In fact, in-vitro, wild type, mutant, and oligomeric tau are all able to reduce RNA translation (Meier et al., 2016), and in primary neurons cultured from transgenic mice overexpressing human mutant tau, P301L, there are both decreases in RNA translation and protein levels (Meier et al., 2016). Specifically, there are marked decreases in the protein PSD-95, a post-synaptic protein important in learning and memory (Meier et al., 2016).
Soluble tau oligomers and truncated tau forms have been specifically implicated in tau induced ER dysfunction. Depleting soluble tau both in cell culture and in P301L mice reduces UPR activation and inhibition of ERAD, suggesting soluble tau oligomers rather than insoluble tau aggregates are responsible for tau-induced ER dysfunction (Abisambra et al., 2013). In regards to truncated tau isoforms, inducing ER stress in immortalized cortical neurons expressing Asp-421 truncated tau, which mimics caspase-3 cleavage, results in increased cellular toxicity and caspase-3 activation compared to cells expressing full length tau (Matthews-Roberson et al., 2008). In fact, ER stress itself has the capability of inducing the caspase-3 activation (Morishima et al., 2002; Song et al., 2002) which leads to tau truncation.
In summary, ER stress is known to occur in several tauopathies, including CTE, and there are several mechanisms by which ER stress can induce tau pathology and by which pathologic tau species can induce ER dysfunction. Therefore, as summarized in Figure 5, a primary event capable inducing ER stress or tau pathology, such as TBI, could set off a chronic and self-propagating cascade of ER dysfunction and pathologic tau formation, which results in protein accumulation, decreased protein synthesis, and cellular dysfunction.
Figure 5. Mechanisms by which endoplasmic reticulum (ER) stress and dysfunction can lead to formation of tau pathology.
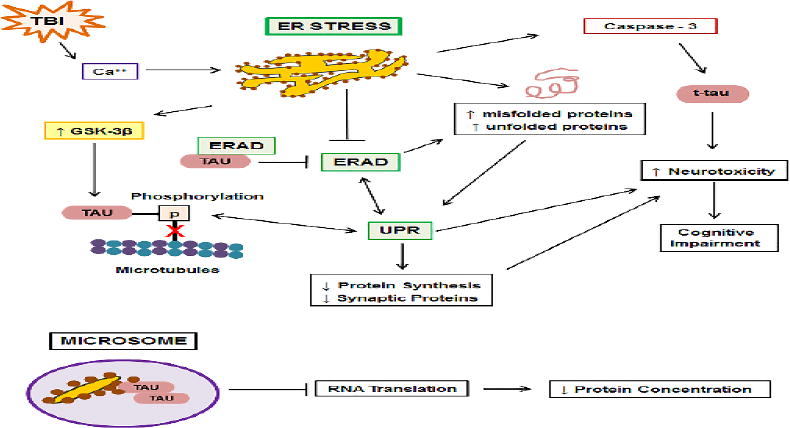
Following TBI increases in intracellular Ca++, derived extracellularly or from dysfunctional mitochondria, induce ER stress. ER stress increases activity of the tau kinase, GSK-3β, resulting in tau hyperphosphorylation, which decreases binding of tau to microtubules (red X). ER stress results in the buildup of misfolded and unfolded proteins, including tau, both directly and through inhibition of ER-associated degradation (ERAD). Tau itself inhibits ERAD through associations with the ERAD heterocomplex. ERAD and buildup of misfolded and unfolded proteins activate the unfolded protein response (UPR), which in turn, activates ERAD. The UPR results in increases in tau phosphorylation, which in turn, activate the UPR. UPR activation increases neurotoxicity and behavioral impairment and decreases synthesis of proteins, including synaptic proteins. ER stress activates the protease, caspase-3, resulting in tau truncation (t-tau) and increased neurotoxicity. Alzheimer's disease-derived microsomes contain ER fragments and ribosomes which strongly associate with oligomeric tau, resulting in decreased RNA translation and decreased protein concentrations.
4.6 Calpain Activated Proteolysis
Tau is a known substrate of the calcium-dependent cysteine protease, calpain (Johnson et al., 1989; Litersky et al., 1993; Yang and Ksiezak-Reding, 1995). Calpain cleavage of tau results in the formation of a 35kD tau fragment (Liu et al., 2011) and a 17kD tau fragment, purported to be between 10-17kD (Garg et al., 2011; Park and Ferreira, 2005). Increased calpain activity and depletion of the endogenous calpain inhibitor, calpastatin, have been found in the tauopathy, AD, (Grynspan et al., 1997; Rao et al., 2008; Saito et al., 1993; Tsuji et al., 1998) and the presence of the 17kD tau fragment correlates with increases in calpain activity in a variety of tauophathies, including AD, FTD and dementia pugilistic (i.e. CTE) (Ferreira and Bigio, 2011).
The 17kD tau fragment has a decreased association with the microtubules and accumulates in the perikarya (Canu et al., 1998). In-vitro, glutamate and thapsigargin induced increases in calcium have been shown to induce formation of calpain-cleaved 17kD tau (Garg et al., 2011). The 17kD tau fragment has been reported for form prior to increases in tau phosphorylation (Park and Ferreira, 2005), and is suspected of being neurotoxic (Park and Ferreira, 2005; Park et al., 2007; Sinjoanu et al., 2008). For example, in-vitro, Aβ induces calpain activation in cultured hippocampal neurons, leading to formation of 17kD tau and neurodegeneration, affects which are attenuated by inhibiting calpain activation (Park and Ferreira, 2005; Park et al., 2007; Sinjoanu et al., 2008). However, other studies have shown that 17kD tau does not induce toxicity in-vitro and is equally expressed in the brains of AD and healthy controls, suggesting that 17kD tau is a marker for calpain activation but does not have a direct role in neurotoxicity (Garg et al., 2011).
In addition to cleaving tau, calpain has also been identified as an upstream activator of extracellular-regulated kinase (ERK) (Veeranna et al., 2004), which is capable of phosphorylating tau (Fang et al., 2010; Qi et al., 2016), In fact, increases in active ERK expression are associated with early tau deposition in neurons and glia in several tauopathies (Ferrer et al., 2001).
Despite controversy surrounding the role of the calpain generated 17kD tau fragment in neurotoxicity, both increases in calpain and 17kD tau have been identified in human tauopathies. Due to increases in intracellular calcium levels, calpain is highly activated following TBI (Deng et al., 2007; Thompson et al., 2006); therefore it is possible that TBI initiates formation of calpain-cleaved 17kD tau, whereas chronic generation of 17kD tau could be the result of calpain activation by calcium derived from chronically dysfunctional calcium buffering organelles suspected of having self-propagating, cyclic interactions with pathologic tau, such as the mitochondria and ER (Figure 6).
Figure 6. The role of calpain in the formation of pathologic tau.
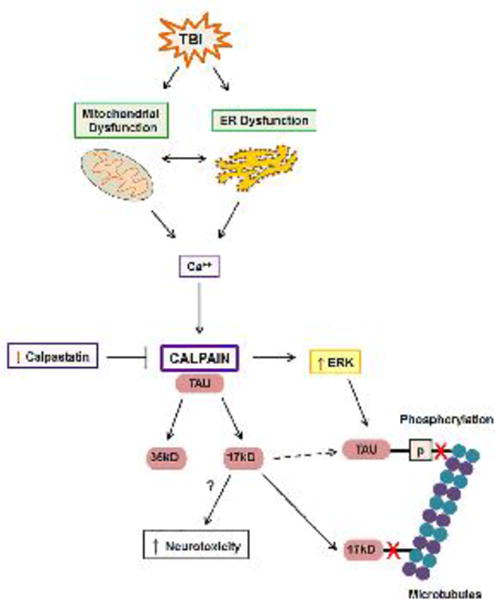
TBI-induced increases in intracellular Ca++ derived extracellularly or from dysfunctional mitochondria and ER, activate the protease calpain. The endogenous calpain inhibitor, calpastatin, is decreased in tauopathies such as Alzheimer's disease. Calpain cleaves tau into 35kD and 17kD fragments. The 17kD tau fragment forms prior to increases in tau phosphorylation and may directly enhance tau phosphorylation (dotted arrow). Calpain induces activation of the tau kinase, ERK, resulting in hyperphosphorylation of tau, which decreases binding of tau to microtubules. 17kD has a decreased affinity for microtubule binding (red X) and may be neurotoxic.
4.7 Caspase Activated Proteolysis
Tau is a known substrate of the caspase proteases (Fasulo et al., 2000; Park et al., 2007), which are well known for their role in apoptosis. The most well characterized caspase cleavage site on tau is Asp421, located on the c-terminus end. Multiple caspases, including caspase 3, can cleave tau at Asp421 (Gamblin et al., 2003), which results in a tau fragment of 50kD (Chung et al., 2001; Park et al., 2007). However, tau can also undergo n-terminal processing by caspases, such as caspase-6 (Horowitz et al., 2004). Caspase cleaved tau is found in several tauopathies (Guillozet-Bongaarts et al., 2007; Newman et al., 2005), and is inversely correlated with cognitive function in the tauopathy, AD (Rissman et al., 2004), with formation of Asp421 truncated tau considered to occur relatively early in the disease process (Guillozet-Bongaarts et al., 2005; Rissman et al., 2004). It is hypothesized that accumulation of Asp421 truncated is the result of dysregulation of autophagy, a process known to occur in the tauopathy, AD (Nixon, 2006). In fact, in immortalized cortical neurons, stably expressed Asp421 truncated tau is preferentially degraded by autophagy, while full length tau is dependent on proteasomal degradation (Dolan and Johnson, 2010).
Several lines of evidence suggest that Asp421 truncated tau is pathologic. In-vitro, the c-terminus of tau is responsible for inhibiting polymerization of tau, suggesting that Asp421 truncated tau has enhanced polymerization properties (Berry et al., 2003). In fact, Asp421 truncated tau assembles into filaments at a faster rate and to a higher degree than full length tau (Gamblin et al., 2003; Rissman et al., 2004). Furthermore, full length tau undergoes enhanced filament formation in the presence of Asp421 truncated tau, suggesting that Asp421 truncated tau may be able to seed filament formation of other tau species (Rissman et al., 2004). In-vitro, overexpression of Asp421 truncated tau, representative of caspase-3 cleaved tau, results in increased apoptosis (Chung et al., 2001; Fasulo et al., 2000). Caspase-3 is an executioner caspase in the apoptotic cascade; therefore, the fact that caspase-3 cleaved tau is capable of enhancing apoptosis suggests that caspase-3 and Asp421 truncated tau may be involved in a chronic, self-propagating apoptotic cascade (Fasulo et al., 2000). Additionally, as previously discussed, caspase cleaved tau fragments are also capable of inducing mitochondrial dysfunction (Quintanilla et al., 2009) and ER stress (Matthews-Roberson et al., 2008).
Evidence suggests that non-truncated tau forms can also initiate caspase activation and tau truncation. For example, overexpressing human mutant tau P301L in mice results in apoptosis, caspase-3 activation, and appearance of caspase truncated tau (de Calignon et al., 2010; Ramalho et al., 2008). Similarly, overexpression of human 4R tau results in caspase activation, although total numbers of caspase bearing neurons are low (de Calignon et al., 2010). Therefore, it is possible that, in-vivo, full-length pathologic tau, particularly soluble pre-tangle tau species, are capable of activating caspase-3, which results in formation of distinct truncated tau species. In fact, suppression of tau in P301L mice decreases caspase activation despite the continued presence of neurofibrillary tangles (de Calignon et al., 2010).
Although formation of caspase truncated tau is considered to occur early in the disease process (Guillozet-Bongaarts et al., 2005; Rissman et al., 2004), Asp421 truncated tau is still capable of inducing tau phosphorylation and conformational change (de Calignon et al., 2010) and can be the subject of phosphorylation and conformation change itself (Rissman et al., 2004). However, similar to human studies, transgenic animal models of tauopathy suggest that formation of caspase truncated tau occurs prior to tau aggregation and correlates with cognitive decline. In fact, in P301L mice, caspase cleavage of tau proceeds neurofibrillary tangle formation (de Calignon et al., 2010; Ramalho et al., 2008), and is coincident with the beginnings of cognitive impairment (Ramalho et al., 2008). Furthermore, animal models confirm in-vitro findings that caspase truncated tau can seed filament formation of full length tau (Gamblin et al., 2003; Rissman et al., 2004). For example, virally overexpressing Asp421 tau in wild type mice results in formation of tau aggregates which contain both Asp421 truncated and full length tau (de Calignon et al., 2010).
As illustrated in Figure 7, tau is a substrate for the apoptotic caspase proteases and caspase truncated tau is present in human tauopathies. Additionally, caspase cleaved can induce cellular dysfunction. Apoptosis and caspase activation occurs following TBI; therefore, it is possible that TBI-induced caspase activation can initiate formation of caspase cleaved tau, whereas long-term generation of caspase truncated tau could be the result of chronic caspase activation due to chronic mitochondrial dysfunction or ER stress, organelles suspected of having self-propagating, cyclic interactions with pathologic tau. In fact, in P301L mice, activated executioner caspases can be found in neurons containing late stage neurofibrillary tangles, despite the absence of apoptosis (Spires-Jones et al., 2008). Furthermore, because caspase truncated tau is capable of activating caspase-3, impairing mitochondrial function, and enhancing ER stress, it is possible that caspase truncated tau itself can self-propagate its formation.
Figure 7. Mechanisms in which caspases play a central role in the formation of pathologic tau.
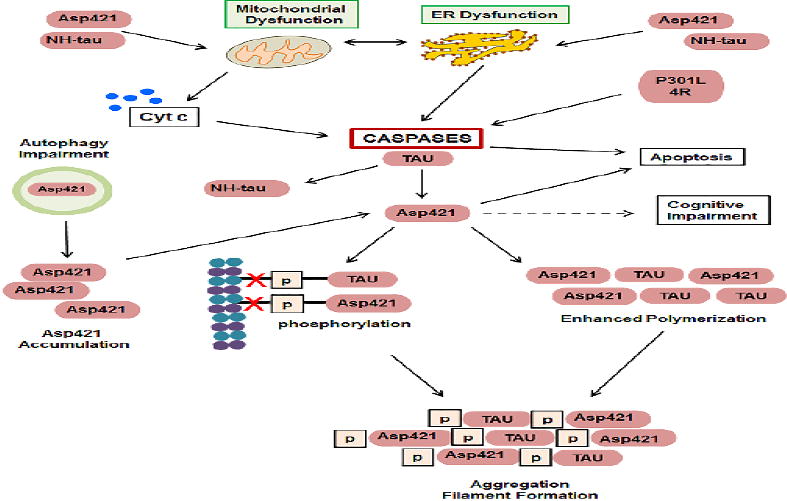
Following TBI, caspases are activated downstream of ER dysfunction and mitochondrial release of cytochrome c. Overexpression of P301L tau or 4R also induce caspases activation. Caspases, including caspase-3, cleave tau at Asp421, but N-terminal caspase-cleavage also occurs (NH-tau). Caspases-cleaved tau can further exacerbate mitochondrial and ER dysfunction. Asp421 is preferentially degraded by autophagy, therefore, impairment to autophagy results in accumulation of Asp421 tau. Asp 421 tau leads to enhanced tau polymerization and phosphorylation, resulting in decreased binding of tau to microtubules (red X) and increased aggregation and filament formation. Caspase activation and Asp421 expression lead to apoptosis. Asp421 precedes cognitive impairment.
4.8 Neuroinflammation
Neuroinflammation occurs in multiple tauopathies including CTE, AD, PSP, Pick's disease, and corticobasal degeneration (Daneshvar et al., 2015; Ishizawa and Dickson, 2001; Sasaki et al., 2008; Zilka et al., 2009a). Astrocytes and microglia make up two of the most important cells of the CNS innate immune system. Astrocytes containing pathologic tau have been identified in several tauopathies, including CTE (Kahlson and Colodner, 2015; McKee et al., 2016). Similarly, microglia are activated in CTE and other tauopathies (Cherry et al., 2016; Faden and Loane, 2015; Ishizawa and Dickson, 2001; Saing et al., 2012; Sasaki et al., 2008; Zilka et al., 2009a). Evidence suggests that neuroinflammation can both induce tau pathology and be induced by pathologic tau species.
Under physiologic conditions astrocytes contain limited amounts of tau protein, and tau is not considered to be a major component of the astrocyte cytoskeleton (Kahlson and Colodner, 2015). However, in CTE and other tauopathies, hyperphosphorylated tau is present in astrocytes (Kahlson and Colodner, 2015; McKee et al., 2016). Although astrocytic tau pathology is not well defined in CTE, supporting neuropathological criteria for the diagnosis of CTE includes p-tau positive thorned astrocytes located at the glial limitans in the subpial and periventricular regions (McKee et al., 2016). Additionally, the astrocytic tau of CTE is known to be composed mainly of the 4R subtype (McKee and Robinson, 2014).
Although the phenotypes of the astrocytes containing hyperphosphorylated tau vary between tauopathies, thorned astrocytes, such as those characteristic of CTE, morphologically resemble reactive astrocytes and therefore may be indicative of a neuroinflammatory response similar to gliosis (Kahlson and Colodner, 2015), a process known to occur following TBI (Burda et al., 2016). The presence of hyperphosphorylated tau within astrocytes is hypothesized to have several consequences for astrocytic function (Kahlson and Colodner, 2015). Astrocytes assist in maintenance of the blood-brain barrier; therefore, it is possible that astrocytic tau pathology can lead to blood-brain barrier dysfunction (Kahlson and Colodner, 2015). For example, overexpressing human tau in the astrocytes of mice results in mild blood-brain barrier disruption in areas of extensive astrocytic tau pathology (Forman et al., 2005). In addition to maintenance of the blood-brain barrier, astrocytes remove excessive extracellular glutamate; therefore, it is possible that astrocytic tau pathology can lead to impairment of extracellular glutamate removal, resulting in neuronal excitotoxicity and synaptic dysfunction (Kahlson and Colodner, 2015). In fact, overexpressing either human wild-type or mutant P301L tau in the astrocytes of mice results in a decrease in astrocyte glutamate transporter expression and activity which correlates with behavioral impairment (Dabir et al., 2006). Furthermore, overexpressing tau in astrocytes leads to astrocyte degeneration both in-vitro and in-vivo (Higuchi et al., 2002; Yoshiyama et al., 2003), and overexpressing human tau in the astrocytes of mice results in neuronal degeneration in regions associated with astrocytic tau pathology (Forman et al., 2005), indicating astrocytic tau pathology may play a direct role in both astrocytic and neuronal cell death (Kahlson and Colodner, 2015).
Although several tauopathies contain astrocytic tau pathology, evidence suggests that astrocytes do not phagocytise pathologic tau species. In the tauopathies, AD and FTD, tau oligomers are found to be surrounded by active astrocytes, but are not co-localized with astrocytes, suggesting the occurrence of an inflammatory response, but that astrocytes do not internalize tau oligomers (Nilson et al., 2016). Conversely, in AD, FTD, and transgenic mice overexpressing human tau (hTau), tau oligomers are found co-localized with active microglia, suggesting active phagocytosis of tau oligomers by microglia (Nilson et al., 2016). In fact, microglia themselves have been implicated in the anatomic spread of tau pathology (Nilson et al., 2016). As shown in Figure 8, microglia are capable of both phagocytosis and exocytosis; therefore, microglial-dependent propagation of tau through phagocytosis and exocytosis may explain non-synaptic spread of tau throughout the brain (Asai et al., 2015). The depletion of microglia has been shown to reduce the propagation of tau in-vivo, both in an adenovirus model of rapid tau propagation and in transgenic mice overexpressing human mutant tau, P301S (Asai et al., 2015). Furthermore, transplanting microglia obtained from the brains of transgenic mice overexpressing human tau (hTau) and null for the microglial receptor, CX3R1, to the brains of wild-type mice results in increases in phosphorylated tau within the wild-type brains (Maphis et al., 2015b).
Figure 8. Proposed role for microglia in the formation of pathologic tau.
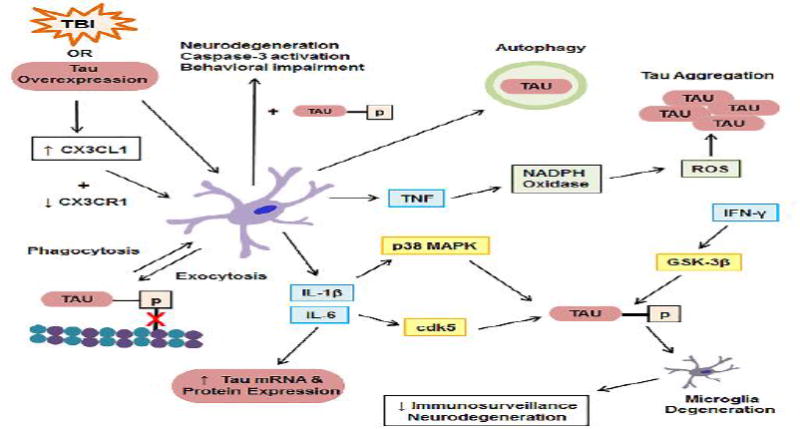
TBI and experimental tau overexpression activate microglia. Microglia can be activated through decreases in the microglia receptor, CX3CR1, and increases in the neuronally-derived ligand, CX3CL1, which is elevated following TBI and experimental tau overexpression. Microglial secretion of IL-6 increases tau mRNA and protein expression. Microglial secretion of IL-1β and IL-6 increases tau kinase activity (p38 MAPK and cdk5), as does the presence of the cytokine ifn-γ (GSK-3β), resulting in tau hyperphosphorylation, which decreases binding of tau to microtubules (red X), and microglial phagocytosis of hyperphosphorylated tau. Microglia spread tau in a prion-like manner through phagocytosis and exocytosis. Hyperphosphorylated tau induces microglia degeneration, resulting in decreased immunosurveillance and increased neurodegeneration. The microglia-derived cytokine, TNF, activates NADPH Oxidase, increasing ROS and formation of tau aggregates. The presence of phosphorylated tau enhances the effect of activated microglia on neurodegeneration, caspase-3 activation, and behavioral impairment. Activated microglia induce autophagy, resulting in autophagic degradation of tau.
The neuronal-microglial chemokine-chemokine receptor signaling pathway, CX3CL1-CX3CR1 has been implicated in both neurodegeneration and neuroprotection following CNS injury (Bhaskar et al., 2010). CX3CL1-CX3CR1 signaling is known to be altered following TBI (Poniatowski et al., 2016), and may play a role in the development of tau pathology. In fact, tau overexpression is capable of increasing CX3CL1 levels, whereas CXCR1 depletion is capable of enhancing microglial activation and tau pathology. Transgenic mice overexpressing human tau (hTau) show increased levels of the neuronally-derived ligand, CX3CL1 (Bhaskar et al., 2010). Knocking out the microglial receptor, CXC3R1, in hTau mice results in increases in microglial activation which are correlated with cognitive impairment (Bhaskar et al., 2010; Maphis et al., 2015b) In fact, hTau/CX3R1-/- mice show earlier tau pathology than hTau/CX3R1+/+ mice (Maphis et al., 2015b).
Alteration in the CX3CLI-CX3CR1 signaling pathway has been shown to enhance tau phosphorylation upon systemic administration of lipopolysaccharide (LPS), a compound capable of activating microglia through the TLR4 receptor (Bhaskar et al., 2010). LPS-induced activation of microglia increases tau phosphorylation in wild-type and hTau mice (Bhaskar et al., 2010). However, the effect is enhanced in CX3R1-/- mice, further enhanced in hTau/CX3R1-/- mice, and is dependent on neuronal activation of the tau kinase, p38 MAPK, and release of the microglial derived cytokine, IL-1β (Bhaskar et al., 2010).
In hTau/CX3R1-/- mice, microglial activation occurs prior to the appearance of tau pathology and correlates with its anatomic spread (Maphis et al., 2015b), suggesting that microglia activation enhances formation of pathologic tau. Additionally, studies suggest that microglial-induced neural toxicity is dependent on the presence of tau. Culturing neurons with LPS-activated CX3R1-/- microglia results in increased neurodegeneration; however this effect is attenuated in tau-/- neurons (Maphis et al., 2015a). Similarly, LPS activation of microglia in CX3R1-/- mice results in caspase-3 activation which is localized to p-tau positive neurons of the dentate gyrus, an effect which is attenuated in tau-/- mice (Maphis et al., 2015a). In non LPS-stimulated CX3R1-/- mice, a reduction in microglial activation and neuronal caspase-3 is also seen in tau-/- mice compared to tau+/+ mice (Maphis et al., 2015a), an effect which correlates with attenuation of behavioral impairment (Maphis et al., 2015a).
In addition to the CX3CLI-CX3CR1 signaling studies, several additional lines of evidence suggest that activation of microglia, as well as secretion of pro-inflammatory cytokines contribute to tau pathology. In fact, in the tauopathy AD, microglial expression of IL-1β is correlated with neuronal tau hyperphosphorylation (Sheng et al., 2001). In-vitro, activation of microglia with LPS results in microglial secretion of IL-β, neuronal activation of the tau kinase, p38 MAPK, increases in neuronal tau phosphorylation, and decreases in the synaptic protein, synaptophysin (Li et al., 2003). In primary hippocampal neurons, the pro-inflammatory cytokine IL-6 has been shown to increase tau phosphorylation, an effect which is attenuated by inhibiting the tau kinase, cdk5 (Quintanilla et al., 2004). Additionally, exposing SY5Y or N-tera2 cells to media conditioned by LPS and IFNγ-activated human microglia results in increased production and release of tau (Lee et al., 2015). In fact, direct treatment of SY5Y or N-tera2 cells with the inflammatory cytokines, IL-1β or IL-6 was sufficient to increase levels of tau mRNA and protein expression (Lee et al., 2015).
In-vivo, systemic administration of the microglial activator, LPS, results in increased tau phosphorylation acutely in the hippocampus of mice though activators of cdk5 and GSK-3β activation (Roe et al., 2011). In regards to cytokine expression, implantation of IL-1β beads into rat cerebral cortex, in simulation of chronic IL-1β exposure, results in increased mRNA expression of tau and p38 MAPK and increased tau phosphorylation (Sheng et al., 2001; Sheng et al., 2000). Similarly, adenovirus expression of the cytokine IFN-γ, a master regulator of inflammation, in two different mouse models of P301L tau overexpression results in increased phosphorylation of soluble tau through activation of the tau kinase, GSK-3β, however, INF-γ induction of tau phosphorylation did not result in increased tangle pathology or behavioral deficits (Li et al., 2015).
Although activation of microglia has been shown to increase tau phosphorylation, the ability of activated microglia to induce tau aggregation or tangle formation is less clear. For example, in rTg4510 mice overexpressing human mutant tau P301L, injection of LPS directly into the anterior cortex and hippocampus, results in ipsilateral and contralateral microglia activation and increases in tau phosphorylation one week following injection; however, pre-tangle and mature tangle formation was not increased (Lee et al., 2010). Interestingly, in this model activated microglia also did not co-localize with p-tau positive neurons (Lee et al., 2010). Conversely, in-vitro, activated microglia are capable of inducing tau aggregation through a mechanism involving production of ROS and the cytokine, TNF. Co-culturing primary hippocampal neurons with microglia pre-activated with LPS and IFN-γ results in aggregation of tau in neurites as determined by FRET imaging, whereas tau aggregation is not seen in neurons cultured in the absence of activated microglia (Gorlovoy et al., 2009). Similarly, direct administration of the cytokine, TNF, to neuronal cultures results in formation of tau aggregates and increases in neuronal ROS production (Gorlovoy et al., 2009). In fact, TNF-induced tau aggregation is attenuated by the scavenging of ROS, suggesting that microglial-induced tau aggregation may be due to microglial secretion of TNF and activation of ROS generating neuronal NADPH oxidase (Gorlovoy et al., 2009).
In addition to activated microglia being shown to induce tau pathology, tau has been shown to activate microglia. In-vitro, transfection of microglia with Tau40, the longest isoform of human tau, results in increases in microglia activation, migration, phagocytosis, and cytokine secretion (Wang et al., 2013b). Additionally, in pure microglial cultures, Tau40 induces accumulation of endogenous phosphorylated tau at the cell membrane, enhances secretion of non-phosphorylated tau, and alters expression of tau phosphatases and kinases (Wang et al., 2013b). In-vivo, overexpression of human mutant tau, P301L, results in increased expression of chemokines and innate immune system components (Li et al., 2015) and activated microglia accumulate near p-tau positive neurons (Sasaki et al., 2008). Similarly, in transgenic rats expressing truncated human tau, reactive microglia are associated with neurofibrillary tangles and axonal degeneration (Zilka et al., 2009b).
Upon further examination, many of the transgenic tau animal models suggest a cyclic relationship between microglial activation and formation of tau pathology. For example, the overexpression of human mutant tau, P301S, in mice is sufficient to increase microglial activation; however, microglial activation, synapse loss and impaired synaptic function all precede filamentous tau formation (Yoshiyama et al., 2007).
It is currently unclear whether neuroinflammation in CTE and other tauopathies is detrimental or protective. In P301S mice, the immunosuppressant, FK506, is able to attenuate neuroinflammation, neurodegeneration and tau pathology (Yoshiyama et al., 2007). Similarly, minocycline, a tetracycline class antibiotic with neuroinflammatory properties, is able to reduce astrocyte activation, the production of inflammatory cytokines, and decrease tau phosphorylation in the cortex of transgenic mice overexpressing human tau (hTau) (Garwood et al., 2010). Importantly, both FK506 and minocycline have been shown to be neuroprotective in TBI (Miyauchi et al., 2013; Singleton et al., 2001; Vink and Nimmo, 2009).
An additional pathway implicated in inflammation, the cholinergic pathway, is known to be impaired in TBI and neurodegenerative disease. In TBI there are deficits in cholinergic transmission and chronic loss of cholinergic neurons (Shin and Dixon, 2015), and recent evidence has demonstrated the occurrence of tau pathology in the cholinergic neurons of the nucleus basalis of Meynert in CTE brains (Mufson et al., 2016). The neurotransmitter, acetylcholine (Ach), is purported to have anti-inflammatory properties and the cholinergic pathway has been implicated in regulation of the immune system (Yoshiyama et al., 2012). In fact, chronic administration of a centrally acting anti-cholinergic to mice overexpressing human mutant tau, P301S, results in increases in tau phosphorylation and insolubility, activation of tau kinases, neurodegeneration, and microglial activation (Yoshiyama et al., 2012). Furthermore, anti-cholinergic administration enhances the effects of LPS-induced microglial activation in P301S mice (Yoshiyama et al., 2012).
Therefore, if neuroinflammation is a detrimental process, acetylcholine esterase inhibitors (AChEI), which have long been used for symptomatic treatment of cognitive impairment in neurodegenerative tauopathies but are also purported to have anti-inflammatory properties due to their ability to upregulate cholinergic pathways (Yoshiyama et al., 2010), should be neuroprotective in animal models of tauopathy. If fact, in transgenic mice overexpressing human mutant tau, P301S, chronic treatment with the AChEI, donepezil, results in decreased astrocyte and microglial activation, decreased tau phosphorylation and insolubility, and decreased synaptic and neuronal loss (Yoshiyama et al., 2010). Furthermore, in P301S mice, donepezil is able to attenuate LPS-induced microglial activation and reduce brain levels of IL-1β and COX-2, suggesting that the neuroprotective effects of AChEI are due to attenuation of neuroinflammation. (Yoshiyama et al., 2010).
Although inhibition of neuroinflammation has been shown to be protective in some tauopathy models, neuroinflammation may also serve a protective role against the development of tau pathology and cognitive dysfunction through promotion of cellular and debris clearance mechanisms. For example, in transgenic mice overexpressing human mutant tau, P301S, LPS-induced activation of microglial also been shown to decrease phosphorylated tau and improve cognitive function through the promotion of authophagy (Qin et al., 2016).
Microglia themselves are also responsible for the clearance of damaged neurons, aberrant synapses and demyelinated axons (Sanchez-Mejias et al., 2016). Therefore, degeneration of microglia leads to neuronal degeneration in areas absent immune surveillance (Sanchez-Mejias et al., 2016). In the temporal cortex of the tauopathy, AD, dystrophic (degenerating, fragmented, senescent, etc.) microglia are co-localized with degenerating neurons positive for tau (Streit et al., 2009). Similarly, degenerating microglia are found in the hippocampus of AD, a region in which phosphorylated tau preferentially accumulates over Aβ plaques (Sanchez-Mejias et al., 2016). Although the appearance of dystrophic microglia, a phenomenon known to increase with aging, precedes the spreading of tau pathology in AD, suggesting that loss of microglial neuroprotection as one ages can lead to neurodegeneration (Streit et al., 2009), additional evidence suggests that soluble phosphorylated tau is capable of inducing microglial degeneration. For example, in-vitro, both phagocytosis of extracellular soluble p-tau and phagocytosis of apoptotic SY5Y cells containing intracellular soluble p-tau induces microglial toxicity, an effect which can be attenuated by immuno-depletion of p-tau (Sanchez-Mejias et al., 2016).
In summary, CTE and TBI share common neuroinflammatory processes such as astrogliosis and activation of microglia. In fact, chronic neuroinflammation occurs in tauopathies and TBI. In CTE, tau positive astrocytes have been hypothesized to have several detrimental consequences, including blood-brain barrier dysfunction, extracellular glutamate dysregulation, and induction of neurodegeneration. In addition to astrocytes, the activation of microglia and the release of inflammatory cytokines have been implicated in induction of tau pathology and neurodegeneration. However, pathologic tau, itself, is capable of inducing both activation and degeneration of microglia, with degeneration of microglia capable of inducing neurodegeneration. Therefore, it is possible that following an initial insult capable of activating microglia and inducing pathologic tau formation, such as TBI, that microglia and tau interact to induce a feedforward cycle of neuroinflammation, pathologic tau formation, and neurodegeneration (Figure 8). In fact, chronic neuroinflammation is found both in tauopathies and decades following TBI.
5. Tau - A Biomarker for TBI
Stretching, shearing and degeneration of axons following TBI causes tau to be released from microtubules, increasing the levels of tau in biofluids. For this reason, there is growing interest in using cerebral spinal fluid (CSF), plasma, or serum tau as a biomarker for TBI, including mild TBI and sports-related head injury (Kulbe and Geddes, 2015). Furthermore, CSF tau already holds diagnostic potential in the tauopathy, AD (Blennow et al., 2010).
Increased levels of serum tau have been found in various models of animal TBI. For example, on the severe end of the spectrum, the cleaved form of tau (c-tau) is acutely elevated 6h post-injury in the serum of rats receiving a severe CCI. However, tissue levels of c-tau remained elevated beyond 6h and were significantly increased for at least 7 days post-injury, both in the hippocampus and the cortex (Gabbita et al., 2005).
More diffuse models of injury, such as mild blast TBI, mild rotational TBI and repetitive closed head injury have shown post-injury increases in serum tau to be sustained for longer periods of time. Following a single, low-intensity blast TBI in mice, serum tau is significantly elevated two hours and one day following injury (Ahmed et al., 2015). However, non-significant elevations in serum tau were sustained for at least one month following injury (Ahmed et al., 2015); a time course which parallels findings in mice receiving a mild blast-injury in which phosphorylated and cleaved forms of neuronal tau were increased 24h and one month post-injury (Huber et al., 2013). Therefore, increases in serum tau following blast TBI are likely to be representative of pathophysiological changes to tau in the brain tissue. Additionally, in rats receiving a mild TBI produced by rotational acceleration forces, serum tau is significantly elevated for at least 14d post-injury (Rostami et al., 2012), and in mice receiving repetitive closed head injuries, serum levels of total tau and phosphorylated tau are significantly elevated one day to one month post-injury, findings which again parallel increases in cortical phosphorylated tau (Yang et al., 2015).
In human TBI, increases in serum tau correlate with injury severity (Wang et al., 2016) and peak two days post-injury (Wang et al., 2016). However, in severe TBI, levels of CSF tau continue to rise beyond two days, peaking between five and 15 days post-injury (Franz et al., 2003). In addition to full-length tau, cleaved tau isoforms have also been found in CSF following severe TBI (Zemlan et al., 2002; Zemlan et al., 1999). Importantly, several studies show that following severe TBI, levels of tau in the serum, cerebrospinal fluid, and brain extracellular fluid correlate with outcomes up to one year post-injury (Liliang et al., 2010; Magnoni et al., 2012; Ost et al., 2006; Wang et al., 2016; Zemlan et al., 2002), suggesting that in severe TBI, acute increases in peripheral tau may be related to pathologic processes that contribute to mortality and morbidity.
The ability of tau to serve as a biomarker for mild TBI in human patients is less clear. Although serum tau is known to be elevated following pediatric mild TBI (Guzel et al., 2010), the clinical utility of serum tau as a biomarker for mild TBI has been questioned. For example, serum tau is unable to distinguish between mild TBI with or without intracranial lesion (Guzel et al., 2010; Kavalci et al., 2007), and serum levels of c-tau are unable to predict the development of post-concussive symptoms following mild TBI (Bazarian et al., 2006; Ma et al., 2008).
Despite the less than promising results in regard to the value of peripheral tau as a biomarker for mild TBI, various tau isoforms have been found to be elevated in the plasma, serum, or CSF of groups at risk for sustaining multiple head injuries, such as athletes and military personnel. In fact, in male athletes, participation in collision sports is associated with elevated levels of plasma tau (Di Battista et al., 2016). In boxing, total tau levels are increased in the CSF for up to ten days and in the plasma for up to six days following a match, with significantly increased levels being seen in the CSF of players sustaining more hits to the head (Neselius et al., 2012; Neselius et al., 2013; Zetterberg et al., 2006). In hockey, total tau is elevated in the serum of concussed players for up to six days following injury and correlates with duration of post-concussive symptoms (Shahim et al., 2014). Similarly, caspase cleaved tau (c-tau), a fragment itself capable of perpetuating tau pathology, is elevated in the serum of hockey players post-concussion, and the tau fragment, Tau-A, correlates with the number of days it takes concussed players to return to play (Shahim et al., 2016). Tau-A is generated by cleavage of tau by ADAM-10 (Shahim et al., 2016), a metalloproteinase which has been shown to be elevated in animal models of TBI (Warren et al., 2012).
In addition to acute and sub-chronic time points, increases in peripheral tau have been shown to be sustained chronically in both athletes and military personnel. Compared to controls, the serum of symptomatic retired NFL players contains a significantly increased number of tau positive exosomes, which correlate with cognitive impairment (Stern et al., 2016). Exosomes are stable cell exocytosed nanovesicles, the content of which is representative of the cell of origin (Stern et al., 2016). Furthermore, plasma levels of total tau are significantly increased in military personnel sustaining three or more TBIs and levels correlate with post-concussive symptoms, despite the majority of participants having sustained TBIs at least 18 months prior (Olivera et al., 2015).
Therefore, these studies suggest that in addition to severe TBI, concussions, sub-concussive hits, and repetitive mild head injuries are sufficient enough to cause acute dissociations of tau from the microtubules, and that repeat injuries can lead to sustained increases in peripheral tau. Furthermore, the correlation that tau positive exosomes and sustained plasma tau levels have with chronic symptoms suggests that increases in peripheral tau may indeed be representative of brain pathology.
6. TBI, Tau, and CTE - The Animal Models
6.1 Experimental TBI-induced Tau pathology
Animal models have been used extensively to model TBI and are able to recapitulate several aspects of human TBI neuropathology and behavioral impairment (Xiong et al., 2013). Currently, animal models are being used to elucidate the effects of single or repetitive TBI on acute and chronic tau pathology. Ojo et al. (Ojo et al., 2016) has extensively covered methodologies, pathologies, and the benefits and challenges of using animals to study repetitive TBI and CTE. A multitude of factors are purported to influence CTE development including injury type, injury severity, impact number, impact frequency, age at injury, time elapsed since injury, genetics, gender, and environment (Ojo et al., 2016). Therefore, examining the relationship between TBI and CTE in animals is challenging; a difficulty which is evidenced throughout the literature. For example, while some animal models of repetitive TBI induce significant increases in tau phosphorylation, others have either found no significant tau pathology or tau pathology in only single animals (Ojo et al., 2016). Furthermore, despite increases in phosphorylated and total tau following injury, most studies fail to recapitulate other pathologic characteristic of CTE such as tangle formation, glial tau, perivascular tau deposition, and deposition of tau at the depths of sulci, a characteristic which would be practically impossible to model in the lissencephalic rodent brain (Ojo et al., 2016).
The anatomic and physiologic differences between humans and rodents (mice being the most common animal used in studies of tau) further complicate matters. Anatomically, human brains contain higher ratios of white to grey matter, higher ratios of glia to neurons, sulci, gyri, longer myelinated axons, and a denser vascular supply (Ojo et al., 2016). The ratio of 3R to 4R tau also differs in rodents and in humans. Healthy adult brains express equal ratios of 3R and 4R tau (Goedert and Jakes, 1990; Ojo et al., 2016), whereas rats and mice express 4R tau, an isoform which is preferentially phosphorylated and prone to aggregation, to a much higher degree than 3R tau (Hanes et al., 2009; McMillan et al., 2008; Ojo et al., 2016). Additionally, animal models of CTE often depend on the use of transgenic mice. For example, many of the transgenic mice used to study pathologic tau utilize FTD-17 mutations, such as P301L, which preferentially favor 4R splicing (Ojo et al., 2013; Ojo et al., 2016). Transgenic tau mice also overexpress tau compared to wild type mice, and are predisposed to the development of tau pathology (Ojo et al., 2016). In fact, in some CTE studies, transgenic tau mice are aged and already display tau pathology prior to injury (Ojo et al., 2013; Ojo et al., 2016).
Despite the difficulty in using animals to investigate the relationship between TBI, tau and CTE, important observations have been made. Several pathologic tau isoforms have been observed in experimental animal models of TBI, including increases in total tau, phosphorylated tau, cleaved tau, tau oligomers, tau aggregates and other pathologic tau confirmations. However, animal models of TBI are diverse, ranging from mild to severe, focal to diffuse, and single to repetitive, which can affect the time course and type of tau pathology seen following injury.
Several different models of TBI, including controlled cortical impact injury (CCI), fluid percussion injury (FPI), and blast injury have shown tau to be increased acutely following injury. CCI, a focal model of injury which includes aspects of contusion, hemorrhage and diffuse injury (Hall et al., 2005), results in severity-dependent increases in cleaved tau (c-tau), with severe injury inducing significant increases in c-tau beginning at 6h in the cortex and 48h in the hippocampus, increases which are sustained for at least seven days following injury and which are attenuated by the neuroprotective drug CsA (Gabbita et al., 2005). Similarly, following a moderate FPI, a mixed model of focal and diffuse injury (Thompson et al., 2005), oligomeric and phosphorylated tau are increased acutely: 4h, 24h and 2 weeks post-injury (Gerson et al., 2016; Hawkins et al., 2013). Mild blast TBI, which encompasses both the blast wave and rotational acceleration-deceleration forces and results in diffuse injury, transient axonal injury, and vascular pathology (Courtney and Courtney, 2015; Kovacs et al., 2014), has also been shown to increase tau acutely following injury. For example, mild blast TBI increases phosphorylated tau in rat hippocampus 6h post-injury (Perez-Polo et al., 2015), and a repetitive blast injury consisting of three closely spaced blasts results in total tau levels being increased 24h post-injury in mouse cerebellum (Arun et al., 2013).
Chronic tau pathology has also been observed following blast injury. Tau phosphorylation and pathologic tau conformations are increased in rat hippocampus and cortex one month following either a single blast or six blasts spaced 48h apart (Turner et al., 2015). Furthermore, rats exposed to repeat injury showed evidence of perivascular tau pathology, a defining feature of human CTE (Turner et al., 2015). Seventy days following a single blast injury in rats, total tau protein is also increased in several brain regions including the amygdala, prefrontal cortex and hippocampus; an affect which can be attenuated by environmental enrichment (Kovesdi et al., 2011). Tau oligomers have also been observed to develop one month following a single blast TBI (Gerson et al., 2016). In fact, tau oligomers extracted from rat brains one month following a single blast TBI have prion-like characteristics, increasing the spread of tau pathology and accelerating the rate of cognitive decline when injected into brains of transgenic mice overexpressing human tau (Gerson et al., 2016). Tau aggregation also occurs following blast TBI, although in a delayed manner. For example, three months following a single blast injury, rats develop tau aggregates in the prefrontal cortex and hippocampus that were not yet present one-month following injury (Sajja et al., 2015).
Chronic tau pathology has also been detected following closed head injury, another animal model of diffuse TBI. Similar to blast injury, increases in tau phosphorylation and pathologic tau conformations have been found one month following a single closed head injury (Turner et al., 2015). However, other studies have shown a requirement for repeat injuries in order to induce tau pathology following closed head injury. For example, in aged, transgenic mice overexpressing human tau and who already showed signs of tau pathology prior to injury, repeat injury (five injuries space 48h apart) but not single injury, resulted in increases in cortical and hippocampal tau phosphorylation three weeks post-injury (Ojo et al., 2013); however, other characteristic CTE pathologies, such as perivascular tau, neuritic threads and astrocytic tangles, were not seen (Ojo et al., 2013). Similar increases in tau phosphorylation following repetitive closed head injury occur in wild-type mice. For example, exposing mice to four closed head injuries spaced three days apart results in increased tau phosphorylation in the hippocampus and cortex, effects which are sustained for at least seven days and 30 days, respectively (Yang et al., 2015). Additionally, chronic increases in tau phosphorylation following repetitive closed head injury have been observed to occur beyond 30 days. For example, exposing mice to three closed head injuries spaced 24h apart, results in increases in tau phosphorylation in the corpus callosum, cortex, hippocampus and amygdala six months following injury (Luo et al., 2014). Furthermore, evidence suggests that the presence of tau following repetitive closed head injury is linked to the development of pathology following injury. In fact, in a two-hit frontal impact model of closed head injury, knocking out tau attenuates chronic axonal degeneration and learning and memory deficits (Cheng et al., 2014).
Newer models of closed head injury, which also incorporate rotational acceleration-deceleration forces in an attempt to more closely model human concussion, also have demonstrated increases in tau pathology following injury. In a model termed CHIMERA, which stands for Closed-Head Impact Model of Engineered Rotational Acceleration, an impact device is used to deliver a closed head injury to mice while allowing for free rotation of the head upon impact (Namjoshi et al., 2014). In CHIMERA, two impacts spaced 24h apart results in increases in tau phosphorylation over the first 48h following injury (Namjoshi et al., 2014). Chronic increases in tau phosphorylation have also been seen in similar models. For example, in another combined model of closed head injury and rotational acceleration, exposing mice to five injuries spaced 24h apart results in increases in tau phosphorylation for at least 30 days following injury (Kane et al., 2012). Similarly, in a closed head injury model which allows for head mobility and the use of non-anaesthetized mice, tau phosphorylation is increased in the cortex, hippocampus, amygdala and dentate gyrus seven days and one month following single or repetitive (42 injuries) injury (Petraglia et al., 2014). Furthermore, increases in tau phosphorylation were sustained out to six months in repetitive injury mice (Petraglia et al., 2014).
Although less studied in isolation, evidence suggests that rotational acceleration-deceleration forces themselves are capable of inducing tau pathology. In fact, head restraint during blast injury is able to attenuate increases in tau phosphorylation, neuroinflammation, microvascular pathology, neurodegeneration, and learning and memory impairment (Goldstein et al., 2012). In swine models of rotational head injury, PHF-tau is reported to accumulate in neuronal perikarya following injury (Smith et al., 1999). Similarly, a model using air pressure to induce a rotational head injury in mice absent a blast wave, results in increases in tau phosphorylation two and eight weeks post-injury; however, similar to other models, additional aspects of tau pathology such as tau aggregation were not seen (Sabbagh et al., 2016).
6.1.1 Select Mechanisms of Experimental TBI-induced Tau Pathology
Multiple animal models of TBI, including diffuse injury, blast injury, closed head injury and rotational acceleration-deceleration injury, have all demonstrated increases in pathologic tau following injury. These models of injury also result in widespread damage to axons and white matter tracts, suggesting the possibility that diffuse axonal damage and pathologic tau formation are related. In fact, in transgenic mice expressing either human mutant tau, P301S, or the six isoforms of wild type human tau, a mild repetitive impact-acceleration injury induces dose-dependent advanced tau hyperphosphorylation and tangle formation ten weeks following injury (Xu et al., 2015). However, tau pathology only develops in regions of the brain associated with transient axonal injury; specifically, tau pathology develops in the retinal ganglion cells as this model induces transient axonal injury to the optic nerve (Xu et al., 2015). In addition to diffuse axonal injury being capable of inducing tau pathology, dendritic damage has also been implicated in increased tau phosphorylation. For example, in rats, epidural cortical compression rapidly results in mechanical disruption of the dendritic cytoskeleton, resulting in increases in phosphorylated tau, alterations in tau kinase and phosphatase activity levels, and dendritic remodeling, implying rapid changes to cortical circuitry that could have long lasting effects on brain function (Chen et al., 2010a).
Several experimental TBI studies have identified additional mechanisms which also may contribute to pathologic tau formation. Several studies have observed that increases in tau phosphorylation following injury are accompanied by alterations in phosphatase or kinase activity. In FPI, increases in tau phosphorylation twelve weeks post-injury are accompanied by decreases in expression and activity of the tau phosphatase, PP2A, neurodegeneration and cognitive impairment, effects which are attenuated by the administration of the PP2A activator, sodium selenite (Shultz et al., 2015). Similarly, in blast and weight-drop models of TBI, acute increases in tau phosphorylation coincide with decreases in activity of tissue non-specific alkaline phosphatase (TNAP), a phosphatase with greater activity towards phosphorylated tau than PP2A (Arun et al., 2015). Additionally, following CCI in mice, increases in tau phosphorylation, cognitive impairment and hippocampal degeneration are all attenuated by administration of lithium, an inhibitor of the tau kinase, GSK-3β (Yu et al., 2012).
In addition to alterations in tau phosphatase and kinase activity, oxidative stress also has been observed to coincide with tau pathology following experimental TBI. Exposing mice to a single blast results in increases in phosphorylated and cleaved tau 24h post-injury in the cortex, hippocampus, and cerebellum, increases which are sustained in the hippocampus 30 days post-injury, and which are accompanied acutely by increases in oxidative stress defense mechanisms in the hippocampus and cerebellum, namely increases in the mitochondrial anti-oxidant, SOD2 (Huber et al., 2013). Oxidative stress has also been seen to accompany tau pathology in repetitive models of TBI. In a repetitive blast injury model, exposing mice to six blasts spaced 48h apart results in increases in pathologic tau, apoptosis, and impulsivity, effects which are attenuated by administration of the antioxidant, lipoic acid (Lucke-Wold et al., 2015). Lipoic acid inhibits NOX4, an enzyme increased in CTE brains which is capable of inducing oxidative stress (Lucke-Wold et al., 2015). Additionally, in a repetitive mild closed head injury model, exposing mice to three impacts spaced 24h apart, results in increases in phosphorylated tau, inflammation, and synaptic and cognitive impairment 30 days post-injury, effects which are attenuated by inhibition of monoacylglycerol lipase (MAGL) (Zhang et al., 2015). MAGL metabolizes endocannabinoid 2-arachidonoylgylcerol (2-AG) to prostaglandins and arachidonic acid (Zhang et al., 2015). Therefore, inhibiting MAGL acts to increase 2-AG, a compound known for having anti-inflammatory and neuroprotective properties (Zhang et al., 2015), while also decreasing the 2-AG metabolite, arachidonic acid, which is itself capable of initiating LP (Bains and Hall, 2012; Hall et al., 2010).
Experimental TBI has also been shown to increase toxic forms of tau by impairing the function of the proline isomerase, Pin1. Pin1, converts tau phosphorylated at Thr231 from the cis isomer to the less toxic trans configuration (Kondo et al., 2015). Phosphorylation of Pin1 increases following experimental TBI, leading to decreased Pin1 activation and, therefore, increased levels of the cis isomer (Kondo et al., 2015). Pin1 can also be inhibited through cysteine oxidation (Chen et al., 2015), an event likely to occur under TBI-induced oxidative stress conditions. Formation of the cis isomer of phosphorylated tau (cis p-tau) is reported to be an early pathogenic event in the tauopathy, AD, (Nakamura et al., 2012), and increases in cis p-tau have been identified in CTE (Kondo et al., 2015), and in experimental models of severe TBI, repetitive mild TBI (rmTBI) and blast TBI (Kondo et al., 2015). In fact, six months following rmTBI cis p-tau which developed acutely at the site of injury has spread to additional brain regions (Kondo et al., 2015). Cis p-tau is suspected of being neurotoxic. In fact, in an experimental model of severe TBI, administration of a cis p-tau antibody prevents development and spread of cis p-tau, attenuates disruptions in microtubule and mitochondrial transport, and improves functional and behavioral outcomes (Kondo et al., 2015).
In addition to dysfunction of enzymes and oxidative stress pathways, dysfunctional tau clearance mechanisms have been observed following experimental TBI. A moderate-severe closed head injury in mice results in impairment to the glymphatic pathway for at least one month following injury and correlates with increases in tau phosphorylation (Iliff et al., 2014). The glymphatic pathway is a perivascular network responsible for CSF recirculation and clearance of interstitial proteins such as tau and Aβ (Iliff et al., 2014; Iliff et al., 2012). The glympathic pathway is highly dependent on the protein, perivascular glial aquaporin-4 (AQP4), which is also chronically impaired following experimental TBI (Iliff et al., 2014; Ren et al., 2013). In fact, knock-out of aquaporin-4 results in increases in tau phosphorylation, axonal degeneration, neuroinflammation, and behavioral deficits following experimental TBI suggesting that chronic glymphatic dysfunction following TBI may contribute to the development of neurodegenerative tauopathies through impairment of tau clearance (Iliff et al., 2014).
6.2 Experimental TBI without Tau Pathology
Although multiple animal models have demonstrated that pathologic tau is increased following acute TBI, pathologic tau can be sustained chronically following TBI, and several possible mechanisms that could lead to tau dysfunction following injury, there have been other animal models which have failed to demonstrate any tau pathology following injury. In fact, even the use of transgenic mice does not always ensure formation of tau pathology following experimental TBI. For example, in a repetitive mild closed head injury in which aged transgenic mice expressing the shortest form of human tau (T44) received a total of 16 injuries over the course of four weeks, only 1 in 9 mice developed neurofibrillary tangles and cerebral atrophy (Yoshiyama et al., 2005). However, except for the individual mouse displaying tau pathology, this model was also unable to induce any cognitive deficits six months post-injury (Yoshiyama et al., 2005).
Several additional examples demonstrate the difficulty of modeling CTE in rodents. In a mouse study that paralleled several clinical and pathological aspects of human TBI, a single mild closed head injury resulted in pathology and behavioral deficits that remained static over time, whereas repeat injury (five injuries spaced 48h apart) resulted in progressive neuroinflammation, white matter degradation, and behavioral impairment over 6-18 months following injury despite no increases in tau phosphorylation (Mouzon et al., 2014). Similarly, in a mouse model combining impact and rotational acceleration, repeating injuries on a daily or weekly basis resulted in increases in astrogliosis and cognitive impairment up to one year following injury, but no increases in tau phosphorylation (Mannix et al., 2013).
A lack of tau pathology following rmTBI has also been seen at more acute time points. For example, in a mouse model of repetitive mild closed head injury, five impacts spaced 24h or 48h apart results in motor and cognitive deficits, axonal degeneration and astrogliosis, particularly in the visual system, ten weeks following injury; however, increases in hyperphosphorylated tau were not seen (Bolton Hall et al., 2016). Similarly, although exposing mice to five injuries spaced 24h apart results in increased hemorrhagic lesions, diffuse axonal injury, neurodegeneration, and astrogliosis 24h post-injury compared to intervals spaced 48h apart, neither injury paradigm results in increased tau phosphorylation (Bolton and Saatman, 2014).
In summary, use of animal models to study CTE and the development of chronic tau pathology following experimental TBI is complicated by a multitude of factors including heterogeneity of human injury, heterogeneity of individuals, and anatomic and physiologic differences between humans and rodents. Although individual studies have failed to recapitulate all aspects of CTE pathology, numerous studies have confirmed the ability of experimental TBI to induce acute and chronic tau pathology, and the ability of repetitive injury to exacerbate that pathology. Furthermore, animal models have confirmed mechanisms suspected of contributing to tau pathology following injury such as tau kinase and phosphatase dysfunction and induction of oxidative stress pathways, and identified additional mechanisms that possibly contribute to tau dysfunction following injury such as Pin1 inhibition and dysfunction of the glympathic pathway.
7. Conclusions and Possible Neuroprotective Strategies to Prevent or Interrupt the Progression of CTE
Traumatic brain injury results in a secondary injury cascade that includes processes such as calpain and caspase activation, mitochondrial and ER dysfunction, increases in oxidative stress including generation of peroxynitrite which trigger, lipid peroxidation and LP-derived breakdown products, most notably the neurotoxic aldehydes 4-HNE and acrolein, and neuroinflammation. While these post-TBI biochemical and cellular pathophysiological events begin within the first minutes, days and hours post-TBI, there is evidence that some of these events may persist into the chronic injury phase exerting negative effects on recovery months and perhaps years after the single of repetitive TBI episodes and contributing to age-related neurodegenerative diseases. Similar processes occur in neurodegenerative tauopathies and many have been shown to occur upstream, downstream, and in a circular cascade with tau dysfunction (Figure 9). A history of TBI, particularly repetitive, is a risk factor for both CTE and AD; therefore, it is possible that several of the above mentioned pathologies, which occur acutely following TBI, lead to chronic neurodegeneration and tau dysfunction. However, the apparent complexities of the mechanistic link between acute post-traumatic secondary injury processes and their contribution to chronic outcomes is yet to be un-raveled. Nevertheless, since CTE has only been documented in individuals with histories of repetitive, and perhaps even single, TBIs, it is entirely possible that early initiation of neuroprotective treatments that will effectively inhibit the acute, subacute and early chronic secondary injury phases will lessen the development of the tauopathy that is the key neuropathology seen in CTE. While a single neuroprotective approach might be able to significantly attenuate the progressive brain damage in CTE, since a variety of cellular dysfunctions can lead to tau dysfunction, which can in turn lead to several cellular dysfunctions, it is likely that a multi-mechanistic neuroprotective combinational approach to CTE prevention and interruption will be needed.
Figure 9. Simplified schematic demonstrating proposed pathological mechanisms that can contribute to CTE development and pathologic mechanisms that can be exacerbated by CTE.
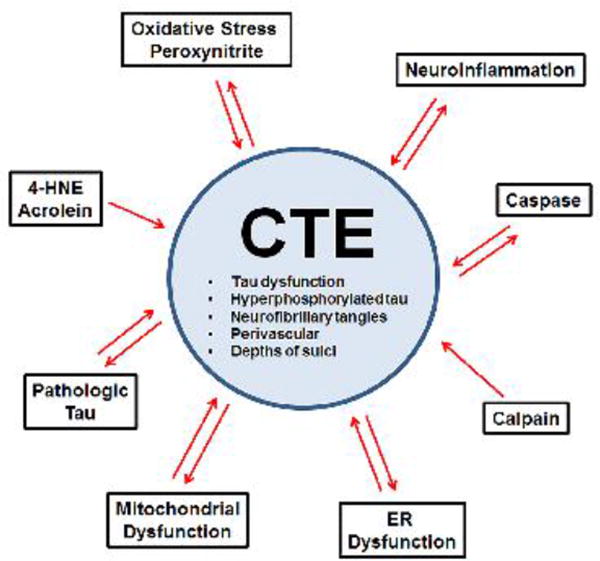
Examples of pathologic tau include hyperphosphorylation, nitration, oxidation, aggregation, filament formation, etc. For simplicity, relationships between pathologic mechanisms are not included in this figure, but are detailed elsewhere (Fig 1– 8).
Highlights.
Chronic traumatic encephalopathy (CTE) is a recently described neurodegenerative tauopathy that is a chronic sequela of traumatic brain injury (TBI).
CTE occurs predominantly after repeated episodes of mild TBI in athletes involved in contact sports or in military combatants exposed to single or repetitive explosive blast-injuries.
This review discusses the possible involvement of the well characterized post-TBI pathophysiological mechanisms in the pathogenesis of tau protein deposition that is distinctly characteristic of CTE.
The analysis of the possible interactions of TBI secondary injury with CTE tauopathy has suggested several neuroprotective approaches that might serve, either singly or in combination, to limit the development of CTE.
Acknowledgments
Some of the work cited in this review from the corresponding author's laboratory was supported by NINDS R01-NS046566; P01 NS058484; R01 NS083405 and R01 NS084847. Additionally, the first author is currently supported by NINDS F30 NS096876.
Definitions
- 2-AG
2-arachidonoylglycerol
- 3-NT
3-nitrotyrosine
- 4-HNE
4-hydroxynonenal
- Ach
acetylcholine
- AChEI
acetylcholine esterase inhibitor
- AD
Alzheimer's disease
- ADAM-10
A disintegrin and metalloproteinase domain-containing protein 10
- ANT
adenine nucleotide translocator
- APP
amyloid precursor protein
- AQP4
aquaporin-4
- CCI
controlled cortical impact injury
- CDK5
cyclin-dependent kinase 5
- CHIMERA
closed-head impact model of engineered rotational acceleration
- COX
cytochrome oxidase
- CsA
cyclosporine A
- c-tau
cleaved tau
- CTE
chronic traumatic encephalopathy
- CX3CL1
C-X3-C motif ligand 1 / fractalkine
- CX3CR1
CX3C chemokine receptor / fractalkine receptor
- DRP1
dynamin-related protein
- ERAD
ER-associated degradation
- ERK
extracellular-regulated kinase
- FPI
fluid percussion injury
- FRET
fluorescence resonance energy transfer
- FTD
frontotemporal dementia
- GSK-3β
glycogen synthase kinase-3β
- hTau
human tau
- IFN
interferon
- IL
interleukin
- JNK
c-Jun-terminal kinase
- LP
lipid peroxidation
- LPS
lipopolysaccharide
- MAGL
monoacylglycerol lipase
- MAP
microtubule-associated protein
- MAPK
mitogen-activated protein kinase
- MAPT
microtubule-associated protein tau
- mPTP
mitochondrial permeability transition pore
- mtNOS
mitochondrial nitric oxide synthase
- NADPH
nicotinamide adenine dinucleotide phosphate
- NFT
neurofibrillary tangle
- NOX4
NADPH oxidase 4 gene
- PHF
paired helical filament
- PIN1
peptidyl-prolyl cis-trans isomerase NIMA-interacting 1
- PN
peroxynitrite
- PP2A
protein phosphatase 2
- PSD-95
postsynaptic density 95
- PSP
progressive supranuclear palsy
- p-tau
phosphorylated tau
- rmTBI
repetitive mild TBI
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SOD2
mitochondrial superoxide dismutase 2, manganese-dependent superoxide dismutase
- TNAP
tissue non-specific alkaline phosphatase
- TBI
traumatic brain injury
- TDP-43
transactive response DNA-binding protein
- TLR4
toll-like receptor 4
- UPR
unfolded protein response
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abisambra JF, Jinwal UK, Blair LJ, O'Leary JC, 3rd, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, Cockman M, Suntharalingham A, Li P, Jin Y, Atkins CA, Dickey CA. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed F, Plantman S, Cernak I, Agoston DV. The Temporal Pattern of Changes in Serum Biomarker Levels Reveals Complex and Dynamically Changing Pathologies after Exposure to a Single Low-Intensity Blast in Mice. Frontiers in neurology. 2015;6:114. doi: 10.3389/fneur.2015.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavi Naini SM, Soussi-Yanicostas N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxidative medicine and cellular longevity. 2015;2015:151979. doi: 10.1155/2015/151979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- Alonso AD, Beharry C, Corbo CP, Cohen LS. Molecular mechanism of prion-like tau-induced neurodegeneration. Alzheimers Dement. 2016 doi: 10.1016/j.jalz.2015.12.014. [DOI] [PubMed] [Google Scholar]
- Amadoro G, Corsetti V, Florenzano F, Atlante A, Ciotti MT, Mongiardi MP, Bussani R, Nicolin V, Nori SL, Campanella M, Calissano P. AD-linked, toxic NH2 human tau affects the quality control of mitochondria in neurons. Neurobiol Dis. 2014;62:489–507. doi: 10.1016/j.nbd.2013.10.018. [DOI] [PubMed] [Google Scholar]
- Amadoro G, Corsetti V, Stringaro A, Colone M, D'Aguanno S, Meli G, Ciotti M, Sancesario G, Cattaneo A, Bussani R, Mercanti D, Calissano P. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. Journal of Alzheimer's disease: JAD. 2010;21:445–470. doi: 10.3233/JAD-2010-100120. [DOI] [PubMed] [Google Scholar]
- Arun P, Abu-Taleb R, Oguntayo S, Tanaka M, Wang Y, Valiyaveettil M, Long JB, Zhang Y, Nambiar MP. Distinct patterns of expression of traumatic brain injury biomarkers after blast exposure: role of compromised cell membrane integrity. Neuroscience letters. 2013;552:87–91. doi: 10.1016/j.neulet.2013.07.047. [DOI] [PubMed] [Google Scholar]
- Arun P, Oguntayo S, Albert SV, Gist I, Wang Y, Nambiar MP, Long JB. Acute decrease in alkaline phosphatase after brain injury: A potential mechanism for tauopathy. Neuroscience letters. 2015;609:152–158. doi: 10.1016/j.neulet.2015.10.036. [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlante A, Amadoro G, Bobba A, de Bari L, Corsetti V, Pappalardo G, Marra E, Calissano P, Passarella S. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochimica et biophysica acta. 2008;1777:1289–1300. doi: 10.1016/j.bbabio.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Bader N, Grune T. Protein oxidation and proteolysis. Biol Chem. 2006;387:1351–1355. doi: 10.1515/BC.2006.169. [DOI] [PubMed] [Google Scholar]
- Bains M, Cebak JE, Gilmer LK, Barnes CC, Thompson SN, Geddes JW, Hall ED. Pharmacological analysis of the cortical neuronal cytoskeletal protective efficacy of the calpain inhibitor SNJ-1945 in a mouse traumatic brain injury model. Journal of neurochemistry. 2013;125:125–132. doi: 10.1111/jnc.12118. [DOI] [PubMed] [Google Scholar]
- Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochimica et biophysica acta. 2012;1822:675–684. doi: 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloyannis SJ. Mitochondrial alterations in Alzheimer's disease. Journal of Alzheimer's disease: JAD. 2006;9:119–126. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, Zhao Q, Zhang XJ, Janesko-Feldman KL, Alexander H, Basova LV, Clark RS, Kochanek PM, Kagan VE. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- Bazarian JJ, Zemlan FP, Mookerjee S, Stigbrand T. Serum S-100B and cleaved-tau are poor predictors of long-term outcome after mild traumatic brain injury. Brain Inj. 2006;20:759–765. doi: 10.1080/02699050500488207. [DOI] [PubMed] [Google Scholar]
- Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:3743–3755. doi: 10.1523/JNEUROSCI.2872-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry RW, Abraha A, Lagalwar S, LaPointe N, Gamblin TC, Cryns VL, Binder LI. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry. 2003;42:8325–8331. doi: 10.1021/bi027348m. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- Bolton AN, Saatman KE. Regional neurodegeneration and gliosis are amplified by mild traumatic brain injury repeated at 24-hour intervals. J Neuropathol Exp Neurol. 2014;73:933–947. doi: 10.1097/NEN.0000000000000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton Hall AN, Joseph B, Brelsfoard JM, Saatman KE. Repeated Closed Head Injury in Mice Results in Sustained Motor and Memory Deficits and Chronic Cellular Changes. PLoS One. 2016;11:e0159442. doi: 10.1371/journal.pone.0159442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free radical biology & medicine. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- Bringold U, Ghafourifar P, Richter C. Peroxynitrite formed by mitochondrial NO synthase promotes mitochondrial Ca2+ release. Free radical biology & medicine. 2000;29:343–348. doi: 10.1016/s0891-5849(00)00318-x. [DOI] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Experimental neurology. 2016;275(Pt 3):305–315. doi: 10.1016/j.expneurol.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer's disease. Biochimica et biophysica acta. 2010;1801:924–929. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canu N, Dus L, Barbato C, Ciotti MT, Brancolini C, Rinaldi AM, Novak M, Cattaneo A, Bradbury A, Calissano P. Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1998;18:7061–7074. doi: 10.1523/JNEUROSCI.18-18-07061.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carron SF, Alwis DS, Rajan R. Traumatic Brain Injury and Neuronal Functionality Changes in Sensory Cortex. Front Syst Neurosci. 2016;10:47. doi: 10.3389/fnsys.2016.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani R, Smith MA, Richey PL, Kalaria R, Gambetti P, Perry G. Evidence for oxidative stress in Pick disease and corticobasal degeneration. Brain research. 1995;696:268–271. doi: 10.1016/0006-8993(95)00535-x. [DOI] [PubMed] [Google Scholar]
- Cebak JE, Singh IN, Hill RL, Wang J, Hall ED. PHENELZINE PROTECTS BRAIN MITOCHONDRIAL FUNCTION In vitro and In vivo FOLLOWING TRAUMATIC BRAIN INJURY BY SCAVENGING THE REACTIVE CARBONYLS 4-HYDROXYNONENAL AND ACROLEIN LEADING TO CORTICAL HISTOLOGICAL NEUROPROTECTION. Journal of neurotrauma. 2016 doi: 10.1089/neu.2016.4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cente M, Filipcik P, Pevalova M, Novak M. Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. The European journal of neuroscience. 2006;24:1085–1090. doi: 10.1111/j.1460-9568.2006.04986.x. [DOI] [PubMed] [Google Scholar]
- Chai X, Dage JL, Citron M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis. 2012;48:356–366. doi: 10.1016/j.nbd.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan NB. Chronic neurodegenerative consequences of traumatic brain injury. Restor Neurol Neurosci. 2014;32:337–365. doi: 10.3233/RNN-130354. [DOI] [PubMed] [Google Scholar]
- Chen CH, Li W, Sultana R, You MH, Kondo A, Shahpasand K, Kim BM, Luo ML, Nechama M, Lin YM, Yao Y, Lee TH, Zhou XZ, Swomley AM, Allan Butterfield D, Zhang Y, Lu KP. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer's disease. Neurobiol Dis. 2015;76:13–23. doi: 10.1016/j.nbd.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LJ, Wang YJ, Tseng GF. Compression alters kinase and phosphatase activity and tau and MAP2 phosphorylation transiently while inducing the fast adaptive dendritic remodeling of underlying cortical neurons. Journal of neurotrauma. 2010a;27:1657–1669. doi: 10.1089/neu.2010.1308. [DOI] [PubMed] [Google Scholar]
- Chen S, Townsend K, Goldberg TE, Davies P, Conejero-Goldberg C. MAPT isoforms: differential transcriptional profiles related to 3R and 4R splice variants. Journal of Alzheimer's disease: JAD. 2010b;22:1313–1329. doi: 10.3233/JAD-2010-101155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Hou Y, Mattson MP. Mitochondria and neuroplasticity. ASN neuro. 2010;2:e00045. doi: 10.1042/AN20100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JS, Craft R, Yu GQ, Ho K, Wang X, Mohan G, Mangnitsky S, Ponnusamy R, Mucke L. Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS One. 2014;9:e115765. doi: 10.1371/journal.pone.0115765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML, Stern RA, McKee AC, Stein TD. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. 2016;4:112. doi: 10.1186/s40478-016-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesser AS, Pritchard SM, Johnson GV. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Frontiers in neurology. 2013;4:122. doi: 10.3389/fneur.2013.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, Woo HN, Kwon YK, Kim HH, Gwag BJ, Mook-Jung IH, Jung YK. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis. 2001;8:162–172. doi: 10.1006/nbdi.2000.0335. [DOI] [PubMed] [Google Scholar]
- Collins-Praino LE, Corrigan F. Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun. 2016 doi: 10.1016/j.bbi.2016.09.027. [DOI] [PubMed] [Google Scholar]
- Conrad C, Zhu J, Conrad C, Schoenfeld D, Fang Z, Ingelsson M, Stamm S, Church G, Hyman BT. Single molecule profiling of tau gene expression in Alzheimer's disease. Journal of neurochemistry. 2007;103:1228–1236. doi: 10.1111/j.1471-4159.2007.04857.x. [DOI] [PubMed] [Google Scholar]
- Coughlin JM, Wang Y, Munro CA, Ma S, Yue C, Chen S, Airan R, Kim PK, Adams AV, Garcia C, Higgs C, Sair HI, Sawa A, Smith G, Lyketsos CG, Caffo B, Kassiou M, Guilarte TR, Pomper MG. Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiol Dis. 2015;74:58–65. doi: 10.1016/j.nbd.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney A, Courtney M. The Complexity of Biomechanics Causing Primary Blast-Induced Traumatic Brain Injury: A Review of Potential Mechanisms. Frontiers in neurology. 2015;6:221. doi: 10.3389/fneur.2015.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchillo-Ibanez I, Seereeram A, Byers HL, Leung KY, Ward MA, Anderton BH, Hanger DP. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:3186–3195. doi: 10.1096/fj.08-109181. [DOI] [PubMed] [Google Scholar]
- Dabir DV, Robinson MB, Swanson E, Zhang B, Trojanowski JQ, Lee VM, Forman MS. Impaired glutamate transport in a mouse model of tau pathology in astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:644–654. doi: 10.1523/JNEUROSCI.3861-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalleau S, Baradat M, Gueraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013;20:1615–1630. doi: 10.1038/cdd.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshvar DH, Goldstein LE, Kiernan PT, Stein TD, McKee AC. Post-traumatic neurodegeneration and chronic traumatic encephalopathy. Mol Cell Neurosci. 2015;66:81–90. doi: 10.1016/j.mcn.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Darios F, Muriel MP, Khondiker ME, Brice A, Ruberg M. Neurotoxic calcium transfer from endoplasmic reticulum to mitochondria is regulated by cyclin-dependent kinase 5-dependent phosphorylation of tau. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:4159–4168. doi: 10.1523/JNEUROSCI.0060-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Drose S, Brandt U, Muller WE, Eckert A, Gotz J. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. The Journal of biological chemistry. 2005;280:23802–23814. doi: 10.1074/jbc.M500356200. [DOI] [PubMed] [Google Scholar]
- Davis DR, Anderton BH, Brion JP, Reynolds CH, Hanger DP. Oxidative stress induces dephosphorylation of tau in rat brain primary neuronal cultures. Journal of neurochemistry. 1997;68:1590–1597. doi: 10.1046/j.1471-4159.1997.68041590.x. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Experimental neurology. 2007;205:154–165. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Battista AP, Rhind SG, Richards D, Churchill N, Baker AJ, Hutchison MG. Altered Blood Biomarker Profiles in Athletes with a History of Repetitive Head Impacts. PLoS One. 2016;11:e0159929. doi: 10.1371/journal.pone.0159929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. The Journal of biological chemistry. 2010;285:21978–21987. doi: 10.1074/jbc.M110.110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doutheil J, Gissel C, Oschlies U, Hossmann KA, Paschen W. Relation of neuronal endoplasmic reticulum calcium homeostasis to ribosomal aggregation and protein synthesis: implications for stress-induced suppression of protein synthesis. Brain research. 1997;775:43–51. doi: 10.1016/s0006-8993(97)00899-8. [DOI] [PubMed] [Google Scholar]
- Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova NN, Yang L, Starkov AA, Beal F. Behavioral deficit, oxidative stress, and mitochondrial dysfunction precede tau pathology in P301S transgenic mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25:4063–4072. doi: 10.1096/fj.11-186650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Khondiker M, Hollerhage M, Muriel MP, Champy P, Bach A, Depienne C, Respondek G, Yamada ES, Lannuzel A, Yagi T, Hirsch EC, Oertel WH, Jacob R, Michel PP, Ruberg M, Hoglinger GU. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:7827–7837. doi: 10.1523/JNEUROSCI.1644-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI, Loane DJ. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics. 2015;12:143–150. doi: 10.1007/s13311-014-0319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Zhang LF, Meng FT, Du X, Zhou JN. Acute hypoxia promote the phosphorylation of tau via ERK pathway. Neuroscience letters. 2010;474:173–177. doi: 10.1016/j.neulet.2010.03.037. [DOI] [PubMed] [Google Scholar]
- Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, Novak M, Cattaneo A. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. Journal of neurochemistry. 2000;75:624–633. doi: 10.1046/j.1471-4159.2000.0750624.x. [DOI] [PubMed] [Google Scholar]
- Faul M, X L, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002-2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- Ferrari R, Hardy J, Momeni P. Frontotemporal dementia: from Mendelian genetics towards genome wide association studies. J Mol Neurosci. 2011;45:500–515. doi: 10.1007/s12031-011-9635-y. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med. 2011;17:676–685. doi: 10.2119/molmed.2010.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, Rey MJ, Cardozo A, Vinals F, Ribalta T. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 2001;11:144–158. doi: 10.1111/j.1750-3639.2001.tb00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer TD, Hylin MJ, Zhao J, Moore AN, Waxham MN, Dash PK. Altered Mitochondrial Dynamics and TBI Pathophysiology. Front Syst Neurosci. 2016;10:29. doi: 10.3389/fnsys.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G. Mitochondrial participation in ischemic and traumatic neural cell death. Journal of neurotrauma. 2000;17:843–855. doi: 10.1089/neu.2000.17.843. [DOI] [PubMed] [Google Scholar]
- Flynn JM, Melov S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free radical biology & medicine. 2013;62:4–12. doi: 10.1016/j.freeradbiomed.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine SN, Sabbagh JJ, Baker J, Martinez-Licha CR, Darling A, Dickey CA. Cellular factors modulating the mechanism of tau protein aggregation. Cell Mol Life Sci. 2015;72:1863–1879. doi: 10.1007/s00018-015-1839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, Trojanowski JQ. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:3539–3550. doi: 10.1523/JNEUROSCI.0081-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz G, Beer R, Kampfl A, Engelhardt K, Schmutzhard E, Ulmer H, Deisenhammer F. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology. 2003;60:1457–1461. doi: 10.1212/01.wnl.0000063313.57292.00. [DOI] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. The Journal of biological chemistry. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu ZQ, Yang Y, Song J, Jiang Q, Lin ZC, Wang Q, Zhu LQ, Wang JZ, Tian Q. LiCl attenuates thapsigargin-induced tau hyperphosphorylation by inhibiting GSK-3beta in vivo and in vitro. Journal of Alzheimer's disease: JAD. 2010;21:1107–1117. doi: 10.3233/jad-2010-100687. [DOI] [PubMed] [Google Scholar]
- Gabbita SP, Scheff SW, Menard RM, Roberts K, Fugaccia I, Zemlan FP. Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. Journal of neurotrauma. 2005;22:83–94. doi: 10.1089/neu.2005.22.83. [DOI] [PubMed] [Google Scholar]
- Galas MC, Dourlen P, Begard S, Ando K, Blum D, Hamdane M, Buee L. The peptidylprolyl cis/trans-isomerase Pin1 modulates stress-induced dephosphorylation of Tau in neurons. Implication in a pathological mechanism related to Alzheimer disease. The Journal of biological chemistry. 2006;281:19296–19304. doi: 10.1074/jbc.M601849200. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nature reviews Neuroscience. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y. Cleavage of Tau by calpain in Alzheimer's disease: the quest for the toxic 17 kD fragment. Neurobiol Aging. 2011;32:1–14. doi: 10.1016/j.neurobiolaging.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Garwood CJ, Cooper JD, Hanger DP, Noble W. Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front Psychiatry. 2010;1:136. doi: 10.3389/fpsyt.2010.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerson J, Castillo-Carranza DL, Sengupta U, Bodani R, Prough DS, DeWitt DS, Hawkins BE, Kayed R. Tau Oligomers Derived from Traumatic Brain Injury Cause Cognitive Impairment and Accelerate Onset of Pathology in Htau Mice. Journal of neurotrauma. 2016 doi: 10.1089/neu.2015.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, Mufson EJ. Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. Journal of neurochemistry. 2006;96:1401–1408. doi: 10.1111/j.1471-4159.2005.03641.x. [DOI] [PubMed] [Google Scholar]
- Giraldo E, Lloret A, Fuchsberger T, Vina J. Abeta and tau toxicities in Alzheimer's are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox biology. 2014;2:873–877. doi: 10.1016/j.redox.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra160. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Ramos A, Diaz-Hernandez M, Cuadros R, Hernandez F, Avila J. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006;580:4842–4850. doi: 10.1016/j.febslet.2006.07.078. [DOI] [PubMed] [Google Scholar]
- Gomez-Ramos A, Diaz-Hernandez M, Rubio A, Miras-Portugal MT, Avila J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci. 2008;37:673–681. doi: 10.1016/j.mcn.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Gomez-Ramos A, Diaz-Nido J, Smith MA, Perry G, Avila J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. Journal of neuroscience research. 2003;71:863–870. doi: 10.1002/jnr.10525. [DOI] [PubMed] [Google Scholar]
- Gorlovoy P, Larionov S, Pham TT, Neumann H. Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23:2502–2513. doi: 10.1096/fj.08-123877. [DOI] [PubMed] [Google Scholar]
- Grynspan F, Griffin WR, Cataldo A, Katayama S, Nixon RA. Active site-directed antibodies identify calpain II as an early-appearing and pervasive component of neurofibrillary pathology in Alzheimer's disease. Brain research. 1997;763:145–158. doi: 10.1016/s0006-8993(97)00384-3. [DOI] [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, Cahill ME, Bigio EH, Berry RW, Binder LI. Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging. 2005;26:1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Glajch KE, Libson EG, Cahill ME, Bigio E, Berry RW, Binder LI. Phosphorylation and cleavage of tau in non-AD tauopathies. Acta Neuropathol. 2007;113:513–520. doi: 10.1007/s00401-007-0209-6. [DOI] [PubMed] [Google Scholar]
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- Guzel A, Karasalihoglu S, Aylanc H, Temizoz O, Hicdonmez T. Validity of serum tau protein levels in pediatric patients with minor head trauma. Am J Emerg Med. 2010;28:399–403. doi: 10.1016/j.ajem.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. Journal of neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. Journal of neurotrauma. 2005;22:252–265. doi: 10.1089/neu.2005.22.252. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Wang JA, Miller DM. Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Experimental neurology. 2012;238:176–182. doi: 10.1016/j.expneurol.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes J, Zilka N, Bartkova M, Caletkova M, Dobrota D, Novak M. Rat tau proteome consists of six tau isoforms: implication for animal models of human tauopathies. Journal of neurochemistry. 2009;108:1167–1176. doi: 10.1111/j.1471-4159.2009.05869.x. [DOI] [PubMed] [Google Scholar]
- Hansson MJ, Mansson R, Morota S, Uchino H, Kallur T, Sumi T, Ishii N, Shimazu M, Keep MF, Jegorov A, Elmer E. Calcium-induced generation of reactive oxygen species in brain mitochondria is mediated by permeability transition. Free radical biology & medicine. 2008;45:284–294. doi: 10.1016/j.freeradbiomed.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998;437:207–210. doi: 10.1016/s0014-5793(98)01217-4. [DOI] [PubMed] [Google Scholar]
- Hawkins BE, Krishnamurthy S, Castillo-Carranza DL, Sengupta U, Prough DS, Jackson GR, DeWitt DS, Kayed R. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. The Journal of biological chemistry. 2013;288:17042–17050. doi: 10.1074/jbc.M113.472746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazrati LN, Tartaglia MC, Diamandis P, Davis KD, Green RE, Wennberg R, Wong JC, Ezerins L, Tator CH. Absence of chronic traumatic encephalopathy in retired football players with multiple concussions and neurological symptomatology. Front Hum Neurosci. 2013;7:222. doi: 10.3389/fnhum.2013.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, Lee VM. Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron. 2002;35:433–446. doi: 10.1016/s0896-6273(02)00789-4. [DOI] [PubMed] [Google Scholar]
- Hill RL, Singh IN, Wang JA, Hall ED. Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochemistry international. doi: 10.1016/j.neuint.2017.03.015. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YS, Yang X, Lau JC, Hung CH, Wuwongse S, Zhang Q, Wang J, Baum L, So KF, Chang RC. Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: implication in Alzheimer's disease pathogenesis. Journal of Alzheimer's disease: JAD. 2012;28:839–854. doi: 10.3233/JAD-2011-111037. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Feger J, Champy P, Prigent A, Medja F, Lombes A, Oertel WH, Ruberg M, Hirsch EC. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. Journal of neurochemistry. 2005;95:930–939. doi: 10.1111/j.1471-4159.2005.03493.x. [DOI] [PubMed] [Google Scholar]
- Holm L, Cassidy JD, Carroll LJ, Borg J Neurotrauma Task Force on Mild Traumatic Brain Injury of the, W.H.O.C.C. Summary of the WHO Collaborating Centre for Neurotrauma Task Force on Mild Traumatic Brain Injury. J Rehabil Med. 2005;37:137–141. doi: 10.1080/16501970510027321. [DOI] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJ, Scheper W. Endoplasmic reticulum: the unfolded protein response is tangled in neurodegeneration. Int J Biochem Cell Biol. 2012;44:1295–1298. doi: 10.1016/j.biocel.2012.04.023. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi T, Uryu K, Giasson BI, Ischiropoulos H, LightFoot R, Bellmann C, Richter-Landsberg C, Lee VM, Trojanowski JQ. Nitration of tau protein is linked to neurodegeneration in tauopathies. Am J Pathol. 2003;163:1021–1031. doi: 10.1016/S0002-9440(10)63462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, Bennett DA, Cryns VL, Berry RW, Binder LI. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:7895–7902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, Cernak I, Petrie EC, Emery MJ, Swenson ER, Mayer C, Mehic E, Peskind ER, Cook DG. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. Journal of Alzheimer's disease: JAD. 2013;37:309–323. doi: 10.3233/JAD-130182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Experimental neurology. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:16180–16193. doi: 10.1523/JNEUROSCI.3020-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147a111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SY, Kim YE, Kim YJ. Genetics of Progressive Supranuclear Palsy. J Mov Disord. 2015;8:122–129. doi: 10.14802/jmd.15033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovino M, Pfisterer U, Holton JL, Lashley T, Swingler RJ, Calo L, Treacy R, Revesz T, Parmar M, Goedert M, Muqit MM, Spillantini MG. The novel MAPT mutation K298E: mechanisms of mutant tau toxicity, brain pathology and tau expression in induced fibroblast-derived neurons. Acta Neuropathol. 2014;127:283–295. doi: 10.1007/s00401-013-1219-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol. 2001;60:647–657. doi: 10.1093/jnen/60.6.647. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. The Journal of biological chemistry. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- Johnson GV, Jope RS, Binder LI. Proteolysis of tau by calpain. Biochemical and biophysical research communications. 1989;163:1505–1511. doi: 10.1016/0006-291x(89)91150-9. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BD. The clinical spectrum of sport-related traumatic brain injury. Nat Rev Neurol. 2013;9:222–230. doi: 10.1038/nrneurol.2013.33. [DOI] [PubMed] [Google Scholar]
- Kahlson MA, Colodner KJ. Glial Tau Pathology in Tauopathies: Functional Consequences. J Exp Neurosci. 2015;9:43–50. doi: 10.4137/JEN.S25515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampfl A, Posmantur RM, Zhao X, Schmutzhard E, Clifton GL, Hayes RL. Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy: a review and update. Journal of neurotrauma. 1997;14:121–134. doi: 10.1089/neu.1997.14.121. [DOI] [PubMed] [Google Scholar]
- Kane MJ, Angoa-Perez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM. A mouse model of human repetitive mild traumatic brain injury. Journal of neuroscience methods. 2012;203:41–49. doi: 10.1016/j.jneumeth.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173:692–702. doi: 10.1111/bph.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalci C, Pekdemir M, Durukan P, Ilhan N, Yildiz M, Serhatlioglu S, Seckin D. The value of serum tau protein for the diagnosis of intracranial injury in minor head trauma. Am J Emerg Med. 2007;25:391–395. doi: 10.1016/j.ajem.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Keogh MJ, Chinnery PF. Mitochondrial DNA mutations in neurodegeneration. Biochimica et biophysica acta. 2015;1847:1401–1411. doi: 10.1016/j.bbabio.2015.05.015. [DOI] [PubMed] [Google Scholar]
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. The Journal of biological chemistry. 2012;287:19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri N, Man HY. Synaptic activity and bioenergy homeostasis: implications in brain trauma and neurodegenerative diseases. Frontiers in neurology. 2013;4:199. doi: 10.3389/fneur.2013.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan PT, Montenigro PH, Solomon TM, McKee AC. Chronic traumatic encephalopathy: a neurodegenerative consequence of repetitive traumatic brain injury. Semin Neurol. 2015;35:20–28. doi: 10.1055/s-0035-1545080. [DOI] [PubMed] [Google Scholar]
- Kilbaugh TJ, Bhandare S, Lorom DH, Saraswati M, Robertson CL, Margulies SS. Cyclosporin A preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. Journal of neurotrauma. 2011;28:763–774. doi: 10.1089/neu.2010.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilinc D, Gallo G, Barbee KA. Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Experimental neurology. 2008;212:422–430. doi: 10.1016/j.expneurol.2008.04.025. [DOI] [PubMed] [Google Scholar]
- Kokjohn TA, Maarouf CL, Daugs ID, Hunter JM, Whiteside CM, Malek-Ahmadi M, Rodriguez E, Kalback W, Jacobson SA, Sabbagh MN, Beach TG, Roher AE. Neurochemical profile of dementia pugilistica. Journal of neurotrauma. 2013;30:981–997. doi: 10.1089/neu.2012.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y, Wei S, Luo ML, Albayram O, Huang P, Rotenberg A, Ryo A, Goldstein LE, Pascual-Leone A, McKee AC, Meehan W, Zhou XZ, Lu KP. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523:431–436. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Bakalis S, Neve RL. Developmentally regulated expression of specific tau sequences. Neuron. 1989;2:1389–1397. doi: 10.1016/0896-6273(89)90077-9. [DOI] [PubMed] [Google Scholar]
- Kovacs SK, Leonessa F, Ling GS. Blast TBI Models, Neuropathology, and Implications for Seizure Risk. Frontiers in neurology. 2014;5:47. doi: 10.3389/fneur.2014.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovesdi E, Gyorgy AB, Kwon SK, Wingo DL, Kamnaksh A, Long JB, Kasper CE, Agoston DV. The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front Neurosci. 2011;5:42. doi: 10.3389/fnins.2011.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewska M, Xu L, Xu W, Krajewski S, Kress CL, Cui J, Yang L, Irie F, Yamaguchi Y, Lipton SA, Reed JC. Endoplasmic reticulum protein BI-1 modulates unfolded protein response signaling and protects against stroke and traumatic brain injury. Brain research. 2011;1370:227–237. doi: 10.1016/j.brainres.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhla B, Haase C, Flach K, Luth HJ, Arendt T, Munch G. Effect of pseudophosphorylation and cross-linking by lipid peroxidation and advanced glycation end product precursors on tau aggregation and filament formation. The Journal of biological chemistry. 2007;282:6984–6991. doi: 10.1074/jbc.M609521200. [DOI] [PubMed] [Google Scholar]
- Kulbe JR, Geddes JW. Current status of fluid biomarkers in mild traumatic brain injury. Experimental neurology. 2015 doi: 10.1016/j.expneurol.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe JR, Hill RL, Singh IN, Wang J, Hall ED. Synaptic Mitochondria Sustain More Damage Than Non-Synaptic Mitochondria Following Traumatic Brain Injury and Are Protected by Cyclosporine A. Journal of neurotrauma. 2016 doi: 10.1089/neu.2016.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulic L, Wollmer MA, Rhein V, Pagani L, Kuehnle K, Cattepoel S, Tracy J, Eckert A, Nitsch RM. Combined expression of tau and the Harlequin mouse mutation leads to increased mitochondrial dysfunction, tau pathology and neurodegeneration. Neurobiol Aging. 2011;32:1827–1838. doi: 10.1016/j.neurobiolaging.2009.10.014. [DOI] [PubMed] [Google Scholar]
- Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. Journal of neurotrauma. 2016;33:1732–1750. doi: 10.1089/neu.2015.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landino LM, Skreslet TE, Alston JA. Cysteine oxidation of tau and microtubule-associated protein-2 by peroxynitrite: modulation of microtubule assembly kinetics by the thioredoxin reductase system. The Journal of biological chemistry. 2004;279:35101–35105. doi: 10.1074/jbc.M405471200. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. Journal of neuroscience research. 2009;87:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larner SF, Hayes RL, McKinsey DM, Pike BR, Wang KK. Increased expression and processing of caspase-12 after traumatic brain injury in rats. Journal of neurochemistry. 2004;88:78–90. doi: 10.1046/j.1471-4159.2003.02141.x. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. Journal of neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Leugers CJ. Tau and tauopathies. Prog Mol Biol Transl Sci. 2012;107:263–293. doi: 10.1016/B978-0-12-385883-2.00004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, McGeer E, McGeer PL. Activated human microglia stimulate neuroblastoma cells to upregulate production of beta amyloid protein and tau: implications for Alzheimer's disease pathogenesis. Neurobiol Aging. 2015;36:42–52. doi: 10.1016/j.neurobiolaging.2014.07.024. [DOI] [PubMed] [Google Scholar]
- Li A, Ceballos-Diaz C, DiNunno N, Levites Y, Cruz PE, Lewis J, Golde TE, Chakrabarty P. IFN-gamma promotes tau phosphorylation without affecting mature tangles. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2015;29:4384–4398. doi: 10.1096/fj.15-275834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:1605–1611. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz J, Friberg H, Neumar RW, Raghupathi R, Welsh FA, Janmey P, Saatman KE, Wieloch T, Grady MS, McIntosh TK. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: evidence for differentially sensitive populations in the cortex and hippocampus. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:219–231. doi: 10.1097/01.WCB.0000040581.43808.03. [DOI] [PubMed] [Google Scholar]
- Liliang PC, Liang CL, Weng HC, Lu K, Wang KW, Chen HJ, Chuang JH. Tau proteins in serum predict outcome after severe traumatic brain injury. J Surg Res. 2010;160:302–307. doi: 10.1016/j.jss.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Lin L, Yang SS, Chu J, Wang L, Ning LN, Zhang T, Jiang Q, Tian Q, Wang JZ. Region-specific expression of tau, amyloid-beta protein precursor, and synaptic proteins at physiological condition or under endoplasmic reticulum stress in rats. Journal of Alzheimer's disease: JAD. 2014;41:1149–1163. doi: 10.3233/JAD-140207. [DOI] [PubMed] [Google Scholar]
- Litersky JM, Scott CW, Johnson GV. Phosphorylation, calpain proteolysis and tubulin binding of recombinant human tau isoforms. Brain research. 1993;604:32–40. doi: 10.1016/0006-8993(93)90349-r. [DOI] [PubMed] [Google Scholar]
- Litvan I. Update on progressive supranuclear palsy. Curr Neurol Neurosci Rep. 2004;4:296–302. doi: 10.1007/s11910-004-0055-z. [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MC, Kobeissy F, Zheng W, Zhang Z, Hayes RL, Wang KK. Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN neuro. 2011;3:e00051. doi: 10.1042/AN20100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Smith MA, Avila J, DeBernardis J, Kansal M, Takeda A, Zhu X, Nunomura A, Honda K, Moreira PI, Oliveira CR, Santos MS, Shimohama S, Aliev G, de la Torre J, Ghanbari HA, Siedlak SL, Harris PL, Sayre LM, Perry G. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free radical biology & medicine. 2005;38:746–754. doi: 10.1016/j.freeradbiomed.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, Matsumoto RR, Huber JD, Rosen CL. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci. 2014;8:421. doi: 10.3389/fncel.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer's disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging. 2001;22:187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xiong S, Xie C, Davies P, Markesbery WR. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. Journal of Alzheimer's disease: JAD. 2004;6:659–671. doi: 10.3233/jad-2004-6610. discussion 673-681. [DOI] [PubMed] [Google Scholar]
- Lucke-Wold BP, Naser ZJ, Logsdon AF, Turner RC, Smith KE, Robson MJ, Bailes JE, Lee JM, Rosen CL, Huber JD. Amelioration of nicotinamide adenine dinucleotide phosphate-oxidase mediated stress reduces cell death after blast-induced traumatic brain injury. Transl Res. 2015;166:509–528 e501. doi: 10.1016/j.trsl.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Lucke-Wold BP, Turner RC, Logsdon AF, Nguyen L, Bailes JE, Lee JM, Robson MJ, Omalu BI, Huber JD, Rosen CL. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. Journal of neurosurgery. 2016;124:687–702. doi: 10.3171/2015.3.JNS141802. [DOI] [PubMed] [Google Scholar]
- Luo J, Nguyen A, Villeda S, Zhang H, Ding Z, Lindsey D, Bieri G, Castellano JM, Beaupre GS, Wyss-Coray T. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Frontiers in neurology. 2014;5:12. doi: 10.3389/fneur.2014.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Lindsell CJ, Rosenberry CM, Shaw GJ, Zemlan FP. Serum cleaved tau does not predict postconcussion syndrome after mild traumatic brain injury. Am J Emerg Med. 2008;26:763–768. doi: 10.1016/j.ajem.2007.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Atkin TA, Kittler JT. Mitochondrial trafficking and the provision of energy and calcium buffering at excitatory synapses. The European journal of neuroscience. 2010;32:231–240. doi: 10.1111/j.1460-9568.2010.07345.x. [DOI] [PubMed] [Google Scholar]
- Maccioni RB, Cambiazo V. Role of microtubule-associated proteins in the control of microtubule assembly. Physiol Rev. 1995;75:835–864. doi: 10.1152/physrev.1995.75.4.835. [DOI] [PubMed] [Google Scholar]
- Magnoni S, Esparza TJ, Conte V, Carbonara M, Carrabba G, Holtzman DM, Zipfel GJ, Stocchetti N, Brody DL. Tau elevations in the brain extracellular space correlate with reduced amyloid-beta levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain: a journal of neurology. 2012;135:1268–1280. doi: 10.1093/brain/awr286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. 2012;21:2538–2547. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix R, Meehan WP, Mandeville J, Grant PE, Gray T, Berglass J, Zhang J, Bryant J, Rezaie S, Chung JY, Peters NV, Lee C, Tien LW, Kaplan DL, Feany M, Whalen M. Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol. 2013;74:65–75. doi: 10.1002/ana.23858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Cardona AE, Ransohoff RM, Lamb BT, Bhaskar K. Loss of tau rescues inflammation-mediated neurodegeneration. Front Neurosci. 2015a;9:196. doi: 10.3389/fnins.2015.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain: a journal of neurology. 2015b;138:1738–1755. doi: 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion DW, Curley KC, Schwab K, Hicks RR, m TBIDW. Proceedings of the military mTBI Diagnostics Workshop, St. Pete Beach, August 2010. Journal of neurotrauma. 2011;28:517–526. doi: 10.1089/neu.2010.1638. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- Marmarou CR, Povlishock JT. Administration of the immunophilin ligand FK506 differentially attenuates neurofilament compaction and impaired axonal transport in injured axons following diffuse traumatic brain injury. Experimental neurology. 2006;197:353–362. doi: 10.1016/j.expneurol.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Maroon JC, Winkelman R, Bost J, Amos A, Mathyssek C, Miele V. Chronic traumatic encephalopathy in contact sports: a systematic review of all reported pathological cases. PLoS One. 2015;10:e0117338. doi: 10.1371/journal.pone.0117338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Terro F. Tau protein phosphatases in Alzheimer's disease: the leading role of PP2A. Ageing Res Rev. 2013;12:39–49. doi: 10.1016/j.arr.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Martinez A, Carmona M, Portero-Otin M, Naudi A, Pamplona R, Ferrer I. Type-dependent oxidative damage in frontotemporal lobar degeneration: cortical astrocytes are targets of oxidative damage. J Neuropathol Exp Neurol. 2008a;67:1122–1136. doi: 10.1097/NEN.0b013e31818e06f3. [DOI] [PubMed] [Google Scholar]
- Martinez A, Dalfo E, Muntane G, Ferrer I. Glycolitic enzymes are targets of oxidation in aged human frontal cortex and oxidative damage of these proteins is increased in progressive supranuclear palsy. J Neural Transm (Vienna) 2008b;115:59–66. doi: 10.1007/s00702-007-0800-y. [DOI] [PubMed] [Google Scholar]
- Matthews-Roberson TA, Quintanilla RA, Ding H, Johnson GV. Immortalized cortical neurons expressing caspase-cleaved tau are sensitized to endoplasmic reticulum stress induced cell death. Brain research. 2008;1234:206–212. doi: 10.1016/j.brainres.2008.07.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Fu W, Waeg G, Uchida K. 4-Hydroxynonenal, a product of lipid peroxidation, inhibits dephosphorylation of the microtubule-associated protein tau. Neuroreport. 1997;8:2275–2281. doi: 10.1097/00001756-199707070-00036. [DOI] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Carrico KM, Saatman KE, Hall ED. Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009;29:87–97. doi: 10.1038/jcbfm.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Experimental neurology. 2008;209:243–253. doi: 10.1016/j.expneurol.2007.09.025. [DOI] [PubMed] [Google Scholar]
- McAllister TW. Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci. 2011;13:287–300. doi: 10.31887/DCNS.2011.13.2/tmcallister. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrory P, Meeuwisse WH, Kutcher JS, Jordan BD, Gardner A. What is the evidence for chronic concussion-related changes in retired athletes: behavioural, pathological and clinical outcomes? Br J Sports Med. 2013;47:327–330. doi: 10.1136/bjsports-2013-092248. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O'Connor K, Alvarez VE, Gordon WA, group TC. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131:75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Daneshvar DH, Alvarez VE, Stein TD. The neuropathology of sport. Acta Neuropathol. 2014;127:29–51. doi: 10.1007/s00401-013-1230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Robinson ME. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 2014;10:S242–253. doi: 10.1016/j.jalz.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC. The spectrum of disease in chronic traumatic encephalopathy. Brain: a journal of neurology. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan P, Korvatska E, Poorkaj P, Evstafjeva Z, Robinson L, Greenup L, Leverenz J, Schellenberg GD, D'Souza I. Tau isoform regulation is region- and cell-specific in mouse brain. J Comp Neurol. 2008;511:788–803. doi: 10.1002/cne.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina M, Avila J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci. 2014;8:113. doi: 10.3389/fncel.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, Mechas E, Chen J, Wolozin B, LeVine H, 3rd, Zhu H, Abisambra JF. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2016;36:1001–1007. doi: 10.1523/JNEUROSCI.3029-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, Schilling B, Mavros C, Masters CL, Volitakis I, Li QX, Laughton K, Hubbard A, Cherny RA, Gibson B, Bush AI. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One. 2007;2:e536. doi: 10.1371/journal.pone.0000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G. Tau protein modifications and interactions: their role in function and dysfunction. Int J Mol Sci. 2014;15:4671–4713. doi: 10.3390/ijms15034671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM, Singh IN, Wang JA, Hall ED. Administration of the Nrf2-ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free radical biology & medicine. 2013;57:1–9. doi: 10.1016/j.freeradbiomed.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DM, Wang JA, Buchanan AK, Hall ED. Temporal and spatial dynamics of nrf2-antioxidant response elements mediated gene targets in cortex and hippocampus after controlled cortical impact traumatic brain injury in mice. Journal of neurotrauma. 2014;31:1194–1201. doi: 10.1089/neu.2013.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Miyauchi T, Wei EP, Povlishock JT. Therapeutic targeting of the axonal and microvascular change associated with repetitive mild traumatic brain injury. Journal of neurotrauma. 2013;30:1664–1671. doi: 10.1089/neu.2013.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenigro PH, Baugh CM, Daneshvar DH, Mez J, Budson AE, Au R, Katz DI, Cantu RC, Stern RA. Clinical subtypes of chronic traumatic encephalopathy: literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther. 2014;6:68. doi: 10.1186/s13195-014-0068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine KS, Kim PJ, Olson SJ, Markesbery WR, Montine TJ. 4-hydroxy-2-nonenal pyrrole adducts in human neurodegenerative disease. J Neuropathol Exp Neurol. 1997;56:866–871. doi: 10.1097/00005072-199708000-00004. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Amarnath V, Martin ME, Strittmatter WJ, Graham DG. E-4-hydroxy-2-nonenal is cytotoxic and cross-links cytoskeletal proteins in P19 neuroglial cultures. Am J Pathol. 1996;148:89–93. [PMC free article] [PubMed] [Google Scholar]
- Morales R, Callegari K, Soto C. Prion-like features of misfolded Abeta and tau aggregates. Virus Res. 2015;207:106–112. doi: 10.1016/j.virusres.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. The Journal of biological chemistry. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75:241–254. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Perez SE, Nadeem M, Mahady L, Kanaan NM, Abrahamson EE, Ikonomovic MD, Crawford F, Alvarez V, Stein T, McKee AC. Progression of tau pathology within cholinergic nucleus basalis neurons in chronic traumatic encephalopathy: A chronic effects of neurotrauma consortium study. Brain Inj. 2016;30:1399–1413. doi: 10.1080/02699052.2016.1219058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntane G, Dalfo E, Martinez A, Rey MJ, Avila J, Perez M, Portero M, Pamplona R, Ayala V, Ferrer I. Glial fibrillary acidic protein is a major target of glycoxidative and lipoxidative damage in Pick's disease. Journal of neurochemistry. 2006;99:177–185. doi: 10.1111/j.1471-4159.2006.04032.x. [DOI] [PubMed] [Google Scholar]
- Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. Journal of neurochemistry. 2010;114:271–280. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AG, Wang JA, Carrico KM, Hall ED. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. Journal of neurochemistry. 2011;117:579–588. doi: 10.1111/j.1471-4159.2011.07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell. 2012;149:232–244. doi: 10.1016/j.cell.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namjoshi DR, Cheng WH, McInnes KA, Martens KM, Carr M, Wilkinson A, Fan J, Robert J, Hayat A, Cripton PA, Wellington CL. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol Neurodegener. 2014;9:55. doi: 10.1186/1750-1326-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT. Traumatic brain injury and time to onset of Alzheimer's disease: a population-based study. Am J Epidemiol. 1999;149:32–40. doi: 10.1093/oxfordjournals.aje.a009724. [DOI] [PubMed] [Google Scholar]
- Neselius S, Brisby H, Theodorsson A, Blennow K, Zetterberg H, Marcusson J. CSF-biomarkers in Olympic boxing: diagnosis and effects of repetitive head trauma. PLoS One. 2012;7:e33606. doi: 10.1371/journal.pone.0033606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neselius S, Zetterberg H, Blennow K, Randall J, Wilson D, Marcusson J, Brisby H. Olympic boxing is associated with elevated levels of the neuronal protein tau in plasma. Brain Inj. 2013;27:425–433. doi: 10.3109/02699052.2012.750752. [DOI] [PubMed] [Google Scholar]
- Newman J, Rissman RA, Sarsoza F, Kim RC, Dick M, Bennett DA, Cotman CW, Rohn TT, Head E. Caspase-cleaved tau accumulation in neurodegenerative diseases associated with tau and alpha-synuclein pathology. Acta Neuropathol. 2005;110:135–144. doi: 10.1007/s00401-005-1027-3. [DOI] [PubMed] [Google Scholar]
- Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol. 2012;226:693–702. doi: 10.1002/path.3969. [DOI] [PubMed] [Google Scholar]
- Nilson AN, English KC, Gerson JE, Whittle TB, Crain CN, Xue J, Sengupta U, Castillo-Carranza DL, Zhang W, Gupta P, Kayed R. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. Journal of Alzheimer's disease: JAD. 2016 doi: 10.3233/JAD-160912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends in neurosciences. 2006;29:528–535. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Odetti P, Garibaldi S, Norese R, Angelini G, Marinelli L, Valentini S, Menini S, Traverso N, Zaccheo D, Siedlak S, Perry G, Smith MA, Tabaton M. Lipoperoxidation is selectively involved in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2000;59:393–397. doi: 10.1093/jnen/59.5.393. [DOI] [PubMed] [Google Scholar]
- Ojo JO, Mouzon B, Greenberg MB, Bachmeier C, Mullan M, Crawford F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J Neuropathol Exp Neurol. 2013;72:137–151. doi: 10.1097/NEN.0b013e3182814cdf. [DOI] [PubMed] [Google Scholar]
- Ojo JO, Mouzon BC, Crawford F. Repetitive head trauma, chronic traumatic encephalopathy and tau: Challenges in translating from mice to men. Experimental neurology. 2016;275(Pt 3):389–404. doi: 10.1016/j.expneurol.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Olivera A, Lejbman N, Jeromin A, French LM, Kim HS, Cashion A, Mysliwiec V, Diaz-Arrastia R, Gill J. Peripheral Total Tau in Military Personnel Who Sustain Traumatic Brain Injuries During Deployment. JAMA Neurol. 2015;72:1109–1116. doi: 10.1001/jamaneurol.2015.1383. [DOI] [PubMed] [Google Scholar]
- Olivieri G, Baysang G, Meier F, Muller-Spahn F, Stahelin HB, Brockhaus M, Brack C. N-acetyl-L-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: effects on beta-amyloid secretion and tau phosphorylation. Journal of neurochemistry. 2001;76:224–233. doi: 10.1046/j.1471-4159.2001.00090.x. [DOI] [PubMed] [Google Scholar]
- Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, Fitzsimmons R. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. 2011a;69:173–183. doi: 10.1227/NEU.0b013e318212bc7b. discussion 183. [DOI] [PubMed] [Google Scholar]
- Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G, Fitzsimmons RP. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus. 2011b;31:E3. doi: 10.3171/2011.9.FOCUS11178. [DOI] [PubMed] [Google Scholar]
- Omalu BI, DeKosky ST, Hamilton RL, Minster RL, Kamboh MI, Shakir AM, Wecht CH. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery. 2006;59:1086–1092. doi: 10.1227/01.NEU.0000245601.69451.27. discussion 1092-1083. [DOI] [PubMed] [Google Scholar]
- Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–134. doi: 10.1227/01.neu.0000163407.92769.ed. discussion 128-134. [DOI] [PubMed] [Google Scholar]
- Ost M, Nylen K, Csajbok L, Ohrfelt AO, Tullberg M, Wikkelso C, Nellgard P, Rosengren L, Blennow K, Nellgard B. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology. 2006;67:1600–1604. doi: 10.1212/01.wnl.0000242732.06714.0f. [DOI] [PubMed] [Google Scholar]
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Tournell C, Sinjoanu RC, Ferreira A. Caspase-3- and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons. Neuroscience. 2007;144:119–127. doi: 10.1016/j.neuroscience.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Polo JR, Rea HC, Johnson KM, Parsley MA, Unabia GC, Xu GY, Prough D, DeWitt DS, Spratt H, Hulsebosch CE. A rodent model of mild traumatic brain blast injury. Journal of neuroscience research. 2015;93:549–561. doi: 10.1002/jnr.23513. [DOI] [PubMed] [Google Scholar]
- Perez M, Cuadros R, Smith MA, Perry G, Avila J. Phosphorylated, but not native, tau protein assembles following reaction with the lipid peroxidation product, 4-hydroxy-2-nonenal. FEBS Lett. 2000;486:270–274. doi: 10.1016/s0014-5793(00)02323-1. [DOI] [PubMed] [Google Scholar]
- Perez M, Hernandez F, Lim F, Diaz-Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. Journal of Alzheimer's disease: JAD. 2003;5:301–308. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- Perreault S, Bousquet O, Lauzon M, Paiement J, Leclerc N. Increased association between rough endoplasmic reticulum membranes and mitochondria in transgenic mice that express P301L tau. J Neuropathol Exp Neurol. 2009;68:503–514. doi: 10.1097/NEN.0b013e3181a1fc49. [DOI] [PubMed] [Google Scholar]
- Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free radical biology & medicine. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, Walker CT, Chen M, Hyrien O, Iliff JJ, Deane R, Huang JH, Nedergaard M. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int. 2014;5:184. doi: 10.4103/2152-7806.147566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picklo MJ, Amarnath V, McIntyre JO, Graham DG, Montine TJ. 4-Hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. Journal of neurochemistry. 1999;72:1617–1624. doi: 10.1046/j.1471-4159.1999.721617.x. [DOI] [PubMed] [Google Scholar]
- Picklo MJ, Montine TJ. Acrolein inhibits respiration in isolated brain mitochondria. Biochimica et biophysica acta. 2001;1535:145–152. doi: 10.1016/s0925-4439(00)00093-4. [DOI] [PubMed] [Google Scholar]
- Piras A, Collin L, Gruninger F, Graff C, Ronnback A. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun. 2016;4:22. doi: 10.1186/s40478-016-0292-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One. 2012;7:e36873. doi: 10.1371/journal.pone.0036873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poniatowski LA, Wojdasiewicz P, Krawczyk M, Szukiewicz D, Gasik R, Kubaszewski L, Kurkowska-Jastrzebska I. Analysis of the Role of CX3CL1 (Fractalkine) and Its Receptor CX3CR1 in Traumatic Brain and Spinal Cord Injury: Insight into Recent Advances in Actions of Neurochemokine Agents. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-9787-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Prabakaran S, Cantrelle FX, Chambraud B, Gunawardena J, Lippens G, Landrieu I. Characterization of Neuronal Tau Protein as a Target of Extracellular Signal-regulated Kinase. The Journal of biological chemistry. 2016;291:7742–7753. doi: 10.1074/jbc.M115.700914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Liu Y, Hao W, Decker Y, Tomic I, Menger MD, Liu C, Fassbender K. Stimulation of TLR4 Attenuates Alzheimer's Disease-Related Symptoms and Pathology in Tau-Transgenic Mice. J Immunol. 2016;197:3281–3292. doi: 10.4049/jimmunol.1600873. [DOI] [PubMed] [Google Scholar]
- Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. The Journal of biological chemistry. 2009;284:18754–18766. doi: 10.1074/jbc.M808908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Orellana DI, Gonzalez-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. 2004;295:245–257. doi: 10.1016/j.yexcr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015;130:633–642. doi: 10.1007/s00401-015-1487-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free radical biology & medicine. 2002;33:1451–1464. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- Ramalho RM, Viana RJ, Castro RE, Steer CJ, Low WC, Rodrigues CM. Apoptosis in transgenic mice expressing the P301L mutated form of human tau. Mol Med. 2008;14:309–317. doi: 10.2119/2007-00133.Ramalho. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Rao MV, Mohan PS, Peterhoff CM, Yang DS, Schmidt SD, Stavrides PH, Campbell J, Chen Y, Jiang Y, Paskevich PA, Cataldo AM, Haroutunian V, Nixon RA. Marked calpastatin (CAST) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, Lee NN, Povlishock JT. Preferential neuroprotective effect of tacrolimus (FK506) on unmyelinated axons following traumatic brain injury. Brain research. 2007;1154:225–236. doi: 10.1016/j.brainres.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, Giese RN, Wang B, Shi X, Nedergaard M. ‘Hit & Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:834–845. doi: 10.1038/jcbfm.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MR, Berry RW, Binder LI. Site-specific nitration and oxidative dityrosine bridging of the tau protein by peroxynitrite: implications for Alzheimer's disease. Biochemistry. 2005;44:1690–1700. doi: 10.1021/bi047982v. [DOI] [PubMed] [Google Scholar]
- Reynolds MR, Lukas TJ, Berry RW, Binder LI. Peroxynitrite-mediated tau modifications stabilize preformed filaments and destabilize microtubules through distinct mechanisms. Biochemistry. 2006;45:4314–4326. doi: 10.1021/bi052142h. [DOI] [PubMed] [Google Scholar]
- Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Drose S, Brandt U, Savaskan E, Czech C, Gotz J, Eckert A. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20057–20062. doi: 10.1073/pnas.0905529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–130. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529(Pt 1):37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Brini M, Chiesa A, Filippin L, Pozzan T. Mitochondria as biosensors of calcium microdomains. Cell Calcium. 1999;26:193–199. doi: 10.1054/ceca.1999.0076. [DOI] [PubMed] [Google Scholar]
- Roe AD, Staup MA, Serrats J, Sawchenko PE, Rissman RA. Lipopolysaccharide-induced tau phosphorylation and kinase activity--modulation, but not mediation, by corticotropin-releasing factor receptors. The European journal of neuroscience. 2011;34:448–456. doi: 10.1111/j.1460-9568.2011.07764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostami E, Davidsson J, Ng KC, Lu J, Gyorgy A, Walker J, Wingo D, Plantman S, Bellander BM, Agoston DV, Risling M. A Model for Mild Traumatic Brain Injury that Induces Limited Transient Memory Impairment and Increased Levels of Axon Related Serum Biomarkers. Frontiers in neurology. 2012;3:115. doi: 10.3389/fneur.2012.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Sabbagh JJ, Fontaine SN, Shelton LB, Blair LJ, Hunt JB, Jr, Zhang B, Gutmann JM, Lee DC, Lloyd JD, Dickey CA. Noncontact Rotational Head Injury Produces Transient Cognitive Deficits but Lasting Neuropathological Changes. Journal of neurotrauma. 2016 doi: 10.1089/neu.2015.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saing T, Dick M, Nelson PT, Kim RC, Cribbs DH, Head E. Frontal cortex neuropathology in dementia pugilistica. Journal of neurotrauma. 2012;29:1054–1070. doi: 10.1089/neu.2011.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajja VS, Hubbard WB, Hall CS, Ghoddoussi F, Galloway MP, VandeVord PJ. Enduring deficits in memory and neuronal pathology after blast-induced traumatic brain injury. Sci Rep. 2015;5:15075. doi: 10.1038/srep15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. The Journal of biological chemistry. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mejias E, Navarro V, Jimenez S, Sanchez-Mico M, Sanchez-Varo R, Nunez-Diaz C, Trujillo-Estrada L, Davila JC, Vizuete M, Gutierrez A, Vitorica J. Soluble phospho-tau from Alzheimer's disease hippocampus drives microglial degeneration. Acta Neuropathol. 2016;132:897–916. doi: 10.1007/s00401-016-1630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar C, Zhao Z, Aungst S, Sabirzhanov B, Faden AI, Lipinski MM. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy. 2014;10:2208–2222. doi: 10.4161/15548627.2014.981787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Kawarabayashi T, Murakami T, Matsubara E, Ikeda M, Hagiwara H, Westaway D, George-Hyslop PS, Shoji M, Nakazato Y. Microglial activation in brain lesions with tau deposits: comparison of human tauopathies and tau transgenic mice TgTauP301L. Brain research. 2008;1214:159–168. doi: 10.1016/j.brainres.2008.02.084. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. Journal of neurochemistry. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathol. 2001;101:518–524. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- Schulz KL, Eckert A, Rhein V, Mai S, Haase W, Reichert AS, Jendrach M, Muller WE, Leuner K. A new link to mitochondrial impairment in tauopathies. Mol Neurobiol. 2012;46:205–216. doi: 10.1007/s12035-012-8308-3. [DOI] [PubMed] [Google Scholar]
- Shahim P, Linemann T, Inekci D, Karsdal MA, Blennow K, Tegner Y, Zetterberg H, Henriksen K. Serum Tau Fragments Predict Return to Play in Concussed Professional Ice Hockey Players. Journal of neurotrauma. 2016 doi: 10.1089/neu.2014.3741. [DOI] [PubMed] [Google Scholar]
- Shahim P, Tegner Y, Wilson DH, Randall J, Skillback T, Pazooki D, Kallberg B, Blennow K, Zetterberg H. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 2014;71:684–692. doi: 10.1001/jamaneurol.2014.367. [DOI] [PubMed] [Google Scholar]
- Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M, Hisanaga S. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WS. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochemistry international. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Zhu SG, Jones RA, Griffin WS, Mrak RE. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Experimental neurology. 2000;163:388–391. doi: 10.1006/exnr.2000.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nature reviews Neuroscience. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SS, Dixon CE. Alterations in Cholinergic Pathways and Therapeutic Strategies Targeting Cholinergic System after Traumatic Brain Injury. Journal of neurotrauma. 2015;32:1429–1440. doi: 10.1089/neu.2014.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz SR, Wright DK, Zheng P, Stuchbery R, Liu SJ, Sashindranath M, Medcalf RL, Johnston LA, Hovens CM, Jones NC, O'Brien TJ. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain: a journal of neurology. 2015;138:1297–1313. doi: 10.1093/brain/awv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D, Garcia-Garcia E, Royo F, Falcon-Perez JM, Avila J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012;586:47–54. doi: 10.1016/j.febslet.2011.11.022. [DOI] [PubMed] [Google Scholar]
- Singh IN, Gilmer LK, Miller DM, Cebak JE, Wang JA, Hall ED. Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:593–599. doi: 10.1038/jcbfm.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Hall ED. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. Journal of neuroscience research. 2007;85:2216–2223. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- Singleton RH, Stone JR, Okonkwo DO, Pellicane AJ, Povlishock JT. The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. Journal of neurotrauma. 2001;18:607–614. doi: 10.1089/089771501750291846. [DOI] [PubMed] [Google Scholar]
- Sinjoanu RC, Kleinschmidt S, Bitner RS, Brioni JD, Moeller A, Ferreira A. The novel calpain inhibitor A-705253 potently inhibits oligomeric beta-amyloid-induced dynamin 1 and tau cleavage in hippocampal neurons. Neurochemistry international. 2008;53:79–88. doi: 10.1016/j.neuint.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. The Journal of biological chemistry. 2002;277:44701–44708. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, de Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, Osetek JD, Jones PB, Bacskai BJ, Feany MB, Carlson GA, Ashe KH, Lewis J, Hyman BT. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:862–867. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Kopeikina KJ, Koffie RM, de Calignon A, Hyman BT. Are tangles as toxic as they look? J Mol Neurosci. 2011;45:438–444. doi: 10.1007/s12031-011-9566-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, Fritts NG, Stamm JM, Robbins CA, McHale L, Simkin I, Stein TD, Alvarez VE, Goldstein LE, Budson AE, Kowall NW, Nowinski CJ, Cantu RC, McKee AC. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81:1122–1129. doi: 10.1212/WNL.0b013e3182a55f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RA, Tripodis Y, Baugh CM, Fritts NG, Martin BM, Chaisson C, Cantu RC, Joyce JA, Shah S, Ikezu T, Zhang J, Gercel-Taylor C, Taylor DD. Preliminary Study of Plasma Exosomal Tau as a Potential Biomarker for Chronic Traumatic Encephalopathy. Journal of Alzheimer's disease: JAD. 2016;51:1099–1109. doi: 10.3233/JAD-151028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JF, Maier CS. Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol Nutr Food Res. 2008;52:7–25. doi: 10.1002/mnfr.200700412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff W, Jones PB, Spires-Jones TL, Joyner D, Chhabra E, Bercury K, Fan Z, Xie H, Bacskai B, Edd J, Irimia D, Hyman BT. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. Journal of neurochemistry. 2009;111:417–427. doi: 10.1111/j.1471-4159.2009.06316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Wang X, Lee HG, Tabaton M, Perry G, Smith MA, Zhu X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neuroscience letters. 2010;468:267–271. doi: 10.1016/j.neulet.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? Journal of neuroscience research. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Sebastian AH, Hall ED. Therapeutic window analysis of the neuroprotective effects of cyclosporine A after traumatic brain injury. Journal of neurotrauma. 2011;28:311–318. doi: 10.1089/neu.2010.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH. Brain aging, Alzheimer's disease, and mitochondria. Biochimica et biophysica acta. 2011;1812:1630–1639. doi: 10.1016/j.bbadis.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Smith MA, Avila J, Nunomura A, Siedlak SL, Zhu X, Perry G, Sayre LM. In Alzheimer's disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. Journal of neurochemistry. 2000;75:1234–1241. doi: 10.1046/j.1471-4159.2000.0751234.x. [DOI] [PubMed] [Google Scholar]
- Tepper K, Biernat J, Kumar S, Wegmann S, Timm T, Hubschmann S, Redecke L, Mandelkow EM, Muller DJ, Mandelkow E. Oligomer formation of tau protein hyperphosphorylated in cells. The Journal of biological chemistry. 2014;289:34389–34407. doi: 10.1074/jbc.M114.611368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. Journal of neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Experimental neurology. 2006;201:253–265. doi: 10.1016/j.expneurol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci. 1999;112(Pt 14):2355–2367. doi: 10.1242/jcs.112.14.2355. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Pathological tau: a loss of normal function or a gain in toxicity? Nat Neurosci. 2005;8:1136–1137. doi: 10.1038/nn0905-1136. [DOI] [PubMed] [Google Scholar]
- Troncoso JC, Costello A, Watson AL, Jr, Johnson GV. In vitro polymerization of oxidized tau into filaments. Brain research. 1993;613:313–316. doi: 10.1016/0006-8993(93)90918-d. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Shimohama S, Kimura J, Shimizu K. m-Calpain (calcium-activated neutral proteinase) in Alzheimer's disease brains. Neuroscience letters. 1998;248:109–112. doi: 10.1016/s0304-3940(98)00348-6. [DOI] [PubMed] [Google Scholar]
- Turner RC, Lucke-Wold BP, Logsdon AF, Robson MJ, Dashnaw ML, Huang JH, Smith KE, Huber JD, Rosen CL, Petraglia AL. The Quest to Model Chronic Traumatic Encephalopathy: A Multiple Model and Injury Paradigm Experience. Frontiers in neurology. 2015;6:222. doi: 10.3389/fneur.2015.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Experimental neurology. 2007;208:185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnav RA, Singh IN, Miller DM, Hall ED. Lipid peroxidation-derived reactive aldehydes directly and differentially impair spinal cord and brain mitochondrial function. Journal of neurotrauma. 2010;27:1311–1320. doi: 10.1089/neu.2009.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranna Kaji T, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA. Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer's disease. Am J Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink R, Nimmo AJ. Multifunctional drugs for head injury. Neurotherapeutics. 2009;6:28–42. doi: 10.1016/j.nurt.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, Liou AK, Leak RK, Gao Y, Chen J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013a;33:1864–1874. doi: 10.1038/jcbfm.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li J, Han L, Guo S, Wang L, Xiong Z, Chen Z, Chen W, Liang J. Serum tau protein as a potential biomarker in the assessment of traumatic brain injury. Exp Ther Med. 2016;11:1147–1151. doi: 10.3892/etm.2016.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Zhang Y. Configuration-specific immunotherapy targeting cis pThr231-Pro232 tau for Alzheimer disease. J Neurol Sci. 2015;348:253–255. doi: 10.1016/j.jns.2014.11.011. [DOI] [PubMed] [Google Scholar]
- Wang KK. Calpain and caspase: can you tell the difference? Trends in neurosciences. 2000;23:20–26. doi: 10.1016/s0166-2236(99)01536-2. [DOI] [PubMed] [Google Scholar]
- Wang L, Jiang Q, Chu J, Lin L, Li XG, Chai GS, Wang Q, Wang JZ, Tian Q. Expression of Tau40 induces activation of cultured rat microglial cells. PLoS One. 2013b;8:e76057. doi: 10.1371/journal.pone.0076057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KM, Reeves TM, Phillips LL. MT5-MMP, ADAM-10, and N-cadherin act in concert to facilitate synapse reorganization after traumatic brain injury. Journal of neurotrauma. 2012;29:1922–1940. doi: 10.1089/neu.2012.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JT. Altered calcium signaling following traumatic brain injury. Front Pharmacol. 2012;3:60. doi: 10.3389/fphar.2012.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weih M, Schmitt M, Gieche J, Harms C, Ruscher K, Dirnagl U, Grune T. Proteolysis of oxidized proteins after oxygen-glucose deprivation in rat cortical neurons is mediated by the proteasome. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2001;21:1090–1096. doi: 10.1097/00004647-200109000-00006. [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M, Kayed R, Zurzolo C, Di Paolo G, Duff KE. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. The Journal of biological chemistry. 2013;288:1856–1870. doi: 10.1074/jbc.M112.394528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. Journal of neurotrauma. 1997;14:23–34. doi: 10.1089/neu.1997.14.23. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nature reviews. Neuroscience. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Ryu J, Nguyen JV, Arena J, Rha E, Vranis P, Hitt D, Marsh-Armstrong N, Koliatsos VE. Evidence for accelerated tauopathy in the retina of transgenic P301S tau mice exposed to repetitive mild traumatic brain injury. Experimental neurology. 2015;273:168–176. doi: 10.1016/j.expneurol.2015.08.014. [DOI] [PubMed] [Google Scholar]
- Yang LS, Ksiezak-Reding H. Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. European journal of biochemistry / FEBS. 1995;233:9–17. doi: 10.1111/j.1432-1033.1995.009_1.x. [DOI] [PubMed] [Google Scholar]
- Yang WJ, Chen W, Chen L, Guo YJ, Zeng JS, Li GY, Tong WS. Involvement of tau phosphorylation in traumatic brain injury patients. Acta Neurol Scand. 2016 doi: 10.1111/ane.12644. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wang P, Morgan D, Lin D, Pan J, Lin F, Strang KH, Selig TM, Perez PD, Febo M, Chang B, Rubenstein R, Wang KK. Temporal MRI characterization, neurobiochemical and neurobehavioral changes in a mouse repetitive concussive head injury model. Sci Rep. 2015;5:11178. doi: 10.1038/srep11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Liu J, McCabe JT. Alterations of cerebral cortex and hippocampal proteasome subunit expression and function in a traumatic brain injury rat model. Journal of neurochemistry. 2008;104:353–363. doi: 10.1111/j.1471-4159.2007.04970.x. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Kojima A, Ishikawa C, Arai K. Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. Journal of Alzheimer's disease: JAD. 2010;22:295–306. doi: 10.3233/JAD-2010-100681. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Kojima A, Itoh K, Uchiyama T, Arai K. Anticholinergics boost the pathological process of neurodegeneration with increased inflammation in a tauopathy mouse model. Neurobiol Dis. 2012;45:329–336. doi: 10.1016/j.nbd.2011.08.017. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Uryu K, Higuchi M, Longhi L, Hoover R, Fujimoto S, McIntosh T, Lee VM, Trojanowski JQ. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. Journal of neurotrauma. 2005;22:1134–1141. doi: 10.1089/neu.2005.22.1134. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Zhang B, Bruce J, Trojanowski JQ, Lee VM. Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau-induced degeneration in astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:10662–10671. doi: 10.1523/JNEUROSCI.23-33-10662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Zhang Y, Chuang DM. Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. Journal of neurotrauma. 2012;29:2342–2351. doi: 10.1089/neu.2012.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano CA, Egana JT, Nunez MT, Maccioni RB, Gonzalez-Billault C. Oxidative stress promotes tau dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free radical biology & medicine. 2004;36:1393–1402. doi: 10.1016/j.freeradbiomed.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Zemlan FP, Jauch EC, Mulchahey JJ, Gabbita SP, Rosenberg WS, Speciale SG, Zuccarello M. C-tau biomarker of neuronal damage in severe brain injured patients: association with elevated intracranial pressure and clinical outcome. Brain research. 2002;947:131–139. doi: 10.1016/s0006-8993(02)02920-7. [DOI] [PubMed] [Google Scholar]
- Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Weiner NE, Cohen JA, Rudick RA, Woo D. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. Journal of neurochemistry. 1999;72:741–750. doi: 10.1046/j.1471-4159.1999.0720741.x. [DOI] [PubMed] [Google Scholar]
- Zetterberg H, Hietala MA, Jonsson M, Andreasen N, Styrud E, Karlsson I, Edman A, Popa C, Rasulzada A, Wahlund LO, Mehta PD, Rosengren L, Blennow K, Wallin A. Neurochemical aftermath of amateur boxing. Arch Neurol. 2006;63:1277–1280. doi: 10.1001/archneur.63.9.1277. [DOI] [PubMed] [Google Scholar]
- Zhang J, Teng Z, Song Y, Hu M, Chen C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:443–453. doi: 10.1038/jcbfm.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Chen XQ, Wang XC, Wang JZ. Nitration and oligomerization of tau induced by peroxynitrite inhibit its microtubule-binding activity. FEBS Lett. 2005;579:2421–2427. doi: 10.1016/j.febslet.2005.03.041. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Liu YH, Yin J, Li HL, Wang Q, Wang JZ. Peroxynitrite induces Alzheimerlike tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:1431–1442. doi: 10.1096/fj.05-5223com. [DOI] [PubMed] [Google Scholar]
- Zilka N, Korenova M, Novak M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol. 2009a;118:71–86. doi: 10.1007/s00401-009-0499-y. [DOI] [PubMed] [Google Scholar]
- Zilka N, Stozicka Z, Kovac A, Pilipcinec E, Bugos O, Novak M. Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J Neuroimmunol. 2009b;209:16–25. doi: 10.1016/j.jneuroim.2009.01.013. [DOI] [PubMed] [Google Scholar]